

Abstract
Resurgent mumps outbreaks have raised questions about the current efficacy of mumps vaccines. We have applied immunoinformatics techniques based on principal component analysis to evaluate patterns in predicted B-cell linear epitopes, MHC binding affinity and cathepsin cleavage in the hemagglutinin neuraminidase protein of vaccine strains and wild-type mumps isolates. We have mapped predicted MHC-peptide binding for 37 MHC-I and 28 MHC-II alleles and predicted cleavage by cathepsin B, L and S. By all measures we applied Jeryl-Lynn JL5 major strain is an outlier with immunomic features arising from a small number of amino acid changes that distinguish it from other virus strains. Individuals vaccinated with Jeryl-Lynn who are not exposed to wild-type virus until their protective antibody titer has waned may be unable to recall a protective immune response when exposed to wild-type virus. Dependence on serology to evaluate mumps vaccines may have overemphasized the conservation of one neutralizing antibody epitope, at the expense of monitoring other related changes in the HN protein that could affect recall responses.
Keywords: mumps, vaccine, immunoinformatics, epitope, MHC, cathepsin, Jeryl-Lynn
Introduction
Attenuated mumps virus vaccines have been effective in reducing the incidence of mumps.1 However, several outbreaks of mumps have occurred among individuals with a history of vaccination.2-9 One such outbreak affected Iowa college students in 2006.2,6 Several attenuated mumps virus strains have been used as vaccines.10 Jeryl-Lynn vaccine remains the most widely used in the United States. This vaccine is known to comprise two strains, JL5 “major” and trace amounts of JL2 “minor”11. In 29 outbreaks of mumps in vaccinated individuals in which the vaccine strain was identified, 14 followed vaccination with Jeryl-Lynn and 14 followed use of the Rubini vaccine in the majority of vaccinees, alone or in conjunction with Jeryl Lynn and/or with Urabe strain vaccine.1,12 Only one outbreak followed use of another vaccine, Torii strain, but the vaccine coverage was only 21.6%. Two other recent reports document outbreaks following MMR Jeryl-Lynn vaccination.7,8 There is a growing consensus that re-evaluation of mumps vaccines may be warranted.1,13,14
Genotyping mumps has been based primarily on differences in the small hydrophobic (SH) protein,15 and the hemagglutinin-neuraminidase (HN).12 It is, however, the HN protein of mumps which functions in receptor recognition and virus release, and which comprises the major neutralizing epitope of mumps mapped to amino acid positions 265 through 360.16-19 Mumps has been considered serologically monotypic.20 Antibodies to Jeryl-Lynn vaccine effectively neutralize virus from the Iowa 2006 outbreak and other recent wild-type strains of different genotypes,20-22 but differences in neutralizing titer with date of isolation points to the gradual evolution of mumps virus.23 Although HN protein is relatively conserved, sequence variants are found within the main epitope region. Of particular note is that amino acid 279 in JL5 is an isoleucine, but is a threonine in wild-type strains. JL5 287 is an isoleucine, but a valine in recent wild-type isolates. Other regions identified as possibly associated with escape mutations are amino acids 113–130, 375–403, and 440–443.23
Antibodies to mumps virus vaccine have been detected up to 21 y post vaccination under circumstances unlikely to provide a boost through exposure to natural infection.24,25 Memory B-cells to mumps virus are detected after MMR vaccination at relatively lower levels compared with measles and rubella, but have not been evaluated over the long-term.26 Anamnestic responses have been documented following re-vaccination of individuals whose antibody titer has waned.27 Re-vaccination has been shown effective in curtailing outbreaks8 indicating a functional immunologic memory to the vaccine strain. In contrast to B-cell epitopes, little information is available on specific T-cell epitopes, on MHC binding to mumps virus peptides, or on the role of CD4+and CD8+ cells in viral clearance. Cell mediated immunity has been shown to persist 10–20 y, as determined by stimulation with whole virus or whole virus lysates.28,29
T-cell epitopes are determined by a number of factors, including the affinity of peptide binding within the MHC molecular groove, their excision from the parent protein sequence by peptidases, and further peptidase trimming to the appropriate length to fit the MHC-I binding groove for their presentation on the surface of a presenting cell. In the case of MHC-II, the relatively open groove permits peptide binding to be followed by exopeptidase trimming to an approximately 15-mer length.30 The processing of peptides for MHC-I loading involves proteasomic cleavage and delivery to the loading compartment by tapasin. Further trimming of delivered peptides is needed and C-terminal cleavage by a cathepsin appears to be a critical initial step necessary for MHC binding.31-33 Endosomal cathepsins have broad sequence specificities and are capable of cutting proteins at many sites thereby releasing the peptides for binding by MHC molecules while simultaneously reducing the number of peptides available for MHC binding. Binding of peptides by MHC molecules prevents this further degradation and enables the peptides to be presented on the cell surface. Cross-correlation pattern analyses have shown a close association among the relative positions of multiple immunologically relevant motifs (BEPI, MHC-I and MHC-II, and cathepsin cleavage) within the primary amino acid sequences of proteins.34 We recently described techniques for applying principal components of amino acids physical properties to predict MHC peptide binding affinity35,36 as well as to prediction of B-cell linear epitope (BEPI) contact points. We have applied similar methodology to extend immunologically relevant predictions to the endosomal cathepsin cleavage.34 Combining these in silico processes provides a means of visualization of complex multivariate inter-relationships among different components of the immune system.
Noting the amino acid differences in mumpsvirus HN, we hypothesized that these mutations might impact not only B-cell epitopes, but also the availability of T-cell epitopes, including those stimulating CD4+ T-helper cells providing memory and enabling a vaccinated individual to recall antibody and cellular immune responses following wild-type virus challenge. We addressed this by comparing 54 mumps virus strains based on their predicted BEPIs and MHC binding affinities. We then compared predicted MHC binding and examined the impact of differential cathepsin cleavage on MHC-peptide binding in seven representative virus strains, paying particular attention to two regions of predicted highest affinity MHC-II binding. This analysis shows that there are differences in both predicted BEPIs and potential T-cell epitopes between JL5 and other virus strains and frames a hypothesis for the circumstances which can lead to a resurgence of mumps cases in previously vaccinated individuals.
