

Introduction
Obesity is associated with leptin resistance (Considine et al., 1996; Frederich et al., 1995; Friedman, 2000; Morton et al., 2006; Myers et al., 2008; Myers et al., 2012), while type 2 diabetes is characterized by insulin resistance in multiple tissues (Guilherme et al., 2008; Kahn et al., 2006; Konner and Bruning, 2012; Weyer et al., 1999). Hypothalamic Pomc neurons are direct targets of both leptin and insulin contributing to their role in regulating energy expenditure, body weight, and glucose homeostasis(Belgardt and Bruning, 2010; Myers and Olson, 2012; Schwartz and Porte, 2005; Spiegelman and Flier, 2001; Williams and Elmquist, 2012; Yeo and Heisler, 2012). Recently, endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) have emerged as a unifying and critical link in the development of cellular leptin and insulin resistance (Ozcan et al., 2009; Ozcan et al., 2006; Shoelson et al., 2006; Wellen and Hotamisligil, 2005; Zhang et al., 2008). In particular, obese mice and mice fed high-fat diets display ER stress in peripheral tissues as well as Pomc neurons within the hypothalamus, suggesting that metabolic disorders associated with obesity and high-fat diets induce ER stress in vivo (Schneeberger et al., 2013; Thaler et al., 2012; Xu et al., 2005). Notably, induction of ER stress or deficiency of the X-box-binding protein 1 (Xbp1) in neurons results in hyperleptinemia, obesity, hyperphagia, and reduced metabolic rate associated with severe hypothalamic leptin resistance (Ozcan et al., 2009). Additionally, ER stress suppresses leptin and insulin signaling in the periphery as well as the CNS via classical inhibitors of cytokine signaling such as the suppressor of cytokine signaling-3 (Socs3) and protein tyrosine phosphatase 1b (Ptp1b) (Howard and Flier, 2006; Myers et al., 2008; Ozcan et al., 2004; White et al., 2009; Zabolotny et al., 2008). Importantly, the neuronal cell type(s) involved in this response remains undefined. To address this issue we assessed the role of Xbp1s in Pomc neurons to regulate glucose metabolism and HFD-induced obesity. Additionally, we examined the cellular mechanisms of Ptp1b, Socs3, and Xbp1s in the ER stress-induced acute leptin and insulin resistance of arcuate Pomc neurons.
Results
Constituitive activation of Xbp1s in Pomc neurons protects against diet-induced obesity
Xbp1s improves leptin and insulin signaling along with metabolism in the periphery as well as the CNS (Deng et al., 2013; Ozcan et al., 2009; Ozcan et al., 2004; Ozcan et al., 2006). We recently developed a mouse model which expresses an inducible “dominant active” Xbp1s transgene via a conventional Tet-On system (Deng et al., 2013). The Xbp1s transgene under the control of a tetracycline-responsive element (TRE) supports inducible expression by the tetracycline reverse transcriptional activator (rtTA) in the presence of doxycycline (Dox). The rtTA transgene is driven by the Rosa26 promoter with a transcriptional stop cassette flanked by 2 loxP sites upstream of rtTA (Belteki et al., 2005). Combined with a Pomc promoter–driven Cre transgene(Balthasar et al., 2004), we obtained a mouse model with Pomc-specific inducible expression of X bp1s (PIXs).
When fed HFD-Dox, male PIXs mice displayed an age-dependent lean body weight compared to wild type mice (Figure 1A), which was reflected by decreases in fat mass (t(11) = 3.965, p<0.05; Figure 1B). The lean phenotype of PIXs was concomitant with significantly lower visceral (t(11) = 3.395, p<0.05) and subcutaneous fat (t(11) = 4.090, p<0.05) distribution than controls (Figures 1C and 1D). PIXs mice fed HFD-Dox also displayed decreased snout-anus length (t(11) = 4.928, p<0.05; Supplemental Figure 1A) and decreased hepatic triglyceride (t(6) = 2.60, p<0.05) and cholesterol (t(6) = 2.571, p<0.05) levels (Figure 1E).
Figure 1.
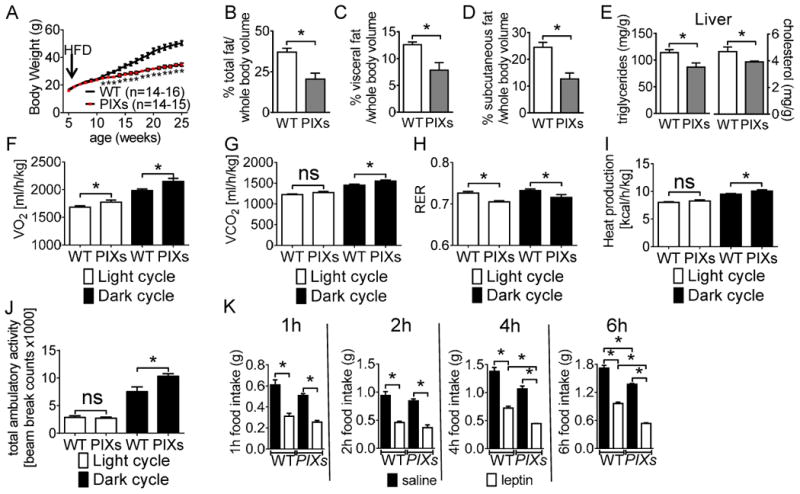
Body weight and metabolic assessment of male WT and PIXs mice on HFD. Body weight curve of (A) male PIXs mice (*p<0.05). Body fat composition: whole body volume (B), visceral (C), and subcutaneous (D). Male PIXs mice display (E) increased hepatic triglyceride and cholesterol, (F) Increased VO2, (G) increased VCO2, (H) decreased RER (I) increased heat production, and (J) increased ambulatory activity. Error bars indicate SEM. (K) Leptin induced hypophagia was observed at 1, 2, 4, and 6 after refeeding. PIXs mice exhibited increased hypophagia in response to pharmacological administration of leptin at 4 and 6 hours after refeeding. (n=10 per group; *p<0.05). (Note: mice used in (E-I) were age-matched male littermates (8 weeks of age), and had comparable body weight and lean mass. (for F-J, n=14-16 per group; *p<0.05).
Age and weight matched PIXs males were hypermetabolic independent of altered food intake, as demonstrated by significant increases in energy expenditure (Figures 1F-1I and Supplemental Figure 1B). Components of total energy expenditure include energy required for physical activities and basal metabolism. In particular, PIXs mice exhibited increased heat production suggestive of higher metabolic rate (Figure 1I). PIXs mice also showed increased ambulatory movements independent of rearing activity (Figure 1J and Supplemental Figure 1C). Although we did not observe changes in ad libitum food intake, PIXs mice were more sensitive to acute leptin-induced hypophagia when compared to littermate controls at 4 and 6 hours after refeeding (Figure 1K).
In support of the hypermetabolic phenotype, PIXs mice displayed increased expression of genes associated with heat production in both brown adipose tissue - BAT (for Ppargc1α: t(9) = 2.957, p<0.05; for Prdm16: t(9) = 3.691, p<0.05; for UCP1: t(9) = 2.527, p<0.05; for Cidea: t(9) = 2.547, p<0.05; for Dio2: t(9) = 1.413, p>0.05; for Elovl6: t(9) = 1.480, p>0.05; Figure 2A) and inguinal white adipose tissue - iWAT (for Ppargc1α: t(9) = 3.289, p<0.05; for Prdm16: t(9) = 4.158, p<0.05; for UCP1: t(9) = 4.573, p<0.05; for Cidea: t(9) = 4.270, p<0.05; for Dio2: t(9) = 4.004, p<0.05; for Elovl6: t(9) = 1.918, p>0.05; Figure 2B). These data are also supported by the apparent decreased multilocular cells in BAT from PIXs mice (Figure 2C and 2D) and increased expression of the browning marker UCP1 in iWAT of PIXs mice (Figure 2E and 2F). Collectively, these results indicate that constitutive expression of Xbp1s in Pomc neurons is sufficient to improve body weight homeostasis in the context of diet-induced obesity. Moreover, Xbp1s in Pomc neurons is sufficient to regulate metabolic rate, locomotor activity and to mediate thermogenesis (both BAT and iWAT).
Figure 2.
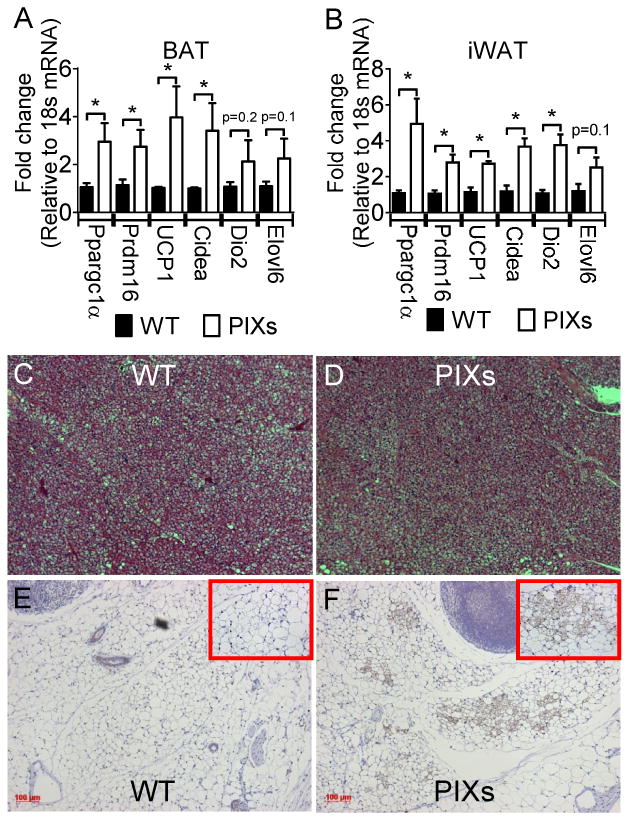
PIXs mice express increased thermogenic markers in BAT and iWAT. Weight matched PIXs mice were fed HFD DOX enriched diet for 2 weeks. qPCR was performed to examine the relative expression Ppargc1α, Prdm16, UCP1, Cidea, Dio2, and Elovl6 which are known genes associated with heat production in (A) BAT and (B) iWAT. *, p < 0.05. Brown adipose tissue (BAT) from (C) WT and (D) PIXs mice was imaged by light microscopy after hematoxylin-eosin staining. Inguinal white adipose tissue from (E) WT and (F) PIXs mice was imaged by light microscopy after UCP-1 immunohistochemistry. Bars, 100μm. (note C and D are the same scale as E and F).
