

Abstract
Virus-like particles (VLPs) are an active area of vaccine research, development and commercialization. Mucosal administration of VLPs provides an attractive avenue for delivery of vaccines with the potential to produce robust immune responses. Nasal and oral delivery routes are particularly intriguing due to differential activation of mucosa-associated lymphoid tissues. We compared both intranasal and oral administration of VLPs with a panel of toll-like receptor (TLR) agonists (TLR3, 5, 7, 7/8, and 9) to determine the mucosal adjuvant activity of these immunomodulators. We selected Norwalk virus (NV) VLPs because it is an effective model antigen and an active area of research and commercialization. To prioritize these adjuvants, VLP-specific antibody production in serum (IgG, IgG1, IgG2a), vaginal lavages (IgG, IgA), and fecal pellets (IgA) were measured across a longitudinal timeseries in vaccinated mice. Additional distal mucosal sites (nasal, brochoalveolar, salivary, and gastrointestinal) were evaluated for VLP-specific responses (IgA). Intranasal co-delivery of VLPs with TLR7 or TLR9 agonists produced the most robust and broad-spectrum immune responses, systemically and at distal mucosal sites inducing VLP-specific antibodies at all sites evaluated. In addition, these VLP-specific antibodies blocked binding of NV VLPs to histo-blood group antigen (H type 1), supporting their functionality. Oral administration and/or other TLR agonists tested in the panel did not consistently enhance VLP-specific immune responses. This study demonstrates that intranasal co-delivery of VLPs with TLR7 or TLR9 agonists provides dose-sparing advantages for induction of specific and functional antibody responses against VLPs (i.e., non-replicating antigens) in the respiratory, gastrointestinal, and reproductive tract.
Keywords: nasal vaccination, oral vaccination, TLR agonists, mucosal adjuvants, norovirus, virus-like particles, mucosal immunology, antibody production
Virus-like particle (VLP)-based vaccines can induce protective immune responses in humans.1-11 Successful VLP-based vaccines include the Hepatitis B virus (HBV) and Human Papillomavirus (HPV) vaccines, both of which have received FDA approval and are commercially available.2,4,8,9,12 In addition, several VLP-based norovirus (NoV) and influenza virus vaccines are currently in the developmental and clinical pipelines.1,3,5-7,10,11 These non-replicating subunit antigens provide a safe alternative to live or attenuated viral vaccines; however, many subunit antigens alone do not elicit an immune response robust enough for a vaccine to be protective. Poor immunogenicity of non-replicating subunit vaccines, however, can be overcome by the addition (or co-delivery) of potent adjuvants.1-4,6,7,9,13-19
One particularly potent group of molecules that can be exploited as mucosal adjuvants are toll-like receptor (TLR) agonists.1,3,14-17,19-24 TLRs are pattern recognition receptors (PRRs) of the innate immune system that have been shown to exhibit tissue or mucosa-specific expression patterns. Each TLR has its own agonist (or set of agonists), known as pathogen associated molecular patterns (PAMP).16,19,21,22,24 These TLR agonists, or PAMP, include bacterial ligands, virus-specific ribonucleotide motifs (i.e., dsRNA), and imidazoquinoline compounds and all are currently studied as adjuvants.1-4,6,14-19 We have previously published results demonstrating that intranasal co-delivery with gardiquimod (GARD; TLR7 agonist) and resiquimod (R848; TLR7/8 agonist) along with Norwalk virus (NV) VLPs can elicit equivalent immune responses relative to VLPs co-delivered with cholera toxin (CT).14 As such, we consider GARD our “gold standard” intranasal adjuvant. In addition, we have demonstrated that a TLR3 agonist (polyinosinic:cytidylic acid (PIC)) co-delivered subcutaneously (s.c.) with Ebola immune complexes (EICs) can induce robust, systemic immunity and protection against lethal challenge with Ebola virus.18 Recently, a TLR4 agonist (MPL, monophosphoryl lipid A) has successfully transitioned from preclinical animal studies to clinical trials and is now used in commercially available, FDA-approved human VLP-based vaccines.1-4,15,16,19
Mucosal administration of VLP-based vaccines is highly desirable for vaccines aimed to protect against sexually transmitted infections (e.g., HPV), respiratory tract infections (e.g., influenza), or non-bacterial gastroenteritis (e.g., norovirus).19,20 Nasal and oral routes of administration are particularly intriguing because they are known to induce immune responses at the delivery site, as well as distal mucosal sites, and provide an easy delivery route for human vaccinations.1,3,5,11,13,14,19,20,25-27 Oral delivery has been shown to be a safe delivery route, however one caveat is that oral vaccination requires a high antigen dose in order to achieve immunogenicity.11,25,26 The nasal vaccination route has been significantly associated with Bell’s palsy following co-delivery with E. coli enterotoxin; likewise, cholera toxin has been shown to transport to the central nervous system via toxin-specific receptors. As such these toxins are no longer being investigated as nasal adjuvants.28-30 The nasal delivery route is an active area of research and preclinical and clinical trials must be conducted to determine the safety and efficacy of any vaccine formulation. One goal of this study is to examine if mucosal adjuvants (i.e., TLR agonists) could lower the amount of VLPs required, resulting in an effective, dose-sparing oral and/or nasal VLP-based vaccine.