Results
Cluster analyses of immunologic patterns in the HN protein of 54 mumps virus strains
Figure 1 shows 3D scatterplots of the first three principal components of the indicated immunological metrics. These graphics constitute a high degree of compression of the underlying data but clearly show the relative distances among the different viruses. Importantly, starting from this type of graphic, the differences can be traced to the effects of amino acid mutations on the binding affinity of specific alleles. Dendrograms supporting the scatterplots and providing the identity for each virus in the clusters are shown in Figure S3. Along with the BEPI contact points, the dendograms further show how the different strains cluster when consolidated by allele subgroups MHC-IA and IB as well as by MHC II DP, DQ, and DR. Clustering based on BEPI prediction approximates to the sequence-based phylogenetic tree.20 In each scatterplot, JL5 is markedly separated from the other closely related clusters. Given its identical HN sequence, RIT4385 overlays JL5 exactly; other vaccine strains are distributed within the larger clusters. To examine the contribution of different HLA alleles to the pattern for both MHC-I and MHC-II binding, we then plotted the hierarchical clusters by each HLA. Figure 2 displays the first three principal components of binding affinity for each virus HN-allele combination individually for all MHC-I and MHC-II alleles for which we have neural network (NN) training sets. A central cluster shows that peptides from a large number of virus HN proteins are very similar to each other and cluster together. Also visible are the JL5, RIT4385 pairs as outliers widely separated from the central cluster of the wild type viruses.
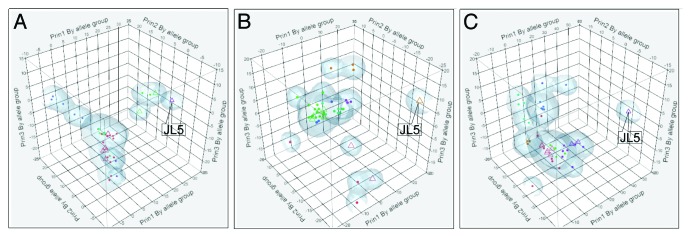
Figure 1. Three dimensional patterns of the first 3 principal components of different immunological metrics within the HN proteins of 54 different mumps virus strains. Axes show principal components (Prin1, Prin2, Prin3) by permuted groups of alleles (all for BEPI, groups as indicated for MHC-I and MHC-II panels). (A) Based on predicted B-cell epitope contact points; (B) Based on permuted average predicted MHC-I binding for all MHC-IA and MHC-IB alleles; (C) Based on permuted average predicted MHC-II binding for all DR, DP, DQ alleles. For panels B and C the permuted averages of a meta-population of all heterozygous and homozygous pairs for a composite of class I and class II alleles was computed as described in the methods. Vaccine strains are indicated by open triangles. Colors are randomly assigned to the clusters by JMP®.

Figure 2. Three dimensional patterns of the first 3 principal components of each (mumps virus)-(HLA allele) pair for the HN protein. Axes show principal components (Prin1, Prin2, Prin3) for each allele. (A) Combined MHC-I A and MHC-I B 54 viruses × 37 HLA alleles. (B) Combined MHC-II DR, DQ and DP 54 viruses × 28 alleles. Vaccine strains are indicated by triangles. Colors are randomly assigned to the clusters by JMP®. JL5 datapoints are indicated by a large triangle containing a small triangle of the identical RIT4385.
Analyses of seven selected viruses
Further analyses were conducted on the HN proteins of seven isolates, comprising representatives of major clusters plus JL5, JL2 and Rubini vaccine. In order to present figures of sufficient size and detail, output of JL5 and Iowa-06 are presented in the main text; equivalent output for the other 5 selected viruses are included in Figures S4-S7.
Population permuted plots of the selected viruses were generated to display predicted high affinity MHC binding regions and predicted BEPI contacts. In Figure 3A (JL5) and Figure 3B (Iowa-06) the principal neutralizing antibody epitope is clearly seen as a strong BEPI signal (orange baseline ribbon) in the region centered at amino acid 275. Notably Iowa-06 shows a strong BEPI centered at 443, which is absent in JL5. The MHC-II trace (blue line) shown in Figure 3 is that for DRB. In the case of JL5, three areas of predicted highest affinity MHC-II DR binding are indicated (blue baseline ribbon) in the transmembrane region (aa 25–60), adjacent to the neutralizing antibody epitope (aa 275–295) and between aa 410 and 445. The latter two regions, considered to be surface exposed, were selected for closer examination. For Iowa-06 and the other selected strains the very marked region of predicted DR high affinity binding in the 275–279 region is absent. This difference arises from the substitution at position 279 of the hydrophobic amino acid isoleucine by threonine between JL5 and Iowa-06, impacting 15 peptide binding frames. In contrast Iowa-06 shows a slight increase in binding affinity at 415–445. Figure S2 includes predicted binding affinities for by DP and DQ alleles; DP alleles follow closely those of DR, whereas for DQ predicted JL5 peptide binding does not differ from the consensus of all strains.

Figure 3. Permuted population profiles of JL5 (A) and Iowa-06 (B). Amino acid positions in HN protein are arrayed N-C on X axis. Red line shows permuted average predicted MHC-IA and IB (37 alleles) binding affinity by index position of sequential 9-mer peptides. Blue line shows permuted average predicted MHC-II DRB allele (16 alleles) binding affinity by index position of sequential 15-mer peptides. Both are plotted in standard deviation units (Y axis). Orange lines show probability of B-cell binding for an amino acid centered in each sequential 9-mer peptide. Note that the standardized B-cell metric has been inverted (multiplied by -1) in order to overlay the standardized probability onto the same scale as the MHC data. Hence, low numbers (valleys) represent predicted high binding affinity of MHC and high BEPI contact probability. Bars (Red:MHC-I, Blue:MHC-II) indicate the top 10% affinity binding. Orange bars indicate top 25% probability B-cell binding. Background shading shows membrane (green) extramembrane (yellow), intramembrane (pink) location. The downward orange spike at 269–275 correlates with the experimentally determined principal neutralizing antibody epitope. The two regions of high density predicted MHC-II binding are indicated by the blue brackets below.