Constitutive activation of Xbp1s in Pomc neurons improves insulin sensitivity and glycemia
Along with the systemic effects on whole-body energy expenditure and body weight, Xbp1s induction in Pomc neurons also leads to profound changes in glucose metabolism. PIXs mice fed a chow-DOX diet showed improved blood glucose levels in the fed and fasted state when compared to littermate controls (for chow dox fed PIXs mice: t(15) = 2.763, p<0.05; for chow-DOX fasted PIXs mice: t(15) = 2.250, p<0.05; Figure 3). Serum insulin were also decreased in PIXs mice during both fed and fasted conditions (for chow dox fed PIXs mice: t(15) = 2.217, p<0.05; for chow dox fasted PIXs mice: t(14) = 2.515, p<0.05; Figure 3).
Figure 3.
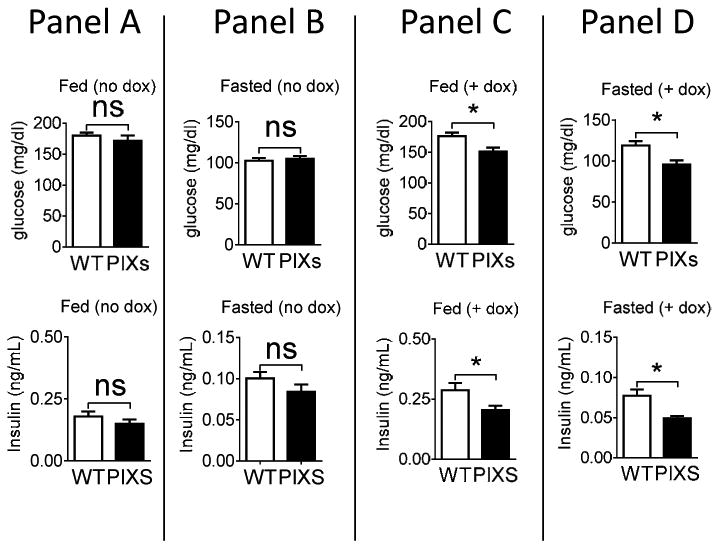
Blood glucose and insulin levels of male WT and PIXs mice on chow diet. Blood glucose and insulin levels were measured from WT and PIXs mice fed chow diet without dox (Panel A), fed on a chow diet without dox then fasted overnight (Panel B), fed a chow diet with dox for 1 week (Panel C), and fed chow diet with dox for 1 week then fasted overnight (Panel D). Blood glucose and insulin levels were decreased in mice fed or fasted after 1 week on a chow diet with dox (n=10-15 per group; *p<0.05).
We next performed hyperinsulinemic-euglycemic clamps to assess whether insulin sensitivity was improved in chow-fed DOX enriched PIXs mice compared with their littermates. Blood glucose was successfully clamped at target levels (150mg/dL; Figure 4A), and the exogenous glucose infusion rate (GIR) was higher in PIXs mice indicating improved insulin sensitivity (Figure 4B). This difference in GIR was due to improved insulin-mediated suppression of endogenous glucose appearance (endo Ra; Figure 4C) and not glucose disappearance (Rd; Figure 4D). Together these data suggest that constitutive expression of Xbp1s in Pomc neurons is sufficient to mimic a postprandial state in the liver – suppressing glucose production and ultimately lowering blood glucose levels.
Figure 4.
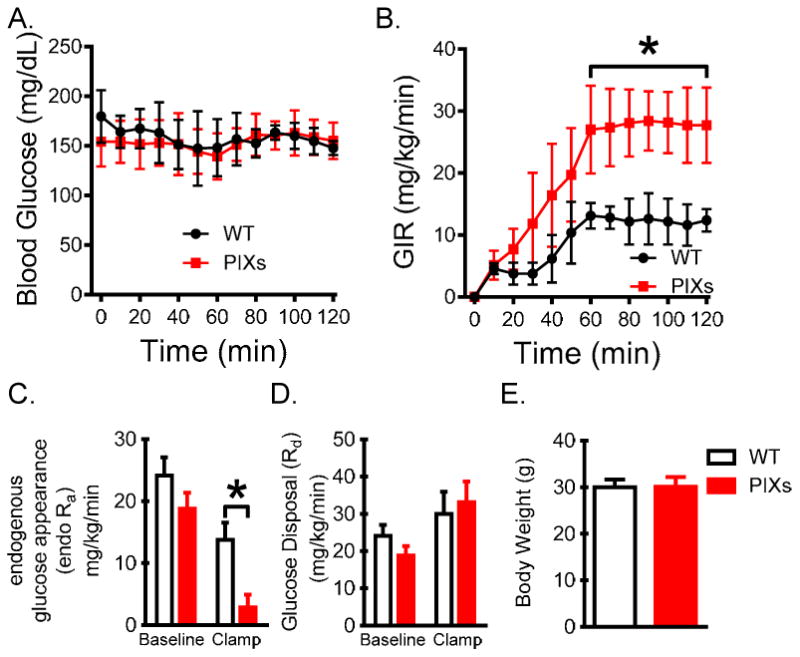
Improved glucoregulation in mice which constitutively express Xbp1s in Pomc neurons. (A) Basal and clamp blood glucose levels during the hyperinsulinemic-euglycemic experiment. (B) Exogenous glucose infusion rate (GIR) needed to clamp blood glucose. (C and D) Endogenous rates of glucose appearance (endo Ra) and disappearance (Rd) were determined using a constant infusion of [3-3H]glucose and Steele's steady-state calculations. (E) Body weight of PIXs mice on day of experiment. (n=7)
Xbp1s is a cell nonautonomous feeding sensor
Upregulation of Xbp1s and the UDP-galactose-4-epimerase (GalE) in the liver maybe indicative of a fed state and contribute to metabolism (Deng et al., 2013). In support of these data, we demonstrated that both Xbp1s and GalE are upregulated in the liver after refeeding (2h) following an 18h fast (for Xbp1s: t(8) = 2.852, p<0.05; for GalE: t(8) = 2.464, p<0.05; Figure 5A). Refeeding (2h) following an 18h fast also readily elevated mRNA for both Xbp1s and GalE in the arcuate nucleus from WT mice, supporting an association of Xbp1s-GalE with feeding or caloric intake (for Xbp1s: t(8) = 3.203, p<0.05; for GalE: t(8) = 3.276, p<0.05; Figure 5B). Similar to our observations in refeeding after an overnight fast, Dox-containing diet induced Xbp1s and GalE mRNA as well as Xbp1s target genes in the Arcuate nucleus from PIXs mice (for Xbp1s: t(9) = 3.094, p<0.05; for GalE: t(9) = 3.328, p<0.05; for Edem1 t(9) = 4.779, p<0.05; for Erdj4 t(9) = 2.860, p<0.05; for Bip t(9) = 3.835, p<0.05; Figure 5B). The enhanced expression of Xbp1s and target genes was also apparent in FACs isolated Pomc neurons subsequent to refeeding or from FACs isolated Pomc from PIXs mice fed a Dox-containing diet (Figures 5C and 5D). Importantly, increased expression of Xbp1s mRNA levels and Xbp1s target genes in Pomc neurons from PIXs mice fed a Dox-containing diet was analogous to the elevated Xbp1s levels observed in Pomc neurons in the refed state, supporting an expression of Xbp1s in PIXs mice which mimicks a physiological post-prandial state. We also found that similar to recent work in C. elegans (Taylor and Dillin, 2013), constitutive expression of Xbp1s in murine Pomc neurons resulted in the transcriptional upregulation of Xbp1s and Xbp1s target genes in the liver (for Xbp1s: t(13) = 2.284, p<0.05; for erdj4: t(13) = 2.477, p<0.05; for erdeml: t(13) = 2.433, p<0.05; for chop: t(13) = 2.545, p<0.05; for bip: t(13) = 1.556, p>0.05; Figure 5E). Although we cannot exclude the involvement of other arms of the UPR in this cell nonautonomous regulation, the Pomc-dependent upregulation of Xbp1s in the liver occurred independent of detectable Cre activity within the liver. Together, these data demonstrate that Xbp1s and presumably activation of Xbp1s transcriptional targets is sufficient to signal a fed state via a cell nonautonomous regulation of the liver which may ultimately contribute to improved liver metabolic homeostasis (Deng et al., 2013).
Figure 5.
