

Abstract
Infection by the human hookworm Necator americanus is a leading cause of anemia and disability in the developing countries of Africa, Asia, and the Americas. In order to prevent childhood hookworm disease in resource poor settings, a recombinant vaccine is under development by the Sabin Vaccine Institute and Texas Children’s Hospital Center for Vaccine Development, a Product Development Partnership (PDP). Previously, we reported on the expression and purification of a highly promising hookworm vaccine candidate, Na-GST-1, an N. americanus glutathione s-transferase expressed in Pichia pastoris (yeast), which led to production of 1.5 g of 95% pure recombinant protein at a 20L scale.1, 2, 3 This yield and purity of Na-GST-1 was sufficient for early pilot manufacturing and initial phase 1 clinical testing. However, based on the number of doses which would be required to allow mass vaccination and a potential goal to deliver a vaccine as inexpensively as possible, a higher yield of expression of the recombinant antigen at the lowest possible cost is highly desirable. Here we report on modifications to the fermentation (upstream process) of the antigen expressed in P. pastoris, and to the purification (downstream process) of the recombinant protein that allowed for a 2–3-fold improvement in the final yield of Na-GST-1 purified protein. The major improvements included upstream process changes such as the addition of a sorbitol pulse and co-feed during methanol induction as well as an extension of the induction stage to approximately 96 hours; downstream process changes included modifying the UFDF to flat sheet with a 10 kDa Molecular Weight cut-off (MWCO), adjusting the capacity of an ion-exchange chromatography step utilizing a gradient elution as opposed to the original step elution, and altering the hydrophobic interaction chromatography conditions. The full process, as well as the purity and stability profiles of the target Na-GST-1, and its formulation on Alhydrogel®, is described.
Keywords: Necator americanus, fermentation, hookworm, purification, vaccine
Introduction
It is estimated that infection by the human hookworm Necator americanus affects hundreds of millions of people living in the poorest regions of Asia, Africa, and Latin America.1,4 N. Americanus hookworms attach to the small intestine causing blood loss that over time leads to iron deficiency anemia. Hookworm anemia and disease in children results in signficant cognitive and intellectual deficits, while in pregnant women it can lead to increased maternal morbidity and mortality as well as poor neonatal outcomes and reduced child survival.5 The recently published Global Burden of Disease Study 20106 reveals that hookworm infection is one of the leading causes of anemia,7 and among the most severe neglected tropical disease infections, as measured in disability adjusted life years.7
An effective vaccine for preventing hookworm infection would therefore be an important public health breakthrough. A recombinant vaccine that targets one of the biochemical pathways necessary for the survival of the parasite within the host is under development by the Sabin Vaccine Institute and Texas Children’s Hospital Center for Vaccine Development, a non-profit product development partnership (PDP).2
One of the leading hookworm antigens currently under development as a recombinant vaccine is a glutathione s-transferase (GST) cloned from N. americanus and expressed in P. pastoris (yeast).4 Hookworm GSTs exhibit characteristic and unique heme binding properties required by the parasite to detoxify host heme released during blood feeding, hemolysis, and hemoglobin degradation.8,9 A vaccine comprised of Na-GST-1 that elicits neutralizing host antibodies, many of which are ingested by blood feeding hookworms, has shown promise in preclinical testing as a protective immunization strategy. Vaccination of dogs with Ac-GST-1, the canine hookworm Ancylostoma caninum Na-GST-1 homolog, was shown to protect dogs and hamsters in hookworm larval challenge studies.9,10 Based on these data, an N. americanus infective larval stage (L3) cDNA expression library was screened with anti-Ac-GST-1 rabbit serum. One positive clone, which had 69% amino acid homology to Ac-GST-1, was selected as a vaccine target (Na-GST-1).11 The full-length Na-GST-1 recombinant protein expressed in P. pastoris was shown to elicit high levels of IgG antibodies in hamsters when formulated with Alhydrogel®. Vaccine protection in hamsters was also confirmed following N. americanus larval challenge studies, thus the molecule was selected for further process development and manufacture. A first generation process developed for the manufacture of P. pastoris-expressed Na-GST-112 yielded approximately 1.5 g of purified recombinant protein from one fermentation run at a 20L scale.3,12 The major features of the original manufacture process included a 6 h glycerol fed-batch phase following a ~20 h batch phase (to increase biomass) followed by approximately 65 h of methanol induction to induce Na-GST-1 expression for the upstream process, and a three-step purification process using ion-exchange, hydrophobic interaction, and gel filtration chromatography for the downstream process. The purified antigen formulated with Alhydrogel®, together with additional adjuvants, including a synthetic Toll-like receptor 4 agonist, is currently undergoing phase 1 clinical testing in both non-endemic (USA) and endemic settings in Brazil and elsewhere.
As the Na-GST-1 antigen progresses through clinical trials, it will be necessary to further optimize its expression and purification in order to achieve a higher yield and to reduce the process cost for future industrial scale manufacturing. Cost effectiveness is especially critical given that the vaccine is intended to be industrially manufactured in the developing countries where hookworm infection is widespread. Here we report on the details of an improved process that almost tripled the yield of our previously reported process at a purity of 95%. We are also reporting new results on the analytical and biochemical characterization of the purified material, and of its formulation with Alhydrogel®.
Results
Fermentation
The original Na-GST-1 fermentation process has been previously published by Goud et al., 2012 and Pickett, et al., 2012.3,12 Fermentation following the new (optimized) procedure (revised BSM formulation, no fed-batch step, sorbitol co-feed, and extended methanol induction time) was performed in triplicate at the 10L scale to: (1) compare the quality and yield of the expressed Na-GST-1 protein to that obtained following the previously published (original) fermentation process (glycerol fed-batch, no sorbitol co-feed, shorter induction time), and (2) to provide multiple lots of clarified fermentation media to assess the repeatability, quality and yield of the revised downstream purification process. SDS-PAGE analysis (reduced) of samples taken during a representative fermentation run comparing the original and optimized fermentation processes is shown in Figure 1A and B, respectively. For the optimized fermentation process, no observable expression was detected in the sample harvested prior to methanol induction (pre-induction sample; Fig. 1B, lane 2). Na-GST-1 protein product of 24 kDa was detectable after 22 h of methanol induction (Fig. 1B, lane 3) and accumulated in the culture supernatant throughout the length of the fermentation (Fig. 1B, lanes 4–6). Clarification of the post-centrifugation fermentation supernatant (FS) using a 0.22 µm polyethersulfone (PES) filter did not result in any significant loss of the target protein (Fig. 1B, lane 7). On a gross scale, the comparison of the accumulated Na-GST-1 protein by SDS-PAGE analysis between the original (Fig. 1A) and optimized (Fig. 1B) fermentation process shows a significant increase in accumulated Na-GST-1 target protein with the optimized fermentation process. In order to evaluate reproducibility of yield from the optimized fermentation process, quantitative SDS-PAGE analysis was performed in triplicate for each of the 3 optimized fermentation runs. A representative quantitative SDS-PAGE gel is shown in Figure 2 and the calculated yield from the SDS-PAGE analysis is presented in Table 1A. The calculated Na-GST-1 yield between fermentation runs ranged from 816 mg/L to 1134 mg/L with a mean just over 1g/L. The coefficient of variation (CV) observed for each triplicate quantitative SDS-PAGE analysis ranged from 13–22% and the CV between the mean of the 3 runs was 17%, which is within the typical variability observed for SDS-PAGE quantitation using Coomassie Blue staining, indicating reproducibility of the optimized fermentation process. The total yield of unpurified Na-GST-1 obtained from the fermentations is shown in Table 1B. The total amount of unpurified target protein from the optimized 10L fermentation ranged from 4.7 to 6.7 g, which is a 2.7 to 3.8-fold greater yield compared with the 1.5 g/10 L yield obtained from the original fermentation process–a significant improvement.