No major differences in predicted MHC-I binding (red line) were noted in the permuted population plots between the seven strains. The permuted population plots show the average predicted MHC binding affinity at each position, a quantitative measure; they do not show which alleles are contributing to the binding and thus do not reflect the qualitative sequence differences which contribute to the separation seen in the scatterplots in Figure 2. Comparable plots for the other five viruses are show in Figure S4
Analysis of predicted cathepsin cleavage was conducted for the complete HN of each selected virus (Fig. S2); the output for the two regions of predicted highest MHC binding outside the transmembrane domain are plotted in Figures 4 and 5. In both regions single amino acid changes are predicted to alter the probability of cathepsin cleavage by one or more of the important endosomal cathepsins. Specifically, in the region aa 265–295 (Fig. 4), for JL5 there is a predicted probability >0.5 of cleavage by cathepsin B at 289–290, cathepsin S at 288–289 and cathepsin L at 279–280, 280–281, and 282–283 whereas for Iowa-06, the probability predicted > 0.5 for cathepsin B lies at 283–284, cathepsin S 282–283, and cathespin L at 279–280, 282–283 and 289–290. These differences arise from the I279T and I287V amino acid differences. Figure 5 shows the multiple changes in predicted cathespin cleavage arising from the six amino acid differences in the region 400–475. Comparable plots for the other five viruses are in Figures S3 and S4.
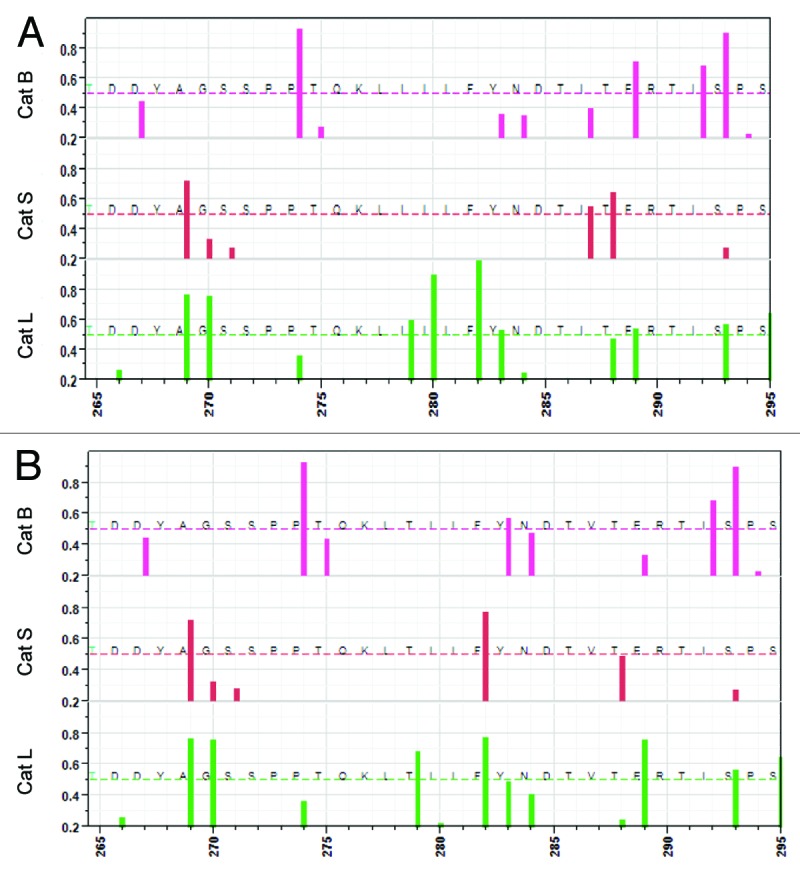
Figure 4. Predicted cleavage sites for cathepsin B, cathepsin S and cathepsin L between amino acid positions 265–295. (A) JL-5, B: Iowa-06. X axis shows the amino acid positions from 265–295, one of the regions of high predicted epitope density. Y axis shows the predicted probability of cleavage by one of three different endosomal cathepsins, top to bottom cathespin (B) (pink), cathepsin S (red) and cathepsin L (green). The bar indicates a cleavage on the C-terminal side of the amino acid at that position. Only probabilities >0.2 are shown. The dotted lines indicate probability of 0.5.

Figure 5. Predicted cleavage sites for cathepsin B, cathepsin S and cathepsin L between amino acid positions 400–475. (A) JL-5, (B) Iowa-06. X axis shows the amino acid positions from 400–475. Y axis shows the predicted probability of cleavage by one of three different endosomal cathepsins, top to bottom cathespin B (pink), cathepsin S (red) and cathepsin L (green). The bar indicates a cleavage on the C-terminal side of the amino acid at that position. Only probabilities >0.2 are shown. The dotted lines indicate probability of 0.5.
The occurrence of cathepsin cleavages can impact whether or not a particular peptide will be generated and thus displayed by an MHC molecule. Figure 6 shows the peptides that are excised by cathepsin B, L, and S and which have predicted high affinity binding for JL5 and Iowa-06 across amino acid sequence 260–475. The complete sequence for these viruses plus the corresponding plots for the other strains are shown in Figure S7. There are many minor differences between JL5 and Iowa 06, however the two regions which exhibit the highest density of predicted MHC-II binding are those which show most notable changes. These are the loss of predicted surviving high affinity peptides in Iowa-06 relative to their presence in JL5 on regions centered at 275 following cleavage by cathepsin B, L, and S and 425 following cleavage by cathepsin L and S, but a gain following cathepsin B. The change at 275 arises as a consequence of the I287V mutation. In the region around 400, where there was less quantitative difference observed in predicted MHC binding, the mutations which invoke a predicted change in cathepsin cleavage have a marked impact on the residual peptides. In this region we also see more variability between the other five virus strains examined in detail (Fig. S7).

Figure 6. Predicted high affinity MHC binding peptides following cleavage by cathepsin. (A) JL-5, (B) Iowa-06. Top left: cathepsin B, top right: cathepsin S, bottom: cathepsin L. Y Axis shows MHC-II alleles colored by cluster but ranked alphabetically. X axis shows peptide index positions from 260–475. The yellow baseline marginal bar shows the areas of high probability BEPI (orange) and high density MHC-II binding (blue) as in Figure 3. Peptides with high predicted binding affinity (-1 to -3 standard deviations below the mean) for each allele, that are between 15 and 20 amino acids in length and that are predicted to be excised by cathepsin cleavage are colored more intensely. The uniform background coloration indicates that there are no high affinity peptides in that region although there are numerous potential cleavages predicted. The difference in background colors are only to aid in identification of the operative peptidase. Peptides might be cut in any length ranging between 15 and 20 amino acids. They may have several different cut sites. The peptide pixel color intensity is indicative of the number of sites with > 0.5 probability of being cleaved i.e., the darkest colors might be cut in any one of six different positions. As the MHC-II binding groove accommodates multiple lengths of peptide any of the peptides is likely to bind. The arrows indicate the regions where most difference is seen at amino acid index positions 275 and 425.
