

Abstract
Hepatic ischemia and reperfusion injury (IRI) occurs in multiple clinical settings including liver transplantation. Cyclic adenosine monophosphate (cAMP)-dependent protein kinase A (PKA) pathway inhibits hepatocellular apoptosis and regulates TLR4-triggered inflammation responses in vitro. Here, we examined the function and therapeutic potential of cAMP-PKA activation in a murine (C57/BL6) model of liver warm ischemia (90 min) followed by reperfusion. Liver IRI triggered cAMP-PKA activation, whereas administration of its specific inhibitor, H-89, exacerbated hepatocellular damage. Conversely, Forskolin therapy, which activates PKA by elevating cAMP levels, protected livers from IRI, evidenced by diminished serum ALT and well-preserved tissue architecture. Liver protection rendered by cAMP-PKA stimulation was accompanied by diminished neutrophil and macrophage infiltration/activation, reduced hepatocyte necrosis/apoptosis, yet increased cAMP response element-binding protein (CREB) and augmented IL-10 expression. Neutralization of IL-10 restored liver damage in otherwise IR-resistant Forskolin-treated mice. In vitro, cAMP-PKA activation diminished macrophage TNF-α/IL-6/IL-12 in an IL-10-dependent manner, and prevented necrosis/apoptosis in primary mouse hepatocyte cultures. Our novel findings in a mouse model of liver IRI document the importance of cAMP-PKA signaling in hepatic homeostasis and cytoprotection in vivo. Activation of cAMP-PKA signaling differentially regulates local inflammation and prevents hepatocyte death, providing the rationale for novel therapeutic approaches to combat liver IRI in transplant recipients.
Keywords: Cyclic Adenosine Monophosphate, Ischemia/Reperfusion Injury, Protein kinase A, Toll-like Receptor 4
Introduction
Liver ischemia and reperfusion injury (IRI) is a common problem that occurs in multiple clinical settings, such as liver transplantation, partial hepatectomy, and trauma. The IR insult, with its unique mechanism of effector tissue damage, combines two distinctive yet interrelated phases of ischemia-trigged hypoxic cellular stress, and inflammation-mediated reperfusion organ injury (1). Reactive oxygen species (ROS)-inflicted tissue damage initiates circulatory disturbances and local inflammation responses, leading to the ultimate hepatocyte death. Our group was among the first to document that activation of the sentinel TLR4 signaling is required in liver IRI (2). We then provided evidence that IR-triggered TLR4, primarily on Kupffer cells, activates downstream “signature” pro-inflammatory cytokine/chemokine programs, i.e., TNF-α, IFN-β, IL-6 and CXCL-10 (3,4).
Cyclic adenosine monophosphate (cAMP) serves as a second messenger that translates a variety of extracellular stimuli into intracellular biological responses. Protein kinase A (PKA) binds cAMP via a complex of catalytic and a mutant regulatory subunits (5). Activation of cAMP-dependent PKA exerts an array of regulatory cell functions, including proliferation, differentiation, apoptosis, migration and immune responses (5-7). Recent studies have documented that TLR4 ligand LPS-facilitated activation of cAMP-PKA signaling increases IL-10 while suppressing macrophage pro-inflammatory TNF-α and MIP-1α levels in vitro (8). The mechanism by which cAMP-PKA axis suppress LPS-induced TNF-α production, involves increasing activity of cAMP response element-binding protein (CREB), and modulation of nuclear factor kappa B (NF-κB) (8,9). Further, cAMP-PKA activation can prevent apoptosis of parenchymal cells, including hepatocytes (10-12). Although PKA-dependent signaling has been shown to mediate anti-apoptotic effects in rat livers preconditioned ex vivo with atrial natriuretic peptides (13), it remains unknown as to whether and how cAMP-PKA activation may affect liver IRI in vivo.
The present study was designed to examine the function of cAMP-PKA pathway in a mouse in-situ model of segmental hepatic “warm” ischemia and reperfusion. This model of acute liver damage is well-established, highly reproducible, and to a certain extend it reflects real-life transplant setting in which donor organ is devoid of blood supply during the harvest and orthotopic liver transplant procedure. However, it does not reflect liver insult suffered due to “cold” storage prior to transplantation. As TLR4 activation induces both pro- and anti-inflammatory gene programs, we need first to determine whether IR does indeed activate cAMP-PKA signaling in vivo. The question then arises whether cAMP-PKA pathway is essential to maintain liver homeostasis after PKA inhibition. Finally, a key issue as to whether or not cAMP-PKA activation can diminish pro-inflammatory response and promote hepatocyte survival warrants critical evaluation while considering manipulation of cAMP-PKA axis as a potential therapeutic concept in the management of IRI in liver transplant recipients.
Materials and Methods
Animals
Male C57BL/6 wild-type (WT) mice (8-12 weeks old) were used (Jackson Laboratory, Bar Harbor, ME). Animals were housed in the University of California Los Angeles animal facility under specific pathogen-free conditions and received humane care according to the criteria outlined in Guide for the Care and Use of Laboratory Animals (prepared by the National Academy of Sciences; National Institutes of Health publication 86-23, revised 1985).
Mouse warm liver IRI model
We have used a well-established mouse model of partial “warm” hepatic IRI (14). In brief, the arterial and portal venous blood supply to the cephalad lobes was interrupted by an atraumatic clip for 90min. Sham-operated mice underwent the same procedure, but without vascular occlusion. In the treatment groups, animals were infused 1h prior to the onset of liver ischemia with a single dose of Forskolin (cAMP-PKA activator) or H-89 (cAMP-PKA inhibitor) (20nmol/mouse i.v., Sigma-Aldrich, St. Louis, MO) dissolved in dimethyl sulfoxide (DMSO). Some recipients were given rat anti-mouse neutralizing IL-10 mAb (JES5-2A5, 0.5mg/mouse i.v.; Bio X Cell, West Lebanon, NH). Mice were sacrificed at various time-points of reperfusion; liver and serum samples were collected for analysis. The UCLA Animal Research Committee approved all animal care and experimental procedures.
The hepatocellular damage
Serum alanine aminotransferase (sALT) levels were measured by IDEXX Laboratory (Westbrook, ME). Culture medium ALT levels were measured by ALT kit (Stanbio, Boerne, TX). Untreated hepatocyte lysates were used to determine total ALT level. Cell death was expressed as ALT released from treated cells (percentage of the total ALT).
