

Abstract
The ability to preserve non-covalent, macromolecular assemblies intact in the gas-phase has paved the way for mass spectrometry to characterize ions of increasing size and become a powerful tool in the field of structural biology. Tandem mass spectrometry experiments have the potential to expand the capabilities of this technique through the gas-phase dissociation of macromolecular complexes, but collisions with small gas atoms currently provide very limited fragmentation. One alternative for dissociating large ions is to collide them into a surface, a more massive target. Here, we demonstrate the ability and benefit of fragmenting large protein complexes and inorganic salt clusters by surface-induced dissociation (SID). SID provides more extensive fragmentation of these systems and shows promise as an activation method for ions of increasing size.
Introduction
Over the past two to three decades, mass spectrometry has expanded significantly, from its early use as a technique for measuring the isotopes of elements and analyzing volatile compounds, to a technique that is now routinely used to study non-volatile molecules and large macromolecular complexes. Increasingly, mass spectrometry and ion mobility-mass spectrometry are described as structural biology tools. Mass spectrometry has recently provided insights on post-translational modifications [1], mono- and polydisperse subunit stoichiometry [2], subunit organization [3,4], as well as non-covalent protein-ligand binding sites [5]. In addition to revealing structural information, mass spectrometry can be used to monitor dynamic processes, such as protein complex assembly [6], protein-substrate and protein-protein interactions [7,8], and substrate-specific conformational changes [9].
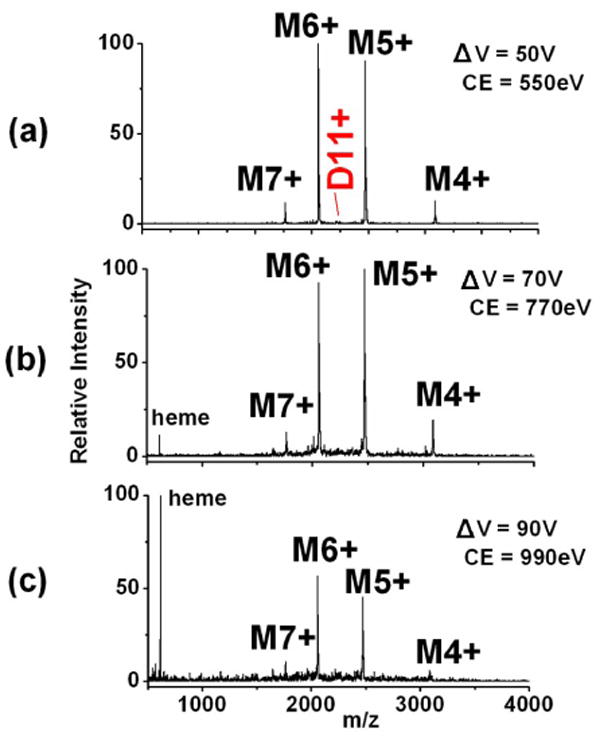
One of the limitations of current technology, however, is the fact that commercial instrumentation is still hampered by the amount of dissociation that can be induced from large biomolecular complexes. Often, MS has to be combined with many solution based experiments (H/D exchange plus digestion, chemical cross-linking plus digestion, limited proteolysis, solution disruption by changes in ionic strength) because the MS instruments commercially available do not provide extensive dissociation of these massive complexes. A typical dissociation result that is achieved is ejection of a monomer subunit as illustrated here (Fig. 1) for a small heat shock protein (sHSP) dodecamer, consisting of 12 subunits each weighing 16.9 kDa [8,10,11]. The 16.9 kDa monomer typically carries away a large percentage of the charge of the original complex with the remainder of the charge on the 11-mer that weighs 186 kDa. Investigators have concluded that this behavior, which has been observed by several research groups for many different non-covalent protein complexes, happens because the low-energy collisions with a gas initiate unfolding of one of the protein subunits [12-14]. As this subunit unfolds, it gains surface area allowing it to accept more charge. Eventually, when the unfolding protein and the remainder of the complex have about the same surface area and charge [15], the two can separate leading to a result such as the one shown in Figure 1. While this type of result does provide structural information, such as confirmation of the oligomeric state of the complex and information on which protein subunit is most easily ejected from the complex, it does not provide as much structural information as desired on the complex substructure. This complex, for example, is known from X-ray crystallography to exist as two stacked rings, each with 6 proteins, and is thought to be in equilibrium in solution with a dimer [16]. The CID spectrum however, reflects neither the presence of dimeric building blocks, nor the stacked ring substructure.
Figure 1.
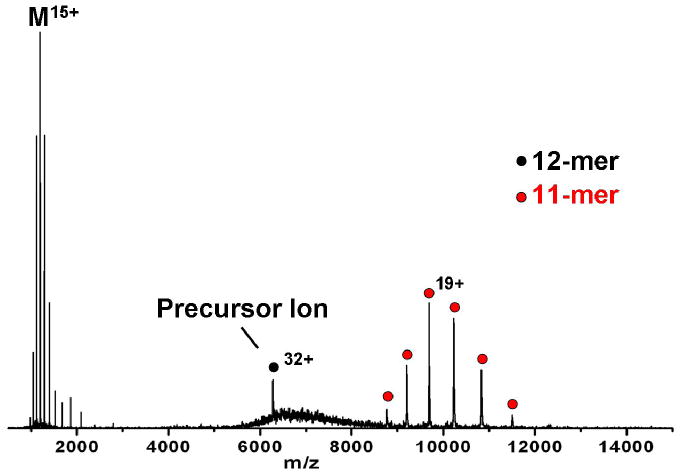
Collision-induced dissociation of dodecameric TaHSP16.9. At a collision energy of 3520 eV, the 32+ dodecamer produces mainly monomer with an average of 15 charges and 11-mer with an average of 19 charges. Collision energy is listed as the product of voltage difference between the source hexapole and the gas-filled collision cell (110 V) and the 32+ precursor ion charge state.
The lack of extensive dissociation is likely the result of the limited amount of energy deposited into the large complex with each collision with the target gas, typically a low mass atomic target such as Ar. The extent of dissociation or even the extent of desolvation of a complex can be increased by using a more massive target gas such as Xe, suggesting that the efficiency of energy transfer is related to the mass of the target as expected [17-19]. It is appealing to be able to directly dissociate large complexes much more extensively than is now possible with collision-induced dissociation (CID) in commercial mass spectrometers. Other activation methods have been attempted, such as electron capture dissociation (ECD) or blackbody infrared radiative dissociation (BIRD) but they have not shown extensive breakup of large complexes [12,20], although ECD has shown charge separation proportional to molecular weight, indicating that the ejected monomer remains mostly folded [20]. An alternative to methods previously studied is to collide the large complexes into a much more massive target, such as a surface. Surface-induced dissociation (SID) provides efficient kinetic to internal energy conversion, which might prove advantageous for fragmenting ions that approach or exceed the MDa size regime. The energy transfer efficiency in SID has been reported to vary with surface properties such as stiffness and mass of the terminal groups, indicating these factors likely contribute to the effective mass of the surface as a collision partner [21,22]. Energy transfer efficiency for fluorinated self-assembled monolayers (FSAMs) on gold has been reported to be between 20-28% [21,23]. The influence of ion size on the SID energy transfer efficiency however, has been debated in the literature and is not entirely understood [24-26], especially for systems as massive as non-covalent proteins. Nonetheless, collision with a surface should still be a more efficient process than collision with a gas for which efficiencies less than 0.01% per collision have been reported for large protein complexes [9]. SID of a few protein complexes has suggested that alternative ion activation methods may provide a way to extract meaningful structural data from the gas-phase fragmentation of protein complexes [27-30].