The region around amino acid 275 is of particular interest given the well documented role of the BEPI as a neutralizing epitope.16-18 The JL5 I279 has a major effect in increasing average predicted MHC-II binding. This mutation also impacts the predicted cleavage pattern of the interferon γ -inducible cathepsin S. When a threonine occurs at 279, as in wild-type viruses, it creates a highly preferred combination for cleavage on the C-terminal side of F282. This would cleave and destroy the peptide immediately downstream of the BEPI, thus precluding MHC binding. The I287 found in JL5 places a hydrophobic amino acid at position P2 of the cleavage site octomer of cathepsin S cleaving on the C-terminal side of T288. A hydrophobic residue at this position enhances cleavage.37 Thus, this JL5 mutation provides for endosomal cleavage to release the high affinity peptide generated by the I279T mutation. The amino acid changes between 400 and 475 lead to predictions of both the lower probability of a BEPI in JL5 and a change in residual high MHC binding peptides post cathepsin cleavage.
The HN protein is thought to be N-glycosylated. A potential glycosylation site has been identified at the N28438 and which we confirmed by submission to N-GlycoSite,39 this is unchanged between JL5 and the wild type strains. In addition there are 3 predicted n-glycosylation sites in the 400–475 region, two of which are unchanged between JL5 and wildtype. Whether in fact these predicted sites are glycosylated is not known. MHC binding peptides have been shown to be disproportionately unglycosylated relative to the parent proteins40 and lysosomes are known to have aggressive enzymatic machinery to deglycosylate proteins.41
Discussion
Mumps vaccine has historically been evaluated entirely based on its ability to generate neutralizing antibody. Our results show that distribution of predicted BEPI contact points in HN protein is consistent with experimental findings of cross reactivity of neutralizing antibody among strains of mumps, given the conservation of the principal neutralizing epitope at 275. Furthermore, it points to additional potential differences in B-cell epitopes at 443 as identified by Santak et al.23 The high probability BEPIs we identify correlate well to the surface exposed residues identified by Kulkarni-Kale et al.16 In addition, our data indicates that there are distinct differences between the predicted MHC-I and MHC-II binding patterns in the HN protein of each mumps virus strain. Changes in a single amino acid result in both quantitative impacts in affinity of MHC binding and qualitative changes in the sequence of MHC-peptide presented for T-cell recognition. A single amino acid change affects 15 or more potential MHC binding frames. Single amino acid changes in the cleavage site octomer can likewise cause a marked change in predicted cathepsin cleavage. The combination of these effects can be substantial. When the cathepsin cleavage pattern is overlaid on the MHC binding pattern, this results in a change in the peptides predicted to be available for presentation as T-cell epitopes. The predicted changes impact each HLA allele differentially and furthermore will depend on the differential cathepsin expression profile in each infected cell type and the cellular responses to interferon gamma.42
By all measures we applied JL5 is an outlier, with immunomic features arising from a small number of amino acid changes that distinguish it from other virus strains. The amino acid changes do not appear to be a necessity for attenuation, although they may have arisen in the process of attenuation. Figure S8 summarizes the sequence alignments for all major attenuated vaccine strains.
The changes in predicted residual high binding peptides surviving cathepsin may indicate that there is a mismatch between JL5 and wildtype of CD4+ helper cells derived from the region adjacent to the neutralizing antibody epitope. The role of cognate CD4+ helper T-cells immediately adjacent to a B-cell epitope, or within the same protein, in enabling a recall response has been noted by others30,43,44 and has been shown to provide contextual stimulation of T-helper cells essential to memory B-cell development. In a recent analysis of proteins of multiple origins, we showed that predicted BEPI and MHC binding peptides have a strong positional interrelationship, suggesting a more specific relationship may exist between BEPI and local T-helper responses.34 The loss of a key T-helper epitope adjacent to the neutralizing BEPI may remove local T-cell help essential for both immunological recognition and recall. Hence, an individual vaccinated with JL5 may not receive appropriate T-helper stimulation to correctly mount an anamnestic response on exposure to wild-type. As a change in a single amino acid impacts MHC-II binding across 15 potential binding frames, multiple alleles are affected, as we show in Figure 4. Even recognizing that heterozygotes benefit from the highest affinity for two alternative HLA alleles, a change in T-helper function is anticipated.
While the role of CD8+ T-cells in recovery from mumps is not well characterized, it is likely that CD8+ play a role in elimination of virus infected cells. Recall of CD8+ function is also predicated upon availability of CD4+ T-helper cells conditioned in the early exposure.45,46 Not only are there changes in potential MHC-II binding in the epitope region impacting the CD4+ role in CD8+ memory recall, but also changes in predicted MHC-I binding necessary for CD8+function.
Dependence on serology to evaluate mumps vaccines may have overemphasized the conservation of one neutralizing antibody epitope, at the expense of monitoring other related changes in the HN protein that might affect recall responses. Noting the unique features of JL5, we posit that differences in both T-cell epitopes as well as other B-cell epitopes in HN may contribute to the mumps vaccine failures now reported. In particular, the two single non-synonymous nucleotide changes resulting in the substitution of I279T and I287V between JL5 HN and wild-type HN protein can, both individually and additively, lead to a mismatch of CD4+ and CD8+ responses on exposure to wild-type virus, and hence potentially to a failure to mount a recall neutralizing antibody titer. Furthermore, the impact of these changes will vary with the immunogenetics of the vaccinee.
We can further speculate that individuals vaccinated in the early years of vaccine availability may have been more frequently exposed to a wild-type boost of immunity, with the correct combination of B and T-cell epitopes, while they still had a protective neutralizing antibody titer from vaccination. As the potential for exposure to wild-type virus has decreased over time, due to the success of vaccination, such boosting has become less common and the likelihood has increased of a person encountering wild-type mumps virus only after vaccinal antibody has waned. This would be consistent with the appearance of mumps outbreaks in adolescents and young adults. A number of recent mumps outbreaks have been reported in ethnic groups which may have less genetic diversity.7,8 While social interactions within such a group may be a contributing factor, the role of immunogenetics should not be overlooked.