Histopathology
Liver specimens (4μm), stained with hematoxylin and eosin (H&E), were analyzed blindly by modified Suzuki's criteria, as described (14). Primary mAb against mouse neutrophils Ly-6G (1A8; BD Biosciences, San Jose, CA) and macrophages CD68 (FA-11; AbD Serotec, Raleigh, NC) were used (14). Liver sections were evaluated blindly by counting labeled cells in 10 high-power fields (HPF).
The cAMP / PKA kinase activity assays
The cAMP levels and PKA activity in liver tissue samples were measured by using cAMP Enzyme Immunoassay and PKA kinase activity kit, respectively (Enzo Life Sciences, Farmingdale, NY). Briefly, liver tissue samples were frozen in liquid nitrogen, and stored in −80°C. In the cAMP activity assay, liver samples were homogenized in 0.1 M HCl to halt endogenous phosphodiesterase activity. Standards and samples were assessed by sandwich ELISA method. In PKA kinase activity assay, a specific synthetic peptide substrate was first phosphorylated by purified active PKA standards or liver sample lysis. The measure of PKA kinase in samples/standards was indirect by assessing the consumption of substrate in an ELISA assay.
Myeloperoxidase activity assay
The presence of myeloperoxidase (MPO) was used as an index of neutrophil accumulation in the liver (14). One absorbance unit (U) of MPO activity was defined as the quantity of enzyme degrading 1mol peroxide/min at 25°C/gram of tissue.
Quantitative RT-PCR
Quantitative PCR was performed with platinum SYBR green quantitative PCR kit (Invitrogen, Carlsbad, CA) by the Chromo 4 detector (MJ Research, Waltham, MA). Primers used to amplify specific gene fragments were published (14). Target gene expressions were calculated by their ratios to the housekeeping gene hypoxanthine-guanine phosphoribosyl transferase (HPRT).
Western blots
Western blots were performed with liver proteins (30μg/sample) and rabbit anti-mouse Bcl-2, Bcl-xl, p-IκBα, p-NF-κB, p-CREB, and β-actin mAbs (Cell Signaling Technology, Danvers, MA), as described (14). Relative quantities of protein were determined by densitometer and expressed in absorbance units (AU).
Terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay
DNA fragments in liver sections, resulting from oncotic necrosis and apoptosis were detected by TUNEL method (Klenow-FragEL DNA Fragmentation Detection Kit, Calbiochem, La Jolla, CA ) (14). TUNEL positive cells were counted in 10 HPF/section under light microscopy (x400).
Caspase-3 activity assay
Caspase-3 activity was performed using Caspase-3 Cellular Activity Assay Kit (Calbiochem). Liver tissue sample and cell lysis were used according to the manufacturer's instruction.
Cell cultures
Bone marrow-derived macrophages (BMM), separated from femurs/tibias of C57BL/6 mice were cultured (5×106/well) with 10% L929 conditioned medium for 6 days. The cell purity was assayed to be 94-99% CD11b+. BMM were activated by LPS (10ng/ml, Sigma-Aldrich) and Forskolin, H-89 (10μM), or DMSO control, and incubated for 24 h. Anti-IL-10 mAb (10μg/ml) was used to neutralize IL-10. Cell-free supernatants were assayed for TNF-α, IL-6, IL-12p40, and IL-10 levels by ELISA (eBioscience, San Diego, CA).
Mouse hepatocytes were isolated by in situ two stage collagenase perfusion method, cultured with complete L-15 medium plus 6.25μg/ml insulin, 1μM dexamethasone, and 10% fetal bovine serum. Hepatocyte viability after isolation was 95-99% as determined by trypan blue dye exclusion. After pretreatment with Forskolin, H-89 (10μM), or DMSO control for 1h, hepatocyte death was induced by hydrogen peroxide (5mM, Sigma-Aldrich), or TNF-α (10ng/ml, R&D systems, Minneapolis, MN) in combination with actinomycin D (0.4μg/ml, Sigma-Aldrich) during 5h incubation period. Cells were processed for flow cytometry and caspase-3 activity, whereas supernatants were assessed for ALT and LDH levels.
Lactate dehydrogenase (LDH) release assay
Culture medium LDH activity was measured by using LDH kit (Stanbio) according to the manufacturer's instruction. Untreated hepatocyte lysates were used to determine total LDH activity. Cell death was expressed as LDH activity released from the treated cells as a percentage of the total LDH activity.
Flow cytometry
Hepatocytes stained with FITC-ANNEXIN V and 7-AAD (BD Biosciences, Mountain View, CA) were analyzed on a FACS-Calibur cytometer (BD Biosciences). Dead cells were identified as ANNEXIN V+7-AAD+.
Statistical analysis
All values are expressed as the mean ± standard deviation (SD). Data were analyzed with an unpaired, two-tailed Student's-t test. P<0.05 was considered to be statistically significant.
Results
cAMP-PKA activation profile in liver IRI
First, we determined whether IR itself triggers cAMP-PKA activation in mouse livers subjected to 90min of warm ischemia, followed by various lengths of reperfusion. Compared with sham controls, cAMP levels increased shortly after the ischemia (0h), then transiently dropped to baseline, and increased progressively thereafter (2-6h), peaking by 12-24h of reperfusion (Fig. 1a). On the other hand, after raising sharply during the first 2h of reperfusion, the PKA activity remained relatively unchanged throughout the observation period (Fig. 1b).
Figure 1.
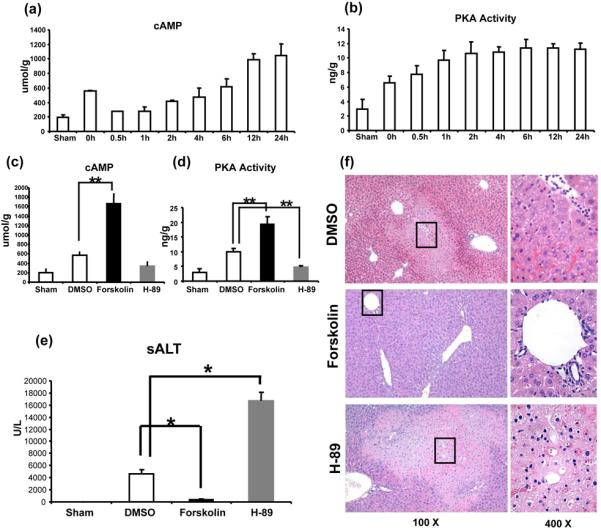
Liver IRI triggers cAMP-PKA activation. Liver samples were harvested from B6 mice that were either sham-operated or subjected to 90min of partial liver warm ischemia, followed by various lengths of reperfusion. (a) cAMP; and (b) PKA activity levels were measured.
