

Abstract
We present a rapid and inexpensive high-throughput screening protocol to identify transcriptional regulators of alpha-synuclein, a gene associated with Parkinson's disease. 293T cells are transiently transfected with plasmids from an arrayed ORF expression library, together with luciferase reporter plasmids, in a one-gene-per-well microplate format. Firefly luciferase activity is assayed after 48 hr to determine the effects of each library gene upon alpha-synuclein transcription, normalized to expression from an internal control construct (a hCMV promoter directing Renilla luciferase). This protocol is facilitated by a bench-top robot enclosed in a biosafety cabinet, which performs aseptic liquid handling in 96-well format. Our automated transfection protocol is readily adaptable to high-throughput lentiviral library production or other functional screening protocols requiring triple-transfections of large numbers of unique library plasmids in conjunction with a common set of helper plasmids. We also present an inexpensive and validated alternative to commercially-available, dual luciferase reagents which employs PTC124, EDTA, and pyrophosphate to suppress firefly luciferase activity prior to measurement of Renilla luciferase. Using these methods, we screened 7,670 human genes and identified 68 regulators of alpha-synuclein. This protocol is easily modifiable to target other genes of interest.
Keywords: Cellular Biology, Issue 88, Luciferases, Gene Transfer Techniques, Transfection, High-Throughput Screening Assays, Transfections, Robotics
Introduction
The ability to identify key genetic regulatory elements and the factors that act on them is fundamental for the exploration of numerous biological processes. However, identifying factors that regulate expression of genes in rare cell types, such as specific neuronal populations, can be challenging. Here, we present a protocol for identifying novel transcriptional regulators of alpha-synuclein (SNCA), a gene associated with Parkinson's disease and expressed in dopaminergic neurons in the substantianigra pars compacta region of the midbrain. We accomplish this using a high-throughput, dual-luciferase, in vitro reporter screen to deconstruct alpha-synuclein expression in 293T cells. The alpha-synuclein promoter is first cloned into a reporter plasmid containing the firefly luciferase gene. A commercially available plasmid containing the Renilla luciferase under the control of a constitutively active promoter serves as an internal control. These reporter constructs are co-transfected into 293T cells in microplates with plasmids from a DNA expression library, such that each well is transfected with a single library plasmid. After 48 hr, luciferase activity for each reporter is measured sequentially using a dual-glow assay. The relative expression of alpha-synuclein in response to each library gene is inferred by the ratio of firefly:Renilla luciferase activity in each well (F:R ratio) after plate-based normalization.
This protocol provides the ability to screen a large number of genes (~2,500 per week) for their ability to transactivate a reporter gene using minimal manpower (1-2 people) and minimal cost (about $3 reagent cost per microplate assay). Transcriptional regulators of neuronal genes (e.g., alpha-synuclein), which are difficult to study in cultured neurons refractory to genetic manipulation, can be compared side-by-side in this direct functional assay. We include in our protocol a detailed method for cloning and growth of plasmids containing the alpha-synuclein promoter, since we found that plasmids containing this region are unstable when grown with conventional procedures (see Discussion). We also include an inexpensive alternative to commercially available dual-luciferase reagents for high-throughput assays. This protocol can be easily adapted to target other regulatory elements of interest, or to any process requiring high-throughput transient transfections.
Protocol
For an experimental overview, see Figure 1.
1. Prepare Reporter Plasmids Containing the Alpha-Synuclein Promoter
Regulatory elements of the alpha-synuclein gene (Figure 2) span approximately 10 kb, from the upstream NACP (non-A beta component of amyloid peptide) dinucleotide repeat sequence1,2 through intron 2 3. We include a portion of this region in our reporter construct. We found that plasmids containing intron 2 required special procedures for growth, as outlined below. The luciferase plasmids, pGL4.10 and pGL4.75 (Figure 3), are commercially available from Promega and can be propagated using conventional molecular biology protocols.
Amplify the 2 components of the alpha-synuclein promoter in separate PCRs using the recipe in Table 1 and the primers in Table 2.
Verify PCR products on an agarose gel and clean using the Qiaquick PCR Purification kit (Qiagen), eluting in 50 μl of TE (Tris-EDTA). Store the eluted 0.9 kb fragment at -20 °C for future use.