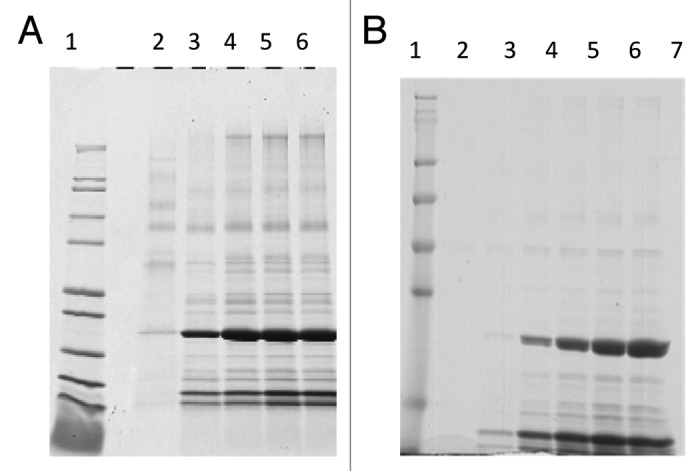
Figure 1. A: Reduced Coomassie Blue stained SDS-PAGE analysis of samples collected at different time points following the previously published (original) fermentation process. Lane 1: Mark12™. Unstained Standard, lane 2: pre-induction; lane 3: 24 h induction; lane 4: 48 h induction; lane 5: 65 h induction; lane 6: post-centrifugation (harvest). B: Reduced Coomassie Blue stained SDS-PAGE analysis of samples collected at different time points following the optimized fermentation process. Lane 1: SeeBlue® Plus2 Pre-stained Protein Standard; lane 2: pre-induction; lane 3: 22 h induction; lane 4: 46 h induction; lane 5: 66 h induction; lane 6: 89 h induction; lane 7: 89 h induction post filter (clarified).
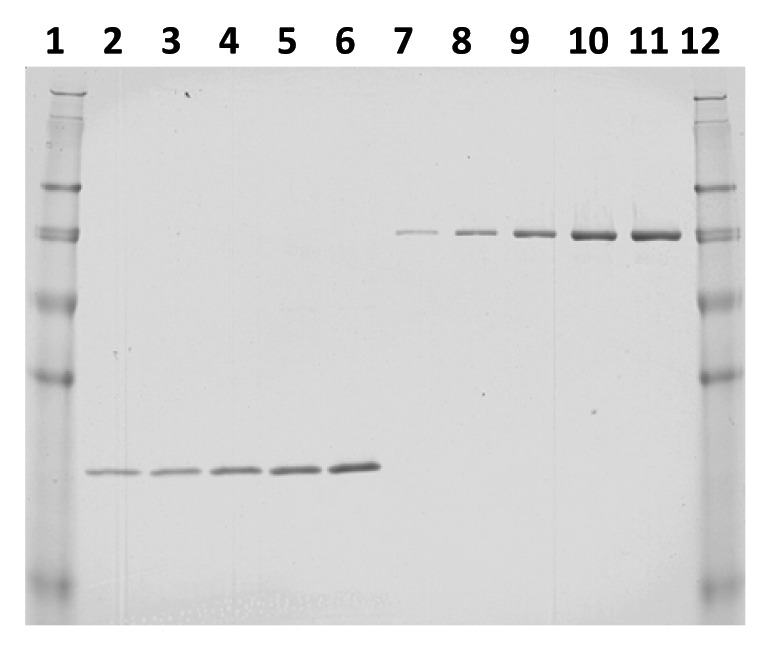
Figure 2. Quantitative SDS-PAGE analysis of a representative optimized fermentation run. Lane 1: SeeBlue® Plus2 Pre-stained Protein Standard; lane 2: fermentation supernatant (FS) 1:30 dilution; lane 3: FS 1:25 dilution; lane 4: FS 1:20 dilution; lane 5: FS 1:15 dilution; lane 6: FS 1:10 dilution; lane 7: 16 µg/mL BSA standard; lane 8: 37 µg/mL BSA standard; lane 9: 65 µg/ml BSA standard; lane 10: 90 µg/mL BSA standard; lane 11: 101 µg/mL BSA standard; lane 12: SeeBlue® Plus2 Pre-stained Protein Standard.
Table 1A. Na-GST-1 yield in fermentation supernatant following optimized fermentation process.
| Na-GST-1 Yield variability | Na-GST-1 Yield variability (between runs) | ||||
|---|---|---|---|---|---|
| Run 1 | Run 2 | Run 3 | |||
| Gel 1 | 999 | 1134 | 1346 | Run 1 | 816 |
| Gel 2 | 740 | 1150 | 1191 | Run 2 | 1065 |
| Gel 3 | 710 | 911 | 864 | Run 3 | 1134 |
| Mean | 816 | 1065 | 1134 | Mean | 1005 |
| SD | 159 | 134 | 246 | SD | 167 |
| CV | 0.19 | 0.13 | 0.22 | CV | 0.17 |
Table 1B. Na-GST-1 yield per run .
| Volume (L) | mg/L | mg Yield | |
|---|---|---|---|
| Run 1 | 5.8 | 816 | 4733 |
| Run 2 | 6.2 | 1065 | 6603 |
| Run 3 | 5.9 | 1134 | 6691 |
Purification
After harvest and clarification, the fermentation supernatant was concentrated and buffer exchanged by tangential flow filtration (TFF) into the appropriate load conditions for the Q Sepharose XL column (QXL). Changing the TFF procedure from a 3 kilo Dalton (kDa) hollow fiber column (original process) to a 10 kDa flat sheet cassette (optimized process) reduced the process time and increased the purity of the sample at this stage of the purification process. The increased purity of the post TFF sample is shown by Sodium Dodecyl Sulfate Polyacrylamide Gel Electrophoresis (SDS-PAGE) analysis (Fig. 3A lane 3 original compared with Fig. 3B lane 3 optimized) and by High Pressure Liquid Chromatography (HPLC), as shown in Figure 4 A and B Following the TFF step, the concentrated and buffer exchanged fermentation supernatant was loaded onto a Q Sepharose XL chromatography column and eluted using a salt gradient as opposed to elution with a step gradient as performed with the original process. Elution using a salt gradient provides additional wash stringency prior to peak elution and results in improved purity. The fractions containing the target protein were pooled and assessed by SDS-PAGE and Size Exclusion High Pressure Liquid Chromatography (SEC-HPLC). Comparison of the elution between the original and optimized Q Sepharose XL purification step is shown by SDS-PAGE analysis in Figure 3A, lane 4, (original process) and Figure 3B, lane 4, (optimized process) and by SEC-HPLC as shown by Figure 4C and D. Based on these data, the purity of Na-GST-1 within the Q Sepharose XL elution pool was 91%, with a recovery yield of 75% (Table 2). Although this recovery yield is slightly lower than the recovery yield of the QXL step observed with the original process (75% vs. 86%), there is a significant improvement in purity following the optimized process (91% vs. 79%). The increased purity observed with the QXL step supports the original rationale for switching from a step elution (original process) to a gradient elution (optimized process). After pooling of the QXL fractions, the ammonium sulfate (NH4)2SO2 concentration was adjusted to 1.5M (original process used 2M) and the protein was loaded onto a Butyl Sepharose High Performance (HP) column at a slower flow rate of 75 cm/hr rather than the 150 cm/hr used for the original process. In addition, the concentration of (NH4)2SO4 used for the elution was reduced from 0.7M to 0.15M. The Butyl Sepharose HP column increased the purity of the protein to greater than 96% and removed the high molecular weight contaminants detected at about 62 kDa with an average recovery rate of approximately 84%, as shown in Figures 3 and 4, and Table 2. Compared with the original purification process, a slight increase of the sample purity after the Butyl step (>96% vs. 94%), but a significant improvement in recovery (84% vs. 64%) was observed. After the Butyl Sepharose HP step, size exclusion chromatography was performed in order to remove the remaining high molecular weight contaminating proteins as well as exchanging the protein into the final formulation buffer. Due to the volume of the pooled Butyl Sepharose HP eluate, the pool was divided into 3 aliquots of approximately 65 mL each, which were loaded in consecutive runs. For all 3 replicate runs, the target protein Na-GST-1 eluted from the HPLC-SEC at approximately 37 min, which is consistent with the elution time of a theoretical molecular weight 48 kDa dimer and identical to the elution time of Na-GST-1 purified following the original method (Fig. 4G [original process] vs. Figure 4H [optimized process]). The purity of the final SEC75 pool was 99% (Table 2) which is greater than the purity of 98% following the original process.