Distinct effects of activation vs. inhibition of cAMP-PKA pathway in liver IRI (6h of reperfusion after 90min of ischemia). (c) cAMP; and (d) PKA activity levels were assessed after Forskolin (activator), H-89 (inhibitor), or DMSO (control) treatment.
The hepatocellular function, as analyzed by (e) sALT levels, and (f) liver histology (representative H&E staining; magnification x100 and x400) of the ischemic liver lobes (**p<0.01, *p<0.001, n=6-8/group).
Activation of cAMP-PKA pathway ameliorates liver IRI
We analyzed the hepatocellular damage after modulation of cAMP-PKA pathway in our principal model of 90min of partial warm ischemia followed by 6h of reperfusion. Treatment with Forskolin activated hepatic PKA by elevating intracellular cAMP levels in vivo (Fig. 1c, d; p<0.01). Unlike control mice given DMSO, those conditioned with Forskolin were resistant against IR-mediated liver injury, as evidenced by reduced sALT levels (IU/L; Fig. 1e: 388±66 vs. 4640±655; p<0.001); well preserved hepatic architecture, with minimal sinusoidal congestion with no edema, vacuolization or necrosis (Fig. 1f); and decreased Suzuki's histological score of IR-liver damage (p<0.001). To document this beneficial effect was dependent on stimulation of cAMP-PKA pathway, a separate group of WT mice was pre-treated with H-89, a specific PKA inhibitor. Indeed, inhibition of PKA activity not only re-created but even exacerbated IR-liver damage, evidenced by increased sALT levels (IU/L; Fig. 1e: 16630±1512 vs. 4640±655; p<0.001); more severe lobular edema, widespread hemorrhage, congestion/hepatocellular necrosis (Fig. 1f); and Suzuki's grading of liver IRI (p<0.001) as compared with controls.
Activation of cAMP-PKA signaling attenuates neutrophil/macrophage sequestration in IR-livers
MPO-based liver neutrophil activity (U/g) was depressed in mice pretreated with Forskolin, compared with controls (Fig. 2a: 0.55±0.28 vs 1.32±0.34; p<0.01). In parallel, PKA activation diminished the frequency of neutrophils (Fig. 2b,c: 1.3±0.5 vs. 29.5±3.7, p<0.001) and macrophages (Fig. 2d,e: 1.2±0.5 vs. 31.0±2.8, P<0.001) in the ischemic liver lobes, compared with controls. In contrast, H-89-facilitated inhibition of PKA not only enhanced neutrophil activity (Fig. 2a; MPO = 2.55±0.34 and 1.32±0.34 U/g, respectively; p<0.01) but also increased neutrophil (Fig. 2b,c: 67.0±6.7 and 29.5±3.7, respectively; p<0.001) and macrophage (Fig. 2d,e: 81.5±5.9 and 31.0±2.8, respectively; p<0.001) infiltration, especially in necrotic areas of IR-livers.
Figure 2.
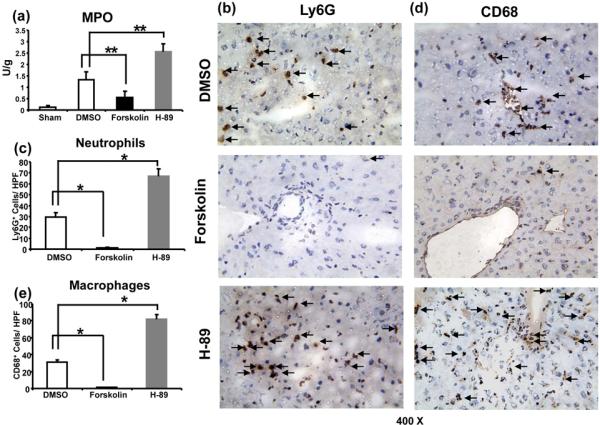
Accumulation of neutrophils and macrophages in IR-livers following activation/inhibition of cAMP-PKA signaling (6h of reperfusion after 90min of ischemia).
(a) Liver MPO levels; (b) and (c) Ly-6G+ neutrophils; (d) and (e) CD68+ macrophages in IR liver lobes (magnification x400) (**p<0.01, *p<0.001, n=6-8/group).
cAMP-PKA signaling regulates IR-induced liver cytokine/chemokine programs
To assess the immunoregulatory function of cAMP-PKA activation, we analyzed hepatic chemokine/ cytokine expression patterns in our model. Pretreatment with Forskolin, a cAMP-PKA activator, suppressed neutrophil/monocyte-derived pro-inflammatory chemokine (CXCL-1, CCL-2, and CXCL-10) and cytokine (TNF-α, IL-1β, IL-6, and IFN-β) programs, as compared with controls (Fig. 3a,c, p<0.01). Strikingly, PKA activation selectively and significantly (p<0.01) increased IL-10 expression (Fig. 3b). In contrast, pretreatment with H-89, a PKA antagonist, uniformly enhanced pro-inflammatory chemokine/cytokine levels (Fig. 3a,c, p<0.01), and simultaneously abolished hepatic IL-10 (Fig. 3b).
Figure 3.
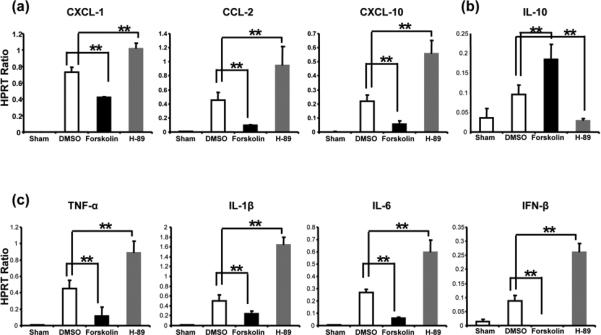
Quantitative RT-PCR-assisted detection of cytokines/chemokines in mouse livers (at 6h of reperfusion after 90min of warm ischemia: (a) CXCL-1, CCL-2, and CXCL-10; (b) IL-10; and (c) TNF-α, IL-1β, IL-6, IFN-β). Data were normalized to HPRT gene expression (**p<0.01, n=6-8/group).