One way to improve our understanding of the dissociation of large, non-covalent molecules may be through the study of other, comparatively simple, and well characterized macromolecular systems, such as inorganic salt clusters. Clusters have received significant experimental and theoretical attention during the past two decades. They are an important group of structures, as they span the chemical and physical gap between discrete atoms or molecules and solid materials, and provide clues to the transitions between the condensed and gaseous states of matter [31]. Furthermore, these systems allow an understanding of several physical/chemical processes including phase transition phenomena, crystal growth, chemical catalysis, and thin film formation [32,33]. In contrast to proteins and their non-covalent complexes, clusters are simple structural moieties with minimum structural complexity. They are structures composed of small numbers of heavy atoms, whereas protein complexes are structures composed of large numbers of light atoms. These represent two extremes, in the sense that, for similar masses of a protein and a salt cluster system, the protein would always possess a higher number of internal degrees of freedom. Their simplicity makes salt clusters ideal for comparing fundamental differences between SID and CID for large molecules. Among the different types of cluster systems, alkali halide clusters have been extensively studied by mass spectrometry, especially singly charged positive ion clusters. The general formula for theses clusters can be given as (MX)nM+ [34-37], where M and X represent the cation and anion of the molecule. The alkali halides are known to generate clusters with extraordinary stability, the most prevalent of which correspond to stable cubic-like crystal structures (magic numbers) [38]. Previous CID studies of these clusters revealed the formation of mass stripped products due to the removal of neutral MX [39-43]. An important question that can be addressed by studying these systems is the influence of cluster size on fragmentation, as CsI has been shown to form clusters consisting of up to 350 CsI molecules [44]. This provides a framework for contrasting different ion activation methods as a function of ion size, and can help to define the advantages of dissociating other large, non-covalent systems via surface collisions.
This article describes recent advances in the application of surface-induced dissociation as a complementary tool for the direct dissociation of large complexes. Several examples are given, including protein complexes with different oligomeric substructures and singly and doubly charged CsI clusters. We also show that the extent of fragmentation increases with increasing collision energy, suggesting that SID of large complexes generally follows the typical expectations of kinetic theory of mass spectrometry [17,45].
Experimental
Materials and methods
Materials
Cesium iodide, equine cytochrome c, human transthyretin (prealbumin), jack bean concanavalin A, and human hemoglobin were purchased from Sigma-Aldrich Co. (St. Louis, MO). Recombinant human C-reactive protein, expressed in E.Coli and serum amyloid P component from human serum were purchased from EMD Chemicals, Calbiochem product brand (Gibbstown, NJ). Small heat shock proteins Ta16.9 (wheat), Ps18.1 (garden pea) and AtHSP18.5 (Arabidopsis) were expressed in E.Coli, and were generously provided by Prof. Elizabeth Vierling and coworkers at the University of Arizona. Ammonium acetate was purchased from Spectrum Chemicals & Laboratory Products (Gardena, CA). Micro Bio-spin columns for buffer exchange were purchased from Bio-Rad (Hercules, CA). Gold surfaces were purchased from Evaporated Metal Films (Ithaca, NY) and 2-(perfluoroctyl)-ethanethiol was synthesized by the Chemical Synthesis Facility at the University of Arizona.
Mass Spectrometry
For the CsI salt cluster experiments, samples were sprayed out of a home-built nano-ESI source. The concentrations of the salt cluster samples were typically in the range of 20-50 mg/ml and sprayed out of 50:50 MeOH:H2O. For the non-covalent protein complexes, samples were buffer exchanged into 200 mM ammonium acetate (pH 7) using micro-centrifuge columns, or in the case of lyophilized samples, dissolved directly into ammonium acetate. The samples were loaded into glass capillaries pulled in-house (Sutter Instruments, P-97 micro-pipette puller) to a final tip diameter of 1-5 μm. A platinum wire was inserted into the glass capillary and a voltage of 1.5 to 2.0 kV was applied to electrospray the ions. The cone voltage was varied between 50-200 V until optimum ion transmission was obtained. No heating or desolvation gas was used with nano-ESI.
All experiments were conducted on a modified Q-ToF II mass spectrometer (Micromass/Waters, Manchester) that has been described previously [28]. Briefly, an in-line SID device was installed in between the quadrupole mass analyzer and hexapole gas collision cell to allow direct comparisons of CID and SID on the same instrument. The SID device, which includes a surface holder, ion beam deflectors, and focusing lenses, is approximately 45 mm in length, and was inserted by removing the transport hexapole following the collision cell. The collision cell was moved closer to the entrance of the ToF to make room for the SID device. For experiments involving small heat shock proteins, a shortened, 13 cm collision cell provided by Waters Corporation replaced the traditional 18.5 cm collision cell to accommodate the SID device without removing the transport hexapole. This allowed the conventional ion optics to remain in place, and facilitated the use of higher gas pressures within the collision cell while limiting detrimental pressure increases in the adjacent ToF region. While both collision cells could be used to transmit product ions over the entire m/z range, transmission efficiency of high m/z product ions generated by either CID or SID was enhanced with the 13 cm collision cell. This is presumably due to the use of higher gas pressures necessary for collisional focusing of high m/z product ions. The instrument was operated in one of two modes; one that allows ion transmission through the SID apparatus without hitting the surface for single-stage MS and CID experiments, or alternatively, surface collision mode in which ions are deflected into the surface for SID experiments. This allowed direct comparison between the two ion activation methods under identical instrument conditions and similar observation time frames.
In CID mode, the experiments were performed using argon (Ar) as the collision target. The pressure was measured in the quadrupole analyzer chamber and typically maintained between 1-6 × 10-4 mbar (the actual pressure in the collision cell is different from the quadrupole chamber pressure because there is no pressure gauge connected directly to the collision cell). For SID experiments, FSAM surfaces were used as the collision target. The FSAM surfaces were prepared on 18 mm × 12 mm glass surfaces, coated with a 10-Å layer of titanium followed by a 1000-Å layer of gold (Evaporated Metal films Corp., Ithaca, NY). A detailed experimental procedure for the surface preparation is given elsewhere [46,47]. The collision cell was operated either without Ar gas with the collision cell serving solely as a hexapole ion guide, or with Ar gas for enhanced collisional focusing of high m/z product ions. The collision voltage for the CID mode was controlled by adjusting the DC offset of the source hexapole between 0 and 200 V, relative to the collision cell (grounded). For SID, the collision voltage was varied between 0 and 190 V by setting the DC offset (10-200 V) of the source hexapole with respect to the surface voltage (∼10-20 V). The actual collision voltage was calculated by subtracting the difference between these two voltages. All collision energies are listed as the product of the voltage difference times the precursor ion charge state.
Mass spectra were acquired under conditions optimized for the transmission of salt clusters and non-covalent protein complexes. Pressure in the source region of the instrument was raised by closing off the vacuum line to the rotary pump as described elsewhere [44]. Ions were selected for tandem mass spectrometry experiments with an extended mass range quadrupole and accelerated into either the surface or the gas-filled collision cell.
Results and Discussion
An extended m/z range QToF was used to perform the experiments described. This general type of instrument has been used for much of the work reported on non-covalent protein and protein-ligand complexes in the literature. The instrument makes use of nanoelectrospray ionization which allows complexes to be sprayed intact into the gas phase. The extended m/z range of the quad allows selection of very large complexes (e.g., ribosomes at 2.3 MDa have a m/z in excess of 20,000 [48-50]).
CsI Clusters
CsI clusters were used to compare CID and SID. From the experimental point of view, clusters are relatively easy to form, and their ionic bonding character has allowed the development of simple models, making them ideal systems for comparing the two activation methods. Furthermore, CsI provides a simpler system than protein complexes because the major losses expected all involve combination of just two atoms. Figure 2 shows surface-induced and collision-induced dissociation spectra of (CsI)13Cs+ at an SID collision energy of 50 eV (a) and at CID collision energies of 30,40, 60, and 80 eV (b). There may be some overlapping doubly-charged ion selected but if so, it is of low relative abundance. The dominant fragments detected in CID and SID correspond to losses of neutral CsI. SID shows more extensive dissociation than does CID, as expected for a more massive target. Another significant difference between the two ion activation methods is that with SID, there is an enhancement of the (CsI)2Cs+ cluster formation. With CID, this fragmentation channel is limited or completely inaccessible. A similar observation was reported for small CsI cluster systems in a sector instrument with CID as the method of activation [51]. In contrast, SID allows the formation of (CsI)2Cs+ via loss of multiple neutral CsI units. This loss is probably associated with higher energy onsets, that only SID could provide under the given instrument operating conditions. Despites these minor differences, CID and SID of small, singly-charged CsI clusters are largely similar with both activation methods providing extensive dissociation.
Figure 2.