The significance of the change in predicted B-cell epitopes at 443 is not known, as a neutralizing antibody epitope has not been experimentally mapped in this region. Memory B-cell relevance and performance may be affected by both changes in BEPIs and changes in MHC binding. Memory B-cells are reported to develop at a low level following mumps infection26,47 which may be an independent risk factor for infection on exposure after vaccination. However, the reactivation of memory B-cells on re-infection may also affect outcome. CD4+ helper cells are needed for development of memory B-cells but may not be essential to their reactivation.48,49 The presence of dual signals of BEPI and MHC binding peptide would be expected to provide optimal stimulation.
This analysis illustrates how a systems biology bioinformatics approach can serve to frame hypotheses for further experimental testing. Final resolution of whether the issues we raise are the basis for lack of protection will depend on field study of T-cell memory in vaccinates vs individuals who have been naturally infected.
Methods
Viruses
Mumpsvirus HN sequences comprising 582 amino acids were retrieved from NCBI and duplicate sequences eliminated. A total of 54 unique sequences were in the final set analyzed comprising wild type isolates spanning 1945–2011 and 11 attenuated vaccine strains. The listing is provided in Table S1 and overlaps those recently studied by others.22,23,50
HN proteins of seven viruses, JL-5 major/USA/63 (JL5) (gi 15077510), JL-2 minor/USA/63 (JL2) (gi 19070176), Iowa.USA/2006 (Iowa-06) (gi 338784253), Calgary.CAN/30.07 (gi 376373394), 4991/Singapore/99–00 (gi 17902221), Lo1/UK/88 (gi 1419691) and Rubini/Switzerland/74 (Rubini) (gi 1071691) were selected for more detailed examination, based on relevance to recent outbreaks and the initial predicted MHC-II binding cluster analysis. These include strains having different patterns of amino acid substitution in the two areas of predicted highest MHC-II binding affinity, within the principal epitope region at amino acid positions 275–28550 and positions 410–445 (Fig. 7).

Figure 7. Epitope region sequences of seven viruses selected as representatives of the different clusters. Mumps virus strains are listed by GenBank accession number (gi), top to bottom JL5, JL2, Lo1/UK/88,. Calgary.CAN/30.07, 4991/Singapore/99–00, Iowa-06, and Rubini. The sequences are shown only for the epitope dense region from 265–294 and from 400–475. Conserved regions between 400 and 475 are omitted. Differences in the amino acid sequence are shaded and underlined.
Statistics and data analysis
Matrix algebra, statistical analysis, and data manipulation was performed with scripts and tools provided with JMP® Version 10 (SAS Institute).
B-cell epitope prediction
Amino acid principal components and neural network (NN) predictions were trained and cross-validated on the output of a large random peptide set submitted to BepiPred 1.0 (cbs.dtu.dk/services/BepiPred).51 This was necessary to provide a workable interconnected set of data processing tools. The predictions of this NN are highly concordant with those of BepiPred 1.0 (r = 0.93). A comparison with BepiPred for the HN protein of mumps virus is shown in Figure S1. The NN was then used to predict sites in the HN proteins associated with antibody binding domains, as described previously.35 Probabilities were then standardized to zero mean and unit variance within each particular HN protein.
The NN provides a structurally-based predictor in which the output is not an “epitope” prediction per se, but a probability that an amino acid (within a window ± 4 amino acids) is on the outside of a protein and thus a likely contact point for a binding antibody. This predicts short linear sequences comprising parts of epitopes based on their biophysical properties. It does not predict whether such sequences participate in three dimensional (3D) epitope configurations, although combinations of the linear epitopes could comprise a 3D epitope.
MHC binding prediction
MHC binding prediction analysis followed methodology described previously35 with several improvements.34 Briefly, each 9-mer and 15-mer indexed by single amino acid positions in HN protein was converted to vectors of principal components of amino acid physical properties, wherein each amino acid is replaced by three z-scale descriptors,9 (z1[aa2],z2[aa2],z3[aa2])… (z1[aa15],z2[aa15],z3[aa15]) that are effectively physical property proxy variables and comprise approximately 90% of the variability in the physical properties of amino acids. With these multi-dimensional descriptors, ensembles of neural network prediction equation sets were used to predict the natural logarithm of 50 percent inhibitory concentration (ln[IC50]) for MHC binding. Publicly available data sets of peptide-MHC binding data for multiple HLAs52,53 were used as training sets. Training of the NN has been enhanced using bootstrap aggregation, a process for using random subsets of the training data for generating ensembles of prediction equations.54 Importantly, this process makes it possible to estimate both a mean and variation in the predicted binding affinity (Fig. S2) that is traceable to the combinatorial amino acid sequence composition of the peptide training set. It also results in a predictor less subject to overfitting than NetMHCIIPan55 and which outperforms NetMHCIIPan outside of the training sets.35 For each of the 9-mers predicted ln(IC50) values were computed for 37 MHC-I alleles, and for each 15-mer for 28 MHC-II alleles. Each peptide was indexed to the N-terminal amino acid and each prediction corresponds to the peptide downstream from that index position. Each MHC allele has a unique range and distribution of predicted binding affinities within a protein. To mitigate against potential scale effects that could lead to spurious relationships, all predictions are converted to a common scale prior to analysis. This is done by converting all metrics to a zero mean unit variance scale within the protein molecule using a Johnson Sb algorithm.56 This creates transformed predictions with standardized normal distributions and also ranks all peptides by affinity within the protein. Using this common scaling system a value of -1 standard deviation below the within protein mean for a particular allele is further used as a threshold for MHC binding and BEPI probability. This has been shown to be an appropriate value for discriminating endpoints in a meta-analysis of influenza epitopes.36 Also, for a number of alleles it corresponds to approximately the 500 nM value that has been used for thresholding purposes. Table S2 provides the matrix of predicted binding affinity to all sequential peptides by allele for the seven virus HN protein subset.
Cathepsin cleavage prediction
In a process similar to prediction of MHC binding affinity, each 8-mer peptide in the protein was converted to z-scale vectors to enable prediction of the probability of cleavage by the endosomal peptidases cathepsin B, cathepsin L, and cathepsin S. Ensembles of NN discriminant equations were applied which had been previously trained on large data sets of cleavage patterns derived from cleavage site labeling experiments for these cathepsins37,57 and shown to have good correlation. A probability was derived for cleavage at the amino acid 4–5 bond within each octomer and was tested in proteins with known cathepsin cleavage patterns.34 Table S2 provides the matrix of all predicted cathepsin cleavage probabilities.