In this study, we systematically evaluated a panel of selected TLR agonists (TLR3, 5, 7, 7/8, and 9) for their ability to induce systemic and mucosa-specific immune responses when co-delivered with norovirus VLPs. While immunological protection against NV may be most desirable in the gastrointestinal (GI) tract, this platform has potential use for presentation of other pathogen-associated epitopes; as such, we evaluated both serum and a variety of mucosal sites for the presence of VLP-specific immunoglobulins.5 We simultaneously tested oral vs. intranasal delivery for optimal induction of VLP-specific antibody responses in the presence or absence TLR agonists. In addition, we evaluated the capability of these VLP-specific antibodies to block NV VLPs binding to their putative carbohydrate receptor.31
Production of NV VLPs was performed in Nicotiana benthamiana using viral vectors derived from tobacco mosaic virus (TMV) as previously described.14,26 NV VLPs were further purified by Ion exchange chromatography with DEAE Sepharose FF resin (GE Healthcare) to remove small molecules, including endotoxin.14 Purified NV VLPs were collected in the DEAE flow-through fraction. Qualitative observations of NV VLPs were made by loading 5µg of vaccination stock, with or without TLR agonists, onto sucrose gradients that were performed as previously described.26,27 VLPs were quantified by sandwich ELISA as previously described.26 VLP structure was not altered by addition of any of the TLR agonists tested (data not shown).
All TLR agonists were purchased from InvivoGen, except CpG-ISS 1018, which was generously provided by Dynavax, Inc. Polyinosinic-polycytidylic acid (PIC; TLR3 agonist) was prepared in PBS at 3.75mg/ml. S. typhimurium flagellin (FLAG; TLR5 agonist), gardiquimod (GARD; TLR7 agonist), CpG oligodeoxynucleotides 1826 (CpG; TLR9 agonist), CpG immunostimulatory sequence 1018 (CpG-ISS; TLR9 agonist), and an imidazoquinoline compound (CL097; TLR7/8 agonist) were resuspended in sterile endotoxin-free water at 0.25, 2.5, 3.2, 1.0, and 2.0 mg/ml, respectively.
All animals were housed in American Association for Laboratory Animal Care-approved quarters and provided unlimited access to food and water. All procedures were approved by the ASU IACUC and performed in accordance with the Animal Welfare Act. Female, 5-wk-old BALB/c mice (Charles River; n = 60) were distributed randomly and acclimated for at least 1 wk prior to any procedures or treatment. Mice (n = 7/group) were immunized intranasally with NV VLPs (25 µg) co-delivered with PIC (10 µg), FLAG (1 µg), GARD (10 µg), CpG (10 µg), CpG-ISS (10 µg) or with NV VLPs alone and compared with mice immunized orally with NV VLPs (100 or 200 µg) co-delivered with FLAG (1 µg), PIC (10 µg), CL097 (100 µg) or with NV VLPs alone and compared with mock-vaccinated (PBS alone) controls. Mice were not anesthetized for mucosal immunization. Intranasal immunization was performed by using a 20 µl pipet to instill half of the vaccine into each nare (~5–10 µl/nare). Intranasal vaccinations were administered at days 0 and 21, while oral vaccinations were given at days 0, 21, and 42.
Serum, vaginal lavages, and fecal pellets were collected during our timecourse as previously described.14 After the animals were humanely euthanized, additional mucosal samples were collected and processed; gastrointestinal lavages, salivary samples, nasal lavages, and bronchoalveolar lavages were collected and processed as previously described.14 All mucosal samples were stored at −80 °C for future analysis of immunoglobulin titers. Serum and mucosal samples were evaluated by ELISA as previously described.14 Sample dilutions included the following ranges: serum (1:100–1:10 000 000), and all other mucosal samples (1:2–1:5000). An absorbance value of 0.1 or higher was considered to be positive and the geometric mean titer (GMT) was calculated as the reciprocal of the highest dilution tested that provided a positive absorbance value.
An H type 1 binding assay was performed to evaluate the neutralization potential of samples derived from our most robust vaccination groups.31,32 Insect cell-derived recombinant VLPs (2 µg/ml) were added to high binding 96-well plates (100 µl/well) (Corning) and incubated at room temperature overnight. Plates were blocked with 5% milk/PBS at 37 °C for 1h. The plates were then washed 4 times in PBS-T. Serum and mucosal samples from immunization groups, as well as the PBS alone group (mock-vaccinated), were pooled using equal volumes per mouse and were then added to the plates (50 µl/well) and incubated at 37 °C for 2h. Mucosal samples were added undiluted, while serum samples were diluted 1:10 in 5% milk in PBS. The plates were then washed 4 times in PBS-T. Synthetic biotinylated H-type 1 (Glycotech), diluted in 5% milk/PBS to a concentration of 10 µg/ml, was added to each well (100 µl/well) and the plates were incubated at 37 °C for 4h. The plates were then washed 6 times in PBS-T. Detection of H type 1 binding was performed by adding 100 µl/well streptavidin peroxidase conjugate (1:1000) (Invitrogen) in 5% milk/PBS, and incubated at 37 °C for 1 h. The plates were then washed 4 times in PBS-T. Plates were developed for 3–10min (depending on sample type) with TMB substrate (KPL) and stopped with 100 µl/well of 1M H3PO4. Absorbance values were measured at 450nm on a MRX microplate reader. To control for substances that would have non-specific blocking activity, we normalized each of the binding assays to the respective mucosal samples from mock-vaccinated mice.