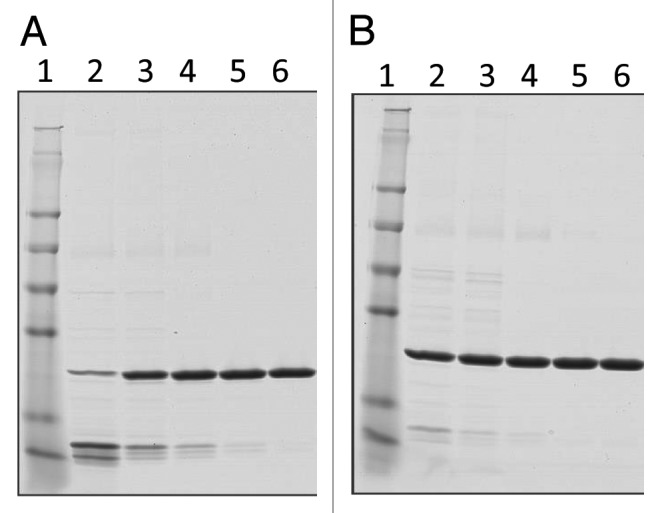
Figure 3. SDS-PAGE analysis of Na-GST-1 fractions collected at different steps of the purification process. (A) Non-reduced Coomassie Blue stained 4–20% Tris-Glycine SDS-PAGE of Na-GST-1 fractions from the original purification process. Lane 1: SeeBlue® Plus2 Pre-stained Protein Standard; lane 2: fermentation supernatant; lane 3: concentrated fermentation supernatant, lane 4: QXL elution; lanes 5: Butyl elution; lane 6: SEC75 elution. (B) Non-reduced Coomassie Blue stained 4–20% Tris-Glycine SDS-PAGE gels of Na-GST-1 fractions from the optimized purification process. Lane 1: SeeBlue® Plus2 Pre-stained Protein Standard; lane 2: Fermentation supernatant; lane 3: Post TFF; lane 4: QXL elution; lane5: Butyl elution; lane 6: SEC75 elution.

Figure 4. SEC-HPLC elution profile comparison of protein samples obtained at different steps of the purification process of the original and optimized purification processes. (A) Original process post TFF; (B) Optimized process post TFF (C) Original process post QXL (D) Optimized process post QXL; (E) Original process post Butyl; (F) Optimized process post Butyl; (G) Original process post SEC; (H) Optimized process post SEC.
Table 2. Comparisons of the yield and purity of Na-GST-1 at each step of the original and optimized purification process.
| Step | Process | Purity (%) | Recovery Yield (%) |
|---|---|---|---|
| QXL | Goud 2012 | 79 | 86 |
| Original | 81 | 83 | |
| Optimized | 91 | 75* | |
| Butyl | Goud 2012 | 94 | 64 |
| Original | 95 | 71 | |
| Optimized | >96 | 84* | |
| SEC | Goud 2012 | 98 | 94 |
| Original | 99 | 98** | |
| Optimized | >98 | 94* |
A “*” indicates that the calculation is based on SDS-PAGE and “**” indicates value is based on A280 measurement.
To ensure repeatability and robustness of the Na-GST-1 downstream process, each purification procedure was performed in triplicate concurrently with the 3 separate fermentations. The coefficient of variation for purity and yield at each purification step was between 2 and 12%, indicating good reproducibility at each step of the optimized process (Tables 3A and B). The Na-GST-1 average yield among the 3 runs was 465 mg for each liter of fermentation supernatant processed, with an SD of 54 mg/L and a CV of 11.5% (SEC elution Table 3A). This yield was approximately 3-fold higher than the ~150 mg per liter fermentation supernatant following the original purification process. The overall recovery was 55% with an SD of 3.2% and a CV of 5.8, which is 3.6% higher recovery than the original process, with an equivalent total purity of 98% (Table 3B).
Table 3A. Yield Comparisons from triplicate processes (mg).
| Step | Run 1 | Run 2 | Run 3 | Average | SD | CV |
|---|---|---|---|---|---|---|
| FS | 897 | 813 | 808 | 839 | 50 | 6.0% |
| TFF | 852 (95%) |
772 (95%) |
768 (95%) |
797 (95%) |
47 (0) |
5.9% (0) |
| QXL | 652 (73%) |
584 (72%) |
544 (67%) |
593 (71%) |
55 (3.2) |
9.2% 4.3% |
| Butyl | 545 (61%) |
494 (61%) |
444 (55/%) |
494 (59%) |
51 (4.6) |
10.2% (4.9%) |
| SEC75 | 527 (59%) |
438 (54%) |
431 (53%) |
465 (55%) |
54 (3.2) |
11.5% (5.8) |
(A) Comparison of the amount of Na-GST-1 in mg/mL obtained from the 3 purification runs. Single yields for each run as well as the average, the standard deviation, the SD, and the coefficient of variation between each run, CV, are reported. The cumulative % of recovery for along the process is presented in brackets.
Table 3B. Comparisons of the Na-GST-1 purity (%) from triplicate processes as determined by HPLC-SEC.
| Step | Run 1 | Run 2 | Run 3 | Average | SD | CV |
|---|---|---|---|---|---|---|
| FS | 10 | 11 | 10 | 10 | 0.6 | 5.6% |
| TFF | 80 | 85 | 85 | 83 | 2.9 | 3.5% |
| QXL | 91 | 92 | 89 | 91 | 1.5 | 1.7% |
| Butyl | 98 | 97 | 96 | 97 | 1.0 | 1.0% |
| SEC75 | 98 | 98 | 98 | 98 | 0 | 0 |
(B) Purity of the protein as determined by SEC-HPLC.
In-process sample stability
In order to address the stability of Na-GST-1 obtained from the optimized process over time, aliquots of in-process samples were collected at each purification step, sterile filtered, divided into 2 fractions, and analyzed up to 7 d after storage at 2–8 °C, 25 °C, and 37 °C. Representative SDS-PAGE analysis of samples stored at the different temperatures is shown in Figure 5B and C. No degradation was observed for any in-process samples throughout the study, suggesting that a 2–8 °C, or even a room temperature, hold step may be incorporated into the process for up to 7 d (data not shown).

Figure 5. Coomassie Blue stained 4–20% Tris Glycine SDS-PAGE gels of Na-GST-1 samples incubated at 2–8 °C, 25 °C, and 37 °C for a total of 30 d. (A) Sample analysis at day 1; (B) Sample analysis at day 3; (C) Sample analysis at day 7; (D) Sample analysis at day 15 and (E) Sample analysis at day 30. For all gels A-E: lane 1: Mark12™ Unstained Standard; lane 2: 2 µg Na-GST-1 at 2–8 °C, non-reduced; lane 3: 2 µg Na-GST-1 at 25 °C, non-reduced; lane 4: 2 µg Na-GST-1 at 37 °C, non-reduced; lane 5: 2 µg Na-GST-1 at 2–8 °C, reduced; lane 6: 2 µg Na-GST-1 at 25 °C, reduced; lane 7: 2 µg Na-GST-1 at 37 °C, reduced.
Na-GST-1 characterization and disulfide bond determination via Liquid Chromatography/Mass Spectrometry (LC/MS)
Purified Na-GST-1 protein, when electrophoresed through denaturing SDS-PAGE gels under non-reducing conditions, resolves as 2 discrete bands migrating at approximately 24kDa. In order to shed light on the nature of the 2 forms, each separate band was characterized by LC/MS analysis as described in the Methods section. LC-MS analysis confirmed that both bands contained identical full-length protein, with no detectable proteolysis between these isoforms. In addition, no detectable N- or O-linked glycosylation was observed in either of the bands. However, LC/MS analysis showed that the lower molecular weight band, under non-reducing conditions, contained an intra-molecular disulfide linkage between Cys-17 and Cys-68 of the protein, which was not present in the higher molecular weight band. Although, there is no indication from the crystal structure of the intact form of this protein that a disulfide bond exists in the molecule,8 the observation of both forms in both the original and optimized processes leads us to assume that the disulfide bond is an intrinsic property of the protein and would not interfere with the purified protein’s ability to act as a suitable vaccine immunogen.