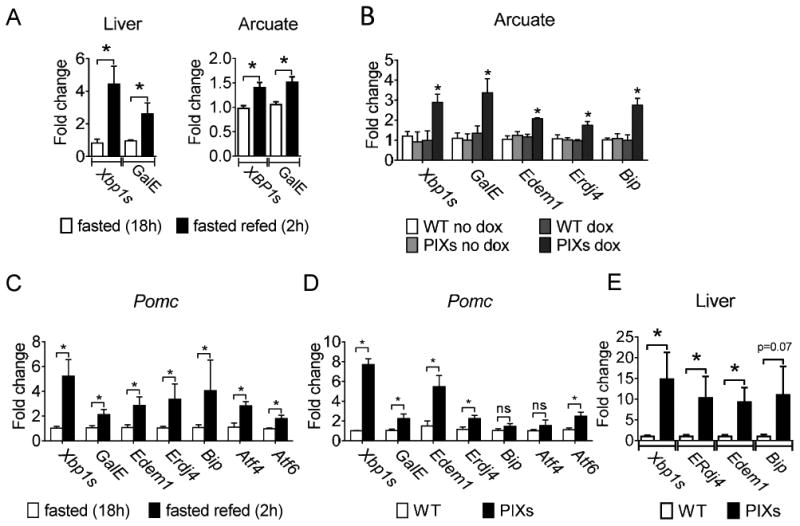
Regulation of Xbp1s and GalE in the arcuate nucleus and the liver. (A) Relative mRNA expression of Xbp1s and GalE in the liver and the arcuate nucleus of mice fasted (18h) and mice re-fed (2h) after fasting (18h). (B) Relative mRNA expression of Xbp1s as well as GalE, Edem1, Erdj4, and Bip, which are which are known Xbp1s target genes, in the arcuate nucleus from PIXs and WT mice fed either non-Dox or Dox containing diet. (C-D) Relative mRNA expression of Xbp1s, GalE, Edem1, Erdj4, Bip, Atf4, and Atf6 in FACS-Pomc neurons. (C) Data represents from WT mice fasted (18h) and WT mice re-fed (2h) after fasting (18h). (D) Data represents PIXs and WT mice chronically fed Dox containing diet. (E) qPCR was performed on mice chronically fed HF-Dox diet to examine the relative expression XBP1s as well as erdj4, erdeml, chop, and bip which are which are known Xbp1s target genes in (L) liver. *P < 0.05 compared with control. A-E. Fold change relative to 18S mRNA; Error bars indicate SEM.
ER stress inhibits leptin and insulin signaling in the arcuate nucleus
In an effort to identify a cellular mechanism underlying the improvements in body weight and glucose homeostasis in PIXs mice, we first utilized a model chronic culture system (organotypic slice preparation) recently developed in our lab (Fukuda et al., 2011; Gahwiler and Llano, 1989). Recent evidence suggests that overnutrition induces ER stress in arcuate Pomc neurons (Schneeberger et al., 2013). We hypothesized that ER stress may blunt leptin and insulin signaling directly in arcuate Pomc neurons. Also, constitutive expression of Xbp1s in Pomc neurons may improve leptin and insulin signaling in times of ER stress. Organotypic slice cultures were exposed to tunicamycin (tm), thapsigargin (tg), or dithiothreitol (dtt) in order to examine the effects of ER stress. Similar to previous observations, tm (15-30μM, 6h), tg (15μM, 6h), and dtt (1mM, 6h) suppressed the leptin-induced phosphorylation of STAT3 in the arcuate nucleus of our organotypic slice preparation (Figures 6A-6C). Notably, at 6 hours tm (30μM) and dtt (1mM) induced robust phosphorylation of the eukaryotic initiation factor 2 alpha (eif2α) and increased the accumulation of the ER resident molecular chaperone (Bip/Grp78), which are both known unfolded protein response (UPR) target genes (Supplemental Figure 2A). Additionally, tm (30μM, 6h) and dtt (1mM, 6h) potently increased mRNA for Bip and CHOP/GADD153 (Supplemental Figure 2B). In addition to leptin signaling, stimulation of DMSO-treated cultures with insulin led to an increase in AKT phosphorylation (Figures 6D and 6E). Pretreatment with tm suppressed the insulin-induced phosphorylation of AKT in the arcuate nucleus (Figures 6D and 6E). Thus, activators of ER stress suppress the activation of multiple signaling cascades activated by leptin and insulin in the arcuate nucleus of the hypothalamus.
Figure 6.
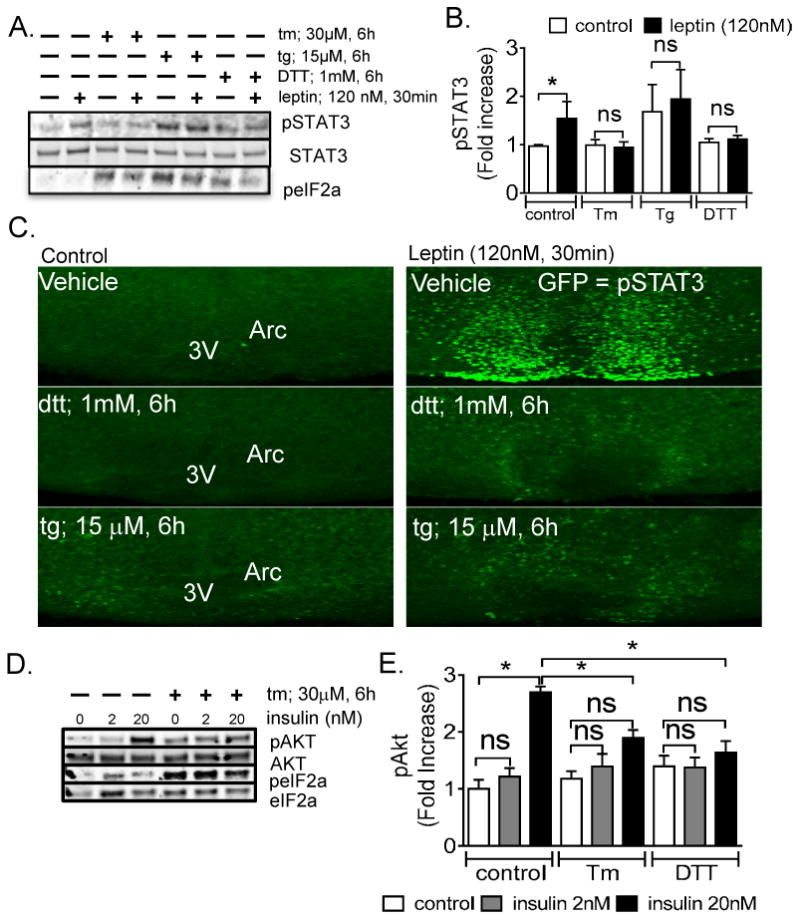
ER stress blunts the leptin and insulin activity in the Arcuate nucleus (A) Blots represent changes in the protein levels for leptin-induced phospho-STAT3 and phospho-eif2α in response to ER stress. (B) Quantitative densitometry for protein expression of leptin-induced pSTAT3 in control and ER stress activators. (C) ER stress blunts leptin-induced pSTAT3 immunoreactivity in the arcuate nucleus of the hypothalamus. (*P < 0.05, values are means ±SEM from 3-6 independent experiments, error bars indicate SEM) (D) Blots represent changes in the protein levels for insulin-induced phospho-AKT and phospho-eif2α in response to ER stress. (E) Quantitative densitometry for the protein expression of insulin-induced pAKT.
ER stress inhibits acute leptin and insulin signaling in arcuate Pomc neurons
Leptin directly activates while insulin directly inhibits arcuate Pomc neurons via a PI3K dependent mechanisms (Al-Qassab et al., 2009; Hill et al., 2008; Morton et al., 2006; Williams et al., 2010). We hypothesized that ER stress may blunt the leptin-induced activation and insulin-induced inhibition of arcuate Pomc neurons, which supports an ER stress-induced cellular resistance to the acute effects of leptin and insulin on metabolism.
Whole-cell recordings were performed on acute hypothalamic slices containing Pomc-GFP neurons within the arcuate nucleus (Parton et al., 2007; Ramadori et al., 2010). To better characterize the effects of ER stress on acute leptin and insulin signaling, we used the cre-loxP technology in order to enrich leptin responsive Pomc neurons in an acute hypothalamic slice preparation (Sohn et al., 2011). As expected, leptin failed to alter the membrane potential of Pomc-hrGFP (green) neurons that did not express Leprs. In current-clamp configuration, 75% of Pomc-hrGFP::Lepr-cre::tdtomato (green/red) neurons from PLT mice(Sohn et al., 2011) were depolarized in response to leptin (Figures 7A and 7B). Notably, none of the Pomc-hrGFP::Lepr-cre::tdtomato (green/red) neurons from PLT mice responded to insulin (supplementary table 1). A subset (40%) of Pomc-hrGFP neurons which did not express Leprs (green cells) were hyperpolarized in response to insulin (Figures 7C and 7D), but were unresponsive to leptin (supplementary table 1). Together, these data support a model in which the acute effects of leptin and insulin are functionally segregated in distinct arcuate Pomc neurons. This novel model enriches the population of leptin responsive neurons in arcuate Pomc neurons which allows for the rapid investigation of acute ER stress-induced leptin and insulin resistance.
Figure 7.