Statistical analysis was performed using Prism software (GraphPad). Geometric mean titer (GMT) values were evaluated statistically at each timepoint using the Kruskal–Wallis One-way analysis of variance (ANOVA), followed by the Dunn post-hoc test. Statistical comparisons between each vaccination group and the PBS delivery (mock-vaccinated) group from the same delivery method (i.e., intranasal or oral) are displayed on each GMT graph. P < 0.05 was considered statistically significant. For H Type 1 binding assays, statistical tests were not performed due to limited power since all these assays were performed using pools (n = 1) of the various sample types. Results of these tests are reported as the percent reduction in VLP:H1 binding in vaccination groups vs. the mock-vaccinated group.
To compare the immunogenicity of several TLR agonists relative to our “gold standard” TLR agonist, gardiquimod (TLR7 agonist), NV VLPs (25 μg) were administered intranasally with or without a TLR agonist.14 TLR agonists tested as nasal adjuvants included PIC (TLR3), FLAG (TLR5), GARD (TLR7), CpG (TLR9, murine motif), and CpG-ISS (TLR9, human motif). All 3 serum immunoglobulins measured (VLP-specific IgG, IgG1, IgG2a) were detected by ELISA when VLPs were administered intranasally, with or without a TLR agonist (Fig. 1A, C, and E). However, One-way ANOVA and the Dunn post-hoc tests between each vaccination group and the PBS alone (mock-vaccinated) group demonstrated that only VLPs co-delivered with TLR7 (GARD) or TLR9 (CpG, CpG-ISS) agonists significantly increased VLP-specific serum antibody titers (Fig. 1A, C, and E). Despite the propensity for induction of a Th1-dominant immune response following co-delivery of TLR7 or TLR9 agonists, we observed a mixed Th1/Th2 response following nasal immunization as reflected by the high levels of both IgG1 and IgG2a isotypes. This is most likely due to the inherent Th2-inducing nature of NV VLPs/antigens, possibly due to their particulate structure with repeating display of epitopes. Our data suggests that intranasal co-delivery of a particulate antigen with this subset of TLR agonists can augment and orchestrate a mixed Th1/Th2 immune response. However, TLR3 and 5 agonists did not significantly augment VLP-specific serum antibody levels. These results support the continued investigation of TLR7 and/or TLR9 agonists for use as potent nasal adjuvants for VLP-based vaccines.

Figure 1. Longitudinal evaluation of VLP-specific immunoglobulins (IgG, IgG1, IgG2a) in serum samples from mice after nasal or oral vaccination. Antibody titers following nasal (A, C, and E) or oral (B, D, and F) co-delivery of VLPs with a panel of TLR agonists. Nasal vaccinations were performed using 25 μg of VLPs per dose, while oral vaccinations were performed using 100–200 μg of VLPs per dose. The panel of TLR agonists included PIC (TLR3), FLAG (TLR5), GARD (TLR7), CpG (TLR9), CpG-ISS (TLR9, alternate CpG motif), and CL097 (TLR7/8). Geometric mean titers (GMT) of all serum samples were determined by ELISA. Immunoglobulins analyzed in serum included IgG (A and B), and the isotypes IgG1 (C and D) and IgG2a (E and F). One-way ANOVA and the Dunn post-hoc tests were performed between each serum sample and samples collected from mock-immunized mice. Statistical significance: ^P < 0.05, *P < 0.01, **P < 0.001.
To determine if specific TLR agonists enhance the oral immunogenicity of VLPs, NV VLPs (100 μg or 200 μg) were co-delivered with or without a TLR agonist to mice by oral gavage. TLR agonists tested as oral adjuvants included PIC (TLR3), FLAG (TLR5), CL097 (TLR7/8), and CpG-ISS (TLR9, human motif). VLP-specific IgG, IgG1, and IgG2a were detected by ELISA in some, but not all of the experimental groups (Fig. 1B, D, and F). Specifically, serum IgG was consistently detected in the oral vaccination group that included VLPs co-delivered with CL097 (TLR 7/8). In addition, serum IgG was minimally detected at our last timepoint (day 57) in test groups using a higher dosage of VLPs alone (200 μg), or in groups co-delivered with FLAG or CpG-ISS as mucosal adjuvants (Fig. 1B). However, One-way ANOVA and the Dunn post-hoc tests comparing each oral vaccination group to the mock-vaccinated group demonstrated that none of the vaccination groups (with or without a TLR agonist) had significantly increased VLP-specific IgG compared with mock vaccination (Fig. 1B). Similar to the IgG levels, the Dunn post-hoc tests demonstrated that none of the oral vaccination groups (with or without a TLR agonist) had significantly increased VLP-specific IgG2a compared with mock vaccination (Fig. 1F). In contrast to total IgG and IgG2a levels, we did consistently detect VLP-specific IgG1 in all of the oral vaccine groups (Fig. 1D). However, One-way ANOVA and the Dunn post-hoc tests demonstrated that only the VLPs co-delivered with CL097 (TLR7/8) had significantly higher titers of VLP-specific IgG1 at multiple timepoints compared with the mock-vaccinated group (Fig. 1D). These results suggest that TLR7/8 agonists may augment systemic immune responses following oral delivery, but the increase in VLP-specific antibody titers is specific to induction of the IgG1 isotype.