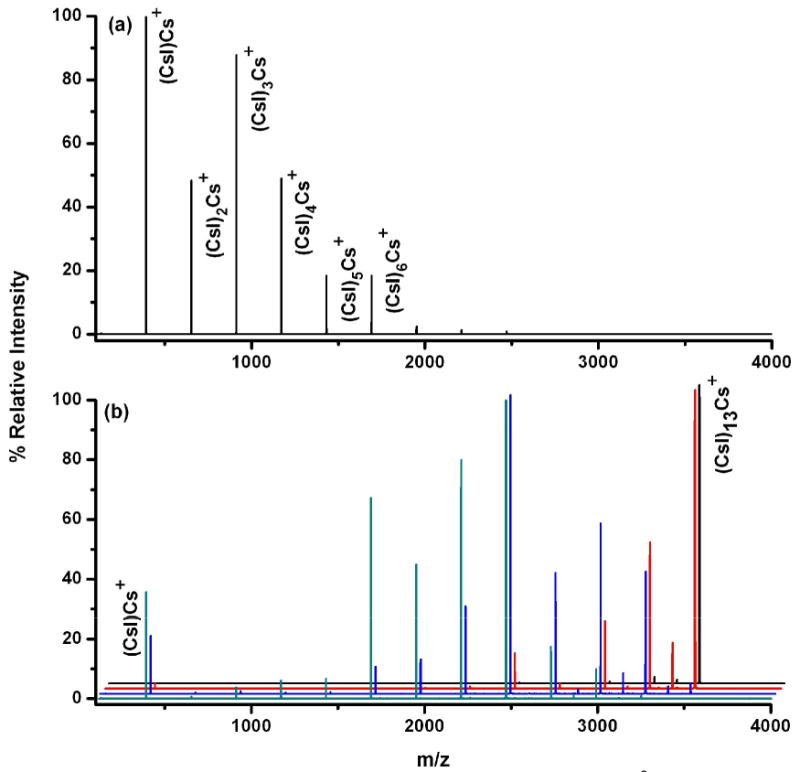
SID (a) and CID (b) spectra of (CsI)n=13Cs+ / (CsI)q=26Cs2+ in a Q-TOF mass spectrometer. The laboratory collision voltages of 30, 40, 60 and 80 V were employed for CID (from back to front of the waterfall plot) at analyzer pressure of 8 × 10-5 mbar. The SID spectrum was collected at collision voltage of 50 V.
Figure 3 shows collision-induced and surface-induced dissociation spectra of (CsI)43Cs2++ at several collision energies. (CsI)43Cs2++ is a doubly-charged cluster with nominal mass of 11,437 Da. This cluster carries significantly higher mass per unit charge, compared to the (CsI)13Cs+ cluster system. As shown in Figure 3 the CID (3a-c) and SID (3d-f) spectra are noticeably different. The CID spectra show a dominant fragmentation pathway by which neutral (CsI)n units are lost (Scheme 1) to produce doubly-charged fragments from the doubly-charged precursor. SID, however, shows clear evidence of charge separation, producing dominantly singly-charged fragments from a doubly charged precursor ion in addition to the loss of neutral (CsI)n units (Figure 3d-f). Although CID shows neutral CsI loss as the most dominant fragmentation pathway, a second fragmentation pathway involving loss of singly-charged (CsI)Cs+ was also evident, but to a very low extent. Neutral loss and fission are two competing processes, where the probability of each is dependent upon energy availability and cluster size. With CID, the neutral loss process happens to be the most dominant and is driven by the size of the cluster and the limited access to high energy pathways. This phenomenon can be explained with the help of the two-body interaction theory [52,53]. The formation of a doubly charged cluster can be considered as a product of a fusion process (fusion of two singly charged clusters). As one would expect there is a long-range repulsive force between two charged clusters. However, as the two fragments are brought within the chemical bonding distance, the cluster should transform from unstable to a kinetically stable (or metastable) cluster [54]. Martin calculated lowest possible energy configurations for doubly-charged NaI clusters and showed a clear trend in which cluster stability increases with cluster size. In other words, the energy barrier preventing the most stable configuration from undergoing Coulombic dissociation increases as the size of the cluster increases [54]. Upon activation, if the added excess energy is sufficient to overcome this energy barrier, the cluster would undergo a fission process producing charge separated products. With (CsI)43Cs2++, multiple-collision CID does not supply sufficient energy to overcome the barrier for the fission process to occur, even at the highest attainable collision energies. However it still provides enough energy to eject neutral (CsI)n units generating doubly-charged mass-stripped product clusters (the doubly-charged products still have large enough masses to be stable product clusters). Previous CID studies published by Cooks and co-workers [55] on doubly-charged NaI cluster systems have also revealed a similar fragmentation pattern, where high mass NaI clusters predominantly dissociated by loss of neutral NaI units and low mass clusters dissociated by charge splitting. For these experiments we propose that SID deposits higher internal energy into the projectile ion, enabling it to overcome the energy barrier for the fission process. This claim is supported by breakdown curves for small CsI clusters, in which it is evident that the dissociation onset for any given fragment ion occurs at a lower laboratory collision energy by SID relative to CID. This explains the observation of abundant charge separated (split) products in the SID spectra. Another striking effect observed in the SID spectra was the observation of ion intensity enhancement of certain fragment ions. These fragments are found to be “magic number” clusters [35,56-60], corresponding to symmetrically stable cubic crystal structures with lattices of 3×3×3, 3×3×5, 5×5×5, etc (Scheme 1). In the event of collision on the surface, clusters relieve the stress of impact by cleaving along the lowest energy plane(s), leading to the formation of stable cubic crystal structures. Previous impact-induced dissociation studies of similar alkali-halide systems with graphite and silicon surfaces by Whetten and co-workers [61-63] also revealed similar fragmentation behavior. With our Q-TOF SID configuration, we were able to fragment CsI clusters larger than those previously reported in the literature, and compare their dissociation by both CID and SID. Overall, CID could readily access the low energy neutral loss dissociation pathway to produce mass-stripped doubly-charged cluster fragments but not the high energy fission process. In contrast SID, could access both competing pathways, offering more extensive fragmentation of the precursor ion cluster.
Figure 3.
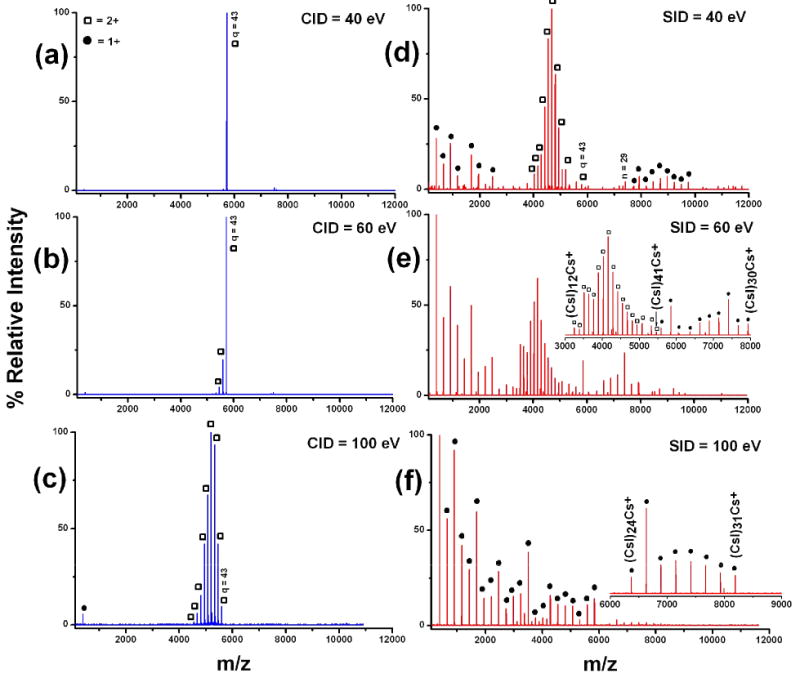
Comparison of CID (left panel) and SID (right panel) spectra of the doubly charged (CsI)43Cs2++ cluster. Inset in SID collision energy 60 eV, shows two distinct charge distributions, doubly and singly-charged. SID at collision energy of 100 eV, shows predominantly singly charged fragments.
● = Singly charged cesium iodide cluster fragments
□ = Doubly charged cesium iodide cluster fragments
Scheme 1.
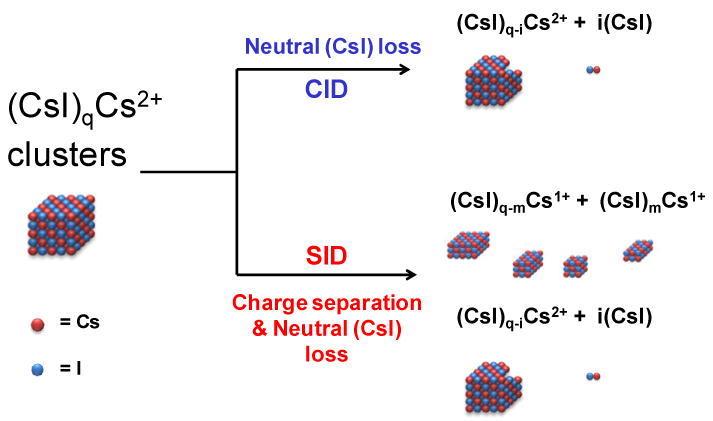
Dissociation channels observed for the fragmentation of (CsI)qCs2+ clusters. CID leads solely to consecutive losses of neutral CsI, while access to an additional charge separation pathway is provided by SID.