Digest the entire volume of the 3.4 kb PCR fragment, as well as 5 μg of pGL4.10, with SacI and XhoI. Treat the vector with Calf Intestine Alkaline Phosphatase (Roche) and gel purify using the PureLink Quick Gel Extraction kit (Invitrogen).
Ligate the fragment and vector using T4 DNA ligase (NEB). Clean the completed reaction using the PureLink PCR Micro kit (Invitrogen), eluting in 10 μl TE buffer.
Transform 2 μl of the cleaned, ligation reaction into 20 μl of ElectroMAX Stbl4 competent cells (Invitrogen) and electroporate according to the manufacturer's instructions. Shake in a 30 °C incubator at 225 rpm for 90 min. Spread 2 volumes on LB plates containing 100 mg/ml ampicillin and incubate at 30 °C for 2 days.
Using a toothpick, select 4-6 small colonies for inoculation into 2 ml of TB (Terrific Broth). Grow these miniprep cultures in a 30 °C shaker O/N.
Purify plasmid DNA from 1.5 ml of each miniprep culture using the PureLink HiPure Plasmid Miniprep kit (Invitrogen).
Digest the minipreps with BamHI and electrophorese on an agarose gel. The correct clone should produce fragments of 2.8 kb and 4.9 kb.
Restreak the remaining miniprep culture from the correct clone on a fresh LB (Luria Broth) plate and grow at 30 °C for 2 days.
Select a small colony and inoculate a 1.5 ml TB starter culture. Shake O/N at 30 °C. Do not let the starter culture grow to saturation.
Dilute the entire starter culture into 150 ml of prewarmed TB and shake at 30 °C for 2 days. Before proceeding to the next step, use 1.5 ml of this culture for an additional miniprep and digestion to confirm that the clone did not mutate during replication.
Purify plasmid DNA from the remaining culture using the PureLink HiPure Plasmid Maxiprep kit (Invitrogen). Expected yields of this intermediate plasmid are 50-150 μg per 150 ml of culture.
Thaw the 0.9 kb fragment frozen in Step 1.2. Repeat Steps 1.3 to 1.12 to clone the 0.9 kb fragment into the intermediate plasmid, digesting instead with KpnI and SacI. Ligate, transform, select, and grow for maxipreps as above. Digestion with BamHI should yield fragments of 5.8 kb and 2.8 kb. This is the plasmid that will be used for the screen.
2. Prepare 293T Cells, Reporter Plasmids, and Library DNA for Screening
293T cells are first seeded into 96-well plates and transfected the following day. Reporter plasmids and library DNAs are diluted into 96-well plates. We obtained plates of transfection-grade library DNA from the DNA Core at Massachusetts General Hospital (dnacore.mgh.harvard.edu). This protocol transfects a single library plate into 2 identical plates of 293T cells (Figure 1), thus 2 plates of cells should be seeded for each library plate to be screened.
Note: Grow cells in media without phenol red or antibiotics, as these interfere with the luciferase assays and increase toxicity during transfection, respectively.
For each library plate to be screened, resuspend trypsinized 293T cells to a concentration of 1.5 x 105 cells/ml in DMEM containing 14% FBS. Pour into a sterile reservoir (Axygen).
Place the reservoir, 2 clear-bottomed, 96-well plates (Corning), and a box of filter pipet tips (Agilent) on the robot deck. Dispense 100 μl of cells into each well of both plates, for a density of 1.5 x 104 cells per well.
Centrifuge the cell plates at 50 x g for 2 min, using low ramp speeds to ensure equal cell density throughout each well. Incubate at 37 °C and 10% CO2 O/N.
Dilute the firefly and Renilla reporter plasmids to 5 ng/μl in serum-free media. Combine the dilutions in a ratio of 5:3 firefly to Renilla plasmid (by volume) in a 15 ml conical tube.
Pipet the combined plasmids into each well of a 96-well plate, dispensing 17 μl per library plate, plus 2-10 μl excess, into each well.
Obtain transfection-grade DNA library plates as above and dilute to 13.3 ng/μl with endotoxin-free water. The minimum volume per well should be 28 μl.