To further evaluate the TLR agonists as mucosal adjuvants, we determined the induction of VLP-specific antibodies locally and at distal mucosal sites following intranasal vaccinations of VLPs (25 μg). VLP-specific mucosal immunoglobulins were detected in all of the sites evaluated, including those analyzed longitudinally (vaginal IgA, vaginal IgG, fecal IgA) (Fig. 2A, C, and E) and those evaluated at the terminal stage of our study (intranasal IgA, bronchoalveolar IgA, salivary IgA, gastrointestinal IgA) (Fig. 3A, C, E, and G). Mucosal sites that were evaluated longitudinally across our timecourse demonstrated that VLP-specific vaginal IgA and fecal IgA were detected in all groups; however, statistical comparisons between these groups and the mock-vaccinated group revealed there was a consistent and significant increase in antibody titers when VLPs were co-delivered with a TLR7 or TLR9 (CpG, CpG-ISS) agonist, but not with the other TLR agonists in the panel (Fig. 2A and E). Vaginal IgG was primarily detected only when a TLR7 or TLR9 agonist was co-delivered intranasally with our model VLPs (Fig. 2C) and these levels corresponded to groups with the highest serum IgG titer. This finding is consistent with passage of IgG from the plasma. Vaccination groups compared with the mock-vaccinated group only showed a significant increase in VLP-specific vaginal IgG at 1 timepoint (day 42) after intranasal co-delivery with a TLR7 agonist (Fig. 2C). These results suggest TLR7 and TLR9 agonists are the most effective intranasal adjuvants for inducing VLP-specific vaginal IgA and fecal IgA, but that vaginal IgG titers are more readily augmented with TLR7 agonists and this response is less sustained than the secretory IgA responses. Fecal IgA titers were equivalent among these intranasal groups and oral groups containing the higher dosage of VLP (Fig. 2E and F). These results support previously published studies that have demonstrated that intranasal co-delivery with TLR7 or TLR9 agonists act as potent inducers of vaginal IgA production.14,33

Figure 2. Longitudinal evaluation of VLP-specific immunoglobulins (IgG, IgA) in vaginal lavages and fecal pellets from mice after nasal or oral vaccination. Antibody titers following nasal (A, C, and E) or oral (B, D, and F) co-delivery of VLPs with a panel of TLR agonists. Nasal vaccinations were performed using 25 μg of VLPs per dose, while oral vaccinations were performed using 100–200 μg of VLPs per dose. The panel of TLR agonists included PIC (TLR3), FLAG (TLR5), GARD (TLR7), CpG (TLR9), CpG-ISS (TLR9, alternate CpG motif), and CL097 (TLR7/8). Geometric mean titers (GMT) of all serum samples were determined by ELISA. Immunoglobulins analyzed in mucosal samples included vaginal IgA (A and B), vaginal IgG (C and D) and fecal IgA (E and F). One-way ANOVA and the Dunn post-hoc tests were performed between each mucosal sample and samples collected from mock-immunized mice. Statistical significance: ^P < 0.05, *P < 0.01, **P < 0.001.

Figure 3. Evaluation of VLP-specific immunoglobulins (IgA) in distal mucosal sites (nasal, bronchoalveolar, salivary, gastrointestinal) from mice after nasal or oral vaccination. Antibody titers following nasal (A, C, E, and G) or oral (B, D, F, and H) co-delivery of VLPs with a panel of TLR agonists. Nasal vaccinations were performed using 25 μg of VLPs per dose, while oral vaccinations were performed using 100–200 μg of VLPs per dose. The panel of TLR agonists included PIC (TLR3), FLAG (TLR5), GARD (TLR7), CpG (TLR9), CpG-ISS (TLR9, alternate CpG motif), and CL097 (TLR7/8). Geometric mean titers (GMT) of all serum samples were determined by ELISA. Immunoglobulins analyzed in mucosal samples included intranasal IgA (A and B), bronchoalveolar IgA (C and D), salivary IgA (E and F), and gastrointestinal IgA (G and H). Nonparametric t tests were performed between each mucosal sample and samples collected from mock-immunized mice. Statistical significance: ^P < 0.05, *P < 0.01, **P < 0.001.