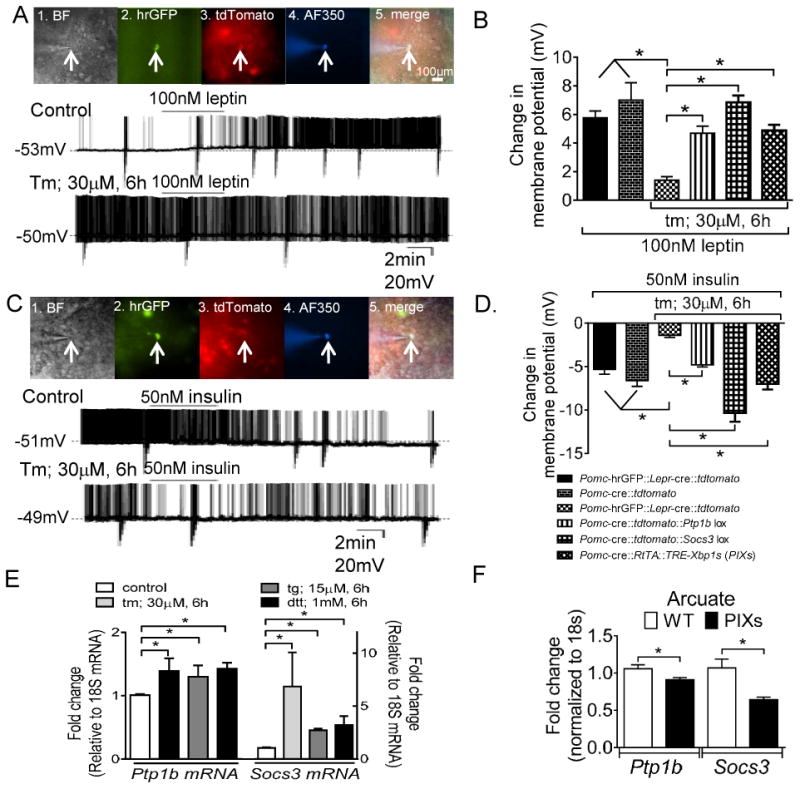
ER stress blunts the leptin-induced activation and the insulin-induced inhibition of Pomc neurons. (A) 1. Brightfield illumination of Pomc-hrGFP::Lepr-cre::tdtomato neuron from PLT mice. 2. and 3. The same neuron under FITC (hrGFP) and Alexafluor 594 (tdtomato) illumination. 4. Complete dialysis of Alexa Fluor 350 from the intracellular pipette. 5. Merge image illustrates colocalization of hr-GFP, tdtomato, and Alexa Fluor 350 indicative of a Pomc neuron which expresses Leprs. Control (above). Electrophysiological study demonstrates a Pomc-hrGFP::Lepr-cre::tdtomato (green/red) neuron that depolarized in response to leptin. Below demonstrates a current clamp recording of a separate Pomc-hrGFP::Lepr-cre::tdtomato (green/red) neuron in which ER stress blunted the leptin induced depolarization. (B) Histogram demonstrating that multiple activators of ER stress blunted the leptin-induced activation of Pomc neurons (n= 8-15 per group). Deletion of either Ptp1b or Socs3 restores the leptin-induced excitation of arcuate Pomc neurons after ER stress induction. Similarly, constitutive expression of Xbp1s in Pomc neurons restores the leptin-induced excitation of arcuate Pomc neurons after ER stress induction. *P < 0.05. Error bars indicate SEM. (C) 1. Brightfield illumination of Pomc-hrGFP neuron from PLT mice. 2. and 3. The same neuron under FITC (hrGFP) and Alexafluor 594 (tdtomato) illumination. 4. Complete dialysis of Alexa Fluor 350 from the intracellular pipette. 5. Merge illustrates colocalization of hr-GFP and Alexa Fluor 350 indicative of a Pomc neuron which does not expresses Leprs. Control (above): Electrophysiological study demonstrates a Pomc-hrGFP (green) neuron is hyperpolarized in response to insulin. Below: A separate Pomc-hrGFP (green) neuron in which ER stress blunts the insulin induced hyperpolarization. (D) Histogram illustrating that chemical activation of ER stress blunts the insulin-induced inhibition of arcuate Pomc neurons (n= 8-18 per group). Deletion of either Ptp1b or Socs3 restores the insulin-induced inhibition of arcuate Pomc neurons after ER stress induction. *P < 0.05, Error bars indicate SEM. (E) Relative mRNA expression of Socs3 and Ptp1b in organotypic slices following pretreatment with ER stress activators. (F) Relative mRNA of Ptp1b and Socs3 in the arcuate nucleus from PIXs and WT mice fed HFD-Dox. (*P < 0.05, values are means ±SEM from 3-6 independent experiments, error bars indicate SEM)
Pretreatment with either tm (30μM, 6h; t(23) = 8.286, p < 0.0001) or tg (15μM, 6h; t(21) = 9.894, p < 0.0001) blunted the ability of leptin to depolarize Pomc-hrGFP::Lepr-cre::tdtomato (green/red) neurons from PLT mice (Figures 7A and 7B). Similar to the observed blunting of acute leptin action in arcuate Pomc neurons pretreatment with tm (30μM, 6h; t(17) = 7.142, p < 0.0001, Figures 7C and 7D) or tg (15μM, 6h; t(11) = 5.169, p < 0.001, Figure 7D) blunted the ability of insulin to hyperpolarize Pomc-hrGFP (green) neurons from PLT mice. Similar results were obtained in Pomc neurons from organotypic hypothalamic slices (Supplemental Figure 3).
Cellular Mechanism for ER-stress induced acute leptin and insulin resistance in Arcuate Pomc neurons includes Ptp1b, Socs3, and Xbp1s
Neuronal protein tyrosine phosphatase 1b (Ptp1b) is required for body weight homeostasis and leptin action (Bence et al., 2006). Mice lacking Ptp1b selectively in Pomc neurons exhibited increased energy expenditure resulting in a resistance to HFD induced obesity (Banno et al., 2010). These mice were also more sensitive to the acute effect of leptin to reduce both food intake and body weight supportive of a role for Ptp1b in the modulation of leptin signaling involved in the acute modulation of Pomc cellular activity. ER stress inhibits leptin signaling via Ptp1b signaling in a dispersed cell culture system and in the liver (Delibegovic et al., 2009; Hosoi et al., 2008). In support of this, we found that pretreatment of hypothalamic slices with activators of ER stress resulted in increased mRNA for both Ptp1b and Socs3 (p < 0.05; Figure 7E). Constitutive expression of Xbp1s in Pomc neurons suppressed the expression of both Ptp1b and Socs3 in the Arcuate nucleus (for Ptp1b: t(9) = 2.371, p<0.05; for Socs3: t(9) = 3.179, p<0.05; Figure 7F). Thus, we hypothesized that Ptp1b and/or Socs3 may be required in Pomc neurons to regulate acute ER stress induced leptin and insulin resistance. Also, Xbp1s alone in Pomc neurons may improve acute leptin and insulin signaling subsequent to ER stress.
In order to first assess the requirement of Ptp1b or Socs3 in the acute ER stress-induced leptin and insulin resistance in Pomc neurons we generated Pomc reporter mice by mating Pomc-cre mice with the tdtomato reporter mouse (Jackson Laboratory, #007908). Similar to previous reports (Al-Qassab et al., 2009; Hill et al., 2008; Williams et al., 2010), Pomc neurons from Pomc-cre::tdtomato mice were activated in response to leptin and inhibited in response to insulin (Figures 7B and 7D). Pomc-cre::tdtomato reporter mice were subsequently mated to either Ptp1b-lox (Pomc-cre::tdtomato::Ptp1b-lox) or Socs3-lox (Pomc-cre::tdtomato::Socs3-lox) mice. Tunicamycin failed to blunt the leptin-induced activation of Pomc neurons selectively deficient for either Ptp1b or Socs3 (for Pomc-cre::tdtomato::Ptp1b-lox t(18) = 1.918, p > 0.05; for Pomc-cre::tdtomato::Socs3-lox t(18) = 1.167, p > 0.05; Figure 7B). Similarly, tunicamycin failed to blunt the insulin-induced inhibition of Pomc neurons selectively deficient for either Ptp1b (for Pomc-cre::tdtomato::Ptp1b-lox t(14) = 0.9115, p > 0.05; Figure 7D). Notably, Pomc neurons deficient for Socs3 were significantly more responsive to insulin compared to Pomc neurons deficient for Ptp1b alone or control mice (for insulin in Pomc-cre::tdtomato::Socs3-lox cells compared to Pomc-cre::tdtomato::Ptp1b-lox mice t(12) = 4.457, p < 0.05; for insulin in Pomc-cre::tdtomato::Socs3-lox cells compared to control PLT mice t(14) = 3.849, p < 0.05 Figure 7D). Although ER stress stimulates both Ptp1b and Socs3 (Figure 7E), these data suggest that Soc3 may have a more potent ER stress induced activity related to the suppression of acute leptin and insulin signaling in arcuate Pomc neurons.
In order to assess the role of Xbp1s in the acute ER stress-induced leptin and insulin resistance, Pomc reporter PIXs mice were generated by mating PIXs mice with tdtomato reporter mouse (Jackson Laboratory, #007908). Tunicamycin failed to blunt the leptin-induced activation of Pomc neurons from PIXs mice fed a doxycycline enriched diet (t(19) = 1.320, p > 0.05; Figure 7B). Similarly, tunicamycin failed to blunt the insulin-induced inhibition of Pomc neurons from PIXs mice fed a doxycycline enriched diet (t(17) = 0.3917, p > 0.05; Figure 7D).
Discussion
Constitutive expression of Xbp1s in Pomc neurons (PIXs mice) increases energy expenditure, desensitizes mice to diet-induced obesity independent of changes in ad-libitum food intake, and directly (i.e. independently of changes in body weight) improves glucose homeostasis with decreases in circulating insulin and blood glucose. In addition to these physiological aberrations, PIXs mice were sensitized the acute effects of leptin pharmacologically to suppress food intake. On a cellular level, PIXs mice exhibited decreased Ptp1b and Socs3 expression concomitant with improved leptin and insulin signaling in Pomc neurons subsequent to ER stress. Increased XBP1s expression only in Pomc neurons was sufficient to upregulate of the XBP1s axis in the liver. Together, these data support a model in which a “fed” signal in Pomc neurons abrogates high-fat diet induced obesity while at the same time induces a “fed” transcriptional program in the liver improving glucose homeostasis.