Secondary pattern analysis
Two types of pattern classification routines, hierarchical clustering and principal component analysis, were conducted to reduce the dimensionality of the data. Cluster analyses were conducted following the method of Ward,58 based on each of predicted B-cell epitope contact points and predicted peptide binding by MHC-I or MHC-II. The resultant hierarchical clusters were displayed as dendrograms. Principal component analysis is a means of identifying patterns in complex multivariate data sets and expressing the data to identify similarities and differences. Covariance matrices within the HN sequences were computed among the intra-protein peptide metrics (various MHC, BEPI and cathepsin) across the 54 virus set. Principal components were computed by eigen decomposition of the matrices. The resulting first three eigenvalues (i.e., the first 3 dimensions) accounted for 34–47% of the variance in the binding affinity patterns in the 54 virus data sets. Thus, principal component analysis achieves substantial dimensional reduction of the data, with the intra-protein patterns being reduced to 3 numbers for each of the HNs while capturing the essence of the variation among the viruses. The 3 dimensional plots of the principal components are thus a means of visualization of patterns showing the distances between the different viruses.
Permuted population analysis
For graphical representation of the distribution of B-cell epitope contact point probability and MHC binding on a population basis, the permuted minimum algorithm was derived as previously described.35 For every peptide locus within the primary amino acid sequence, the predicted binding affinity of two HLA alleles, comprising every possible heterozygous or homozygous pair is considered. The alleles offering the highest binding affinity at that locus are selected and averaged to provide a prediction of affinity at each locus for the metapopulation in which all alleles are given equal weight. The graphical output provides an overview of the high and low epitope density components of the protein.
Residual high affinity binding following cathepsin cleavage
The output of the predicted MHC binding analysis and the predicted cathepsin analysis were overlaid to determine the distribution of predicted high affinity MHC-II binding peptides which are predicted to survive cleavage by cathepsin B, L, or S. These are the peptides most likely to be presented as T-cell epitopes, depending on the cathepsin profile of the host cell.
The open binding groove of MHC-II molecules can bind peptides of variable sizes.30 All of our NN predictions are built on training sets of 15-mers. It is thought that the binding affinity is the result of molecular contacts within the 15-mer and that the longer peptides will bind to the molecule in a register with the most favorable contacts. To accommodate this possibility we use a consistent N-terminal indexing, but for the cathepsin cleavages assess the possibility of a cleavage occurring beyond the 15-mer in the training set (up to 20 amino acids in length). Further, several mouse alleles, routinely computed, are shown in some of the graphics. They are included only because of the importance of comparisons to human alleles for many experimental purposes.
Supplementary Material
Disclosure of Potential Conflicts of Interest
Both authors are equity holders and employees of ioGenetics LLC, a commercial company.
Supplementary Marterial
References
- 1.Dayan GH, Rubin S. Mumps outbreaks in vaccinated populations: are available mumps vaccines effective enough to prevent outbreaks? Clin Infect Dis. 2008;47:1458–67. doi: 10.1086/591196. [DOI] [PubMed] [Google Scholar]
- 2.Cortese MM, Barskey AE, Tegtmeier GE, Zhang C, Ngo L, Kyaw MH, Baughman AL, Menitove JE, Hickman CJ, Bellini WJ, et al. Mumps antibody levels among students before a mumps outbreak: in search of a correlate of immunity. J Infect Dis. 2011;204:1413–22. doi: 10.1093/infdis/jir526. [DOI] [PubMed] [Google Scholar]
- 3.Barskey AE, Glasser JW, LeBaron CW. Mumps resurgences in the United States: A historical perspective on unexpected elements. Vaccine. 2009;27:6186–95. doi: 10.1016/j.vaccine.2009.06.109. [DOI] [PubMed] [Google Scholar]
- 4.Kaaijk P, van der Zeijst B, Boog M, Hoitink C. Increased mumps incidence in the Netherlands: review on the possible role of vaccine strain and genotype. Euro Surveill. 2008;13:18914. [PubMed] [Google Scholar]
- 5.Eriksen J, Davidkin I, Kafatos G, Andrews N, Barbara C, Cohen D, Duks A, Griskevicius A, Johansen K, Bartha K, et al. Seroepidemiology of mumps in Europe (1996-2008): why do outbreaks occur in highly vaccinated populations? Epidemiol Infect. 2012:1–16. doi: 10.1017/S0950268812001136. Forthcoming. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cohen C, White JM, Savage EJ, Glynn JR, Choi Y, Andrews N, Brown D, Ramsay ME. Vaccine effectiveness estimates, 2004-2005 mumps outbreak, England. Emerg Infect Dis. 2007;13:12–7. doi: 10.3201/eid1301.060649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barskey AE, Schulte C, Rosen JB, Handschur EF, Rausch-Phung E, Doll MK, Cummings KP, Alleyne EO, High P, Lawler J, et al. Mumps outbreak in Orthodox Jewish communities in the United States. N Engl J Med. 2012;367:1704–13. doi: 10.1056/NEJMoa1202865. [DOI] [PubMed] [Google Scholar]
- 8.Nelson GE, Aguon A, Valencia E, Oliva R, Guerrero ML, Reyes R, Lizama A, Diras D, Mathew A, Camacho EJ, et al. Epidemiology of a mumps outbreak in a highly vaccinated island population and use of a third dose of measles-mumps-rubella vaccine for outbreak control--Guam 2009 to 2010. Pediatr Infect Dis J. 2013;32:374–80. doi: 10.1097/INF.0b013e318279f593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Centers for Disease Control and Prevention (CDC) Mumps outbreak on a university campus--California, 2011. MMWR Morb Mortal Wkly Rep. 2012;61:986–9. [PubMed] [Google Scholar]