Mucosal sites evaluated at the terminal stage of our study (day 57) demonstrated that VLP-specific immunoglobulins were detected in all sites, but not in all groups (Fig. 3A, C, E, and G). Statistical comparisons between each vaccination group (with or without a TLR agonist) relative to the mock-vaccinated group revealed that TLR7 and/or TLR9 agonists significantly increased the titers of VLP-specific IgA at these sites (Fig. 3C, E, G). Specifically, both salivary IgA and gastrointestinal IgA had significantly higher VLP-specific antibody titers compared with the mock-vaccinated group when VLPs were co-delivered with either TLR7 (GARD) or TLR9 (CpG, CpG-ISS) agonists (Fig. 3E, G). Bronchoalveolar IgA only significantly increased using 1 of the TLR9 (CpG) agonists (Fig. 3C). Lastly, intranasal IgA had higher antibody titers in groups using TLR7 or TLR9 agonists as mucosal adjuvants, but these differences in antibody titers were not statistically significant (Fig. 3A). These results suggest that both TLR7 and TLR9 agonists may be useful for inducing VLP-specific immune responses at distal mucosal sites following intranasal vaccination, but that TLR9 agonists may be preferable for effector sites in the respiratory tract.
In addition to evaluating distal and mucosal antibody induction following intranasal delivery, mucosal immunoglobulins were evaluated both longitudinally and at the terminal stage of our study following oral vaccination. Sites evaluated longitudinally (vaginal IgA, vaginal IgG, fecal IgA) demonstrated that both vaginal IgA and fecal IgA were detectable in all vaccination groups (Fig. 2B and F). Statistical comparisons between our vaccination groups and the mock-vaccinated group, however, revealed that there was only a significant increase in VLP-specific vaginal IgA at 1 timepoint (day 42) after oral co-delivery with a TLR5 agonist (Fig. 2B), and VLP-specific fecal IgA was only significantly augmented in our higher dose of VLPs alone or when VLPs were co-delivered with a TLR7/8 or TLR9 (CpG-ISS) agonist (Fig. 2F). These data are consistent with previously published results demonstrating high-dose (200 μg or higher) oral administration of VLPs induces VLP-specific fecal IgA.11,25,26 Interestingly, vaginal IgG was not detected in any vaccination group, with or without a TLR agonist/mucosal adjuvant (Fig. 2D), most likely correlating to the low serum IgG response.
Mucosal sites evaluated at the terminal stage of our study (day 57) demonstrated that VLP-specific immunoglobulins were only detected in sites associated with the GALT (i.e., salivary IgA and gastrointestinal IgA) (Fig. 3B, D, F, and H). However, One-way ANOVA and the Dunn post-hoc tests showed that none of these antibody titers were significantly higher than the mock-vaccinated group. These data suggest that while fecal IgA titers were equivalent between intranasal and oral groups, the intranasal delivery route may provide an advantage for norovirus vaccines because VLP-specific antibodies were induced at additional gastrointestinal sites (i.e., salivary and gastrointestinal) (Fig. 3E, F, G, and H). In addition, no VLP-specific immunoglobulins were detected in the sites associated with the nasal-associated lymphoid tissue (NALT) (i.e., intranasal IgA and bronchoalveolar IgA) (Fig. 3B and D). These results suggest that oral co-delivery of VLPs with TLR7/8 (CL097) or TLR9 (CpG-ISS) agonists may augment local VLP-specific antibody titers specific to the gastrointestinal tract, particularly fecal IgA.
TLR3 and TLR5 agonists did not consistently elicit a VLP-specific immune response following nasal or oral administration; however, for TLR3 agonist at least, we have previously demonstrated PIC to be highly effective at generating antigen-specific antibodies when delivered subcutaneously with another subunit vaccine.18 As such these compounds may be more susceptible to gut-induced degradation/inactivity (e.g., due to pH, stomach acid content, gastrointestinal microflora, poor absorption, etc.) thereby affecting their potency and activity following oral delivery. Future studies may evaluate these TLR agonists co-delivered orally with VLPs using formulations designed to promote stability within the gastrointestinal microenvironment (i.e., dry powder formulations, VLPs and adjuvant cocktails with a mucoadhesive agent, or conjugation of TLR agonists to the antigen) and/or at different concentrations.27 Our results suggest that unstabilized forms of these agonists do not work well and a higher dosage (>200 μg) of VLPs combined with TLR7/8 agonists provides the most promising approach for oral vaccination.