Protein Complexes
We previously reported the CID and SID spectra of cytochrome c dimers [30]. Other investigators had shown that CID of the 11+ charge state of cytochrome c leads to dominant 8+ and 3+ monomers [13,64,65]. As shown in Figure 4, SID over a broad range of collision energies instead leads to formation of 5+ and 6+ monomers, the charge states expected if 11 charges are split between two equivalent subunits. The different charge distributions detected for CID and SID are consistent with an unfolding mechanism for CID (monomer unfolding leads to charge enrichment of one of the subunits) and break-up of the complex by SID with greatly reduced unfolding, leading to a monomer with approximately the number of charges that would be obtained if the complex simply dissociates and shares the charge equally among subunits. This has been observed for several complexes investigated, including the 25+ pentamer of serum amyloid P component (SAP) shown in Figure 5; when activated by CID this protein produces monomer with an average charge of 10.3 and tetramer with an average charge of 14.4. When activated by SID however, only monomer is observed with an average charge state of 5.3, or approximately 1/5 of the precursor ion charge. This charge state distribution is consistent with dissociation of the 25+ pentamer into 5 charge-equivalent monomers, allowing for slight variations in the charge uniformity of subunits within the pentameric precursor. Dimeric product ions are also observed in the SID spectrum. Again, the charge states of these products are proportional to their molecular weight, retaining approximately 2/5 of the precursor ion charge. In addition, both the CID and SID spectra show losses corresponding to the sialic acid group of the nonasaccharide, attached to Asn-32 of each subunit.
Figure 4.
Energy-resolved SID of the 11+ dimer of cytochrome c. The dimer dissociates into monomers with an approximately equal partitioning of the precursor ion charge, regardless of the SID collision energy. (Figure modified from supplemental figure in Ref [22]).
Figure 5.

MS/MS of the serum amyloid P component (SAP) pentamer. (a) CID of the 25+ pentamer at 2500 eV produces monomer and tetramer fragments, with the monomer retaining approximately 40% of the charge. (b) SID of the same precursor ion at 1750 eV produces monomers and dimers with 1/5 and 2/5 of the precursor charge, respectively.
Interestingly, CID of SAP shows unusual covalent fragmentation of the protein backbone at the same collision energy employed to remove a single, non-covalently bound subunit. Around 1500 m/z several protein backbone fragments corresponding to y403+, y302+, and y282+ are observed. While we have previously observed covalent fragment ions at sufficiently high collision energies (see Figure 6 and discussion thereof), these fragments have always appeared to correspond to further fragmentation of the ejected monomer after expulsion from the oligomeric protein. In this case however, covalent cleavage of the protein appears to be a competitive process to non-covalent subunit ejection. This is evident from the observation of complementary fragment ions between 5300-6600 m/z that correspond to [pentamer-y40]22+, [pentamer-(2y40)]19+, and [pentamer-y30]23+. This process is summarized in Scheme 2, and is an unusual example of backbone fragmentation from oligomeric proteins by CID, while preserving some of the non-covalent interactions between subunits as well as the covalently bound saccharide. Table 1 lists each of the complementary covalent fragment ions observed by CID and the site of backbone cleavage. Each covalent bond cleavage occurs at a site known to be enhanced in peptides with a mobile or partially mobile proton, such as N-terminal to proline or glycine [66]. Equally noteworthy is that SID at 1750 eV reveals dissociation of the pentamer into monomers and dimers, but shows no evidence of covalent backbone fragmentation. This may signify that covalent cleavage directly from the pentamer is facilitated by charge transfer to the unfolding subunit during CID. The charge-enriched environment created as a SAP subunit unfolds during CID leads to a situation in which the monomer contains more protons (∼10 judging from the charge state of the dissociating monomer) than Arg residues (9), the most basic, and thus most tightly proton sequestering residue. Selective cleavages N-terminal to Pro have been extensively documented in the peptide fragmentation literature [66-69], especially in cases where the number of protons exceeds the number of Arg, and are free to initiate charge-directed cleavage of the protein backbone [66]. For the SAP pentamer, the fact that this cleavage appears to compete with removal of a non-covalent subunit may be useful in elucidating mechanistic aspects of the unfolding and charge transfer process that occurs during CID, although it is not currently understood why this is observed for CID of SAP, but not other oligomeric protein systems.
Figure 6.
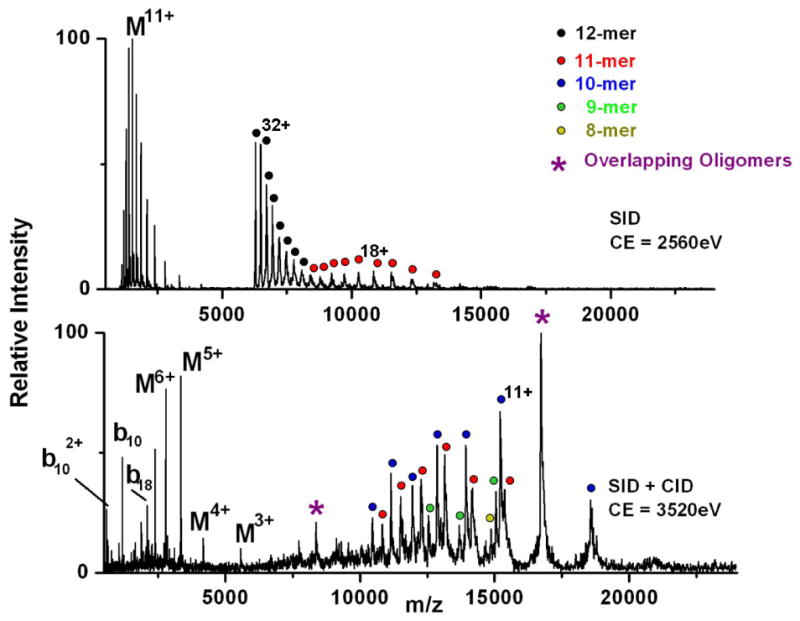
MS/MS of the 32+ dodecamer of TaHSP16.9. SID at a collision energy of 2560 eV (top) produces several charge-stripped dodecameric ions along with monomer and undecamer fragments. SID of TaHSP16.9 at a collision energy of 3520 eV with Ar gas also present in the collision cell (bottom) produces more extensive fragmentation to give octamers through undecamers, in addition to covalent fragmentation of the monomer population along the protein backbone.
Scheme 2.
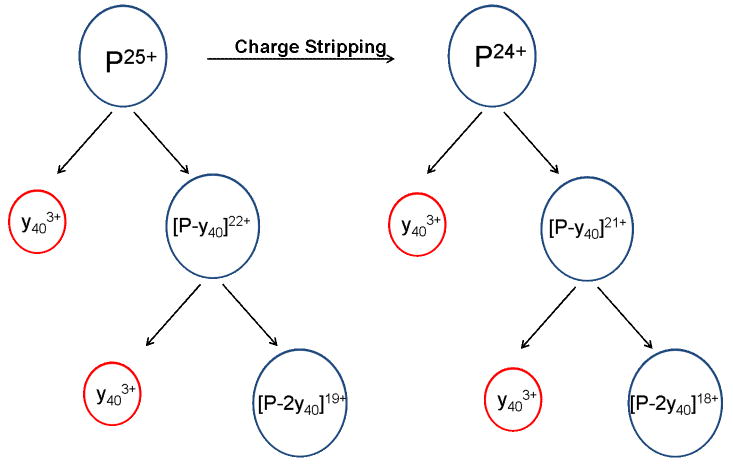
Representation of covalent backbone fragmentation directly from the pentamer of serum amyloid P component. The y403+ ions are complemented by ions corresponding to the loss of y403+ from the 25+ pentamer, and are in competition with the ejection of an entire non-covalently bound subunit.
Table 1.