- 10.World Health Organization Mumps virus vaccines. Wkly Epidemiol Rec. 2007;82:51–60. [PubMed] [Google Scholar]
- 11.Afzal MA, Pickford AR, Forsey T, Heath AB, Minor PD. The Jeryl Lynn vaccine strain of mumps virus is a mixture of two distinct isolates. J Gen Virol. 1993;74:917–20. doi: 10.1099/0022-1317-74-5-917. [DOI] [PubMed] [Google Scholar]
- 12.World Health Organization Mumps virus nomenclature update. Wkly Epidemiol Rec. 2012;87:217–24. [PubMed] [Google Scholar]
- 13.Peltola H, Kulkarni PS, Kapre SV, Paunio M, Jadhav SS, Dhere RM. Mumps outbreaks in Canada and the United States: time for new thinking on mumps vaccines. Clin Infect Dis. 2007;45:459–66. doi: 10.1086/520028. [DOI] [PubMed] [Google Scholar]
- 14.Plotkin SA. Commentary: Mumps vaccines: do we need a new one? Pediatr Infect Dis J. 2013;32:381–2. doi: 10.1097/INF.0b013e3182809dda. [DOI] [PubMed] [Google Scholar]
- 15.Jin L, Rima B, Brown D, Orvell C, Tecle T, Afzal M, Uchida K, Nakayama T, Song JW, Kang C, et al. Proposal for genetic characterisation of wild-type mumps strains: preliminary standardisation of the nomenclature. Arch Virol. 2005;150:1903–9. doi: 10.1007/s00705-005-0563-4. [DOI] [PubMed] [Google Scholar]
- 16.Kulkarni-Kale U, Ojha J, Manjari GS, Deobagkar DD, Mallya AD, Dhere RM, Kapre SV. Mapping antigenic diversity and strain specificity of mumps virus: a bioinformatics approach. Virology. 2007;359:436–46. doi: 10.1016/j.virol.2006.09.040. [DOI] [PubMed] [Google Scholar]
- 17.Kövamees J, Rydbeck R, Orvell C, Norrby E. Hemagglutinin-neuraminidase (HN) amino acid alterations in neutralization escape mutants of Kilham mumps virus. Virus Res. 1990;17:119–29. doi: 10.1016/0168-1702(90)90073-K. [DOI] [PubMed] [Google Scholar]
- 18.Orvell C. The reactions of monoclonal antibodies with structural proteins of mumps virus. J Immunol. 1984;132:2622–9. [PubMed] [Google Scholar]
- 19.Cusi MG, Fischer S, Sedlmeier R, Valassina M, Valensin PE, Donati M, Neubert WJ. Localization of a new neutralizing epitope on the mumps virus hemagglutinin-neuraminidase protein. Virus Res. 2001;74:133–7. doi: 10.1016/S0168-1702(00)00254-9. [DOI] [PubMed] [Google Scholar]
- 20.Rubin S, Mauldin J, Chumakov K, Vanderzanden J, Iskow R, Carbone K. Serological and phylogenetic evidence of monotypic immune responses to different mumps virus strains. Vaccine. 2006;24:2662–8. doi: 10.1016/j.vaccine.2005.10.050. [DOI] [PubMed] [Google Scholar]
- 21.Rubin SA, Qi L, Audet SA, Sullivan B, Carbone KM, Bellini WJ, Rota PA, Sirota L, Beeler J. Antibody induced by immunization with the Jeryl Lynn mumps vaccine strain effectively neutralizes a heterologous wild-type mumps virus associated with a large outbreak. J Infect Dis. 2008;198:508–15. doi: 10.1086/590115. [DOI] [PubMed] [Google Scholar]
- 22.Rubin SA, Link MA, Sauder CJ, Zhang C, Ngo L, Rima BK, Duprex WP. Recent mumps outbreaks in vaccinated populations: no evidence of immune escape. J Virol. 2012;86:615–20. doi: 10.1128/JVI.06125-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Šantak M, Lang-Balija M, Ivancic-Jelecki J, Košutić-Gulija T, Ljubin-Sternak S, Forcic D. Antigenic differences between vaccine and circulating wild-type mumps viruses decreases neutralization capacity of vaccine-induced antibodies. Epidemiol Infect. 2013;141:1298–309. doi: 10.1017/S0950268812001896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Davidkin I, Jokinen S, Broman M, Leinikki P, Peltola H. Persistence of measles, mumps, and rubella antibodies in an MMR-vaccinated cohort: a 20-year follow-up. J Infect Dis. 2008;197:950–6. doi: 10.1086/528993. [DOI] [PubMed] [Google Scholar]
- 25.LeBaron CW, Forghani B, Matter L, Reef SE, Beck C, Bi D, Cossen C, Sullivan BJ. Persistence of rubella antibodies after 2 doses of measles-mumps-rubella vaccine. J Infect Dis. 2009;200:888–99. doi: 10.1086/605410. [DOI] [PubMed] [Google Scholar]
- 26.Latner DR, McGrew M, Williams N, Lowe L, Werman R, Warnock E, Gallagher K, Doyle P, Smole S, Lett S, et al. Enzyme-linked immunospot assay detection of mumps-specific antibody-secreting B cells as an alternative method of laboratory diagnosis. Clin Vaccine Immunol. 2011;18:35–42. doi: 10.1128/CVI.00284-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Date AA, Kyaw MH, Rue AM, Klahn J, Obrecht L, Krohn T, Rowland J, Rubin S, Safranek TJ, Bellini WJ, et al. Long-term persistence of mumps antibody after receipt of 2 measles-mumps-rubella (MMR) vaccinations and antibody response after a third MMR vaccination among a university population. J Infect Dis. 2008;197:1662–8. doi: 10.1086/588197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jokinen S, Osterlund P, Julkunen I, Davidkin I. Cellular immunity to mumps virus in young adults 21 years after measles-mumps-rubella vaccination. J Infect Dis. 2007;196:861–7. doi: 10.1086/521029. [DOI] [PubMed] [Google Scholar]
- 29.Hanna-Wakim R, Yasukawa LL, Sung P, Arvin AM, Gans HA. Immune responses to mumps vaccine in adults who were vaccinated in childhood. J Infect Dis. 2008;197:1669–75. doi: 10.1086/588195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moss CX, Tree TI, Watts C. Reconstruction of a pathway of antigen processing and class II MHC peptide capture. EMBO J. 2007;26:2137–47. doi: 10.1038/sj.emboj.7601660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chapiro J, Claverol S, Piette F, Ma W, Stroobant V, Guillaume B, Gairin JE, Morel S, Burlet-Schiltz O, Monsarrat B, et al. Destructive cleavage of antigenic peptides either by the immunoproteasome or by the standard proteasome results in differential antigen presentation. J Immunol. 2006;176:1053–61. doi: 10.4049/jimmunol.176.2.1053. [DOI] [PubMed] [Google Scholar]