ER stress pathways in Pomc neurons regulates hepatic glucose production. It is of particular interest that Xbp1s expression in Pomc neurons has profound effects on body weight and glucose homeostasis. Cellular stress and inflammatory pathways including ER stress and the UPR have been linked to leptin and insulin resistance as well as obesity and diabetes (Kaneto et al., 2005; Tsiotra and Tsigos, 2006; Wellen and Hotamisligil, 2005). Notably, Xbp-1+/− heterozygous mice are more sensitive to diabetes caused by obesity and high-fat diet (Ozcan et al., 2004). Similar results were obtained in mice deficient for Xbp1s selectively in neurons (Ozcan et al., 2009). The decreased body weight observed in mice which constitutively express Xbp1s in Pomc neurons was dependent upon a hypermetabolic phenotype (increased energy expenditure and heat production) independent of altered food intake. Notably, these responses are phenotypic signatures of improved leptin action within Pomc neurons (Balthasar et al., 2004; Berglund et al., 2012; Williams et al., 2011). The hypermetabolic phenotype was also supported by increased markers for thermogenesis in BAT as well as iWAT (Cypess et al., 2009; van Marken Lichtenbelt et al., 2009; Virtanen et al., 2009; Wu et al., 2012). The increased thermogenesis of iWAT suggests that Xbp1s expression in Pomc neurons promotes browning/beiging of white adipose tissues contributing to improvements in body weight. Similar to Xbp1s activity in the liver (Deng et al., 2013), we found that constitutive expression of Xbp1s in Pomc neurons alone was also sufficient to improve blood glucose and insulin levels, supportive of improved insulin signaling and reminiscent of a fed state even in the absence of caloric intake.
A neuronal cell non-autonomous regulation of Xbp1 transcription in the intestinal cell of C. elegans has recently been described (Taylor and Dillin Cell 2013). Notably, the improved glucoregulation in the current study may be due to elevated Xbp1s levels in the liver via a similar cell nonautonomous mechanism. Hypothalamic neuronal regulation of peripheral tissues such as liver and pancreas is likely due to hypothalamic regulation of the activity of key autonomic control neurons in the brainstem and spinal cord. Although, this occurs via multisynaptic connections that may involve melanocortin 4 receptor expressing neurons in key autonomic circuits (Rossi et al., 2011; Sohn et al., 2013), it's currently unclear how Pomc neurons propagate the Xbp1s transcriptional signal to the liver.
Another salient finding is the relationship between Xbp1s and both Ptp1b and Socs3 in the ER stress induced acute leptin and insulin resistance of Arcuate Pomc neurons. Recently, ER stress has been shown to induce leptin resistance within the hypothalamus as well as peripheral insulin resistance in the liver and muscle (Ozcan et al., 2009; Ozcan et al., 2006; Thaler and Schwartz, 2010; Thaler et al., 2012). In the current study, our findings point to a fundamental role of ER-stress in regulating leptin and insulin signaling in hypothalamic Pomc neurons. Classical suppressors of cytokine signaling have been likely candidates in ER stress induced leptin and insulin resistance (Howard and Flier, 2006; Myers et al., 2008; White et al., 2008; Zabolotny et al., 2008) . In particular, both Socs3 and Ptp1b are increased within the hypothalamus in a state of excess nutrition or obesity (Bjorbaek et al., 1998; Enriori et al., 2007; Munzberg et al., 2004; White et al., 2008; Zabolotny et al., 2008). Brain specific Socs3 knockout mice or haploinsufficient mice were significantly protected against the development of DIO associated with leptin resistance (Howard et al., 2004; Mori et al., 2004). Ptp1b knockout mice were similarly resistant to DIO (Bence et al., 2006; Cook and Unger, 2002; Zabolotny et al., 2002). Notably, both Ptp1b and Socs3 can be induced by non-cytokine stimuli, including ER stress (Hosoi et al., 2008; Ozcan et al., 2009; Yoshimura et al., 2007; Zhang et al., 2008). Ptp1b also mediates ER stress-induced hypothalamic leptin resistance independent of Socs3 activity (Hosoi et al., 2008). Recently, liver specific deficiency of Ptp1b attenuated the induction of ER stress in response to high-fat diet (Delibegovic et al., 2009). These data are supported in the current study with the demonstration that ER stress stimulates Ptp1b and Socs3 in the arcuate nucleus of the hypothalamus. Pomc neurons selectively deficient for either Ptp1b or Socs3 demonstrated improved acute leptin and insulin signaling subsequent to ER stress activation. Constitutive expression of Xbp1s in Pomc neurons lowered mRNA levels of Ptp1b and Socs3 at the same time blunting the ability of strong inducers of ER stress to induce cellular leptin and insulin resistance in Pomc neurons. It should be noted that several groups have reported that ER stress stimulates the production and phosphatase activity of PTP1B and Socs3 in multiple tissues (Agouni et al., 2011; Bettaieb et al., 2011; Hosoi et al., 2008; Panzhinskiy et al., 2013). However, it has been unclear how ER stress induced PTP1B or SOCS3 activity may affect acute hypothalamic leptin and insulin signaling. In particular, PTP1B has been suggested to operate as a component of the UPRosome, selectively controlling IRE1 activation and signaling (Gu et al., 2004). These data suggest the PTP1b dependent ER stress induced acute leptin and insulin resistance in the current study may be explained by a lack of IRE1-XBP1 activation. In contrast, ER stress-induced sulfhydration of PTP1B inhibits it's native activity and thereby promotes PERK activity during the response to ER stress(Krishnan et al., 2011). Thus although it is apparent that PTP1B activity is intertwined with ER stress and the UPR, the current study extends previous observations and supports a requirement of PTP1B and SOCS3 in ER stress-induced acute leptin and insulin resistance of POMC neurons.
Overall, these observations underscore a physiologically important role for ER stress and the UPR to alter the ability of arcuate Pomc neurons to properly respond to humoral signals. ultimately abrogating diet-induced obesity and diabetes. Notably, the improvements in leptin and insulin signaling observed in the current study during times of ER stress link Xbp1s with both Ptp1b and Socs3 in Pomc neurons. These data also demonstrate that Pomc neurons induce changes in the metabolic flux in the liver in a cell nonautonomous mechanism which contributes to improved glucose homeostasis independent of altered body weight. In addition, these data clarify the roles of these molecules in the regulation of metabolism and may highlight useful targets for regulating obesity and related metabolic disorders.
Experimental Procedures
Animals
Male (4- 16-week-old) pathogen-free POMC-hrGFP mice(Parton et al., 2007; Ramadori et al., 2010) were used for all experiments. To identify POMC neurons with or without leptin receptors, we generated PLT mice as previously described(Sohn et al., 2011). Briefly, LepR reporter mice were made by mating LepR-cre mice(Scott et al., 2009) with the tdtomato reporter mouse (Jackson Laboratory, #007908). LepR-cre::tdtomato reporter mice were subsequently mated with POMC-hrGFP mice to produce POMC::LepR-cre::tdtomato (PLT) mice. To identify Pomc neurons with or without Ptp1b or Socs3, we generated Pomc-cre::tdtomato::Ptp1b lox or Pomc-cre::tdtomato::Socs3 lox mice, respectively. Briefly, Pomc-cre reporter mice were made by mating Pomc-cre mice with the tdtomato reporter mouse. Pomc-cre::tdtomato mice were subsequently mated with either Ptp1b lox or Socs3 lox mice. Subsequent matings generated mice that were deficient for either Ptp1b or Socs3 in Pomc neurons.
All mice were housed under standard laboratory conditions (12 hr on/off; lights on at 7:00 a.m.) and temperature-controlled environment with food and water available ad libitum. All experiments were performed in accordance with the guidelines established by the National Institute of Health Guide for the Care and Use of Laboratory Animals, and approved by the University of Texas Institutional Animal Care and Use Committee.
Western Blot Analysis
The arcuate nucleus was microdissected with a scalpel under a microscope age matched male mice. Whole cell proteins were extracted by homogenizing the hypothalamic blocks in IP Lysis buffer [ 25mM Tris-HCl pH 7.4, 150mM NaCl, 1mM EDTA, 1% NP-40 and 5% glycerol (87787 Pierce)], with protease and phosphatase inhibitors cocktails (1:100, 78440, Pierce). Equal amounts of the samples (10 μg) were separated by SDS-PAGE and transferred to a nitrocellulose membrane by electroblotting. Antibodies used here are the following: A phospho-STAT3 antibody (1:1,000, Cell Signaling Technology, 9131), a phospho-ERK antibody (1:1,000, Cell Signaling Technology, 4370), an antibody against STAT3 (1:1,000, Cell Signaling Technology, 9139), pAKT (1:1,000, Cell Signaling, # 4060), AKT (1:1,000, Cell Signaling, # 4691), an antibody against SOCS3 (1:600, Abcam, ab16030), a PTP1B antibody (1:333, Abcam, ab52650), Bip (1:1,000, Chop Cell Signaling Technology, 3177), eif2α (1:1,000, Cell Signaling, # 3398), XBP1 (1:200, Santa Cruz Biotechnology Inc, SC-7160), and anti β-actin antibodies (1:10,000, beta Actin antibody, mAbcam 8226). After incubation in primary antibodies for 72 h, the membranes were incubated for 1 h in HRP-conjugated secondary antibodies (1:7,000, Southern Biotech, 1010-05 and 4050-05), followed by chemiluminescent detection using West Pico Chemiluminescent Substrate (Thermo Fisher Scientific Inc.). To measure the fluorescent intensity, the Odyssey IR imaging system (LI-COR Biosciences) was used. After incubation for primary antibodies, the membrane was incubated in the secondary antibody conjugated to a fluorescent entity: IRDye 800-conjugated goat antirabbit IgG and/or Alexa Fluor 680-conjugated goat antimouse IgG (dilution 1:10,000) with gentle agitation for 1 h at room temperature. At the end of the incubation period, membranes were washed twice with phosphate -buffered saline (PBS) with 0.05% Tween (PBS-T). The membrane was visualized and analyzed on the Odyssey IR imaging system (LI-COR Biosciences). Phospho-proteins were normalized to the levels of the corresponding total protein.