PKA activation inhibits IR-mediated liver necrosis and apoptosis
We used TUNEL and caspase-3 activity assays to screen for IR-induced oncotic necrosis and apoptosis. Forskolin treatment diminished otherwise abundant hepatocellular necrosis/apoptosis in IR-livers, as evidenced by reduced frequency of TUNEL+ cells (Fig. 4a,b: 1.5±0.6 vs. 29.5±3.7; p<0.001) and caspase-3 activity (Fig. 4c: 5.7±1.0 vs. 20.2±4.7 pmol/min/mg; p<0.01). Western blot analysis has revealed selectively increased expression (AU) of Bcl-2, Bcl-xl, and p-CREB, yet suppressed phosphorylation of IκBα and NF-κB proteins in Forskolin treatment group (Fig. 4d). In contrast, pretreatment with H-89 increased frequency of TUNEL+ cells (Fig. 4a,b: 63.8±6.7 and 29.5±3.7, respectively; p<0.001) and enhanced caspase-3 activity (Fig. 4c: 53.2±14.6 and 20.2±4.7 pmol/min/mg, respectively; p<0.01). In addition, phosphorylated IκBα and NF-κB levels were enhanced, whereas those of Bcl-2, Bcl-xl, and p-CREB remained diminished after H-89-facilitated PKA inhibition (Fig. 4d).
Figure 4.
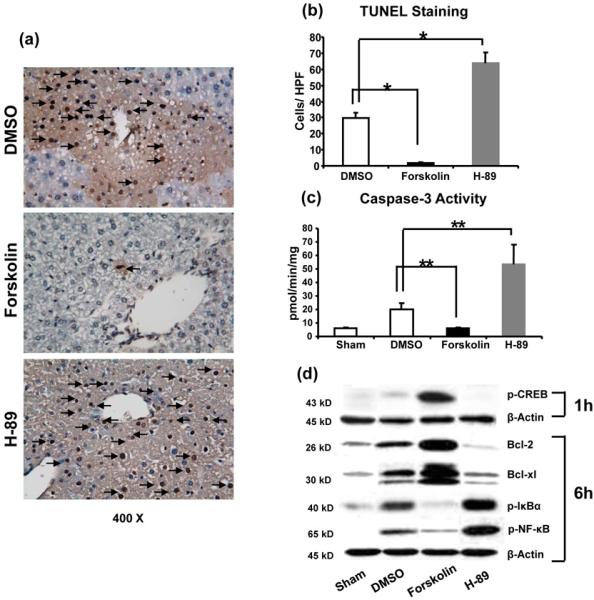
Necrosis/apoptosis in IR livers following activation/inhibition of cAMP-PKA (6h of reperfusion after 90min of ischemia). (a) and (b) TUNEL-assisted detection of hepatic necrosis/apoptosis (dark arrows) in ischemic liver lobes (magnification x400); (c) caspase-3 activity levels; (d) Western blot-assisted detection of p-CREB protein (1 h of reperfusion), anti-necrosis/apoptotic Bcl-2 and Bcl-xl protein; p-IκBα, and p-NF-κB levels (6h of reperfusion). (**p<0.01, *p<0.001, n=6-8/group).
IL-10 neutralization restores liver IRI in Forskolin-treated mice
Having shown that IR-triggered liver inflammation was accompanied by preferentially increased IL-10 expression in Forskolin-pretreated mice (Fig. 3b) we then asked whether cAMP-PKA-induced IL-10 is indeed essential for cytoprotection. Interestingly, neutralization of IL-10 readily restored hepatic damage in Forskolin-treated mice, evidenced by sALT levels (Fig. 5a: 5008±655 vs. 388±66 U/L after Forskolin monotherapy, p<0.001); and liver histology (Fig. 5b). Livers after adjunctive PKA activation and IL-10 neutralization were characterized by zonal/pan-lobular parenchyma necrosis, combined with widespread sinusoidal congestion and severe edema (Fig. 5b: Suzuki's score = 2.8±0.3), comparable with untreated controls. Concomitant infusion of anti-IL-10 mAb significantly increased (p<0.001) liver expression of CXCL-10, TNF-α, and IL-1β (Fig. 5c). Recreation of liver IRI pathology after neutralizing IL-10 in Forskolin-treated mice implies the essential cytoprotective function of IL-10 in cAMP-PKA activation pathway.
Figure 5.
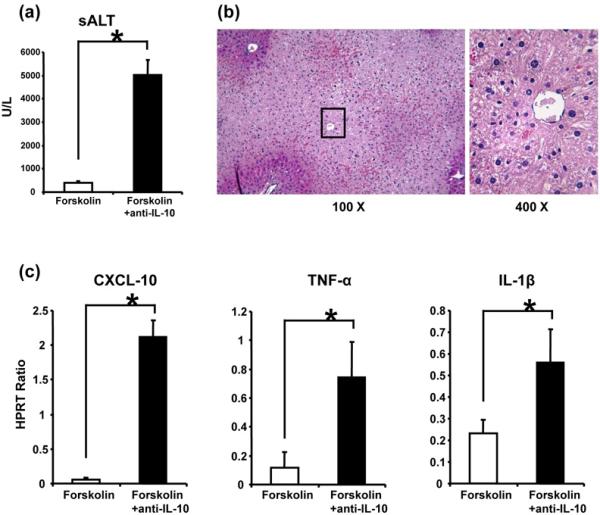
The functional significance of IL-10 in liver cytoprotection following activation of cAMP-PKA pathway. Adjunctive neutralization of IL-10 prior to the onset of ischemia restored liver injury in Forskolin (PKA activator)-pretreated mice, as evidenced by (a) sALT levels (*p<0.001, n=6/group); (b) liver histology (representative H&E staining; magnification x100 and x400); and (c) CXCL-10, TNF-α, and IL-1β gene expression pattern (*p<0.001).