Protein stability after freeze/thaw
Freezing, storage, and thawing of proteins are routinely evaluated in the pharmaceutical industry as a conservative approach to establish the risks of freezing on stability. To this end a freeze/thaw study was conducted. As shown in Table 4 once subjected to 3 freeze/thaw cycles, the purified protein did not show any variations in color and appearance, pH, protein content (as measured by absorbance at 280 nm, data not shown), SEC-HPLC elution profile (Fig. 6A) and SDS-PAGE analysis (Figs. 6B and C), indicating that the protein did not undergo degradation or loss of integrity. After 3 freeze/thaw cycles, the protein eluted from the SEC-HPLC column at 26.04, 26.03, and 26.03 min, for an average of 26.03 ± 0.01 min, as expected for Na-GST-1. Similarly, on reduced SDS-PAGE the target protein ran as a predominant band at 24 kDa without evidence of aggregate formation or degradation products. Similar data were obtained for all 3 reproducibility runs, showing consistency among the three lots produced (data not shown).
Table 4. Comparisons of the Na-GST-1 protein samples before and after 3 freeze/thaw cycles.
| Parameter | Freeze/thaw cycles | |||
|---|---|---|---|---|
| Prior F/T | 1 | 2 | 3 | |
| Particles observed | - | - | - | - |
| pH | 7.46 | 7.46 | 7.48 | 7.47 |
| Protein content (mg/mL) | 1.07 | 1.07 | 1.087 | 1.09 |
Evaluation of color and appearance, and measurement of pH and protein content by absorbance at 280nm for the Na-GST-1 protein samples before and after 3 freeze/thaw cycles.
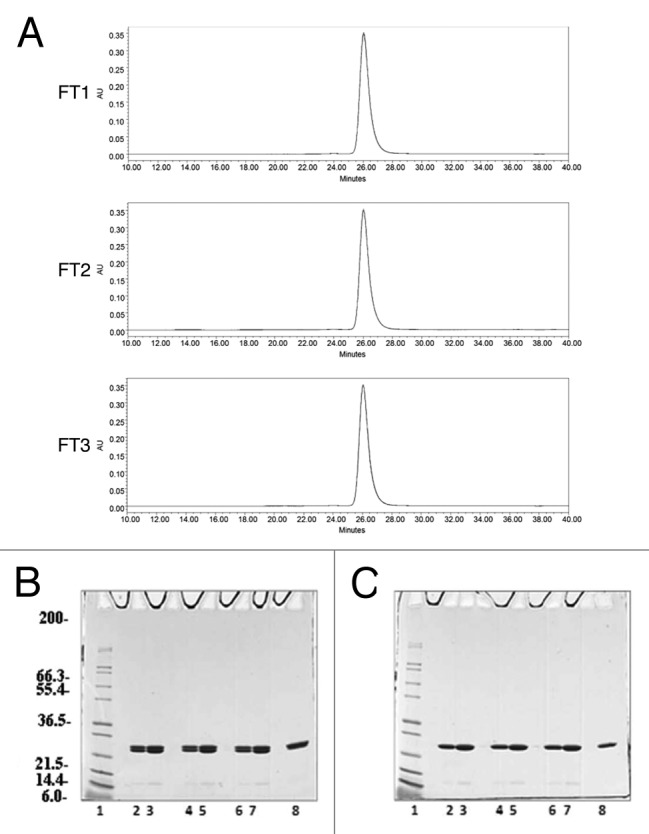
Figure 6. Analysis of the Na-GST-1 purified protein after 3 freeze/thaw cycles. (A) Representative SEC-HPLC elution chromatograms of the Na-GST-1 sample after 1 freeze/thaw cycle (FT1), 2 freeze/thaw cycles (FT2), and 3 freeze/thaw cycles (FT3), respectively. (B and C) Non-reduced and Reduced Coomassie Blue stained 4–20% Tris-Glycine SDS-PAGE gels of Na-GST-1 purified samples after 1, 2, and 3 freeze/thaw cycles, respectively. Lane 1: Mark12™ Unstained Standard; lane 2: 2 µg Na-GST-1 (FT1); lane 3: 4 µg Na-GST-1 (FT1); lane 4: 2 µg Na-GST-1 (FT2); lanes 5: 4 µg Na-GST-1 (FT2); lane 6: 2 µg Na-GST-1 (FT3); lane 7: 4 µg Na-GST-1 (FT3); lane 8: 2 µg Na-GST-1 (time 0).
30 d stability of Na-GST-1 protein at 2–8 °C, RT, and 37 °C
Studies were performed to test the stability of the protein upon incubation at different temperatures for a period up to 30 d. The stability data describes how the protein qualities vary with time under the influence of a variety of environmental factors such as temperature. The testing was performed over a period of 30 d and included storage of the molecule at three different temperatures. The purified protein did not show any variations in pH or protein content as measured by absorbance at 280 nm at all temperatures after 30 d (Table 5). Sample integrity was maintained at 2–8 °C for the entire 30 d. However, after 7 d of incubation at 25 °C, or higher temperatures, the presence of particulates could be noticed in the protein solution indicative of protein instability (Table 5). When incubated for 7 d at 37 °C, protein degradation, while not evident on Coomassie Blue Stained gels, (Fig. 5C) was detectable by Silver stain (Fig. 7). The SEC-HPLC analysis was only partially successful in detecting protein instability in terms of peak purity and peak area. While all samples tested eluted at the 26 min with a variance of only 0.03 min, the peak purity and the peak area decreased only 2% to 3% in the samples stored at 37 °C for 30 d and did not change for the samples stored at room temperature (data not shown).
Table 5. Comparisons of the Na-GST-1 protein samples stored at 2–8 °C, 25 °C, and 37 °C for up to 30 d.
| Time | Test | 2–8 °C | 25 °C | 37 °C |
|---|---|---|---|---|
| Day 0 | Particles observed | - | - | - |
| pH | 7.46 | 7.54 | 7.46 | |
| Protein content (mg/mL) | 1.067 | 1.039 | 1.067 | |
| Day 1 | Particles observed | - | - | - |
| pH | 7.54 | 7.60 | 7.57 | |
| Protein content (mg/mL) | 1.086 | 1.057 | 1.074 | |
| Day 3 | Particles observed | - | - | + |
| pH | 7.50 | 7.60 | 7.54 | |
| Protein content (mg/mL) | 1.082 | 1.055 | 1.08 | |
| Day 7 | Particles observed | - | + | + |
| pH | 7.52 | 7.51 | 7.52 | |
| Protein content (mg/mL) | 1.087 | 1.063 | 1.086 | |
| Day 15 | Particles observed | + | ++ | +++ |
| pH | 7.53 | 7.56 | 7.55 | |
| Protein content (mg/mL) | 1.102 | 1.071 | 1.086 | |
| Day 30 | Particles observed | + | ++ | +++ |
| pH | 7.49 | 7.55 | 7.52 | |
| Protein content (mg/mL) | 1.098 | 1.052 | 1.067 |
Evaluation of color, and appearance, pH and protein content by absorbance at 280nm for Na-GST-1 samples stored at 2–8 °C, 25 °C, and 37 °C for up to 30 d. – indicates lacks of precipitation, + indicates 1–2 particles, ++ indicates 2–5 particles, +++ indicates more than 5 particles.