3. Perform Transfections
On day 2 of the screen, reporter plasmids and library DNAs are transfected into 293T cells.
For each library plate, mix 107.5 μl of RT Optifect (Invitrogen) with 4.3 ml serum-free media (1:40 dilution) in a polystyrene tube. Incubate for 5 min at RT, and then dispense 44 μl (per library plate) into each well of a 96-well, polystyrene V-bottomed plate (Greiner).
Place the plate of diluted Optifect, reporter plasmids (Step 2.5), library DNAs (Step 2.6), and a box of pipet tips on the robot deck.
Aspirate 17 μl of reporter plasmids, 43 μl of diluted Optifect, and 26 μl of library DNA, inserting a 5 μl air gap between each aspiration. The library DNA should be aspirated last to prevent cross-contamination. Dispense the contents of the tips into a new polystyrene V-bottomed plate, mix, cover, and set aside for 20 min to allow lipid/DNA complexes to form. If transfecting multiple library plates, replace the pipet tips and library plate, and repeat.
Uncover the Optifect/DNA mixture plate and place it on the robot deck with 2 plates of 293T cells. Using a single box of pipet tips, aspirate 80 μl of the Optifect:DNA mixture and slowly dispense 40 μl onto each plate of cells. If transfecting multiple library plates, repeat for all Optifect:DNA plates. Cover cells.
Centrifuge the cell plates at 1,200 x g for 30 min. Cover and return to the incubator.
Incubate the cells for 4-6 hr. During this incubation, prepare fresh media by mixing 12 ml DMEM containing 10% FBS and 10 μg/ml ciprofloxacin (CellGro) for each transfected library plate. Pour fresh media into a sterile reservoir.
Place 2 boxes of robot tips, a single plate of cells, the reservoir containing fresh media, and a waste reservoir onto the robot deck. Aspirate 100 μl of media from the cells and dispense into the waste reservoir partially filled with 70% ethanol. Using fresh tips, aspirate 100 μl of fresh media and slowly dispense onto the cells. Repeat for all plates of cells.
Return plates to the incubator for 48 hr.
4. Perform Dual-Luciferase Assays
On day 4 of the screen, the firefly and Renilla luciferase assays are performed.
Note: The dual-luciferase assay requires the sequential addition of 2 assay buffers, 3X firefly assay buffer and 3X Renilla assay buffer, as described in Table 3. Firefly and Renilla luciferases are not secreted, thus cells must be lysed prior to measuring luciferase activity. Firefly assay buffer lyses cells and provides substrate for the firefly luciferase; Renilla assay buffer quenches the firefly signal and provides substrate for the Renilla luciferase.
For each library plate, prepare 8 ml of 3X firefly assay buffer from stock solutions as described in Table 3, and dispense 82 μl into each well of a 96-well V-bottomed plate.
For each library plate, also prepare 12 ml of 3X Renilla assay buffer and dispense 122 μl into each well of a 96-well V-bottomed plate. Set aside both the firefly and Renilla assay buffers.
Place a box of pipet tips, a waste reservoir, and 2 plates of cells (transfected from the same library plate) on the robot deck.
Aspirate 60 μl of media from each plate and discard in the waste reservoir partially filled with 70% ethanol. Cover plates and set aside. If transfecting multiple library plates, repeat for all sets of plates.
Place 2 boxes of pipet tips, the plate of 3X firefly assay buffer, and a set of cell plates on the robot deck.
Aspirate 40 μl of 3X firefly assay buffer and add it to the first plate of cells, mixing thoroughly. Switching to new pipet tips, repeat for the second cell plate. Place cells at RT for 10 min to complete lysis. If transfecting multiple library plates, replace the boxes of tips and cell plates, and repeat.
Record luminescence from each well of each cell plate on a luminometer, recording for 1 sec per well. Return plates to the robot deck.
Place 2 boxes of pipet tips, the plate of 3X Renilla assay buffer, and a set of cell plates on the robot deck.
Aspirate 60 μl of 3X Renilla assay buffer, and add it to the first plate of cells, mixing thoroughly. Switching to a new box of pipet tips, repeat for the second cell plate. If transfecting multiple library plates, repeat for all sets of cell plates.