Activation of cAMP-PKA directly regulates macrophage TLR4 response
TLR4 activation is the pivotal step in IR-mediated liver inflammation (2). To investigate the regulatory mechanism of PKA activation, we assessed as to whether and how cAMP-PKA signaling may affect macrophage TLR4 responses in vitro. BMM were stimulated with LPS in the presence or absence of Forskolin (PKA activator) or H-89 (PKA inhibitor). As shown in Fig. 6, Forskolin depressed otherwise enhanced LPS-induced pro-inflammatory cytokine program (pg/ml) (Fig. 6a: 392.5±7.3 vs. 909.8±42.4 [TNF-α]; 739.7±95.3 vs. 2138.1±214.1 [IL-6]; and 2850.1±415.7 vs. 6793.0±729.1 [IL-12p40]; p<0.01), but augmented IL-10 (Fig. 6b: 1746.3±202.6 vs. 1101.0±100.1; p<0.05). In contrast, H-89-facilitated PKA inhibition did enhance TNF-α, IL-6, and IL-12p40 (Fig. 6a: 1534.2±122.6, 4004.4±439.7, and 12150.0±1046.2; p<0.01), compared with LPS-treated cultures. IL-10 levels were comparable in LPS cultures without/with H-89 (Fig. 6b: 1101.0±100.1 vs. 720.1±76.6). Interestingly, adjunctive anti-IL-10 mAb readily re-created BMM activation, evidenced by increased expression of TNF-α (1135.6±68.3), IL-6 (2425.4±212.3) and IL-12p40 (7095.3±800.5) (Fig. 6a). These results document the direct IL-10-dependent regulatory function of cAMP-PKA signaling in macrophage TLR4 responses in vitro.
Figure 6.
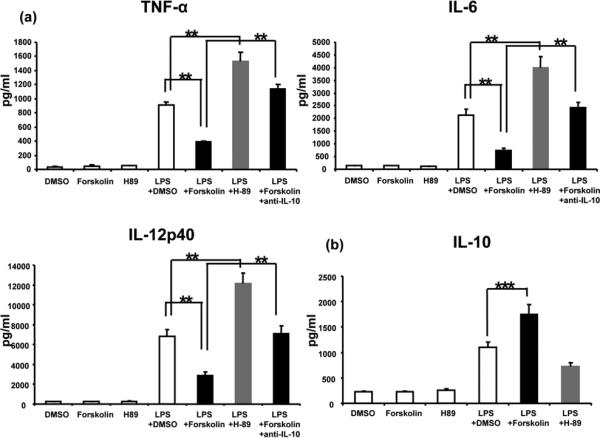
The effects of cAMP-PKA activation/inhibition upon macrophage TLR4 activation in vitro. BMM were stimulated with LPS in the absence or presence of Forskolin (PKA activator), H-89 (PKA inhibitor), or DMSO (control). The expression of (a) TNF-α, IL-6, IL-12p40, and (b) IL-10 are shown. (**p<0.01, ***p<0.05, n=6/group).
Activation of cAMP-PKA prevents hepatocyte death
We analyzed immunomodulatory function of cAMP-PKA signaling in well-controlled in vitro primary hepatocyte cultures, designed to mimic liver IR-mediated hepatocellular damage in vivo. Since necrosis and apoptosis are essential in the mechanism of liver IRI, we used hydrogen peroxide (H2O2) to mimic in vivo ROS-triggered necrosis, and TNF-α/actinomycin D (ActD) to induce apoptosis. Native mouse hepatocytes were cultured in the presence of Forskolin (PKA agonist), H-89 (PKA antagonist), or DMSO (control). Addition of Forskolin consistently suppressed hepatocyte death, assessed by FACS-assisted screening frequency (%) of Annexin V+7-AAD+ cells (Fig. 7a: 4.5±0.9 vs. 13.2±3.0 [H2O2]; 3.5±0.2 vs. 15.9±2.7 [TNF-α+ActD]; p<0.01); diminished cellular caspase-3 activity (pmol/min/5X10E4 cells) (Fig. 7b: 0.11±0.08 vs. 0.28±0.18 [H2O2]; 0.74±0.15 vs. 1.92±0.33 [TNF-α+ActD]; p<0.01); LDH release (%) (Fig. 7c: 10.72±2.37 vs. 19.29±5.27 [H2O2]; 16.08±4.37 vs. 37.51±9.66 [TNF-α+ActD]; p<0.01); and ALT release (%) (Fig. 7d: 11.34±2.11 vs. 22.68±4.69 [H2O2]; 14.42±3.89 vs. 36.28±8.60 [TNF-α+ActD]; p<0.01), as compared with controls. In contrast, inhibition of PKA enhanced hepatocyte death (Fig. 7a: 27.3±3.7 [H2O2]; 31.9±4.7 [TNF-α+ActD];); caspase-3 activity (Fig. 7b: 0.41±0.32 [H2O2]; 3.53±0.63 [TNF-α+ActD];); LDH release (Fig. 7c: 27.53±9.37 [H2O2]; 81.03±18.37 [TNF-α+ActD]); as well as ALT release (Fig. 7d: 34.29±8.34 [H2O2]; 76.19±16.35 [TNF-α+ActD]) in hepatocyte cultures.
Figure 7.
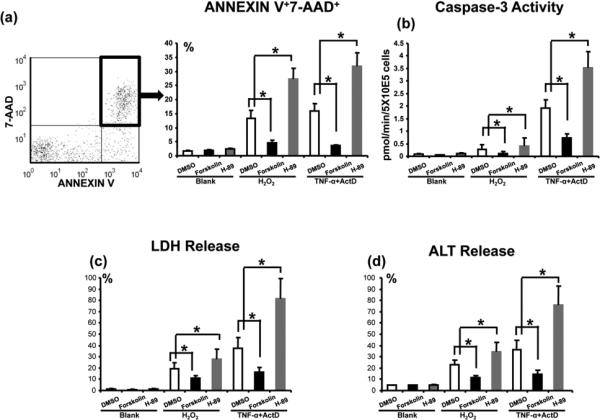
The effects of cAMP-PKA activation/inhibition upon hepatocyte death in vitro. Hydrogen peroxide (H2O2), or TNF-α+actinomycin D (ActD) were used to induce primary murine hepatocyte necrosis/apoptosis, in the absence or presence of Forskolin (PKA activator), H-89 (PKA inhibitor), or DMSO (control). (a) FACS-assisted detection of ANNEXIN V+7-AAD+ dead cells; (b) Hepatocellular caspase-3 activity levels; (c) LDH release; and (d) ALT levels in supernatants (*p<0.001, n=6/group).
Discussion
This study is the first to document that specific cAMP-PKA activation in vivo ameliorates liver damage in a mouse model of warm ischemia and reperfusion, by depressing TLR4-driven inflammation in an IL-10-dependent manner, and preventing the hepatocyte death. Hence, engaging negative cAMP-PKA signaling is essential to maintain liver homeostasis in IR-insult. Many cellular effects of cAMP can be mediated by PKA, including cell differentiation, proliferation, and apoptosis (5, 6). Triggering cAMP-PKA activation has been shown to regulate immune responses (7-9) and promote parenchymal cell survival (10-12). In addition, recent study has revealed that activation of cAMP-PKA has blocked mitochondrial permeability transition and protected cultured rat hepatocytes from anoxia-induced cell death in vitro (15).