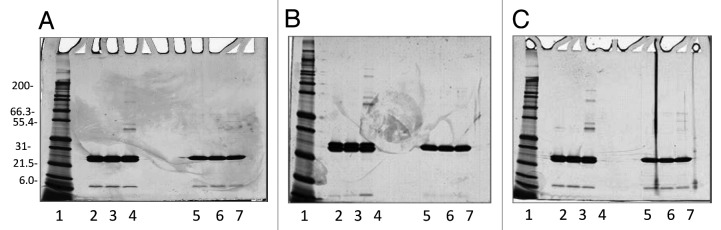
Figure 7. Silver stained 4–20% Tris-Glycine SDS-PAGE gels of Na-GST-1 sample incubated at 2–8 °C, 25 °C, and 37 °C for a total of 30 d. (A) Samples analysis at day 7; (B) Samples analysis at day 15 and (C) Samples analysis at day 30. For all gels, A-C, lane 1: Mark12™ Unstained Standard; lane 2: 2 µg Na-GST-1 at 2–8 °C, non-reduced; lane 3: 2 µg Na-GST-1 at 25 °C, non-reduced; lane 4: 2 µg Na-GST-1 at 37 °C, non-reduced; lane 5: 2 µg Na-GST-1 at 2–8 °C, reduced; lane 6: 2 µg Na-GST-1 at 25 °C, reduced; lane 7: 2 µg Na-GST-1 at 37 °C, reduced.
Three months stability of the Na-GST-1 formulation
Na-GST-1 was formulated with Alhydrogel® as described by Plieskatt et al., 2012.12 The formulation was stored at 2–8 °C for a period of 3 mo and its stability was investigated by assessing the color and appearance, the pH, the protein content by the o-phthaldialdehyde (OPA) assay, and the identity by SDS-PAGE followed by Coomassie Blue staining. Throughout the study there were no variations observed in the color and appearance of the formulated samples and all aliquots tested maintained the expected characteristics for Na-GST-1/ Alhydrogel® formulations. The pH was stable at 7.4 ± 0.1, and no pH variation was observed from the time of formulation to the end of the study. As shown in Figure 8 when the Na-GST-1/ Alhydrogel® formulation was centrifuged to separate the pellet fraction from the supernatant, the Na-GST-1 protein segregated in the pellet fraction and the Na-GST-1 protein was not detected in the supernatant fraction, indicating a tight binding of the molecule to the surface of the Alhydrogel®. Similar conclusions were drawn from the data collected by the OPA assay, which indicate that 98% of the protein was bound to the surface of the Alhydrogel® by a total protein content assay using o-phthaldialdehyde (data not shown).
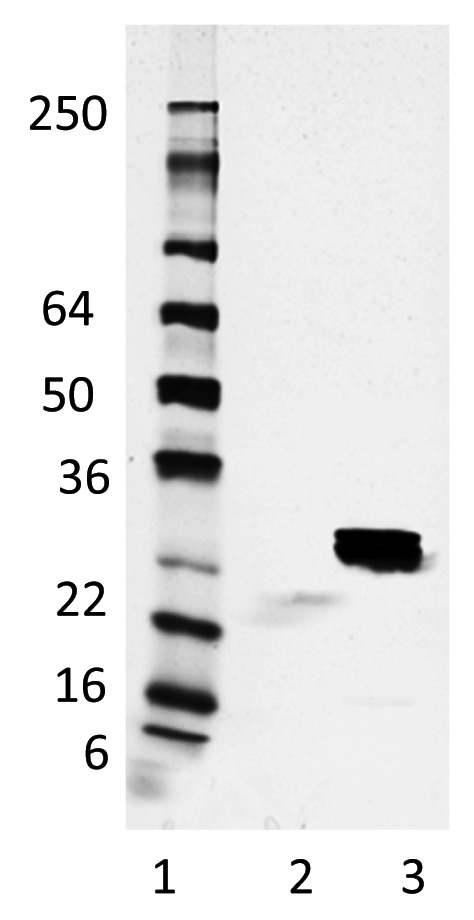
Figure 8. Non-reduced Coomassie Blue stained 4–20% Tris-Glycine SDS-PAGE gels of the Na-GST-1/Alhydrogel® vaccine samples 3 mo post formulation and storage at 2–8 °C. Lane 1: SeeBlue® Plus2 Pre-stained Protein Standard; lane 2: formulation supernatant fraction; lane 3: formulation pellet fraction.
Discussion
A recombinant human hookworm subunit vaccine is being developed by the Sabin Vaccine Institute and Texas Children’s Hospital Center for Vaccine Development, a non-profit PDP.4,13,14 Na-GST-1, one of the vaccine antigens, has been previously manufactured at pilot scale according to current Good Manufacturing Practices (cGMP) using a process described by Goud et al., and Plieskatt, et al.3,12 Following regulatory submissions in the United States and Brazil, the Na-GST-1 Hookworm Vaccine is currently in phase 1 clinical trials in healthy adult volunteers in the United States and in non-endemic and endemic areas of Minas Gerais State, Brazil.2 In order to design a more mature production process suitable for downstream industrial manufacturing, additional process development was performed with the goal to achieve higher yields of expression of the recombinant antigen as well as higher overall recovery and purity after purification. Therefore, the protein expression yields and the percentage of recovery of the manufacturing process were further optimized. For the optimization of the fermentation process, several different parameters were evaluated including various alternative media formulations, the necessity of the fed-batch phase, mixed carbon feeds, and length of induction time. Performing side-by-side fermentations with and without the glycerol fed-batch phase provided evidence that this 6 h step had little to no impact on the Na-GST-1 yield; therefore this step was omitted to reduce overall fermentation time. Alternate media formulations were tested from completely defined media to complex rich media containing yeast extract, casamino acids, and/or phytone, but these alternate media formulations had a minimal effect on Na-GST-1 yield. Addition of a sorbitol co-feed has been previously shown to improve overall yields of proteins expressed in Pichia during methanol induction.15-19 Performing side-by-side experiments with and without sorbitol co-feed during methanol induction showed a dramatic improvement in Na-GST-1 accumulation and final yield. Finally, several fermentation time course experiments were performed to determine whether the original ~72 h induction was optimal or if extending the induction time would result in a higher yield of Na-GST-1. Extending the induction period to ~96 h resulted in a significant improvement in Na-GST-1 yield with a moderate improvement when the induction period was extended to 120 h. To make the process more amenable to manufacturing, the 96-h induction time was selected for the optimized process. Together, these changes provide an increase in the Na-GST-1 fermentation yield from 0.3 g/L to approximately 1 g/L. For the downstream process, and in order to accommodate the increased yield from the optimized fermentation process, the column capacity of the Q Sepharose XL step was adjusted and optimized. In addition, changing the Q Sepharose XL elution from a step elution to a gradient elution increased the protein purity without introducing a completely new variable so as to keep the changes minimal. A significant improvement (64% to 84%) in protein recovery at the Butyl Sepharose HP step, while maintaining similar purity, was achieved by reducing the ammonium sulfate concentration in the load (from 2M to 1.5M) as well as in the elution buffer (0.7M to 0.15M), and by increasing the amount of Butyl HP resin used and decreasing the linear flow rate. We believe that the reduced salt in the elution buffer allowed for the improvement in yield at this step. Although using size exclusion may not be entirely optimal for large scale processes, it does serve as a polishing step for our process since it (1) prevents any carryover of high molecular weight contaminating proteins which are seen at full manufacturing scale and (2) keeps the number of purification process changes to a minimum. While changing from a final size exclusion step to a less costly or time consuming step, such as a second ion exchange step, may be more manufacturing friendly, it would likely be more difficult to transition the new product into the clinic since it probably would require several comparability or bridging clinical studies. Stability studies were performed on the in-process samples and final product of the optimized process to ensure biochemical and analytical comparability with material generated during the original process. Our stability data for the in-process samples, as well as the final purified protein, indicate that the Na-GST-1 is stable at 2–8 °C for at least 30 d, and at temperatures from 25 °C to 37 °C for at least 7 d before the first signs of degradation. These data indicate that refrigerated hold steps could be incorporated into the manufacturing process without any detrimental impact on the integrity of the protein. Integrity at 2–8 °C for the 3 mon was also maintained upon formulation with Alhydrogel®. No variation in pH, concentration of protein absorbed to Alhydrogel®, or protein identity was detected within the 3-mon period. These results are concordant to our previous and ongoing stability studies with the protein and formulated protein generated with the original process.
In conclusion, starting from an original manufacturing process that was used to generate pilot phase 1 clinical material, we have now made minimal changes to the upstream and downstream processes achieving a significant increase in yield and purity while retaining stability and integrity of the molecule. These improvements are essential steps during the continuum development of vaccines targets and show that the revised process used to manufacture Na-GST-1 has great promise to become a mature industrial scale manufacturing process.