Record luminescence from all plates on a luminometer, recording each well for 1 sec as above. Discard plates.
5. Data Analysis
Calculate the firefly:Renilla luminescence ratio ("F:R ratio") for each well by dividing the counts of firefly signal by the counts of Renilla signal.
Calculate the induction value by dividing the F:R ratio of each well by the average F:R ratio of the plate, excluding the highest and lowest 25% of wells.
Average the induction values across replicates for a single transfected library plate. Genes inducing or repressing alpha-synuclein more than three-folds are considered "hits" in this screen and are subject to further validation and secondary screens.
Representative Results
Typical luciferase values, F:R ratios and induction values for a single half-plate are shown in Figure 4. Note the hit in well H2, a 32-fold inducer. Genes causing excessive toxicity (for example, well E3), or wells that were poorly transfected, will produce low values for both firefly and Renilla luciferases, but an average induction value. Genes causing non-specific induction of luciferase activity, perhaps via interaction with the pGL4 backbone, will induce firefly and Renilla luciferases equally, resulting in an average induction value. Large discrepancies in induction values between replicates should raise suspicion for execution errors. It is important to verify that the screen is performed within the linear range of the reporter assays so that clones that cause increased expression will be measured as increased luciferase activity. We transfected increasing amount of each reporter gene to validate the linearity of the reporter assay under our experimental conditions (Figure 5). It is also important to perform the assays promptly after the incubation step to avoid non-linearity due to substrate depletion. We observed significant firefly luciferase activity up to 1 hr after addition of reagents (Figure 6).
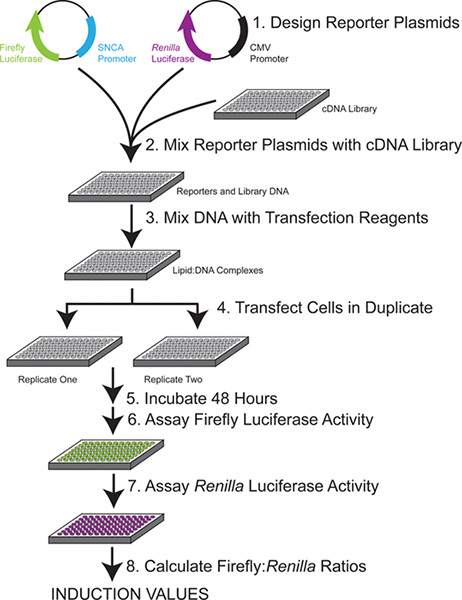 Figure 1. Experimental overview. Each single plate of library DNA is combined with 2 reporter constructs and used to transfect duplicate plates of cells. After 48 hr, the activity of each luciferase reporter is measured sequentially. The specific induction of the SNCA promoter plasmid by each library plasmid is calculated by the ratio of firefly to Renilla luciferase after plate-based normalization.
Figure 1. Experimental overview. Each single plate of library DNA is combined with 2 reporter constructs and used to transfect duplicate plates of cells. After 48 hr, the activity of each luciferase reporter is measured sequentially. The specific induction of the SNCA promoter plasmid by each library plasmid is calculated by the ratio of firefly to Renilla luciferase after plate-based normalization.
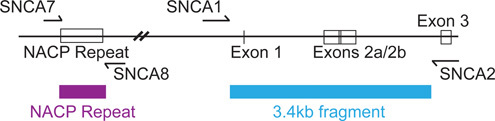 Figure 2. Schematic of the 5' region of alpha-synuclein (SNCA), located on chromosome 4, used in the firefly reporter plasmid. Primer names correspond to sequences in Table 2. Hatch marks indicate an approximately 8 kb segment eliminated from the figure, otherwise to scale. The ATG start codon for SNCA is located in exon 3.
Figure 2. Schematic of the 5' region of alpha-synuclein (SNCA), located on chromosome 4, used in the firefly reporter plasmid. Primer names correspond to sequences in Table 2. Hatch marks indicate an approximately 8 kb segment eliminated from the figure, otherwise to scale. The ATG start codon for SNCA is located in exon 3.