Fragment ions observed in the CID spectrum of SAP corresponding to covalent backbone fragmentation directly from the SAP pentamer. Δm/z corresponds to the difference between the theoretical m/z as determined from the amino acid sequence and the experimentally measured m/z. The measured values for the intact pentamer and the fragment ions for which at least a portion of all 5 subunits are still bound (i.e. Pentamer-covalents) are heavy by ∼2-2.5 m/z due to the presence of solvent adducts.
| Fragment Ions Observed by CID | Charge State | Theoretical m/z | Measured m/z | Δ m/z | Cleavage: N-term./C-term. Residue | Complementary Fragment | Complementary Charge State |
|---|---|---|---|---|---|---|---|
| Pentamer (precursor) | 25+ | 5093.48 | 5095.95 | -2.47 | ---- | ---- | ---- |
| Pentamer | 24+ | 5305.67 | 5308.14 | -2.47 | ---- | ---- | ---- |
| y40-NH3 | 3+ | 1517.45 | 1517.38 | 0.07 | Leu/Pro | ---- | ---- |
| y40 | 3+ | 1523.13 | 1523.40 | -0.27 | Leu/Pro | Pentamer-y40 | 22+ |
| y28 | 2+ | 1648.49 | 1648.46 | 0.03 | Thr/Pro | ---- | ---- |
| y30 | 2+ | 1727.56 | 1727.73 | -0.17 | Glu/Gly | Penatmer-y30 | 23+ |
| Pentamer-y40 | 22+ | 5580.35 | 5582.71 | -2.36 | Leu/Pro | y40 | 3+ |
| Pentamer-y40 | 21+ | 5846.03 | 5848.62 | -2.59 | Leu/Pro | y40 | 3+ |
| Pentamer-(2y40) | 19+ | 6220.96 | 6223.03 | -2.07 | Leu/Pro | 2y40 | 3+ |
| Pentamer-(2y40) | 18+ | 6566.52 | 6568.90 | -2.38 | Leu/Pro | 2y40 | 3+ |
| Penatmer-y30 | 23+ | 5386.17 | 5387.14 | -0.97 | Glu/Gly | y30 | 2+ |
Table 2 shows the average number of charges retained by monomers produced by CID and SID from several mass-selected oligomers [27]. The average charge state is higher in all cases for CID than for SID, consistent with more unfolding, allowing higher charging by CID. The monomer ejected during CID, takes away close to 50% of the charge from the original precursor in every case [15]. The percentage of the precursor ion charge retained by SID-generated monomers however, approaches the statistical mass percent of each subunit within the intact complex. For example, monomers produced from pentameric C-reactive protein constitute 20% of the overall mass of the intact complex, and SID yields monomers that retain 21% of the charge. Similarly, SID of tetrameric transthyretin (TTR) yields monomers that retain 29% of the charge compared to 25% composition by mass. It is possible that the minor discrepancies between the charge and mass percent of SID-generated monomers is due to the presence of low abundance charge asymmetric products in the SID spectra of each complex. Energy-resolved studies of TTR have shown that at low collision energies, the SID products are similar in mass and charge partitioning to those produced by CID [27]. As collision energy increases, the monomer distribution becomes more charge symmetric with respect to mass, though some of the asymmetric pathways are still observed to a minor extent. Consequently, the rapid deposition of higher internal energy may serve to better access dissociation pathways that lead to charge symmetric dissociation. To date, there is little data available regarding the conformation of SID-generated product ions, but the evidence of charge-symmetric monomers implies a large degree of conformational similarity among the dissociating subunits. It is presumed here that the monomers remain mostly folded upon dissociating from one another, leading to their enhanced charge symmetry. It is not clear whether unfolding may occur after dissociation from the intact complex, but the dissociating subunits would not be capable of charge transfer after separating from one another. Clearly additional work, such as the combination of SID with ion mobility, is needed to test the validity of this presumption.
Table 2.
Charge state statistics for monomer product ions ejected during collision- and surface-induced dissociation of the listed charge state of the precursor protein complex. In all cases, the average charge state of the ejected monomers is lower for SID than for CID and is within a few percent of the % charge that would be expected if the charge is distributed evenly across the subunits (e.g., for a homotetramer, the monomer is 25% of the total mass and could be expected to carry away 25% of the total charge).
| Transthyretin 15+ Tetramer | Concanavalin A 22+ Tetramer | Hemoglobin (α-subunit) 17+ Tetramer | Hemoglobin (β-subunit) 17+ Tetramer | C-Reactive Protein | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CID | SID | CID | SID | CID | SID | CID | SID | CID | SID | |
| Avg. Charge State | 7.7 | 4.4 | 12.5 | 6.4 | 8.5 | 5.3 | 9.0 | 5.6 | 11.5 | 5.5 |
| Expected Charge State
(Based on Mass) |
3.8 | 5.5 | 4.0 | 4.2 | 5.2 | |||||
| CID | SID | CID | SID | CID | SID | CID | SID | CID | SID | |
| % Charge of Precursor | 51% | 29% | 57% | 29% | 50% | 31% | 53% | 33% | 44% | 21% |
| % Mass of Precursor | 25.0% | 25.0% | 24% | 25% | 20.0% | |||||
We have previously shown that SID can be used to fragment protein complexes over 200kD [28,29]. Surface-induced dissociation of the TaHSP16.9 dodecamer with 32 charges is shown in the top panel of Figure 6, for comparison with the CID spectrum from Figure 1. SID of the dodecamer at a collision energy of 2560eV looks similar to the CID spectrum at 3520 eV, with respect to the mass of the product ions. In both cases, the dodecamer fragments into monomers and undecamers, with relatively asymmetric partitioning of the charge. One notable difference is a higher extent of charge stripped dodecamer following surface collision. Charge stripping has been attributed to the loss of positively charged solvent adducts, such as ammonium, from the intact complex, and in the case of SID might also involve charge transfer to the surface [70]. Proton transfer from the projectile ion to the surface has been observed in some soft-landing experiments, including fluorinated self-assembled monolayers as employed here [71, 72]. Although the CID and SID product ions are similar in these cases, the SID of smaller complexes has been observed to be highly dependent on collision energy, as stated above. For this reason, higher SID collision energies were explored to see if fragmentation could be achieved without significant monomer unfolding. The bottom panel of Figure 6 shows SID at a collision energy of 3520eV. In this case, argon was also used in the gas cell following the surface collision to assist in ion transmission and provide further activation. The high m/z region above 10,000 shows products ranging from 8-mers to 11-mers, while much of the monomer population in the low m/z region has further dissociated to give covalent fragmentation along the protein backbone. As in the case of SAP, the covalent fragments are predominantly low charge b- and y-ions, dominated by cleavage at select sites, such as N-terminal to Pro-11 and Pro-19. Table 3 provides a list of the covalent fragments identified from the bottom spectrum in Figure 6. Benesch et al. have shown that at the highest CID collision energies available in a QToF 2 instrument, up to two monomers can be sequentially ejected from the intact TaHSP16.9 dodecamer (i.e. the 12-mer dissociates to 11-mer, which in turn further fragments to 10-mer) [10]. While 10-mers dominate the high m/z end of the SID spectrum, 11-mers, 9-mers and 8-mers are also observed. The peaks marked with an asterisk at m/z 16,274 and 8,362 correspond to overlapping oligomers with one and two charges per subunit, respectively (i.e. where the 11+ undecamer, 10+ decamer, etc. would overlap in mass-to-charge ratio). Whether the 8-, 9-, and 10-mers are the products of sequential monomer stripping, or form directly from the 12-mer, is still under investigation, but in either case, colliding the intact complex into a surface prior to sending it through the gas-filled collision cell provides more extensive fragmentation of the complex than CID alone.
Table 3.
Covalent fragments identified from the SID spectrum of TaHSP16.9 shown in the bottom panel of Figure 6.
| Covalent Fragment Ion | Theoretical m/z | Measured m/z | Δm/z | Cleavage: N-terminal Residue/C-terminal Residue |
|---|---|---|---|---|
| b102+ | 587.8200 | 587.8406 | -0.0206 | Asp/Pro |
| b5+ | 612.3940 | 612.4098 | -0.0158 | Arg/Ser |
| Y7+ | 716.4301 | 716.5059 | -0.0758 | Lys/Ala |
| y10+ | 1041.5952 | 1041.6503 | -0.0551 | Lys/Pro |
| bl82+ | 1045.5263 | 1045.5457 | -0.0194 | Asp/Pro |
| [b10-H2O]+ | 1156.6222 | 1156.6248 | -0.0026 | Asp/Pro |
| y11+ | 1169.6888 | 1169.6998 | -0.0110 | Lys/Lys |
| b10+ | 1174.6327 | 1174.6403 | -0.0076 | Asp/Pro |
| b14+ | 1604.8180 | 1604.7990 | 0.0190 | Asp/Leu |
| b18+ | 2090.0454 | 2090.0776 | -0.0322 | Asp/Pro |
Another important observation in comparison of SID and CID results is the fact that lower kinetic energies can be used for SID, with its more massive target, than for CID where the target is substantially lower in mass. This is illustrated by plots of fragmentation efficiency vs. collision energy in Figure 7 for CID and SID of TaHSP16.9. When colliding TaHSP16.9 into a surface, the point corresponding to 50% fragmentation of the dodecamer, occurs at a laboratory collision energy that is more than 1000eV lower than when colliding the protein into argon, despite the fact that the complex undergoes several thousand collisions via CID. This clearly demonstrates the potential for more extensive fragmentation at lower energies for SID than for CID.