Materials and Methods
Generation of Research Seed stock
Cloning, small-scale expression, and selection of clone of Na-GST-1 for production scale up have been reported previously.11 The research seed stock was generated from a single clone identified by Western blot analysis to be a highly expressing clone.3
Fermentation
The Na-GST-1 production and purification was optimized since originally reported.3 Elimination of the 6-hr fed-batch step, addition of a sorbitol pulse and sorbitol co-feed during methanol induction, and extension of the induction stage from approximately 72 h to approximately 96 h are the main changes introduced into the new revised fermentation process. The details of the original fermentation process are described in Goud et al., 2012. For the optimized fermentation procedure, the process was initiated by the preparation of a 1 L seed stock to be used for inoculation of the fermenter (New Brunswick). 1–3 mL of the research cell bank was added to 1 L BMG media and the flask incubated at 29 +/−1 °C with agitation at 250 +/−10 rpm overnight. After reaching the target OD600 (1.0–5.0), 250–500 mL of the seed culture was inoculated into 4 L of sterile basal salt media (85% Phosphoric Acid (H3PO4) 26.7 mL/L, Calcium Sulfate (CaSO4 2H2O) 0.93 g/L, Potassium Sulfate (K2SO4) 18.2 g/L, Magnesium Sulfate (MgSO4 7H2O) 14.9 g/L, Potassium Hydroxide (KOH) 4.13 g/L, Glycerol 40 g/L) containing 4.35 mL/L PTM1 Trace Salts (Copper Sulfate (CuSO4 5H2O) 6.0 g/L, Sodium Iodide (NaI) 0.08 g/L, Manganese Sulfate (MnSO4 H2O) 3.0 g/L), Sodium Molybdate (MoNa2O4 2H2O) 0.2 g/L, Boric Acid (H3BO3) 0.02 g/L, Copper Chloride (CoCl2) 0.5 g/L, Zinc Chloride (ZnCl2) 20.0 g/L, Iron (II) Sulfate ((FeSO4) 7H2O) 65.0 g/L, Sulfuric Acid (H2SO4) 5 mL/L), and 4.35 mL/L 0.02% d-Biotin in a 14 L fermentation vessel. The pH of the BSM was adjusted to 5.00 +/− 0.2 prior to inoculation and the pH maintained at this set point by the addition of 14% NH4OH. Fermentation set points used were 30 +/− 1 °C, dissolved oxygen 30%, air flow 0.5–1.0 SPLM, and agitation 500 rpm. Antifoam 204 was added to minimize foaming. Approximately 16 +/− 4 h after inoculation of the fermenter, a DO spike was observed due to the depletion of glycerol within the media. After observation of the DO spike, a bolus of 50% sorbitol (~9.0 g/L) was added to the fermenter over a 30 min period. After the 30 min sorbitol feed, the pH of the culture was ramped from 5.0 to 6.0 and the temperature from 30 °C to 26 °C within a one hour period. During this period, no feed was added to the fermenter (starvation phase). At the end of the starvation phase, the agitation was increased to 700 rpm and a methanol feed (containing 12 mL/L PTM1 Trace Salts) was ramped from 1–11 mL/L/hr over an 8-h period. 50% sorbitol was fed concurrently during the methanol ramp at a rate of 3 mL/L/hr. After the ramp period, the methanol feed was maintained at 11 mL/L/hr and the 50% sorbitol feed was maintained at 3 mL/L/hr until the end of fermentation (after approximately 96 h of methanol induction).
Purification clarification and tangential flow filtration (TFF)
After approximately 96 h of methanol induction, the cell culture was harvested and centrifuged at 7000 rpm (12 227 x g) for 45 min at 4 °C. Following clarification, the supernatant was filtered using a 0.22 µm PES filtration unit and transferred to a vessel for buffer exchange. Tangential flow filtration was used to concentrate the fermentation supernatant by approximately 50% and to buffer exchange it into Q Sepharose XL (Q SepharoseE XL) binding buffer 30 mM Tris, 20mM NaCl pH 8.0 (3.9 mS/cm at 25 °C). The buffer exchange was performed using 2 flat sheet Pellicon 2 Biomax 10 modified PES mini cassettes with a 10 kDa MWCO and a filtration area of 0.1 m2 per cassette. The starting conductivity of the clarified/filtered fermentation supernatant was approximately 25 mS/cm and the pH was approximately 6.0. The target conductivity of 4.2 +/− 0.5 mS/cm and pH of 8.0 +/− 0.05 was achieved after approximately 4–5 volumes of Q Sepharose XL binding buffer had been used.
Q Sepharose XL chromatography
Anion exchange chromatography was used as the first step to concentrate the target protein and remove host cell protein contaminants, pyrogens, and DNA present within the concentrated/buffer exchanged fermentation broth. Q Sepharose XL resin (GE Healthcare) was packed into a Millipore Vantage L column to a bed height of 22.7 cm and column volume of 350 mL. The column was equilibrated with 30 mM Tris, 20 mM NaCl, pH 8.0, followed by loading the buffer exchanged fermentation supernatant at a flow rate of 150 cm/hr. After loading the sample, the column was washed with 30 mM Tris, 20 mM NaCl pH 8.0 for 8–10 column volumes, and then the bound protein was eluted with a NaCl gradient from 20 mM to 250 mM over 7.5 column volumes. The Q Sepharose XL step was improved upon the original process by adjusting the capacity to accommodate the increased yields from the improved fermentation process as well as by increasing the purity obtained by utilizing a gradient elution as opposed to the original step elution.
Butyl Sepharose high performance chromatography column (Butyl HP)
To prepare for loading onto a Butyl HP column, the Q Sepahrose XL elution pool was diluted with an equal volume of 3M (NH4)2SO4, 30mM Tris pH 8.0 to bring the final concentration of (NH4)2SO4 to 1.5 M. Butyl Sepharose HP resin was packed into a Millipore Vantage Column to a bed height of 22.0 cm and a column volume of 335 mL. The column was equilibrated with 30 mM Tris, 1.5 M (NH4)2SO4 pH 8.0, and the diluted Q Sepharose XL elution pool was loaded at a flow rate of 75 cm/hr. After loading, the column was washed for 5 column volumes with 30 mM Tris, 1.5M (NH4)2SO4 pH 8.0, followed by a wash with 30 mM Tris, 1.3 M (NH4)2SO4, pH 8.0 for 3 column volumes to remove loosely bound contaminants and a high molecular weight contaminant of approximately 62 kDa. After the second wash step, an isocratic (step) elution was performed with 30 mM Tris, 0.15 M (NH4)2SO4 pH 8.0. This Butyl Sepharose HP process improves upon the original process by adjusting the (NH4)2SO4 concentration in the load and elution as well as adjusting the amount of resin to significantly increase the recovery while maintaining similar purity.
SEC75 (size exclusion SEC75, prep grade chromatography)
SEC75 chromatography was performed to remove the remaining contaminants from the target Na-GST-1 protein. Superdex75 prep grade resin (GE Healthcare) was packed in a GE Healthcare Index70 column to a bed height of 56.0 cm and column volume of 2155 mL. A total of 3–4 cycles were required to process the entire Butyl Sepharose elution pool by loading aliquots up to 3% of the column volume (approximately 65 mL). The column was equilibrated with 10 mM Imidazole, 10% Sucrose pH 7.4, and the Butyl Sepharose HP elution pool was loaded at a flow rate of 30 cm/hr. The elution was performed in the same buffer at the same flow rate for 1.5 column volumes.