In the present study, we first found that both cAMP and PKA were triggered by IR insult, yet PKA activity did not increase while liver damage subsided between 6-24h of reperfusion. The requirement for cAMP-PKA signaling in liver IRI became evident when inhibition of cAMP-PKA activity amplified the hepatocellular damage. We have reported similarly exacerbated IRI in livers deficient of PD-1 (14) and TIM-3 (16) negative T cell co-stimulation. In analogy with cytoprotection rendered by stimulating PD-1–B7-H1 negative pathway (14), we then asked whether activation of cAMP-PKA may improve liver function. We have chosen a chemical approach by using a specific PKA agonist, Forskolin, which elevates cAMP levels. Indeed, stimulating cAMP-PKA decreased sALT levels and ameliorated liver IR-damage.
As in our previous study (17), we detected increased activation/recruitment of CD68+ macrophages in IR-livers, consistent with preferential pro-inflammatory chemotactic gene expression programs in the initial phase of IR-inflammation (2-4). As cAMP-PKA elevation may suppress macrophage function (8,18), others have suggested cAMP-PKA may function as a “master switch” in innate immune regulation (19,20). Of note, PKA activation further diminished hepatic pro-inflammatory macrophage infiltration/activation, evidenced by immunohistology and decreased expression of TNF-α, IL-1β, IL-6, CXCL-10 and CCL-2 (MCP-1).
TLR4 activation promotes innate immune responses through adaptor myeloid differentiation factor 88 (MyD88), or Toll/IL-1 receptor domain containing adaptor inducing IFN-β (TRIF) dependent pathway (21). We have shown that TRIF-IFN regulatory factor 3 (IRF3) is instrumental for downstream activation of NF-κB and ultimate hepatocellular damage (2,4). There have been, however, conflicting reports (22) on the functional role of cAMP-PKA - NF-κB crosstalk in various cell types, including monocytes (23), lymphocytes (24), epithelial (25) and endothelial cells (26). In macrophages (Fig. 8) PKA inhibits NF-κB directly by modulating the phosphorylation of p65 or by stabilizing IκB (27); regulating the transactivation/stability of NF-κB complexes; and suppressing NF-κB-triggered pro-inflammatory genes (28). On the other hand, PKA may also inhibit NF-κB indirectly by enhancing CREB phosphorylation, which has higher affinity for CREB-binding protein (CBP) and results in sequestration of p65/CBP complexes. As a consequence, CBP becomes unavailable for NF-κB binding, further reducing NF-κB transactivation (29). Here, we show that cAMP-PKA activation did increase CREB expression in the very early phase of liver IR, decreased phosphorylation/proteolytic degradation of IκB subunit, and suppressed the phosphorylation of NF-κB p65. Consistently, cAMP-PKA activation blunted TLR4-NF-κB downstream pro-inflammatory expression program, abolished de novo activation of TNFR/IL-1R, and decreased hepatic cell recruitment. In agreement with our in vivo findings, PKA activation diminished pro-inflammatory cytokine elaboration profile in LPS-activated BMM cultures.
Figure 8.
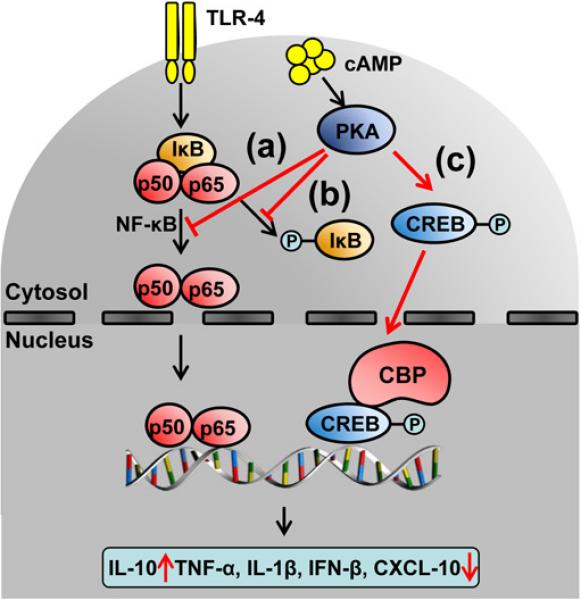
The molecular mechanisms of cAMP-PKA mediated inhibition of TLR-4 – NF-κB axis. The activation of cAMP-PKA: (a) directly prevents NF-κB translocation; (b) stabilizes IκB inhibitor; (c) induces phosphorylation of the CREB, which owing to its high affinity for the co-activator CREB-binding protein (CBP), suppresses the association of CBP with p65. Such transcriptional machinery is perfectly assembled to selectively promote IL-10, but not pro-inflammatory TNF-α, IL-1β, IFN-β and CXCL-10.
In parallel, PKA-triggered CREB/CBP complexes not only inhibit NF-κB transcription, but also induce anti-inflammatory IL-10 (30), which in turn suppresses TLR4 - NF-κB axis to promote hepatocyte survival. Indeed, PKA activation preferentially enhanced macrophage IL-10 production both in vivo and in vitro in the present study. This is consistent with our findings where IL-10 was required for liver protection against IR in CXCL-10 deficient mice (3), and that vIL-10 gene transfer prevented hepatic IRI in WT recipients (31). It is plausible that by selectively overexpressing IL-10, activation of PKA raises the defensive threshold to inflammatory response in IR-livers. Neutralization of IL-10 re-created pro-inflammatory cytokine programs in PKA activated cultures, confirming altered local inflammation phenotype to be responsible for liver protection in our model. Strikingly, in vivo neutralization of IL-10 not only rendered otherwise IR-resistant Forskolin pre-treated hosts susceptible to the panoply of hepatic pro-inflammatory events, but also readily restored liver IRI.