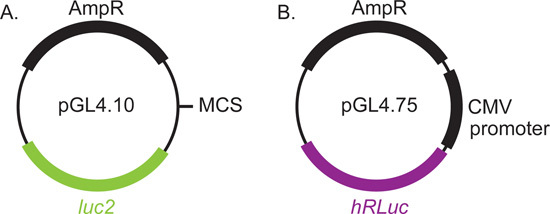 Figure 3. Luciferase reporter plasmids used in this screening protocol. The SNCA genomic regions are cloned into the multiple cloning site of pGL4.10 immediately upstream of the firefly luciferase. A plasmid with the hCMV-IE1 (human cytomegalovirus immediate early 1) promoter directing expression of Renilla luciferase was used as an internal control. Both plasmids are commercially available from Promega.
Figure 3. Luciferase reporter plasmids used in this screening protocol. The SNCA genomic regions are cloned into the multiple cloning site of pGL4.10 immediately upstream of the firefly luciferase. A plasmid with the hCMV-IE1 (human cytomegalovirus immediate early 1) promoter directing expression of Renilla luciferase was used as an internal control. Both plasmids are commercially available from Promega.
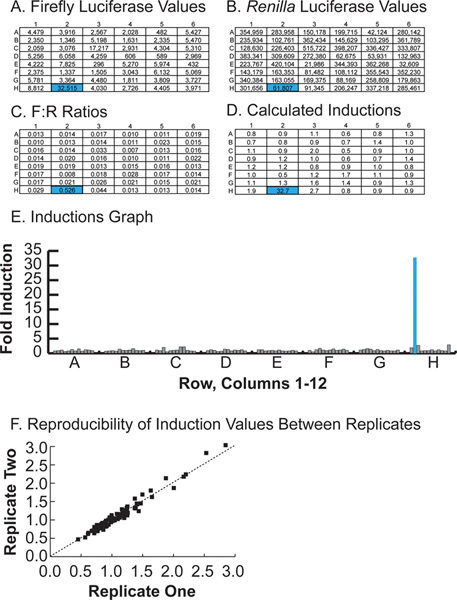 Figure 4. Representative results for half of a single 96-well plate. A. Raw firefly luciferase counts. B. Raw Renilla luciferase counts. C. F:R ratios, calculated by dividing the value in A by the value in B. D. Induction values for each well, calculated by dividing each F:R ratio by the average F:R ratio of the plate, excluding the top and bottom 25% of wells. E. Graphical representation of induction values for a single plate, with well H2, a positive hit, highlighted. Hit H2 is a neuronal transcription factor not previously associated with SNCA expression. Click here to view larger figure.
Figure 4. Representative results for half of a single 96-well plate. A. Raw firefly luciferase counts. B. Raw Renilla luciferase counts. C. F:R ratios, calculated by dividing the value in A by the value in B. D. Induction values for each well, calculated by dividing each F:R ratio by the average F:R ratio of the plate, excluding the top and bottom 25% of wells. E. Graphical representation of induction values for a single plate, with well H2, a positive hit, highlighted. Hit H2 is a neuronal transcription factor not previously associated with SNCA expression. Click here to view larger figure.
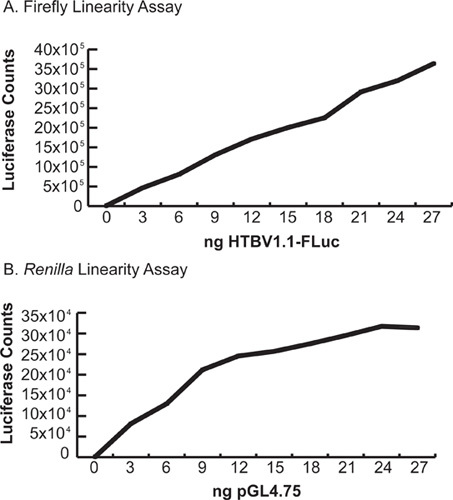 Figure 5. Linearity assays. Cells were transfected with increasing amounts of firefly and Renilla luciferase plasmids under the control of the strong hCMV-IE1 promoter and assayed with the protocol above. (Total amount of DNA transfected was kept constant by use of a neutral balancer plasmid). A. Firefly linearity assay. Note that firefly activity remains linear through a broad range of values. B.