Electrophysiology
Whole-cell patch-clamp recordings from POMC-hrGFP neurons maintained in hypothalamic slice preparations and data analysis were performed as previously described(Hill et al., 2008). Briefly, 4- to 16-week-old male mice were anesthetized and transcardially perfused with a modified ice-cold artificial CSF (ACSF) (described below), in which an equiosmolar amount of sucrose was substituted for NaCl. The mice were then decapitated, and the entire brain was removed, and immediately submerged in ice-cold, carbogen-saturated (95% O2 and 5% CO2) ACSF (126 mM NaCl, 2.8 mM KCl, 1.2 mM MgCl2, 2.5 mM CaCl2, 1.25 mM NaH2PO4, 26 mM NaHCO3, and 5 mM glucose). Coronal sections (250 μm) were cut with a Leica VT1000S Vibratome and then incubated in oxygenated ACSF at room temperature for at least 1 hr before recording. Slices were transferred to the recording chamber and allowed to equilibrate for 10–20 min before recording. The slices were bathed in oxygenated ACSF (32°C–34°C) at a flow rate of ∼2 ml/min.
The pipette solution for whole-cell recording was modified to include an intracellular dye (Alexa Fluor 594 or Alexa Fluor 350) for whole-cell recording: 120 mM K-gluconate, 10 mM KCl, 10 mM HEPES, 5 mM EGTA, 1 mM CaCl2, 1 mM MgCl2, and 2 mM MgATP, 0.03 mM Alexa Fluor 594 or Alexa Fluor 350 hydrazide dye (pH 7.3). Epifluorescence was briefly used to target fluorescent cells, at which time the light source was switched to infrared differential interference contrast imaging to obtain the whole-cell recording (Zeiss Axioskop FS2 Plus equipped with a fixed stage and a QuantEM:512SC electron-multiplying charge-coupled device camera). Electrophysiological signals were recorded using an Axopatch 700B amplifier (Molecular Devices), low-pass filtered at 2–5 kHz, and analyzed offline on a PC with pCLAMP programs (Molecular Devices). Recording electrodes had resistances of 2.5–5 MΩ when filled with the K-gluconate internal solution. Input resistance was assessed by measuring voltage deflection at the end of the response to a hyperpolarizing rectangular current pulse steps (500 ms of −10 to −50 pA).
Leptin (100 nM; provided by A.F. Parlow, through the National Hormone and Peptide Program) or insulin (50 nM, Humulin-R 100 U/ml; Lilly), were added to the ACSF for specific experiments. Solutions containing leptin or insulin were typically perfused for 2–4 min. A drug effect was required to be associated temporally with peptide application, and the response had to be stable within a few minutes. A neuron was considered depolarized or hyperpolarized if a change in membrane potential was at least 2 mV in amplitude.
Energy expenditure and locomotor activity
Weight- and body composition–matched 8-week-old WT and PIXs mice were used for metabolic assessment. Three separate cohorts of animals were used to produce the metabolic data, with measurements sorted into 12-hour-light/dark periods. To exclude the possibility that changes in body weight or composition would contribute to energy expenditure measurements, metabolic assessment was done using weight- and body composition–matched mice(Butler and Kozak, 2010; Tschop et al., 2012). Data, where applicable, were also normalized to lean body mass, by raising metabolic data to the power of ×0.75 (or by expressing data on a per animal basis)(Butler and Kozak, 2010; Tschop et al., 2012).
Mice were first acclimatized to the metabolic cages and housed individually for 4 days before measurements were taken. Mice were analyzed in the metabolic chambers for 4 days and were provided with food ad libitum. Energy expenditure was measured by indirect calorimetry, while locomotor activity was assessed using an infrared light beam detection system (Labmaster, TSE Systems GmbH). Data was collected using a TSE Labmaster monitoring system (TSE Systems GmbH). Locomotor activity and energy expenditure were determined for both the 12-hour-light and 12-hour-dark cycle as well as for the whole 24-hour period. Data were averaged over the 4-day period of measurement.
Body weight/composition, and fat distribution
Animals were weaned and maintained on a high-fat diet enriched with doxycycline (600mg/kg) at 4 to 5 weeks of age. Body weight was measured weekly up to 25 weeks. Body fat composition of 25-week-old ad libitum fed mice was assessed using nuclear magnetic resonance spectroscopy using an NMR spectrometer (EchoMRI). For fat distribution, mice were anesthetized with 1% isoflurane inhalation and then the trunk (from base of the skull as the spinal canal begins to widen and the distal end of the tibia) of each mouse was scanned at an isotropic voxel size of 93 μm (80 kV, 450 μA, and 100 ms integration time) using the eXplore Locus micro-CT scanner (GE Health Care). Three-dimensional images were reconstructed from two-dimensional grayscale image slices and visualized using Microview Software (GE Medical System). Density values for soft tissue and bone were calibrated from a phantom (GE Health Care) containing air bubble, water, and hydroxyl apatite rod. The separation of fat regions was obtained from the appropriate grayscale value (upper threshold, −165; lower threshold, −360). The abdominal muscular wall was used as the differentiation line to separate visceral adipose tissue from subcutaneous adipose tissue. The contour lines were drawn around the viscera and three-dimensional ROI was generated. The visceral fat was determined from the histogram of these segmented viscera using the same thresholds. Subcutaneous fat was obtained by subtracting visceral fat from the total body fat.
Data Analysis
Statistical analysis was carried out using GraphPad 5 (GraphPad) software. All data were evaluated using a 2-tailed Student's t test with a P value of less than 0.05 being considered significant. In all instances, data are presented as mean ± SEM. Body weight curves were compared using a linear regression analysis. . Degrees of freedom (DF) for t statistics are marked as t(DF).
Supplementary Material
Article Highlights.
Xbp1s in Pomc neurons protects against ER stress induced leptin/insulin resistance
Xbp1s in Pomc neurons improves glucose levels and hepatic insulin sensitivity
Pomc-specific xbp1s protects against high-fat diet induced obesity
Pomc-specific xbp1s regulates the UPRER in the liver
Acknowledgments
We thank Dr. Jeffrey Friedman (Rockefeller University) for kindly providing us with the Lepr-cre mice. We also thank Dr. Bradford Lowell (Beth Israel Deaconess Medical Center) for kindly providing us with the Pomc-hrGFP mice. This work was supported by grants to K.W.W. (K01DK087780), T.L. (American Diabetes Association 7-11-MN-16), M.F. (American Heart Association – 9SDG2080223), J.-W.S. (American Heart Association 12POST8860007), Y.D. (American Diabetes Association 7-08-MN-53), E.D.B. (NIH F32 DK092083 and K01 DK098317), X.K. (American Heart Association 13POST16710016), T.F. (Juvenile Diabetes Research Foundation – 3-2011-405), and J.K.E. (R01DK53301, R01DK088423, and RL1DK081185). This work was also supported by PL1 DK081182 and UL1RR024923, as well as P01DK088761 (P.E.S. and J.K.E.) and R01DK55758 (P.E.S.).