Liver parenchymal cytodestruction, including oncotic necrosis and apoptosis, both proceed via DNA degradation that can be detected by TUNEL assay (32). Consistent with the essential role of cAMP-PKA signaling in hepatic homeostasis, inhibition of PKA increased frequency of TUNEL+ cells and augmented caspase-3 activity. Conversely, PKA activation did inhibit necrosis/apoptosis in IR livers. It is plausible that cellular mechanism by which PKA activation exerts cytoprotection involves the induction of anti-necrotic/apoptotic proteins. Indeed, enhanced expression of Bcl-2/Bcl-xl can prevent hepatodestruction by modifying proapoptotic/anti-apoptotic ratio; decreasing the release of apoptogenic factors, such as cytochrome c and apoptosis inducing factor from mitochondria into the cytosol; maintaining mitochondria integrity; or promoting ATP generation (33). We confirmed our in vivo findings in well-controlled primary murine hepatocyte cultures. As hepatocyte damage in liver IRI encompasses both necrosis and apoptosis pathways, we employed hydrogen peroxide (H2O2) to mimic in vivo ROS-triggered necrosis; and TNF-α to induce cell apoptosis. Interestingly, PKA inhibition exacerbated the hepatocyte death induced by all three promoters, whereas activation of cAMP-PKA diminished hepatocyte death, reduced capase-3 activity, and ameliorated sALT release in the cultures. NF-κB pathway is involved in ROS-induced necrosis (34), whereas TNF-α triggers apoptosis (35). In agreement with our in vivo Western blot data, these results reinforce the role of cAMP-PKA to depress NF-κB not only in nonparenchymal cells but also in hepatocytes, with resultant improvement of liver function.
In summary, this study documents the essential role of cAMP-PKA pathway in local inflammation leading to liver damage due to warm IR. Stimulating PKA negative signaling ameliorated liver IRI by inhibiting macrophage function in IL-10 dependent manner, and by improving hepatocyte survival. Our results provide evidence that harnessing physiological mechanisms of cAMP-PKA stimulation may be instrumental in hepatic homeostasis in vivo by minimizing organ damage and promoting IL-10 dependent cytoprotection.
Acknowledgements
This study was funded by NIH Grants RO1 DK062357; DK 062357-06S1 (JWKW) ; The Diann Kim Foundation; The Dumont Research Foundation.
Abbreviations
- cAMP
cyclic adenosine monophosphate
- BMM
bone marrow-derived macrophages
- CREB
cAMP response element-binding protein
- IRI
ischemia and reperfusion injury
- LDH
lactate dehydrogenase
- mAb
monoclonal antibody
- MPO
myeloperoxidase
- PKA
protein kinase A
- ROS
reactive oxygen species
- sALT
serum alanine aminotransferase
- TUNEL
terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling
- WT
wide type
References
- 1.Zhai Y, Busuttil RW, Kupiec-Weglinski JW. Liver ischemia and reperfusion injury: new insights into mechanisms of innate-adaptive immune-mediated tissue inflammation. Am J Transplant. 2011;11(8):1563–1569. doi: 10.1111/j.1600-6143.2011.03579.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhai Y, Shen XD, O'Connell R, Gao F, Lassman C, Busuttil RW, et al. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol. 2004;173(12):7115–7119. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- 3.Zhai Y, Shen XD, Gao F, Zhao A, Freitas MC, Lassman C, et al. CXCL-10 regulates liver innate immune response against ischemia and reperfusion injury. Hepatology. 2008;47(1):207–214. doi: 10.1002/hep.21986. [DOI] [PubMed] [Google Scholar]
- 4.Zhai Y, Qiao B, Gao F, Shen X, Vardanian A, Busuttil RW, et al. Type I, but not type II, interferon is critical in liver injury induced after ischemia and reperfusion. Hepatology. 2008;47(1):199–206. doi: 10.1002/hep.21970. [DOI] [PubMed] [Google Scholar]
- 5.Kim C, Cheng CY, Saldanha SA, Taylor SS. PKA-I holoenzyme structure reveals a mechanism for cAMP-dependent activation. Cell. 2007;130(6):1032–1043. doi: 10.1016/j.cell.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 6.Dorsa KK, Santos MV, Silva MR. Enhancing T3 and cAMP responsive gene participation in the thermogenic regulation of fuel oxidation pathways. Arq Bras Endocrinol Metabol. 2010;54(4):381–389. doi: 10.1590/s0004-27302010000400007. [DOI] [PubMed] [Google Scholar]
- 7.Castro A, Jerez MJ, Gil C, Martinez A. Cyclic nucleotide phosphodiesterases and their role in immunomodulatory responses: advances in the development of specific phosphodiesterase inhibitors. Med Res Rev. 2005;25(2):229–244. doi: 10.1002/med.20020. [DOI] [PubMed] [Google Scholar]
- 8.Wall EA, Zavzavadjian JR, Chang MS, Randhawa B, Zhu X, Hsueh RC, et al. Suppression of LPS-induced TNF-alpha production in macrophages by cAMP is mediated by PKAAKAP95-p105. Sci Signal. 2009;2(75):ra28. doi: 10.1126/scisignal.2000202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Natarajan M, Lin KM, Hsueh RC, Sternweis PC, Ranganathan R. A global analysis of crosstalk in a mammalian cellular signalling network. Nat Cell Biol. 2006;8(6):571–580. doi: 10.1038/ncb1418. [DOI] [PubMed] [Google Scholar]
- 10.Wang Y, Kim PK, Peng X, Loughran P, Vodovotz Y, Zhang B, et al. Cyclic AMP and cyclic GMP suppress TNFalpha-induced hepatocyte apoptosis by inhibiting FADD up-regulation via a protein kinase A-dependent pathway. Apoptosis. 2006;11(3):441–451. doi: 10.1007/s10495-005-4293-6. [DOI] [PubMed] [Google Scholar]
- 11.Torella D, Gasparri C, Ellison GM, Curcio A, Leone A, Vicinanza C, et al. Differential regulation of vascular smooth muscle and endothelial cell proliferation in vitro and in vivo by cAMP/PKA-activated p85alphaPI3K. Am J Physiol Heart Circ Physiol. 2009;297(6):H2015–2025. doi: 10.1152/ajpheart.00738.2009. [DOI] [PubMed] [Google Scholar]
- 12.Schildberg FA, Schulz S, Dombrowski F, Minor T. Cyclic AMP alleviates endoplasmic stress and programmed cell death induced by lipopolysaccharides in human endothelial cells. Cell Tissue Res. 2005;320(1):91–98. doi: 10.1007/s00441-004-1066-4. [DOI] [PubMed] [Google Scholar]