Renilla linearity assay. Note the flattening of the curve above 15 ng/well.
Figure 5. Linearity assays. Cells were transfected with increasing amounts of firefly and Renilla luciferase plasmids under the control of the strong hCMV-IE1 promoter and assayed with the protocol above. (Total amount of DNA transfected was kept constant by use of a neutral balancer plasmid). A. Firefly linearity assay. Note that firefly activity remains linear through a broad range of values. B.
Renilla linearity assay. Note the flattening of the curve above 15 ng/well.
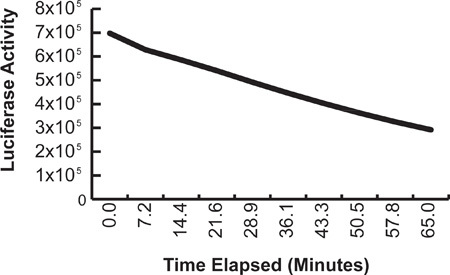 Figure 6. Time course of decay signal for firefly luciferase assay. Luciferase reagents were added to a single plate transfected with a CMV-luciferase construct at time 0; serial measurements were made without the addition of further reagents.
Figure 6. Time course of decay signal for firefly luciferase assay. Luciferase reagents were added to a single plate transfected with a CMV-luciferase construct at time 0; serial measurements were made without the addition of further reagents.
| Component | Volume | Final Concentration |
| BAC 2002-D6 at 5 ng/μl | 4 μl | -- |
| 5X Phusion GC Buffer | 20 μl | 1X |
| DMSO | 6 μl | 6% |
| dNTPs (10 mm) | 2 μl | 200 μM |
| Forward Primer (100 μM) | 1 μl | 10 μM |
| Reverse Primer (100 μM) | 1 μl | 10 μM |
| ddH2O | 65 μl | -- |
| Phusion DNA Polymerase | 1 μl | -- |
| Total: 100 μl |
Table 1. PCR set-up for amplifying the alpha-synuclein promoter.
| Name | Sequence | Restriction Site |
| SNCA7 | 5'-gtcggtacctgaagttaacctcccctcaatacc-3' | KpnI |
| SNCA8 | 5'-tgcgagctcaagaagacagccatctgcaagcc-3' | SacI |
| SNCA1 | 5'-tctgagctctctgagcatttccctaggtg-3' | SacI |
| SNCA2 | 5'-tagctcgagggctaatgaattcctttaca-3' | XhoI |
Table 2. Primers for amplifying the alpha-synuclein promoter. The NACP Repeat amplicon should produce a 0.9 kb fragment; the other amplicon length is 3.4 kb. For primer locations, please refer to Figure 2.
| A: 3X Firefly Assay Buffer | ||
| Stock Solutions (store at -80 °C) | Volume per 100 ml 3X Firefly Assay Buffer | Final Concentration (3X) |
| 500 mM DTT | 3.0 ml | 15 mM |
| 10 mM coenzyme A | 6.0 ml | 0.6 mM |
| 100 mM ATP | 0.45 ml | 0.45 mM |
| 80 mg/mlluciferin | 0.525 ml | 4.2 mg/ml |
| Triton lysis buffer | 90.025 ml | -- |
| B. Triton Lysis Buffer | ||
| Component (store at RT) | Volume per 100 ml Triton Lysis Buffer | Final Concentration |
| Tris-HCl powder | 1.705 g | 0.1082 M |
| Tris-base powder | 0.508 g | 0.0419 M |
| 5 M NaCl | 1.50 ml | 75 mM |
| 1 M MgCl2 | 0.30 ml | 3 mM |
| Triton X-100 pure liquid | 0.75 ml | 0.25% |
| Water | to 100 ml total volume | -- |
| C. 3X Renilla Assay Buffer | ||
| Stock Solutions (store at -80 °C) | Volume per ~100 ml 3X Renilla Assay Buffer | Final Concentration (3X) |
| 10 mM PTC124 in DMSO | 0.6 ml | 0.06 mM |
| 2 mM h-CTZ in ethanol | 0.5 ml | 0.01 mM |
| Renilla Salts | 100 ml | -- |
| D. Renilla Salts | ||
| Component (store at RT) | Volume per 100 ml Renilla Salts | Final Concentration |
| 0.5 M Na2EDTA | 9.0 ml | 45 mM |
| Na Pyrophosphate | 1.34 g | 30 mM |
| NaCl | 8.33 g | 1.425 M |
| Water | to 100 ml total volume | -- |
Table 3. Stock solutions and assay buffers for the firefly and Renilla luciferase assays. A. 3X firefly assay buffer recipe. DTT, dithiothreitol. Unless noted, all components are soluble in water. B. Triton lysis buffer recipe. C. 3X Renilla assay buffer recipe. h-CTZ, h-coelenterazine. Make 2 mM h-CTZ by adding 10 mg h-CTZ to 12.2 ml of 100% EtOH and 120 μl of concentrated HCl. PTC124 is soluble in DMSO. D. Renilla salts recipe. Triton lysis buffer and Renilla salts are stable for several weeks at 4 °C and RT, respectively. 3X firefly assay buffer and 3X Renilla assay buffer should be mixed within 3-4 hr of performing the luciferase assay.