Footnotes
Author Contributions: K.W.W., T.L., X.K. and M.F. are co-first authors. K.W.W. designed all experiments and performed all experiments except gene expression and immunoblotting, analyzed the data, and wrote the manuscript. T.L. designed and performed experiments except gene expression and immunoblotting, analyzed the data, and wrote the manuscript. X.K. analyzed gene expression in adipose depots, performed immunohistochemistry in BAT and iWAT, analyzed the data, and reviewed the manuscript. M.F. designed and performed organotypic slice experiments including immunoblotting and gene expression, analyzed data, and reviewed the manuscript. E.D.B. designed and performed hyperinsulinemic euglycemic clamp experiments, analyzed the data, and reviewed the manuscript. Y.F., J-W S., Z.D., Y.G., T.L., L.J., T.F., D.K., M.M.S., S.L., C.E.L., K.S., and Y.C. assisted performing experiments. P.E.S. and J.K.E. are co-senior authors – they supervised development of the mouse models, designed experiments, and edited the manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agouni A, Mody N, Owen C, Czopek A, Zimmer D, Bentires-Alj M, Bence KK, Delibegovic M. Liver-specific deletion of protein tyrosine phosphatase (PTP) 1B improves obesity- and pharmacologically induced endoplasmic reticulum stress. Biochem J. 2011;438:369–378. doi: 10.1042/BJ20110373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Qassab H, Smith MA, Irvine EE, Guillermet-Guibert J, Claret M, Choudhury AI, Selman C, Piipari K, Clements M, Lingard S, et al. Dominant role of the p110beta isoform of PI3K over p110alpha in energy homeostasis regulation by POMC and AgRP neurons. Cell Metab. 2009;10:343–354. doi: 10.1016/j.cmet.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balthasar N, Coppari R, McMinn J, Liu SM, Lee CE, Tang V, Kenny CD, McGovern RA, Chua SC, Jr, Elmquist JK, et al. Leptin receptor signaling in POMC neurons is required for normal body weight homeostasis. Neuron. 2004;42:983–991. doi: 10.1016/j.neuron.2004.06.004. [DOI] [PubMed] [Google Scholar]
- Banno R, Zimmer D, De Jonghe BC, Atienza M, Rak K, Yang W, Bence KK. PTP1B and SHP2 in POMC neurons reciprocally regulate energy balance in mice. The Journal of clinical investigation. 2010;120:720–734. doi: 10.1172/JCI39620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belgardt BF, Bruning JC. CNS leptin and insulin action in the control of energy homeostasis. Annals of the New York Academy of Sciences. 2010;1212:97–113. doi: 10.1111/j.1749-6632.2010.05799.x. [DOI] [PubMed] [Google Scholar]
- Belteki G, Haigh J, Kabacs N, Haigh K, Sison K, Costantini F, Whitsett J, Quaggin SE, Nagy A. Conditional and inducible transgene expression in mice through the combinatorial use of Cre-mediated recombination and tetracycline induction. Nucleic Acids Res. 2005;33:e51. doi: 10.1093/nar/gni051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bence KK, Delibegovic M, Xue B, Gorgun CZ, Hotamisligil GS, Neel BG, Kahn BB. Neuronal PTP1B regulates body weight, adiposity and leptin action. Nat Med. 2006;12:917–924. doi: 10.1038/nm1435. [DOI] [PubMed] [Google Scholar]
- Berglund ED, Vianna CR, Donato J, Jr, Kim MH, Chuang JC, Lee CE, Lauzon DA, Lin P, Brule LJ, Scott MM, et al. Direct leptin action on POMC neurons regulates glucose homeostasis and hepatic insulin sensitivity in mice. The Journal of clinical investigation. 2012;122:1000–1009. doi: 10.1172/JCI59816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettaieb A, Liu S, Xi Y, Nagata N, Matsuo K, Matsuo I, Chahed S, Bakke J, Keilhack H, Tiganis T, et al. Differential regulation of endoplasmic reticulum stress by protein tyrosine phosphatase 1B and T cell protein tyrosine phosphatase. J Biol Chem. 2011;286:9225–9235. doi: 10.1074/jbc.M110.186148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjorbaek C, Elmquist JK, Frantz JD, Shoelson SE, Flier JS. Identification of SOCS-3 as a potential mediator of central leptin resistance. Mol Cell. 1998;1:619–625. doi: 10.1016/s1097-2765(00)80062-3. [DOI] [PubMed] [Google Scholar]
- Bookout AL, Mangelsdorf DJ. Quantitative real-time PCR protocol for analysis of nuclear receptor signaling pathways. Nucl Recept Signal. 2003;1:e012. doi: 10.1621/nrs.01012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler AA, Kozak LP. A recurring problem with the analysis of energy expenditure in genetic models expressing lean and obese phenotypes. Diabetes. 2010;59:323–329. doi: 10.2337/db09-1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Considine RV, Sinha MK, Heiman ML, Kriauciunas A, Stephens TW, Nyce MR, Ohannesian JP, Marco CC, McKee LJ, Bauer TL, et al. Serum immunoreactive-leptin concentrations in normal-weight and obese humans. The New England journal of medicine. 1996;334:292–295. doi: 10.1056/NEJM199602013340503. [DOI] [PubMed] [Google Scholar]
- Cook WS, Unger RH. Protein tyrosine phosphatase 1B: a potential leptin resistance factor of obesity. Dev Cell. 2002;2:385–387. doi: 10.1016/s1534-5807(02)00158-2. [DOI] [PubMed] [Google Scholar]
- Cravo RM, Margatho LO, Osborne-Lawrence S, Donato J, Jr, Atkin S, Bookout AL, Rovinsky S, Frazao R, Lee CE, Gautron L, et al. Characterization of Kiss1 neurons using transgenic mouse models. Neuroscience. 2011;173:37–56. doi: 10.1016/j.neuroscience.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB, Kuo FC, Palmer EL, Tseng YH, Doria A, et al. Identification and importance of brown adipose tissue in adult humans. The New England journal of medicine. 2009;360:1509–1517. doi: 10.1056/NEJMoa0810780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delibegovic M, Zimmer D, Kauffman C, Rak K, Hong EG, Cho YR, Kim JK, Kahn BB, Neel BG, Bence KK. Liver-specific deletion of protein-tyrosine phosphatase 1B (PTP1B) improves metabolic syndrome and attenuates diet-induced endoplasmic reticulum stress. Diabetes. 2009;58:590–599. doi: 10.2337/db08-0913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Y, Wang ZV, Tao C, Gao N, Holland WL, Ferdous A, Repa JJ, Liang G, Ye J, Lehrman MA, et al. The Xbp1s/GalE axis links ER stress to postprandial hepatic metabolism. The Journal of clinical investigation. 2013;123:455–468. doi: 10.1172/JCI62819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enriori PJ, Evans AE, Sinnayah P, Jobst EE, Tonelli-Lemos L, Billes SK, Glavas MM, Grayson BE, Perello M, Nillni EA, et al. Diet-induced obesity causes severe but reversible leptin resistance in arcuate melanocortin neurons. Cell Metab. 2007;5:181–194. doi: 10.1016/j.cmet.2007.02.004. [DOI] [PubMed] [Google Scholar]
- Frederich RC, Hamann A, Anderson S, Lollmann B, Lowell BB, Flier JS. Leptin levels reflect body lipid content in mice: evidence for diet-induced resistance to leptin action. Nat Med. 1995;1:1311–1314. doi: 10.1038/nm1295-1311. [DOI] [PubMed] [Google Scholar]
- Friedman JM. Obesity in the new millennium. Nature. 2000;404:632–634. doi: 10.1038/35007504. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Jones JE, Olson D, Hill J, Lee CE, Gautron L, Choi M, Zigman JM, Lowell BB, Elmquist JK. Monitoring FoxO1 localization in chemically identified neurons. J Neurosci. 2008;28:13640–13648. doi: 10.1523/JNEUROSCI.4023-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Williams KW, Gautron L, Elmquist JK. Induction of leptin resistance by activation of cAMP-Epac signaling. Cell Metab. 2011;13:331–339. doi: 10.1016/j.cmet.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahwiler BH, Llano I. Sodium and potassium conductances in somatic membranes of rat Purkinje cells from organotypic cerebellar cultures. J Physiol. 1989;417:105–122. doi: 10.1113/jphysiol.1989.sp017793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu F, Nguyen DT, Stuible M, Dube N, Tremblay ML, Chevet E. Protein-tyrosine phosphatase 1B potentiates IRE1 signaling during endoplasmic reticulum stress. J Biol Chem. 2004;279:49689–49693. doi: 10.1074/jbc.C400261200. [DOI] [PubMed] [Google Scholar]
- Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:367–377. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JW, Elias CF, Fukuda M, Williams KW, Berglund ED, Holland WL, Cho YR, Chuang JC, Xu Y, Choi M, et al. Direct insulin and leptin action on pro-opiomelanocortin neurons is required for normal glucose homeostasis and fertility. Cell Metab. 2010;11:286–297. doi: 10.1016/j.cmet.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JW, Williams KW, Ye C, Luo J, Balthasar N, Coppari R, Cowley MA, Cantley LC, Lowell BB, Elmquist JK. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. The Journal of clinical investigation. 2008;118:1796–1805. doi: 10.1172/JCI32964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoi T, Sasaki M, Miyahara T, Hashimoto C, Matsuo S, Yoshii M, Ozawa K. Endoplasmic reticulum stress induces leptin resistance. Mol Pharmacol. 2008;74:1610–1619. doi: 10.1124/mol.108.050070. [DOI] [PubMed] [Google Scholar]
- Howard JK, Cave BJ, Oksanen LJ, Tzameli I, Bjorbaek C, Flier JS. Enhanced leptin sensitivity and attenuation of diet-induced obesity in mice with haploinsufficiency of Socs3. Nat Med. 2004;10:734–738. doi: 10.1038/nm1072. [DOI] [PubMed] [Google Scholar]
- Howard JK, Flier JS. Attenuation of leptin and insulin signaling by SOCS proteins. Trends Endocrinol Metab. 2006;17:365–371. doi: 10.1016/j.tem.2006.09.007. [DOI] [PubMed] [Google Scholar]
- Kahn SE, Hull RL, Utzschneider KM. Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006;444:840–846. doi: 10.1038/nature05482. [DOI] [PubMed] [Google Scholar]
- Kaneto H, Nakatani Y, Kawamori D, Miyatsuka T, Matsuoka TA, Matsuhisa M, Yamasaki Y. Role of oxidative stress, endoplasmic reticulum stress, and c-Jun N-terminal kinase in pancreatic beta-cell dysfunction and insulin resistance. The international journal of biochemistry & cell biology. 2005;37:1595–1608. doi: 10.1016/j.biocel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Konner AC, Bruning JC. Selective insulin and leptin resistance in metabolic disorders. Cell Metab. 2012;16:144–152. doi: 10.1016/j.cmet.2012.07.004. [DOI] [PubMed] [Google Scholar]