- 13.Kulhanek-Heinze S, Gerbes AL, Gerwig T, Vollmar AM, Kiemer AK. Protein kinase A dependent signalling mediates anti-apoptotic effects of the atrial natriuretic peptide in ischemic livers. J Hepatol. 2004;41(3):414–420. doi: 10.1016/j.jhep.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 14.Ji H, Shen X, Gao F, Ke B, Freitas MC, Uchida Y, et al. Programmed death-1/B7-H1 negative costimulation protects mouse liver against ischemia and reperfusion injury. Hepatology. 2010;52(4):1380–1389. doi: 10.1002/hep.23843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pediaditakis P, Kim JS, He L, Zhang X, Graves LM, Lemasters JJ. Inhibition of the mitochondrial permeability transition by protein kinase A in rat liver mitochondria and hepatocytes. Biochem J. 2010;431(3):411–421. doi: 10.1042/BJ20091741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uchida Y, Ke B, Freitas MC, Yagita H, Akiba H, Busuttil RW, et al. T-cell immunoglobulin mucin-3 determines severity of liver ischemia/reperfusion injury in mice in a TLR4-dependent manner. Gastroenterology. 2010;139(6):2195–2206. doi: 10.1053/j.gastro.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Uchida Y, Ke B, Freitas MC, Ji H, Zhao D, Benjamin ER, et al. The emerging role of T cell immunoglobulin mucin-1 in the mechanism of liver ischemia and reperfusion injury in the mouse. Hepatology. 2010;51(4):1363–1372. doi: 10.1002/hep.23442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aronoff DM, Canetti C, Serezani CH, Luo M, Peters-Golden M. Cutting edge: macrophage inhibition by cyclic AMP (cAMP): differential roles of protein kinase A and exchange protein directly activated by cAMP-1. J Immunol. 2005;174(2):595–599. doi: 10.4049/jimmunol.174.2.595. [DOI] [PubMed] [Google Scholar]
- 19.Serezani CH, Ballinger MN, Aronoff DM, Peters-Golden M. Cyclic AMP: master regulator of innate immune cell function. Am J Respir Cell Mol Biol. 2008;39(2):127–132. doi: 10.1165/rcmb.2008-0091TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peters-Golden M. Putting on the brakes: cyclic AMP as a multipronged controller of macrophage function. Sci Signal. 2009;2(75):pe37. doi: 10.1126/scisignal.275pe37. [DOI] [PubMed] [Google Scholar]
- 21.Seki E, Brenner DA. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48(1):322–335. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- 22.Gerlo S, Kooijman R, Beck IM, et al. Cyclic AMP: a selective modulator of NF-κB action. Cell Mol Life Sci. 2011 doi: 10.1007/s00018-011-0757-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mandrika I, Muceniece R, Wikberg JE. Effects of melanocortin peptides on lipopolysaccharide/interferon-gamma-induced NF-kappaB DNA binding and nitric oxide production in macrophage-like RAW 264.7 cells: evidence for dual mechanisms of action. Biochem Pharmacol. 2001;61(5):613–621. doi: 10.1016/s0006-2952(00)00583-9. [DOI] [PubMed] [Google Scholar]
- 24.Minguet S, Huber M, Rosenkranz L, Schamel WW, Reth M, Brummer T. Adenosine and cAMP are potent inhibitors of the NF-kappa B pathway downstream of immunoreceptors. Eur J Immunol. 2005;35(1):31–41. doi: 10.1002/eji.200425524. [DOI] [PubMed] [Google Scholar]
- 25.Takahashi N, Tetsuka T, Uranishi H, Okamoto T. Inhibition of the NF-kappaB transcriptional activity by protein kinase A. Eur J Biochem. 2002;269(18):4559–4565. doi: 10.1046/j.1432-1033.2002.03157.x. [DOI] [PubMed] [Google Scholar]
- 26.Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, et al. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation. 2000;102(11):1296–1301. doi: 10.1161/01.cir.102.11.1296. [DOI] [PubMed] [Google Scholar]
- 27.Mustafa SB, Olson MS. Expression of nitric-oxide synthase in rat Kupffer cells is regulated by cAMP. J Biol Chem. 1998;273(9):5073–5080. doi: 10.1074/jbc.273.9.5073. [DOI] [PubMed] [Google Scholar]
- 28.Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene. 2006;25(51):6717–6730. doi: 10.1038/sj.onc.1209937. [DOI] [PubMed] [Google Scholar]
- 29.Delgado M, Ganea D. Inhibition of endotoxin-induced macrophage chemokine production by vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide in vitro and in vivo. J Immunol. 2001;167(2):966–975. doi: 10.4049/jimmunol.167.2.966. [DOI] [PubMed] [Google Scholar]
- 30.Avni D, Ernst O, Philosoph A, Zor T. Role of CREB in modulation of TNFalpha and IL-10 expression in LPS-stimulated RAW264.7 macrophages. Mol Immunol. 2010;47(7-8):1396–1403. doi: 10.1016/j.molimm.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 31.Ke B, Shen XD, Tsuchihashi S, Gao F, Araujo JA, Busuttil RW, et al. Viral interleukin-10 gene transfer prevents liver ischemia-reperfusion injury: Toll-like receptor-4 and heme oxygenase-1 signaling in innate and adaptive immunity. Hum Gene Ther. 2007;18(4):355–366. doi: 10.1089/hum.2007.181. [DOI] [PubMed] [Google Scholar]
- 32.Jaeschke H, Lemasters JJ. Apoptosis versus oncotic necrosis in hepatic ischemia/ reperfusion injury. Gastroenterology. 2003;125(4):1246–1257. doi: 10.1016/s0016-5085(03)01209-5. [DOI] [PubMed] [Google Scholar]
- 33.Zhou F, Yang Y, Xing D. Bcl-2 and Bcl-xL play important roles in the crosstalk between autophagy and apoptosis. FEBS J. 2011;278(3):403–413. doi: 10.1111/j.1742-4658.2010.07965.x. [DOI] [PubMed] [Google Scholar]
- 34.Jones BE, Lo CR, Liu H, Pradhan Z, Garcia L, Srinivasan A, et al. Role of caspases and NF-kappaB signaling in hydrogen peroxide- and superoxide-induced hepatocyte apoptosis. Am J Physiol Gastrointest Liver Physiol. 2000;278(5):G693–699. doi: 10.1152/ajpgi.2000.278.5.G693. [DOI] [PubMed] [Google Scholar]
- 35.Xu Y, Bialik S, Jones BE, Iimuro Y, Kitsis RN, Srinivasan A, et al. NF-kappaB inactivation converts a hepatocyte cell line TNF-alpha response from proliferation to apoptosis. Am J Physiol. 1998;275(4 Pt 1):C1058–1066. doi: 10.1152/ajpcell.1998.275.4.C1058. [DOI] [PubMed] [Google Scholar]