Discussion
Alpha-synuclein has been implicated in Parkinson's disease (PD) as a component of Lewy bodies4, intracellular inclusions considered pathognomonic for the disease. Numerous genome-wide association studies have linked single nucleotide polymorphisms in alpha-synuclein with increased risk for sporadic PD5,6,7. Although less common than sporadic PD, familial PD may be also caused by mutations in alpha-synuclein8, as well as duplication and triplication of the alpha-synuclein locus9,10,11, a phenomenon also seen in sporadic PD12. Taken together, these studies reinforce the centrality of alpha-synuclein to the pathogenesis of PD. The aim of this screen was to identify genes that regulate alpha-synuclein and therefore may play a role in the pathogenesis of Parkinson's disease. Using this protocol, we screened 7,670 human genes and identified 154 preliminary hits (at least 3-fold inducers), representing 140 unique genes. These hits were subject to secondary assays using new DNA preparations to determine hit reproducibility. Hits in the 3-fold range reproduced in 28% of subsequent luciferase assays. Reproducibility increased to 59%, 69%, and 78% in 4-, 5-, and 5-fold inducers, respectively. Hits with inductions over 7-fold reproduced 100% of the time. We ultimately identified 68 novel regulators of alpha-synuclein transcription.
Several lines of evidence suggest that the 68 hits we obtained are real regulators of alpha-synuclein. Our list of identified hits includes GATA3, a member of the GATA family of transcription factors previously shown to regulate SNCA expression3. Scoring one of the few known SNCA regulators gives great confidence in the reliability of our screen. In addition, we independently scored as hits several members of the same family of neuronal transcription factors, suggesting that the screen is reproducible and not identifying genes at random. Most importantly, in additional follow-up assays, the ability of our top hits to regulate endogenous SNCA expression in neuronal cells was validated in overexpression and knockdown assays. We overexpressed a few of the top hits using BacMamvectors3 in SY5Y dopaminergic cells and were able to increase endogenous SNCA levels 2-8 fold, while shRNA knockdown of these hits resulted in decreased SNCA mRNA levels. These studies conclusively demonstrate that our top hits are regulators of endogenous SNCA levels and are not simply artifacts of the deconstructed luciferase system. Additional follow-up studies for other hits will be required to determine the true false positive and false negative rates for the full list of hits in our screen. A complete description of the hits and their relationship to Parkinson's Disease is in preparation.