- Krishnan N, Fu C, Pappin DJ, Tonks NK. H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci Signal. 2011;4:ra86. doi: 10.1126/scisignal.2002329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao X, Fujiwara Y, Chapdelaine A, Yang H, Orkin SH. Activation of EGFP expression by Cre-mediated excision in a new ROSA26 reporter mouse strain. Blood. 2001;97:324–326. doi: 10.1182/blood.v97.1.324. [DOI] [PubMed] [Google Scholar]
- Mori H, Hanada R, Hanada T, Aki D, Mashima R, Nishinakamura H, Torisu T, Chien KR, Yasukawa H, Yoshimura A. Socs3 deficiency in the brain elevates leptin sensitivity and confers resistance to diet-induced obesity. Nat Med. 2004;10:739–743. doi: 10.1038/nm1071. [DOI] [PubMed] [Google Scholar]
- Morton GJ, Cummings DE, Baskin DG, Barsh GS, Schwartz MW. Central nervous system control of food intake and body weight. Nature. 2006;443:289–295. doi: 10.1038/nature05026. [DOI] [PubMed] [Google Scholar]
- Munzberg H, Flier JS, Bjorbaek C. Region-specific leptin resistance within the hypothalamus of diet-induced obese mice. Endocrinology. 2004;145:4880–4889. doi: 10.1210/en.2004-0726. [DOI] [PubMed] [Google Scholar]
- Myers MG, Cowley MA, Munzberg H. Mechanisms of leptin action and leptin resistance. Annu Rev Physiol. 2008;70:537–556. doi: 10.1146/annurev.physiol.70.113006.100707. [DOI] [PubMed] [Google Scholar]
- Myers MG, Jr, Heymsfield SB, Haft C, Kahn BB, Laughlin M, Leibel RL, Tschop MH, Yanovski JA. Challenges and opportunities of defining clinical leptin resistance. Cell Metab. 2012;15:150–156. doi: 10.1016/j.cmet.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers MG, Jr, Olson DP. Central nervous system control of metabolism. Nature. 2012;491:357–363. doi: 10.1038/nature11705. [DOI] [PubMed] [Google Scholar]
- Ozcan L, Ergin AS, Lu A, Chung J, Sarkar S, Nie D, Myers MG, Jr, Ozcan U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009;9:35–51. doi: 10.1016/j.cmet.2008.12.004. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science (New York, NY) 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science (New York, NY) 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panzhinskiy E, Hua Y, Culver B, Ren J, Nair S. Endoplasmic reticulum stress upregulates protein tyrosine phosphatase 1B and impairs glucose uptake in cultured myotubes. Diabetologia. 2013;56:598–607. doi: 10.1007/s00125-012-2782-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton LE, Ye CP, Coppari R, Enriori PJ, Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, et al. Glucose sensing by POMC neurons regulates glucose homeostasis and is impaired in obesity. Nature. 2007;449:228–232. doi: 10.1038/nature06098. [DOI] [PubMed] [Google Scholar]
- Ramadori G, Fujikawa T, Fukuda M, Anderson J, Morgan DA, Mostoslavsky R, Stuart RC, Perello M, Vianna CR, Nillni EA, et al. SIRT1 deacetylase in POMC neurons is required for homeostatic defenses against diet-induced obesity. Cell Metab. 2010;12:78–87. doi: 10.1016/j.cmet.2010.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi J, Balthasar N, Olson D, Scott M, Berglund E, Lee CE, Choi MJ, Lauzon D, Lowell BB, Elmquist JK. Melanocortin-4 receptors expressed by cholinergic neurons regulate energy balance and glucose homeostasis. Cell Metabolism. 2011;13:195–204. doi: 10.1016/j.cmet.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeberger M, Dietrich MO, Sebastian D, Imbernon M, Castano C, Garcia A, Esteban Y, Gonzalez-Franquesa A, Rodriguez IC, Bortolozzi A, et al. Mitofusin 2 in POMC neurons connects ER stress with leptin resistance and energy imbalance. Cell. 2013;155:172–187. doi: 10.1016/j.cell.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz MW, Porte D., Jr Diabetes, obesity, and the brain. Science (New York, NY) 2005;307:375–379. doi: 10.1126/science.1104344. [DOI] [PubMed] [Google Scholar]
- Scott MM, Lachey JL, Sternson SM, Lee CE, Elias CF, Friedman JM, Elmquist JK. Leptin targets in the mouse brain. J Comp Neurol. 2009;514:518–532. doi: 10.1002/cne.22025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, Goldfine AB. Inflammation and insulin resistance. The Journal of clinical investigation. 2006;116:1793–1801. doi: 10.1172/JCI29069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn JW, Harris LE, Berglund ED, Liu T, Vong L, Lowell BB, Balthasar N, Williams KW, Elmquist JK. Melanocortin 4 receptors reciprocally regulate sympathetic and parasympathetic preganglionic neurons. Cell. 2013;152:612–619. doi: 10.1016/j.cell.2012.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn JW, Xu Y, Jones JE, Wickman K, Williams KW, Elmquist JK. Serotonin 2C receptor activates a distinct population of arcuate pro-opiomelanocortin neurons via TRPC channels. Neuron. 2011;71:488–497. doi: 10.1016/j.neuron.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiegelman BM, Flier JS. Obesity and the regulation of energy balance. Cell. 2001;104:531–543. doi: 10.1016/s0092-8674(01)00240-9. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Dillin A. XBP-1 is a cell-nonautonomous regulator of stress resistance and longevity. Cell. 2013;153:1435–1447. doi: 10.1016/j.cell.2013.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler JP, Schwartz MW. Minireview: Inflammation and obesity pathogenesis: the hypothalamus heats up. Endocrinology. 2010;151:4109–4115. doi: 10.1210/en.2010-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaler JP, Yi CX, Schur EA, Guyenet SJ, Hwang BH, Dietrich MO, Zhao X, Sarruf DA, Izgur V, Maravilla KR, et al. Obesity is associated with hypothalamic injury in rodents and humans. The Journal of clinical investigation. 2012;122:153–162. doi: 10.1172/JCI59660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschop MH, Speakman JR, Arch JR, Auwerx J, Bruning JC, Chan L, Eckel RH, Farese RV, Jr, Galgani JE, Hambly C, et al. A guide to analysis of mouse energy metabolism. Nat Methods. 2012;9:57–63. doi: 10.1038/nmeth.1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsiotra PC, Tsigos C. Stress, the endoplasmic reticulum, and insulin resistance. Annals of the New York Academy of Sciences. 2006;1083:63–76. doi: 10.1196/annals.1367.007. [DOI] [PubMed] [Google Scholar]
- van Marken Lichtenbelt WD, Vanhommerig JW, Smulders NM, Drossaerts JM, Kemerink GJ, Bouvy ND, Schrauwen P, Teule GJ. Cold-activated brown adipose tissue in healthy men. The New England journal of medicine. 2009;360:1500–1508. doi: 10.1056/NEJMoa0808718. [DOI] [PubMed] [Google Scholar]
- Virtanen KA, Lidell ME, Orava J, Heglind M, Westergren R, Niemi T, Taittonen M, Laine J, Savisto NJ, Enerback S, et al. Functional brown adipose tissue in healthy adults. The New England journal of medicine. 2009;360:1518–1525. doi: 10.1056/NEJMoa0808949. [DOI] [PubMed] [Google Scholar]
- Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. The Journal of clinical investigation. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. The Journal of clinical investigation. 1999;104:787–794. doi: 10.1172/JCI7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CL, Whittington A, Barnes MJ, Wang Z, Bray GA, Morrison CD. HF diets increase hypothalamic PTP1B and induce leptin resistance through both leptin-dependent and -independent mechanisms. Am J Physiol Endocrinol Metab. 2009;296:E291–299. doi: 10.1152/ajpendo.90513.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CL, Whittington A, Barnes MJ, Wang ZQ, Bray G, Morrison CD. HF diets increase hypothalamic PTP1B and induce leptin resistance through both leptin-dependent and independent mechanisms. Am J Physiol Endocrinol Metab. 2008 doi: 10.1152/ajpendo.90513.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KW, Elmquist JK. From neuroanatomy to behavior: central integration of peripheral signals regulating feeding behavior. Nat Neurosci. 2012;15:1350–1355. doi: 10.1038/nn.3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KW, Margatho LO, Lee CE, Choi M, Lee S, Scott MM, Elias CF, Elmquist JK. Segregation of acute leptin and insulin effects in distinct populations of arcuate proopiomelanocortin neurons. J Neurosci. 2010;30:2472–2479. doi: 10.1523/JNEUROSCI.3118-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams KW, Scott MM, Elmquist JK. Modulation of the central melanocortin system by leptin, insulin, and serotonin: co-ordinated actions in a dispersed neuronal network. Eur J Pharmacol. 2011;660:2–12. doi: 10.1016/j.ejphar.2010.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Bostrom P, Sparks LM, Ye L, Choi JH, Giang AH, Khandekar M, Virtanen KA, Nuutila P, Schaart G, et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell. 2012;150:366–376. doi: 10.1016/j.cell.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: cell life and death decisions. The Journal of clinical investigation. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo GS, Heisler LK. Unraveling the brain regulation of appetite: lessons from genetics. Nat Neurosci. 2012;15:1343–1349. doi: 10.1038/nn.3211. [DOI] [PubMed] [Google Scholar]
- Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol. 2007;7:454–465. doi: 10.1038/nri2093. [DOI] [PubMed] [Google Scholar]
- Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, Kim YB, Elmquist JK, Tartaglia LA, Kahn BB, et al. PTP1B regulates leptin signal transduction in vivo. Dev Cell. 2002;2:489–495. doi: 10.1016/s1534-5807(02)00148-x. [DOI] [PubMed] [Google Scholar]
- Zabolotny JM, Kim YB, Welsh LA, Kershaw EE, Neel BG, Kahn BB. Protein-tyrosine phosphatase 1B expression is induced by inflammation in vivo. J Biol Chem. 2008;283:14230–14241. doi: 10.1074/jbc.M800061200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Zhang G, Zhang H, Karin M, Bai H, Cai D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell. 2008;135:61–73. doi: 10.1016/j.cell.2008.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.