Figure 7.
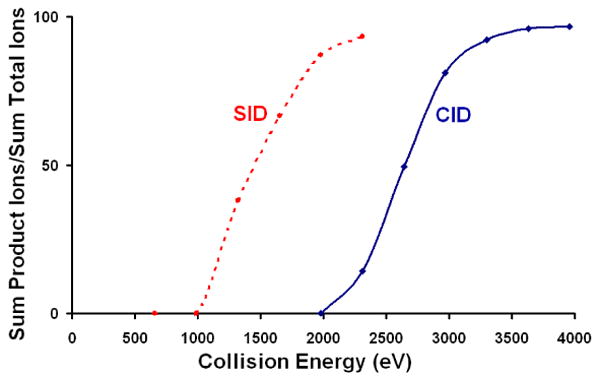
Fragmentation efficiency curves for dodecamers of TaHSP16.9 dissociated via SID or CID. Much lower laboratory collision energy is needed to fragment TaHSP16.9 when a surface is used as the collision partner.
An important question that arises in surface-induced dissociation studies of large complexes is whether the fragmentation conforms to expected kinetic theory of mass spectrometry, where it is expected that fragmentation rates will increase when kinetic energy of collision is increased and that greater amounts of internal energy will be needed to fragment larger precursor ions [17,18,45]. Both expectations have been shown to be met by SID of large non-covalent protein complexes, as illustrated in Figure 8 which shows fragmentation efficiency curves for a sHSP dimer (AtHSP18.5 dimer, ∼37 kDa), transthyretin tetramer (∼55.5 kDa), and two sHSP dodecamers at ∼201 kDa (TaHSP16.9) and ∼216 kDa (PsHSP18.1). The amount of fragmentation varies with collision energy and the more massive complexes require higher collision energies for onset of fragmentation.
Figure 8.
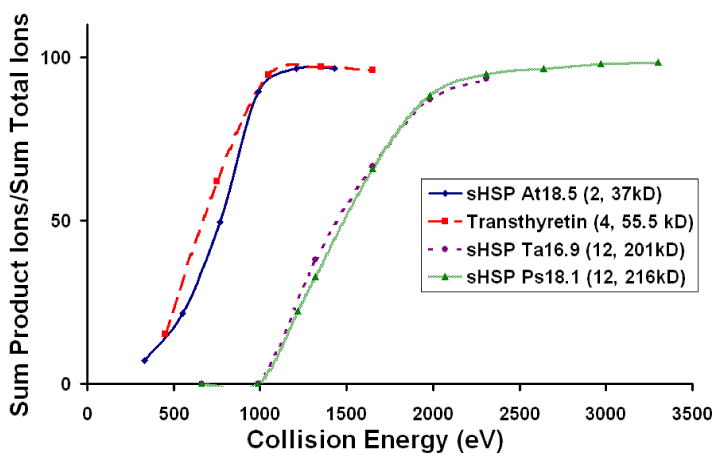
Fragmentation efficiency curves for four different protein-protein complexes. The laboratory collision energy shifts with the mass of the complex, indicating statistical fragmentation behavior. The legend lists each complex, the number of subunits, and the molecular weight.
Conclusions
Surface-induced dissociation shows much more extensive dissociation of protein complexes and inorganic CsI clusters than is observed by CID. In the case of CsI clusters, CID generates consecutive neutral losses of CsI moieties, whereas SID provides access not only to this pathway, but also to a higher energy charge fission process. This process results in a splitting of the CsI clusters in such a way that favors the formation of symmetrically stable magic number cluster products.
Upon SID of protein-protein complexes, monomers are ejected from oligomers with a charge state distribution that is much closer than that of CID to the statistical prediction made by taking the total charge and dividing by the oligomer number. Energies required to promote dissociation on the timescale of the instrument have been found to be lower for SID than for CID and the extent of dissociation increases with an increase in collision energy. Thus far, the majority of the SID data has involved homooligomers. Clearly, more work is needed on heterogeneous protein complexes where SID has the potential to provide substructure information. It is also desirable to combine the SID technique with ion mobility so that ions can be shape-selected prior to surface collision or so that shape can be monitored after surface collision. SID has great potential to contribute to structural biology studies and studies of other large macromolecular complexes.
Acknowledgments
The authors acknowledge Dr. Elizabeth Vierling, Eman Basha, and Nomalie Jaya for providing samples of small heat shock proteins and Dr. Richard Beardsley for helpful discussions. We also thank the NSF DBI grant CHE 024447 for support for the development of the QToF instrument and NIH grant 5R01 GM 051387 for support of experiments on fragmentation. CMJ thanks Pfizer for a Graduate Research Fellowship in Analytical Chemistry and AEB thanks Science Foundation Arizona.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Damoc E, Fraser CS, Zhou M, Videler H, Mayeur GL, Hershey JWB, Doudna JA, Robinson CV, Leary JA. Structural characterization of the human eukaryotic initiation factor 3 protein complex by mass spectrometry. Molecular & Cellular Proteomics. 2007;6:1135–1146. doi: 10.1074/mcp.M600399-MCP200. [DOI] [PubMed] [Google Scholar]
- 2.Aquilina JA, Benesch JLP, Bateman OA, Slingsby C, Robinson CV. Polydispersity of a mammalian chaperone: Mass spectrometry reveals the population of oligomers in alpha B-crystallin. Proc Natl Acad Sci U S A. 2003;100:10611–10616. doi: 10.1073/pnas.1932958100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boskovic J, Coloma J, Aparicio T, Zhou M, Robinson CV, Mendez J, Montoya G. Molecular architecture of the human GINS complex. EMBO Rep. 2007;8:678–684. doi: 10.1038/sj.embor.7401002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taverner T, H Helena, Sharon Michal, Ruotolo Brandon T, Matak-Vinkovic Jijana, Devos Damien, Russell Robert B, Robinson Carol V. Subunit Architecture of Intact Protein Complexes from Mass Spectrometry and Homology Modeling. Acc Chem Res. 2008 doi: 10.1021/ar700218q. [DOI] [PubMed] [Google Scholar]
- 5.Xie YM, Zhang J, Yin S, Loo JA. Top-down ESI-ECD-FT-ICR mass spectrometry localizes noncovalent protein-ligand binding sites. J Am Chem Soc. 2006;128:14432–14433. doi: 10.1021/ja063197p. [DOI] [PubMed] [Google Scholar]
- 6.Bernstein SL, Wyttenbach T, Baumketner A, Shea JE, Bitan G, Teplow DB, Bowers MT. Amyloid beta-protein: Monomer structure and early aggregation states of A beta 42 and its Pro(19) alloform. J Am Chem Soc. 2005;127:2075–2084. doi: 10.1021/ja044531p. [DOI] [PubMed] [Google Scholar]
- 7.van den Heuvel RHH, Gato S, Versluis C, Gerbaux P, Kleanthous C, Heck AJR. Real-time monitoring of enzymatic DNA hydrolysis by electrospray ionization mass spectrometry. Nucleic Acids Res. 2005;33 doi: 10.1093/nar/gni099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sobott F, Benesch JLP, Vierling E, Robinson CV. Subunit exchange of multimeric protein complexes - Real-time monitoring of subunit exchange between small heat shock proteins by using electrospray mass spectrometry. J Biol Chem. 2002;277:38921–38929. doi: 10.1074/jbc.M206060200. [DOI] [PubMed] [Google Scholar]
- 9.van Duijn E, Simmons DA, van den Heuvel RHH, Bakkes PJ, van Heerikhuizen H, Heeren RMA, Robinson CV, van der Vies SM, Heck AJR. Tandem mass spectrometry of intact GroEL-substrate complexes reveals substrate-specific conformational changes in the trans ring. J Am Chem Soc. 2006;128:4694–4702. doi: 10.1021/ja056756l. [DOI] [PubMed] [Google Scholar]
- 10.Benesch JLP, Aquilina JA, Ruotolo BT, Sobott F, Robinson CV. Tandem mass spectrometry reveals the quaternary organization of macromolecular assemblies. Chem Biol. 2006;13:597–605. doi: 10.1016/j.chembiol.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 11.Benesch JLP, Sobott F, Robinson CV. Thermal dissociation of multimeric protein complexes by using nanoelectrospray mass spectrometry. Anal Chem. 2003;75:2208–2214. doi: 10.1021/ac034132x. [DOI] [PubMed] [Google Scholar]