Quantitative SDS-PAGE for mass balance
The concentration of Na-GST-1 in the fermentation supernatant as well as in the in process intermediate fractions was determined in triplicate by SDS-PAGE on 14% Tris Glycine gels (Invitrogen) using manufacturer’s instructions. Beta-mercaptoethanol was added to the 2X sample-loading buffer to a final concentration of 10%. BSA standards (16–101 µg/mL) were included on the gels for quantitative analysis by densitometry. After electrophoresis at 125V for approximately 1.5–2 h, gels were stained with Coomassie Blue, de-stained and scanned using either an ImageScanner II (manufacturer and quantified with the Image Quant v 8.1 software (GE Healthcare) or a GS-800 Self-Calibrating Densitometer (Bio-Rad), and quantified with Quantity One v. 4.6 software (Bio-Rad). The BSA standards run on each gel (16 µg/mL to 101 µg/mL) were used to establish a linear quantitation curve for the Na-GST-1 dilutions also run on the same gels. The concentration of each Na-GST-1 dilution was determined by plotting the Na-GST-1 dilution on the standard curve and then multiplying by the dilution factor used.
Purity by SDS-PAGE
4–20% Tris glycine gels were used for evaluation of the sample’s purity. Gels were run following the manufacturer’s recommendations. For the reduced samples, β-mercaptoethanol was added to the 2X Tris-glycine sample-loading buffer to a final concentration of 10%. Approximately 2 µg of non-reduced and reduced protein was loaded for each lane. Gels were stained, de-stained, and scanned as described above.
Purity by SEC-HPLC
Purity of all in-process samples was analyzed by SEC-HPLC. The fermentation supernatant and the final purified protein were not diluted prior to injection, while all remaining in-process samples were diluted with SEC running buffer (50 mM sodium acetate pH 4.8) prior to injection in order to standardize concentration of target protein with the fermentation supernatant. All samples were injected on the column in a 50 µL volume at a flow rate of 0.25 mL/min onto a Tosoh TSK gel G2000SWxl column using a Shimadzu Prominence UFLC with a PDA detector. The column was run at room temperature for 75 min in 50 mM sodium acetate, 200 ppm sodium azide pH 4.8. To determine the percent purity, the area of the main peak was compared with the area of all other peaks on the chromatograph (excluding sample buffer peaks).
Protein characterization and disulfide bond determination via LC/MS
The Na-GST-1 protein separates into 2 discrete bands when resolved by SDS-PAGE electrophoresis under non-reducing conditions. Each gel band (labeled as “top” and “bottom” band respectively) was isolated from the gel individually and split into multiple ~3mm sections. Each gel section isolated from each individual band was destained in 50% methanol/50mM ammonium bicarbonate, then subjected to protease digestion with one or more enzymes, either after alkylation alone with iodoacetamide, or after both reduction and alkylation of cysteine residues by DTT and iodoacetamide, essentially as previously described,20 to map sites of potential disulfide bond formation. Multiple enzymatic reactions were used to increase amino acid sequence coverage of the Na-GST-1 protein in the mass spectrometer, and consisted of 4 separate digestion schemes. Digest reactions included the use of trypsin digestion alone; trypsin digestion followed by Glu-C digestion; Asp-N digestion followed by Arg-C digestion; or Asp-N/Arg-C digestion followed by Glu-C digestion. Digestions were performed at a substrate-to-enzyme ratio of 50:1 at 37 °C in 50mM ammonium bicarbonate buffer for at least 8 h for each enzyme, and stopped by acidification to 0.2% formic acid. Resulting peptides were injected onto the LC column equilibrated in 0.1% formic/99.9% water and separated by reverse phase chromatography by increasing acetonitrile gradient formation (0–40% over 30min) across a C-18 column (75 μm × 250 mm BEH130 C18 column; 1.7μm particle size) using nanoflow chromatography at a flow rate of 300 nL/min and column temperature of 35°C using a nanoAcquity Ultra Pressure Liquid Chromatography system (UPLC), Waters Corp. Eluting peptides were transferred directly into a Synapt HDMS mass spectrometer (Waters Corp.) via a nanoelectrospray union operated under positive ion mode, at a capillary voltage of 3kV. The mass spectrometer was operated in the LC/MSE mode (parallel-ion fragmentation conditions) in positive-ion mode). LC/MS instrumentation operation and data collection was accomplished using MassLynx software (v4.1: Waters Corp), and all resulting spectra were processed and analyzed using ProteinLynx Global Server software (v2.4; Waters Corp.). A protein database of the Na-GST-1 protein sequence concatenated to the Pichia complete proteome was used to search the mass spectra against, with the false discovery rate set to 0% (reverse and randomized decoy database FDR method). Protein spectra derived from each SDS-gel band were interrogated for full-length amino acid sequence by looking at the sequence coverage maps from each of the individual enzymatic digestion reactions, N- and O-glycosylation status, disulfide bond formation, and for evidence of any Pichia protein contamination of the purified Na-GST-1 protein bands. Manual interpretation of all spectra was used for verification, and for de novo sequence analysis of peptides containing disulfide bonds.
Stability of in -process samples by SDS-PAGE
The stability of the in-process samples (FS, TFF, QXL, Butyl, SEC75) at 2–8 °C and room temperature was evaluated up to 7 d by SDS-PAGE. Two aliquots of each sample were taken at each purification step and placed at the respective temperatures. Time zero samples were immediately prepared for each in process step while the remaining samples were prepared at the defined time points by first diluting with 30 mM Tris/20 mM NaCl pH 8.0 and then with 2X sample buffer to achieve a final concentration of approximately 5 µg protein/12 µL loading volume. Diluted samples were loaded onto 4–20% Tris-Glycine gels followed by Coomassie Blue staining as previously described.
Freeze/thaw analysis of the Na-GST-1 protein
As protein biologics can be susceptible to instabilities, degradation, unfolding, and aggregation upon storage, which can influence their effectiveness and immunogenicity, a stability study was designed to include freeze/thaw cycles. To simulate the time and manner by which proteins could be exposed to repetitive cycles of freezing during typical handling and testing processes, the purified protein was subjected to 3 freeze/thaw cycles while being exposed to ambient temperature after each thaw cycle. Three 1 mL aliquots of Na-GST-1 were placed in liquid nitrogen for 10 min followed by a thaw step at room temperature. The 3 vials were exposed respectively to 1, 2, or 3 freeze/thaw cycles. After each cycle, samples were tested for color and appearance by visual inspection on a white and black background, for pH using a Thermo Scientific Orion Versa Star™ pH meter following standard manufacturer recommendations, and for protein content by the absorbance at 280 nm with Ultrospec 6300pro. The identity and purity of the sample were also tested by SEC-HPLC using a Waters Alliance® HPLC system with the Empower® 2 software for data analysis. 50 µg of Na-GST-1 at 1 µg/ µL were loaded on a TSK gel G2000SWxl column with 1XPBS, 10% Sucrose and 100 mM NaCl at pH 7.4 as mobile phase and with a flow rate of 0.29 mL/min and run time of 75 min. SDS-PAGE analysis was also performed on 4–20% Tris-glycine gels, with a protein load of 2 µg of each sample per lane under reducing and non-reducing conditions. Gels were stained with Coomassie Blue or Silver stained, following conventional procedures.
Short-term accelerated stability studies of the Na-GST-1 protein
Accelerated stability is a useful tool to shorten a process development time and to gain some insights in the shelf life of biologics. A 3-d short-term study was designed to evaluate the protein’s ability to maintain its properties if exposed to different temperatures in advance of further manufacturing situations where hold periods could be required prior to formulation. To this end, the protein was stored at 3 different temperatures: 2–8 °C, 25 °C, and 37 °C, for a maximum time period of 30 d and then characterized. At time zero, 1 mL aliquots of the protein were placed at the specified temperatures and analyzed after 3, 7, 15, and 30 d for color and appearance, pH measurement, and protein content by the absorbance at 280 nm, as described above. Identity, and purity by size exclusion HPLC, as well as identity by SDS-PAGE followed by Coomassie Blue staining and silver staining were also performed following the same procedures as described above.
Preparation of the Na-GST-1 formulation
Similarly to the formulation already in use in Phase I studies, Alhydrogel® was selected as Na-GST-1 adjuvant. The protein stock solution and the adjuvant were combined such that the final protein concentration was 0.1 mg/mL and the final aluminum concentration was 0.8 mg/mL. The desired concentrations were achieved by adding 10 mM Imidazole buffer, 10% sucrose pH of 7.4, to the protein and alum stock solutions. The formulations were mixed at room temperature for a minimum of one hour to ensure adsorption. The degree of antigen adsorption was assessed by centrifuging the vaccine at 13 000 rpm for 2 min, and evaluating the concentration of the antigen in the pellet and supernatant, respectively, by SDS-PAGE and by OPA assay as described below.