Intranasal co-delivery of VLPs with TLR7 or TLR9 agonists resulted in the most robust immune responses; as such, we evaluated serum and mucosal samples from these 2 groups along with our mock-vaccinated group for their ability to block NV VLPs binding to the histo-blood group antigen (HGBA), H type 1 (Fig. 4).31,32 Pools from serum samples, as well as gastrointestinal, vaginal, intranasal, bronchoalveolar, and salivary samples all robustly blocked binding of VLPs to H type 1 for the 2 test groups (VLPs co-delivered with GARD and VLPs co-delivered with CpG) compared with the mock-vaccinated group. Specifically, VLPs co-delivered with GARD resulted in a mean binding reduction of 82.0%, 61.7%, 58.5%, 85.1%, 48.2%, and 34.6%, for the serum, vaginal, intranasal, bronchoalveolar, salivary, and gastrointestinal samples, respectively. VLPs co-delivered with CpG resulted in a mean binding reduction of 88.1%, 54.1%, 25.6%, 57.0%, 47.8%, and 41.5%, for the serum, vaginal, intranasal, bronchoalveolar, salivary, and gastrointestinal samples, respectively. These data suggest that our intranasal vaccine strategy is capable of generating high levels of functional serum and mucosal antibody production, as well as secretion at the sites, which may provide protection against NoV infection. The ability to generate VLP-specific antibodies that block H type I has been defined as the correlate of protection for NV in human challenge studies.31,32
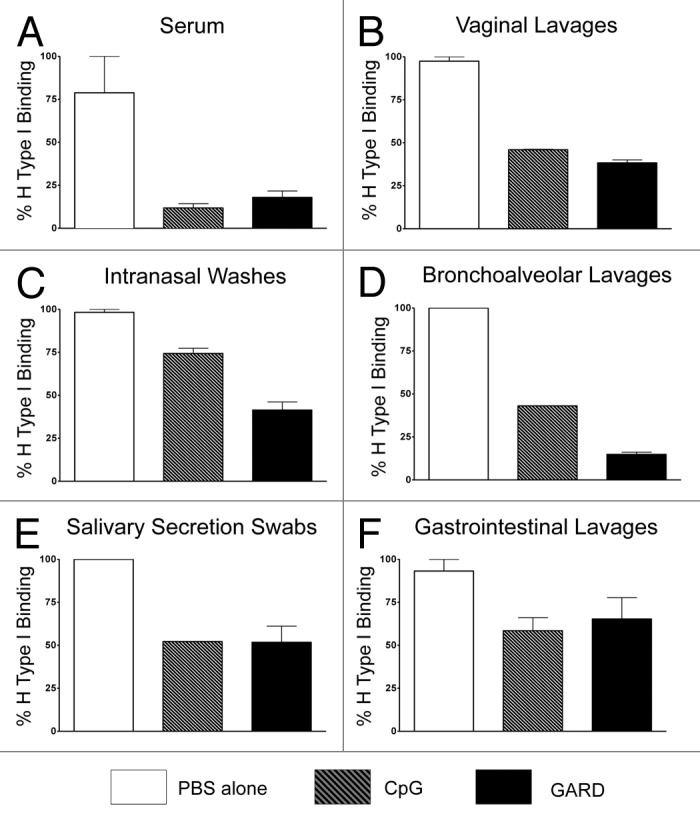
Figure 4. Functional evaluation of VLP-specific antibodies in both serum and mucosal samples from mice after nasal vaccination with TLR7 and TLR9 agonists. The human blood group antigen, H type 1, binds our model NV VLPs; pools from serum and mucosal samples were evaluated for functionality by analysis of their ability to block this interaction. Samples from serum (A), vaginal lavages (B), intranasal washes (C), broncholaveolar lavages (D), salivary secretion swabs (E), and gastrointestinal lavages (F) were evaluated between the highest responding groups (i.e., intranasal co-delivery of VLPs and GARD (TLR7 agonist; black bar) or VLPs and CpG (TLR9 agonist; gray striped bar) compared with mock-immunized mice (PBS; white bar).
We conclude from this study that nasal administration of NV VLPs, in conjunction with TLR7 or TLR9 adjuvants, offers highly significant dose-sparing advantages for induction of specific and functional antibody responses against a non-replicating antigen in the respiratory, gastrointestinal, and reproductive tracts. This data suggests the intranasal route of delivery combined with TLR agonists is a more potent inducer of systemic and mucosal immune responses to VLPs relative to oral administration at the doses tested in our study. In addition, TLR7 or TLR9 agonists co-delivered intranasally with NV VLPs induced significant levels of VLP-specific antibody that effectively blocked HBGA binding, a correlate of NV protection.31,32 By induction of both mucosal and systemic VLP-specific protective immunity, a 2-tiered immunological barrier is created. Furthermore, these data have particular implications for vaccine development targeted to prevent mucosal infections including respiratory, GI and sexually transmitted infection pathogens.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported in part by NIH grant 1 U19 AI062150-01. We would like to thank Dynavax for providing us with the CpG-ISS 1018. We are greatly appreciative to Drs Hugh Mason and Charles Arntzen for their support, and for providing us with the vector and reagents used to generate the plant-derived NV VLPs. Lastly, we are thankful to Daaimah LaVigne for her assistance during the animal procedures and sample processing.