- 12.Felitsyn N, Kitova EN, Klassen JS. Thermal decomposition of a gaseous multiprotein complex studied by blackbody infrared radiative dissociation. Investigating the origin of the asymmetric dissociation behavior. Anal Chem. 2001;73:4647–4661. doi: 10.1021/ac0103975. [DOI] [PubMed] [Google Scholar]
- 13.Jurchen JC, Williams ER. Origin of asymmetric charge partitioning in the dissociation of gas-phase protein homodimers. J Am Chem Soc. 2003;125:2817–2826. doi: 10.1021/ja0211508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ruotolo BT, Hyung SJ, Robinson PM, Giles K, Bateman RH, Robinson CV. Ion mobility-mass spectrometry reveals long-lived, unfolded intermediates in the dissociation of protein complexes. Angewe Chem Int Ed. 2007;46:8001–8004. doi: 10.1002/anie.200702161. [DOI] [PubMed] [Google Scholar]
- 15.Wanasundara SN, Thachuk M. Theoretical investigations of the dissociation of charged protein complexes in the gas phase. J Am Soc Mass Spectrom. 2007;18:2242–2253. doi: 10.1016/j.jasms.2007.09.022. [DOI] [PubMed] [Google Scholar]
- 16.van Montfort RLM, Basha E, Friedrich KL, Slingsby C, Vierling E. Crystal structure and assembly of a eukaryotic small heat shock protein. Nat Struct Biol. 2001;8:1025–1030. doi: 10.1038/nsb722. [DOI] [PubMed] [Google Scholar]
- 17.McLuckey SA. Principles of Collisional Activation in Analytical Mass-Spectrometry. J Am Soc Mass Spectrom. 1992;3:599–614. doi: 10.1016/1044-0305(92)85001-Z. [DOI] [PubMed] [Google Scholar]
- 18.Shukla AK, Futrell JH. Tandem mass spectrometry: dissociation of ions by collisional activation. J Mass Spectrom. 2000;35:1069–1090. doi: 10.1002/1096-9888(200009)35:9<1069::AID-JMS54>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 19.Lorenzen K, Versluis C, van Duijn E, van den Heuvel RHH, Heck AJR. Optimizing macromolecular tandem mass spectrometry of large non-covalent complexes using heavy collision gases. Int J Mass Spectrom. 2007;268:198–206. [Google Scholar]
- 20.Geels RBJ, van der Vies SM, Heck AJR, Heeren RMA. Electron capture dissociation as structural probe for noncovalent gas-phase protein assemblies. Anal Chem. 2006;78:7191–7196. doi: 10.1021/ac060960p. [DOI] [PubMed] [Google Scholar]
- 21.Laskin J, Futrell JH. Energy Transfer in Collisions of Peptide Ions with Surfaces. J Chem Phys. 2003;119:341–3420. [Google Scholar]
- 22.Smith DL, Wysocki VH, Colorado R, Jr, Shmakova OE, Graupe M, Lee RT. Low-Energy Ion-Surface Collisions Characterize Alkyl- and Fluoroalkyl-Terminated Self-Assembled Monolayers on Gold. Langmuir. 2002;18:3895–3902. [Google Scholar]
- 23.Vekey K, Somogyi A, Wysocki VH. Internal energy Distribution of Benzene Molecular Ions in Surface-Induced Dissociation. J Mass Spectrom. 1995;30:212–217. [Google Scholar]
- 24.Vekey K, Somogyi A, Wysocki VH. Average Activation Energies of Low-Energy Fragmentation Processes of Protonated Peptides Determined by a New Approach. Rapid Commun Mass Spectrom. 1996;10:911–918. doi: 10.1002/(SICI)1097-0231(19960610)10:8<911::AID-RCM593>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 25.Lim HJ, Schultz DG, Yu CW, Hanley L. Relative Dissociation Energies of Protonated Peptides by Electrospray Ionization Suface-Induced Dissociation. Anal Chem. 1999;71:2307–2317. doi: 10.1021/ac981411p. [DOI] [PubMed] [Google Scholar]
- 26.Laskin J, Futrell JH. Collisional Activation of Peptide Ions in FT-ICR Mass Spectrometry. Mass Spectrom Rev. 2003;22:158–181. doi: 10.1002/mas.10041. [DOI] [PubMed] [Google Scholar]
- 27.Beardsley RL, Jones CM, Galhena AS, Wysocki VH. Investigations of the Surface-Induced Dissociation of Non-Covalent Protein Complexes in the Gas Phase. J Am Chem Soc. In Preparation. [Google Scholar]
- 28.Galhena AS, Dagan S, Jones CM, Beardsley RL, Wysocki VH. Surface-Induced Dissociation of Peptides and Protein Complexes in a Quadrupole/Time-of-Flight Mass Spectrometer. Anal Chem. 2008;80:1425–1436. doi: 10.1021/ac701782q. [DOI] [PubMed] [Google Scholar]
- 29.Wysocki VH, Joyce Karen E, Jones Christopher M, Beardsley Richard L. Surface-Induced Dissociation of Small Molecules, Peptides, and Non-Covalent Protein Complexes. J Am Soc Mass Spectrom. 2008;19:190–208. doi: 10.1016/j.jasms.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jones CM, Beardsley RL, Galhena AS, Dagan S, Cheng GL, Wysocki VH. Symmetrical gas-phase dissociation of noncovalent protein complexes via surface collisions. J Am Chem Soc. 2006;128:15044–15045. doi: 10.1021/ja064586m. [DOI] [PubMed] [Google Scholar]
- 31.Stace AJ. Metal ion solvation in the gas phase: The quest for higher oxidation states. J Phys Chem A. 2002;106:7993–8005. [Google Scholar]
- 32.Mark TD. Cluster Ions - Production, Detection and Stability. Int J Mass Spectrom Ion Processes. 1987;79:1–59. [Google Scholar]
- 33.Castleman AW. Reactions and Properties of Clusters. Int J Mass Spectrom Ion Processes. 1992;118:167–189. [Google Scholar]
- 34.Busch KL, Cooks RG. Mass-Spectrometry of Large, Fragile, and Involatile Molecules. Science. 1982;218:247–254. doi: 10.1126/science.218.4569.247. [DOI] [PubMed] [Google Scholar]
- 35.Barlak TM, Wyatt JR, Colton RJ, Decorpo JJ, Campana JE. Secondary Ion Mass-Spectrometry of Metal-Halides .2. Evidence for Structure in Alkali Iodide Clusters. J Am Chem Soc. 1982;104:1212–1215. [Google Scholar]
- 36.Baldwin MA, Proctor CJ, Amster IJ, Mclafferty FW. The Behavior of Cesium Iodide Cluster Ions Produced by Fast-Atom Bombardment. Int J Mass Spectrom Ion Processes. 1983;54:97–107. [Google Scholar]
- 37.Campana JE, Green BN. Unicluster Dissociation of Large Alkali Iodide Cluster Ions. J Am Chem Soc. 1984;106:531–535. [Google Scholar]
- 38.Honea EC, Homer ML, Whetten RL. Electron-Binding and Stability of Excess-Electron Alkali-Halide Clusters - Localization and Surface-States. Phys Rev B. 1993;47:7480–7493. doi: 10.1103/physrevb.47.7480. [DOI] [PubMed] [Google Scholar]
- 39.Morgan TG, Rabrenovic M, Harris FM, Beynon JH. Investigation of the Relative Stability of Positive Alkali-Halide Cluster Ions Generated by Fast Atom Bombardment. Org Mass Spectrom. 1984;19:315–320. [Google Scholar]
- 40.Drewello T, Herzschuh R, Stach J. Direct Fission Versus Sequential Evaporation Mechanism of Sputtered Cesium Iodide Cluster Ions. Z Phys D Atom Mol Cl. 1993;28:339–343. [Google Scholar]
- 41.Derrick PJ, Colburn AW, Sheil MM, Uggerud E. Collisional Excitation and Fragmentation of Alkali-Metal Halide Cluster Ions. J Chem Soc Faraday T. 1990;86:2533–2535. [Google Scholar]