- 32.Blanchard N, Shastri N. Coping with loss of perfection in the MHC class I peptide repertoire. Curr Opin Immunol. 2008;20:82–8. doi: 10.1016/j.coi.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sijts EJ, Kloetzel PM. The role of the proteasome in the generation of MHC class I ligands and immune responses. Cell Mol Life Sci. 2011;68:1491–502. doi: 10.1007/s00018-011-0657-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bremel RD, Homan EJ. Recognition of higher order patterns in proteins: immunologic kernels. PLoS One. 2013;8:e70115. doi: 10.1371/journal.pone.0070115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bremel RD, Homan EJ. An integrated approach to epitope analysis II: A system for proteomic-scale prediction of immunological characteristics. Immunome Res. 2010;6:8. doi: 10.1186/1745-7580-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Homan EJ, Bremel RD. Patterns of predicted T-cell epitopes associated with antigenic drift in influenza H3N2 hemagglutinin. PLoS One. 2011;6:e26711. doi: 10.1371/journal.pone.0026711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biniossek ML, Nägler DK, Becker-Pauly C, Schilling O. Proteomic identification of protease cleavage sites characterizes prime and non-prime specificity of cysteine cathepsins B, L, and S. J Proteome Res. 2011;10:5363–73. doi: 10.1021/pr200621z. [DOI] [PubMed] [Google Scholar]
- 38.Yates PJ, Afzal MA, Minor PD. Antigenic and genetic variation of the HN protein of mumps virus strains. J Gen Virol. 1996;77:2491–7. doi: 10.1099/0022-1317-77-10-2491. [DOI] [PubMed] [Google Scholar]
- 39.Zhang M, Gaschen B, Blay W, Foley B, Haigwood N, Kuiken C, Korber B. Tracking global patterns of N-linked glycosylation site variation in highly variable viral glycoproteins: HIV, SIV, and HCV envelopes and influenza hemagglutinin. Glycobiology. 2004;14:1229–46. doi: 10.1093/glycob/cwh106. [DOI] [PubMed] [Google Scholar]
- 40.Szabó TG, Palotai R, Antal P, Tokatly I, Tóthfalusi L, Lund O, Nagy G, Falus A, Buzás EI. Critical role of glycosylation in determining the length and structure of T cell epitopes. Immunome Res. 2009;5:4. doi: 10.1186/1745-7580-5-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science. 2005;307:1630–4. doi: 10.1126/science.1108003. [DOI] [PubMed] [Google Scholar]
- 42.Beers C, Honey K, Fink S, Forbush K, Rudensky A. Differential regulation of cathepsin S and cathepsin L in interferon gamma-treated macrophages. J Exp Med. 2003;197:169–79. doi: 10.1084/jem.20020978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aiba Y, Kometani K, Hamadate M, Moriyama S, Sakaue-Sawano A, Tomura M, Luche H, Fehling HJ, Casellas R, Kanagawa O, et al. Preferential localization of IgG memory B cells adjacent to contracted germinal centers. Proc Natl Acad Sci U S A. 2010;107:12192–7. doi: 10.1073/pnas.1005443107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sette A, Moutaftsi M, Moyron-Quiroz J, McCausland MM, Davies DH, Johnston RJ, Peters B, Rafii-El-Idrissi Benhnia M, Hoffmann J, Su HP, et al. Selective CD4+ T cell help for antibody responses to a large viral pathogen: deterministic linkage of specificities. Immunity. 2008;28:847–58. doi: 10.1016/j.immuni.2008.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanchot C, Rocha B. CD8 and B cell memory: same strategy, same signals. Nat Immunol. 2003;4:431–2. doi: 10.1038/ni0503-431. [DOI] [PubMed] [Google Scholar]
- 46.Janssen EM, Lemmens EE, Wolfe T, Christen U, von Herrath MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes. Nature. 2003;421:852–6. doi: 10.1038/nature01441. [DOI] [PubMed] [Google Scholar]
- 47.Amanna IJ, Slifka MK. Quantitation of rare memory B cell populations by two independent and complementary approaches. J Immunol Methods. 2006;317:175–85. doi: 10.1016/j.jim.2006.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hebeis BJ, Klenovsek K, Rohwer P, Ritter U, Schneider A, Mach M, Winkler TH. Activation of virus-specific memory B cells in the absence of T cell help. J Exp Med. 2004;199:593–602. doi: 10.1084/jem.20030091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McHeyzer-Williams M, Okitsu S, Wang N, McHeyzer-Williams L. Molecular programming of B cell memory. Nat Rev Immunol. 2012;12:24–34. doi: 10.1038/nri3128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ivancic-Jelecki J, Santak M, Forcic D. Variability of hemagglutinin-neuraminidase and nucleocapsid protein of vaccine and wild-type mumps virus strains. Infect Genet Evol. 2008;8:603–13. doi: 10.1016/j.meegid.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 51.Larsen JE, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006;2:2. doi: 10.1186/1745-7580-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wold S, Sjorstrom M, Eriksson L. PLS-regression: a basic tool of chemometrics. Chemom Intell Lab Syst. 2001;58:109–30. doi: 10.1016/S0169-7439(01)00155-1. [DOI] [Google Scholar]
- 53.Greenbaum J, Sidney J, Chung J, Brander C, Peters B, Sette A. Functional classification of class II human leukocyte antigen (HLA) molecules reveals seven different supertypes and a surprising degree of repertoire sharing across supertypes. Immunogenetics. 2011;63:325–35. doi: 10.1007/s00251-011-0513-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Breiman L. Bagging predictors. Mach Learn. 1996;24:123–40. doi: 10.1007/BF00058655. [DOI] [Google Scholar]
- 55.Nielsen M, Lundegaard C, Blicher T, Peters B, Sette A, Justesen S, Buus S, Lund O. Quantitative predictions of peptide binding to any HLA-DR molecule of known sequence: NetMHCIIpan. PLoS Comput Biol. 2008;4:e1000107. doi: 10.1371/journal.pcbi.1000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Johnson NL. Systems of frequency curves generated by methods of translation. Biometrika. 1949;36:149–76. [PubMed] [Google Scholar]
- 57.Tholen S, Biniossek ML, Gessler AL, Müller S, Weisser J, Kizhakkedathu JN, Reinheckel T, Schilling O. Contribution of cathepsin L to secretome composition and cleavage pattern of mouse embryonic fibroblasts. Biol Chem. 2011;392:961–71. doi: 10.1515/BC.2011.162. [DOI] [PubMed] [Google Scholar]
- 58.Ward JH. Hierarchical Grouping to Optimize an Objective Function. J Am Stat Assoc. 1963;48:236–44. doi: 10.1080/01621459.1963.10500845. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.