Glossary
Abbreviations:
- GALT
gastrointestinal-associated lymphoid tissue
- NALT
nasal-associated lymphoid tissue
- NoV
norovirus
- NV
Norwalk virus
- PAMP
pathogen-associated molecular patterns
- PRR
pattern recognition receptors
- TLR
toll-like receptor
- VLPs
virus-like particles
References
- 1.Atmar RL, Bernstein DI, Harro CD, Al-Ibrahim MS, Chen WH, Ferreira J, Estes MK, Graham DY, Opekun AR, Richardson C, et al. Norovirus vaccine against experimental human Norwalk Virus illness. N Engl J Med. 2011;365:2178–87. doi: 10.1056/NEJMoa1101245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Deschuyteneer M, Elouahabi A, Plainchamp D, Plisnier M, Soete D, Corazza Y, Lockman L, Giannini S, Deschamps M. Molecular and structural characterization of the L1 virus-like particles that are used as vaccine antigens in Cervarix™, the AS04-adjuvanted HPV-16 and -18 cervical cancer vaccine. Hum Vaccin. 2010;6:407–19. doi: 10.4161/hv.6.5.11023. [DOI] [PubMed] [Google Scholar]
- 3.El-Kamary SS, Pasetti MF, Mendelman PM, Frey SE, Bernstein DI, Treanor JJ, Ferreira J, Chen WH, Sublett R, Richardson C, et al. Adjuvanted intranasal Norwalk virus-like particle vaccine elicits antibodies and antibody-secreting cells that express homing receptors for mucosal and peripheral lymphoid tissues. J Infect Dis. 2010;202:1649–58. doi: 10.1086/657087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giannini SL, Hanon E, Moris P, Van Mechelen M, Morel S, Dessy F, Fourneau MA, Colau B, Suzich J, Losonksy G, et al. Enhanced humoral and memory B cellular immunity using HPV16/18 L1 VLP vaccine formulated with the MPL/aluminium salt combination (AS04) compared to aluminium salt only. Vaccine. 2006;24:5937–49. doi: 10.1016/j.vaccine.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 5.Herbst-Kralovetz M, Mason HS, Chen Q. Norwalk virus-like particles as vaccines. Expert Rev Vaccines. 2010;9:299–307. doi: 10.1586/erv.09.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Landry N, Ward BJ, Trépanier S, Montomoli E, Dargis M, Lapini G, Vézina LP. Preclinical and clinical development of plant-made virus-like particle vaccine against avian H5N1 influenza. PLoS One. 2010;5:e15559. doi: 10.1371/journal.pone.0015559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.López-Macías C. Virus-like particle (VLP)-based vaccines for pandemic influenza: performance of a VLP vaccine during the 2009 influenza pandemic. Hum Vaccin Immunother. 2012;8:411–4. doi: 10.4161/hv.18757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ludwig C, Wagner R. Virus-like particles-universal molecular toolboxes. Curr Opin Biotechnol. 2007;18:537–45. doi: 10.1016/j.copbio.2007.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paavonen J, Jenkins D, Bosch FX, Naud P, Salmerón J, Wheeler CM, Chow SN, Apter DL, Kitchener HC, Castellsague X, et al. HPV PATRICIA study group Efficacy of a prophylactic adjuvanted bivalent L1 virus-like-particle vaccine against infection with human papillomavirus types 16 and 18 in young women: an interim analysis of a phase III double-blind, randomised controlled trial. Lancet. 2007;369:2161–70. doi: 10.1016/S0140-6736(07)60946-5. [DOI] [PubMed] [Google Scholar]
- 10.Roy P, Noad R. Virus-like particles as a vaccine delivery system: myths and facts. Hum Vaccin. 2008;4:5–12. doi: 10.4161/hv.4.1.5559. [DOI] [PubMed] [Google Scholar]
- 11.Tacket CO, Sztein MB, Losonsky GA, Wasserman SS, Estes MK. Humoral, mucosal, and cellular immune responses to oral Norwalk virus-like particles in volunteers. Clin Immunol. 2003;108:241–7. doi: 10.1016/S1521-6616(03)00120-7. [DOI] [PubMed] [Google Scholar]
- 12.Wheeler CM, Bautista OM, Tomassini JE, Nelson M, Sattler CA, Barr E, Protocol 11 study Investigators Safety and immunogenicity of co-administered quadrivalent human papillomavirus (HPV)-6/11/16/18 L1 virus-like particle (VLP) and hepatitis B (HBV) vaccines. Vaccine. 2008;26:686–96. doi: 10.1016/j.vaccine.2007.11.043. [DOI] [PubMed] [Google Scholar]
- 13.Jackson EM, Herbst-Kralovetz MM. Intranasal vaccination with murabutide enhances humoral and mucosal immune responses to a virus-like particle vaccine. PLoS One. 2012;7:e41529. doi: 10.1371/journal.pone.0041529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Velasquez LS, Hjelm BE, Arntzen CJ, Herbst-Kralovetz MM. An intranasally delivered Toll-like receptor 7 agonist elicits robust systemic and mucosal responses to Norwalk virus-like particles. Clin Vaccine Immunol. 2010;17:1850–8. doi: 10.1128/CVI.00230-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harandi AM, Davies G, Olesen OF. Vaccine adjuvants: scientific challenges and strategic initiatives. Expert Rev Vaccines. 2009;8:293–8. doi: 10.1586/14760584.8.3.293. [DOI] [PubMed] [Google Scholar]