- 42.Vekey K, Czira G. Large Translational Energy-Loss and Scattering in Collision-Induced Dissociation Processes. Org Mass Spectrom. 1993;28:546–551. [Google Scholar]
- 43.Vekey K, Ludanyi K. Translational Energy-Losses of Lif Cluster Ions. Org Mass Spectrom. 1994;29:615–618. [Google Scholar]
- 44.Sobott F, Hernandez H, McCammon MG, Tito MA, Robinson CV. A tandem mass spectrometer for improved transmission and analysis of large macromolecular assemblies. Anal Chem. 2002;74:1402–1407. doi: 10.1021/ac0110552. [DOI] [PubMed] [Google Scholar]
- 45.Griffin LL, McAdoo DJ. The Effect of Ion Size on Rate of Dissociation - RRKM Calculations on Model Large Polypeptide Ions. J Am Soc Mass Spectrom. 1993;4:11–15. doi: 10.1016/1044-0305(93)85037-X. [DOI] [PubMed] [Google Scholar]
- 46.Callahan JH, Somogyi A, Wysocki VH. Collisions of C-60(+.) and C-60(2+) at Fluorinated and Non-Fluorinated Self-Assembled Monolayer Films. Rapid Commun Mass Spectrom. 1993;7:693–699. [Google Scholar]
- 47.Kane TE, Somogyi A, Wysocki VH. Reactive Ion-Surface Collisions - Application of Ionized Acetone-D(6), Dmso-D(6) and Pyridine-D(5) as Probes for the Characterization of Self-Assembled Monolayer Films on Gold. Org Mass Spectrom. 1993;28:1665–1673. [Google Scholar]
- 48.Mckay AR, Ruotolo BT, Ilag LL, Robinson CV. Mass measurements of increased accuracy resolve heterogeneous populations of intact ribosomes. J Am Chem Soc. 2006;128:11433–11442. doi: 10.1021/ja061468q. [DOI] [PubMed] [Google Scholar]
- 49.Ilag LL, Videler H, McKay AR, Sobott F, Fucini P, Nierhaus KH, Robinson CV. Heptameric (L12)(6)/L10 rather than canonical pentameric complexes are found by tandem MS of intact ribosomes from thermophilic bacteria. Proc Natl Acad Sci U S A. 2005;102:8192–8197. doi: 10.1073/pnas.0502193102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rostom AA, Fucini P, Benjamin DR, Juenemann R, Nierhaus KH, Hartl FU, Dobson CM, Robinson CV. Detection and selective dissociation of intact ribosomes in a mass spectrometer. Proc Natl Acad Sci U S A. 2000;97:5185–5190. doi: 10.1073/pnas.97.10.5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Herzschuh R, Drewello T. The fragmentation dynamics of small Cs(CsI)(n)(+) cluster ions under low-energy multiple collision conditions. Int J Mass Spectrom. 2004;233:355–359. [Google Scholar]
- 52.Martin TP. Structure and Vibrational Frequencies of Ionic Clusters. J Chem Phys. 1978;69:2036–2042. [Google Scholar]
- 53.Martin TP. The Structure of Ionic Clusters - Thermodynamic Functions, Energy Surfaces, and Sims. J Chem Phys. 1980;72:3506–3510. [Google Scholar]
- 54.Martin TP. Stability of Doubly Charged Alkali-Halide Clusters. J Chem Phys. 1982;76:5467–5469. [Google Scholar]
- 55.Zhang DX, Cooks RG. Doubly charged cluster ions [(NaCl)(m)(Na)(2)](2+): magic numbers, dissociation, and structure. Int J Mass Spectrom. 2000;196:667–684. [Google Scholar]
- 56.Echt O, Flotte AR, Knapp M, Sattler K, Recknagel E. Magic Numbers in Mass-Spectra of Xe, C2f4cl2 and Sf6 Clusters. Phys Chem Chem Phys. 1982;86:860–865. [Google Scholar]
- 57.Echt O, Sattler K, Recknagel E. Magic Numbers for Sphere Packings - Experimental-Verification in Free Xenon Clusters. Phys Rev Lett. 1981;47:1121–1124. [Google Scholar]
- 58.Barlak TM, Campana JE, Colton RJ, Decorpo JJ, Wyatt JR. Secondary Ion Mass-Spectrometry of Metal-Halides .1. Stability of Alkali Iodide Clusters. J Phys Chem. 1981;85:3840–3844. [Google Scholar]
- 59.Muhlbach J, Sattler K, Pfau P, Recknagel E. Evidence for Magic Numbers of Free Lead-Clusters. Phys Lett A. 1982;87:415–417. [Google Scholar]
- 60.Campana JE, Colton RJ, Wyatt JR, Bateman RH, Green BN. Ultrahigh Mass-Spectrometry. Appl Spectrosc. 1984;38:430–432. [Google Scholar]
- 61.Stjohn PM, Beck RD, Whetten RL. Fragmentation and Reaction Processes in Cluster-Surface Collisions. Z Phys D Atom Mol Cl. 1993;26:226–228. [Google Scholar]
- 62.Beck RD, Stjohn P, Homer ML, Whetten RL. Impact-Induced Cleaving and Melting of Alkali-Halide Nanocrystals. Science. 1991;253:879–883. doi: 10.1126/science.253.5022.879. [DOI] [PubMed] [Google Scholar]
- 63.Beck RD, John PS, Homer ML, Yeretzian C, Whetten RL. Scattering of Mass-Selected Cluster Beams from Solid-Surfaces. Abstracts of Papers of the American Chemical Society. 1992;204:264-PHYS. [Google Scholar]
- 64.Jurchen JC, Garcia DE, Williams ER. Further studies on the origins of asymmetric charge partitioning in protein homodimers. J Am Soc Mass Spectrom. 2004;15:1408–1415. doi: 10.1016/j.jasms.2004.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Versluis C, van der Staaij A, Stokvis E, Heck AJR, de Craene B. Metastable ion formation and disparate charge separation in the gas-phase dissection of protein assemblies studied by orthogonal time-of-flight mass spectrometry. J Am Soc Mass Spectrom. 2001;12:329–336. doi: 10.1016/S1044-0305(00)00227-0. [DOI] [PubMed] [Google Scholar]
- 66.Huang YY, Triscari JM, Tseng GC, Pasa-Tolic L, Lipton MS, Smith RD, Wysocki VH. Statistical characterization of the charge state and residue dependence of low-energy CID peptide dissociation patterns. Anal Chem. 2005;77:5800–581359. doi: 10.1021/ac0480949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wysocki VH, Tsaprailis G, Smith LL, Breci LA. Mobile and Localized Protons: A Framework for Understanding Peptide Dissociation. J Mass Spectrom. 2000;35:1399–1406. doi: 10.1002/1096-9888(200012)35:12<1399::AID-JMS86>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 68.Breci LA, Tabb DL, Yates JR, III, Wysocki VH. Cleavage N-Terminal to Proline: Analysis of a Database of Peptide Tandem Mass Spectra. Anal Chem. 2003;75:1963–1971. doi: 10.1021/ac026359i. [DOI] [PubMed] [Google Scholar]
- 69.Leymarie N, Berg EA, McComb ME, O'Connor PB, Grogan J, Oppenheim FG, Costello CE. Tandem Mass Spectrometry for Structural Characterization of Proline-Rich Proteins: Application to Salivary PRP-3. Anal Chem. 2002;74:4124–4132. doi: 10.1021/ac0255835. [DOI] [PubMed] [Google Scholar]
- 70.Sobott F, McCammon MG, Robinson CV. Gas-phase dissociation pathways of a tetrameric protein complex. Int J Mass Spectrom. 2003;230:193–200. [Google Scholar]
- 71.Volny M, Elam WT, Ratner BD, Turecek F. Preparative soft and reactive landing of gas-phase ions on plasma-treated metal surfaces. Anal Chem. 2005;77:4846–4853. doi: 10.1021/ac0505019. [DOI] [PubMed] [Google Scholar]
- 72.Alvarez J, Futrell JH, Laskin J. Soft-landing of peptides onto self-assembled monolayer surfaces. J Phys Chem A. 2006;110:1678–1687. doi: 10.1021/jp0555044. [DOI] [PubMed] [Google Scholar]