Stability testing of the formulated protein
Similar to the recombinant protein, formulations are subject to changes over time, making the prediction of the product shelf life difficult. Alteration of pharmaceutical properties caused by different degradation routes must be determined on a case-by-case basis for each protein. To this end the stability of the Na-GST-1 formulation was monitored at day 3, 5, 7, 15, and 30, and then every month for a total period of 3 mo. Color and appearance, pH measurement, protein content by OPA and identity by SDS-PAGE followed by Coomassie Blue staining were evaluated at each time point. While the color and appearance and pH were evaluated as previously described, the OPA assay was used to quantify the total protein content of the vaccine, after formulation of the Na-GST-1 with Alhydrogel®. The OPA assay was performed using Victor 3 multilabel counter with the Wallac 1420 software (PerkinElmer). A series of dilutions of non-formulated Na-GST-1 protein ranging from 0 to 250 µg/mL were made in PBS pH 7.4 as the diluent. Over this range the fluorescence intensity increased in a linear fashion with the concentration of the protein. 40 µl of each test sample and standard was dispensed in a 96 wells microplate to which 200 µL of OPA reagent solution was added. Samples were allowed to incubate for approximately 2 min with moderate shaking at room temperature and the fluorescence signal was measured using a Victor 3 multilabel counter plate reader with a 355 nm, 40 bandwidth, excitation filter and a 460 nm, 40 nm bandwidth, emission filter. Using the Wallac 1420 software the least means squared linear equation describing the standard curve was generated. The same assay was used to evaluate the Percentage Protein Adsorbed to Alhydrogel®.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Hotez P. Enlarging the “Audacious Goal”: elimination of the world’s high prevalence neglected tropical diseases. Vaccine. 2011;29(Suppl 4):D104–10. doi: 10.1016/j.vaccine.2011.06.024. [DOI] [PubMed] [Google Scholar]
- 2.Hotez PJ, Diemert D, Bacon KM, Beaumier C, Bethony JM, Bottazzi ME, Brooker S, Couto AR, Freire MdaS, Homma A, et al. The Human Hookworm Vaccine. Vaccine. 2013;31(Suppl 2):B227–32. doi: 10.1016/j.vaccine.2012.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goud GN, Deumic V, Gupta R, Brelsford J, Zhan B, Gillespie P, Plieskatt JL, Tsao EI, Hotez PJ, Bottazzi ME. Expression, purification, and molecular analysis of the Necator americanus glutathione S-transferase 1 (Na-GST-1): a production process developed for a lead candidate recombinant hookworm vaccine antigen. Protein Expr Purif. 2012;83:145–51. doi: 10.1016/j.pep.2012.03.013. [DOI] [PubMed] [Google Scholar]
- 4.Hotez PJ, Diemert D, Bacon KM, Beaumier C, Bethony JM, Bottazzi ME, Brooker S, Couto AR, Freire MdaS, Homma A, et al. The Human Hookworm Vaccine. Vaccine. 2013;31(Suppl 2):B227–32. doi: 10.1016/j.vaccine.2012.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kassebaum NJ, Jasrasaria R, Naghavi M, Wulf SK, Johns N, Lozano R, et al. A systematic analysis of global anemia burden from 1990 to 2010. Blood 2014;http:// [DOI] [PMC free article] [PubMed]
- 6.Murray CJ, Vos T, Lozano R, Naghavi M, Flaxman AD, Michaud C, Ezzati M, Shibuya K, Salomon JA, Abdalla S, et al. Disability-adjusted life years (DALYs) for 291 diseases and injuries in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2197–223. doi: 10.1016/S0140-6736(12)61689-4. [DOI] [PubMed] [Google Scholar]
- 7.Vos T, Flaxman AD, Naghavi M, Lozano R, Michaud C, Ezzati M, Shibuya K, Salomon JA, Abdalla S, Aboyans V, et al. Years lived with disability (YLDs) for 1160 sequelae of 289 diseases and injuries 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012;380:2163–96. doi: 10.1016/S0140-6736(12)61729-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Asojo OA, Homma K, Sedlacek M, Ngamelue M, Goud GN, Zhan B, Deumic V, Asojo O, Hotez PJ. X-ray structures of Na-GST-1 and Na-GST-2 two glutathione S-transferase from the human hookworm Necator americanus. BMC Struct Biol. 2007;7:42. doi: 10.1186/1472-6807-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhan B, Liu S, Perally S, Xue J, Fujiwara R, Brophy P, Xiao S, Liu Y, Feng J, Williamson A, et al. Biochemical characterization and vaccine potential of a heme-binding glutathione transferase from the adult hookworm Ancylostoma caninum. Infect Immun. 2005;73:6903–11. doi: 10.1128/IAI.73.10.6903-6911.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xiao S, Zhan B, Xue J, Goud GN, Loukas A, Liu Y, Williamson A, Liu S, Deumic V, Hotez P. The evaluation of recombinant hookworm antigens as vaccines in hamsters (Mesocricetus auratus) challenged with human hookworm, Necator americanus. Exp Parasitol. 2008;118:32–40. doi: 10.1016/j.exppara.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 11.Zhan B, Perally S, Brophy PM, Xue J, Goud G, Liu S, Deumic V, de Oliveira LM, Bethony J, Bottazzi ME, et al. Molecular cloning, biochemical characterization, and partial protective immunity of the heme-binding glutathione S-transferases from the human hookworm Necator americanus. Infect Immun. 2010;78:1552–63. doi: 10.1128/IAI.00848-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Plieskatt JL, Rezende WC, Olsen CM, Trefethen JM, Joshi SB, Middaugh CR, Hotez PJ, Bottazzi ME. Advances in vaccines against neglected tropical diseases: enhancing physical stability of a recombinant hookworm vaccine through biophysical and formulation studies. Hum Vaccin Immunother. 2012;8:765–76. doi: 10.4161/hv.19726. [DOI] [PubMed] [Google Scholar]
- 13.WHO. Deworming for health and development. Report of the third global meeting of the partners for parasite control. World Health Organization, Geneva, Switzerland. 2005.
- 14.Hotez P. A handful of ‘antipoverty’ vaccines exist for neglected diseases, but the world’s poorest billion people need more. Health Aff (Millwood) 2011;30:1080–7. doi: 10.1377/hlthaff.2011.0317. [DOI] [PubMed] [Google Scholar]
- 15.Celik E, Calik P, Oliver SG. Fed-batch methanol feeding strategy for recombinant protein production by Pichia pastoris in the presence of co-substrate sorbitol. Yeast. 2009;26:473–84. doi: 10.1002/yea.1679. [DOI] [PubMed] [Google Scholar]
- 16.Jungo C, Schenk J, Pasquier M, Marison IW, von Stockar U. A quantitative analysis of the benefits of mixed feeds of sorbitol and methanol for the production of recombinant avidin with Pichia pastoris. J Biotechnol. 2007;131:57–66. doi: 10.1016/j.jbiotec.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 17.Ramón R, Ferrer P, Valero F. Sorbitol co-feeding reduces metabolic burden caused by the overexpression of a Rhizopus oryzae lipase in Pichia pastoris. J Biotechnol. 2007;130:39–46. doi: 10.1016/j.jbiotec.2007.02.025. [DOI] [PubMed] [Google Scholar]
- 18.Shen Y, Gu L, Zhang J, Chen J, Du G. [Effects of mixed carbon sources on glucose oxidase production by recombinant Pichia pastoris] Sheng Wu Gong Cheng Xue Bao. 2013;29:927–36. [PubMed] [Google Scholar]
- 19.Wang Z, Wang Y, Zhang D, Li J, Hua Z, Du G, Chen J. Enhancement of cell viability and alkaline polygalacturonate lyase production by sorbitol co-feeding with methanol in Pichia pastoris fermentation. Bioresour Technol. 2010;101:1318–23. doi: 10.1016/j.biortech.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 20.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat Protoc. 2006;1:2856–60. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]