- 16.Kanzler H, Barrat FJ, Hessel EM, Coffman RL. Therapeutic targeting of innate immunity with Toll-like receptor agonists and antagonists. Nat Med. 2007;13:552–9. doi: 10.1038/nm1589. [DOI] [PubMed] [Google Scholar]
- 17.Knuschke T, Epple M, Westendorf AM. The type of adjuvant strongly influences the T-cell response during nanoparticle-based immunization. Hum Vaccin Immunother. 2013;10:10. doi: 10.4161/hv.26203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Phoolcharoen W, Dye JM, Kilbourne J, Piensook K, Pratt WD, Arntzen CJ, Chen Q, Mason HS, Herbst-Kralovetz MM. A nonreplicating subunit vaccine protects mice against lethal Ebola virus challenge. Proc Natl Acad Sci U S A. 2011;108:20695–700. doi: 10.1073/pnas.1117715108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rhee JH, Lee SE, Kim SY. Mucosal vaccine adjuvants update. Clin Exp Vaccine Res. 2012;1:50–63. doi: 10.7774/cevr.2012.1.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Holmgren J, Czerkinsky C. Mucosal immunity and vaccines. Nat Med. 2005;11(Suppl):S45–53. doi: 10.1038/nm1213. [DOI] [PubMed] [Google Scholar]
- 21.Akira S, Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 2004;4:499–511. doi: 10.1038/nri1391. [DOI] [PubMed] [Google Scholar]
- 22.Beutler BA. TLRs and innate immunity. Blood. 2009;113:1399–407. doi: 10.1182/blood-2008-07-019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chilton PM, Embry CA, Mitchell TC. Effects of Differences in Lipid A Structure on TLR4 Pro-Inflammatory Signaling and Inflammasome Activation. Front Immunol. 2012;3:154. doi: 10.3389/fimmu.2012.00154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pulendran B, Ahmed R. Translating innate immunity into immunological memory: implications for vaccine development. Cell. 2006;124:849–63. doi: 10.1016/j.cell.2006.02.019. [DOI] [PubMed] [Google Scholar]
- 25.Ball JM, Hardy ME, Atmar RL, Conner ME, Estes MK. Oral immunization with recombinant Norwalk virus-like particles induces a systemic and mucosal immune response in mice. J Virol. 1998;72:1345–53. doi: 10.1128/jvi.72.2.1345-1353.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Santi L, Batchelor L, Huang Z, Hjelm B, Kilbourne J, Arntzen CJ, Chen Q, Mason HS. An efficient plant viral expression system generating orally immunogenic Norwalk virus-like particles. Vaccine. 2008;26:1846–54. doi: 10.1016/j.vaccine.2008.01.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Velasquez LS, Shira S, Berta AN, Kilbourne J, Medi BM, Tizard I, Ni Y, Arntzen CJ, Herbst-Kralovetz MM. Intranasal delivery of Norwalk virus-like particles formulated in an in situ gelling, dry powder vaccine. Vaccine. 2011;29:5221–31. doi: 10.1016/j.vaccine.2011.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mutsch M, Zhou W, Rhodes P, Bopp M, Chen RT, Linder T, Spyr C, Steffen R. Use of the inactivated intranasal influenza vaccine and the risk of Bell’s palsy in Switzerland. N Engl J Med. 2004;350:896–903. doi: 10.1056/NEJMoa030595. [DOI] [PubMed] [Google Scholar]
- 29.Pedersen GK, Cox RJ. The mucosal vaccine quandary: intranasal vs. sublingual immunization against influenza. Hum Vaccin Immunother. 2012;8:689–93. doi: 10.4161/hv.19568. [DOI] [PubMed] [Google Scholar]
- 30.van Ginkel FW, Jackson RJ, Yuki Y, McGhee JR. Cutting edge: the mucosal adjuvant cholera toxin redirects vaccine proteins into olfactory tissues. J Immunol. 2000;165:4778–82. doi: 10.4049/jimmunol.165.9.4778. [DOI] [PubMed] [Google Scholar]
- 31.Reeck A, Kavanagh O, Estes MK, Opekun AR, Gilger MA, Graham DY, Atmar RL. Serological correlate of protection against norovirus-induced gastroenteritis. J Infect Dis. 2010;202:1212–8. doi: 10.1086/656364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harrington PR, Lindesmith L, Yount B, Moe CL, Baric RS. Binding of Norwalk virus-like particles to ABH histo-blood group antigens is blocked by antisera from infected human volunteers or experimentally vaccinated mice. J Virol. 2002;76:12335–43. doi: 10.1128/JVI.76.23.12335-12343.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cong Y, Jupelli M, Guentzel MN, Zhong G, Murthy AK, Arulanandam BP. Intranasal immunization with chlamydial protease-like activity factor and CpG deoxynucleotides enhances protective immunity against genital Chlamydia muridarum infection. Vaccine. 2007;25:3773–80. doi: 10.1016/j.vaccine.2007.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]