It is important to consider the limitations of our screening methodology. It should be obvious that the screen would not identify any regulator not present in the screening library. While our screening library was substantial, containing about one-fourth of the human genome, it was not comprehensive. Given the simplicity of our assay, additional libraries could be screened with ease. We increased the number of genes screened in a targeted manner by including paralogs of hits, when available, in our secondary assays. We also would not score any hit which bound to genomic regions outside of those included in our reporter construct. To maximize the chance of including the proper region, our reporter contained the immediate flanking sequences for the multiple transcription start sites found in the SNCA promoter. It is likely that additional factors bind to regions far outside the immediate promoter region to regulate SNCA expression. These could be detected by performing screens with additional upstream and downstream regions, if desired, though only ~10 kb at a time could be assessed in this manner due to limitations on efficient cloning into plasmid vectors and the stability of the high copy number pGL4 vectors. Alternatively, the luciferase construct could be retrofitted into BAC DNAs containing the SNCA locus. One drawback to the latter approach is that transfection of BACs even under ideal conditions is >100-fold less efficient then plasmids14. Another approach would be to "knock-in" the luciferase reporter into the endogenous SNCA locus, a method that is far more laborious then our approach. We would also not score any hit which is already expressed at saturating levels so that overexpression does not result in increased luciferase activity. For this reason we chose to employ non-neuronal 293T cells, which might lack neuronal factors regulating SNCA, and indeed we did score a number of neuronal transcriptional factors. One potential disadvantage of 293T cells, however, is that we might not score any gene as a hit if it required additional neuronal factors to function properly. Thus, like all screens, our study could miss a number of potential SNCA regulators. However, in practical terms, the 68 hits we obtained increase the number of known regulators of SNCA by more than 10-fold and will require years of work to follow up, so additional hits may not be necessary in our case.
It is also important to realize the potential artifacts that might occur using this methodology. In chemical screens, luciferase is notorious for artifactually scoring compounds by stabilization of the luciferase protein rather than by induction of transcription. To rule out protein stabilization artifacts, we screened our hits against several other promoter-luciferase constructs and did not find any hits which appeared to act post-transcriptionally by elevating expression from constitutive promoters. Another potential artifact could be a transactivating factor which binds to the pGL4 vector backbone rather than the SNCA promoter. We employed the pGL4 vector which has been extensively mutagenized to remove transcription factor binding sites to avoid this issue. To assess whether any of our hits required the vector backbone for induction, we used PCR to amplify the SNCA-luciferase vector region devoid of any backbone and used this linear fragment as a reporter for secondary assays. We found no significant difference in induction values between our complete plasmid reporter and the linear backbone-free construct with one exception. GATA2 showed ~15-fold induction with the plasmid reporter, but only ~2.7 fold with the linear reporter, suggesting that some of the GATA2 induction may be due to binding to backbone components.
A number of other methods to identify transcriptional regulators have been described. Nearly 30 years ago, the mapping of cis-acting transcriptional control elements in strong viral promoters via transfected reporter gene expression was established15. This method, sometimes termed "promoter bashing," has fallen out of favor for mapping of cell-type specific promoters since it is difficult to obtain and transfect suitable primary cell lines, and such lines may not accurately represent their tissue of origin. The yeast one-hybrid system16 circumvents this issue by providing a deconstructed reporter system to detect interactions between a library of fusion genes and a specific cis-acting element. Our approach is similar to the yeast one-hybrid system in its deconstructed design, but in our system we employ normal expression clones rather than gene fusions, we employ mammalian cells rather than yeast, and we employ robotics which allows side-by-side quantitative functional readouts for each test gene. A promising method termed proteomics of isolated chromatin segments (PICh) which allows identification of proteins bound to specific genome regions has been described17, but has not yet been successfully applied to single-copy human genes. Genome-wide chromatin immunoprecipitation (ChIP-seq and ChIP-chip) studies have been used to identify cis-acting regions on a large scale18. This method is potentially powerful, but, like PICh, may be less useful for cell types such as specific neuronal populations which are not easily obtained in homogenous culture. Moreover, when we have checked ChIP databases for our validated hits, they have not shown binding of these hits to the SNCA promoter. The reason for this is not clear, since we have been able to demonstrate binding of these factors to the SNCA promoter by conventional ChIP; but it may be due to the lack of the entire SNCA promoter region being employed in ChIP studies using microarray readouts (ChIP-chip). PICh, ChIP-seq, and other proteomic approaches may therefore be most useful when coupled to the direct functional readouts that our method generates.
Dual-luciferase assays employing firefly and Renilla luciferases offer a sensitive method for studying numerous intracellular processes in high-throughput format 19. The fusion of a target promoter to firefly luciferase has been used to study the transcriptional regulation and promoter structure of numerous targets in vitro, including alpha-synuclein1,2. We developed an inexpensive alternative to commercially available dual-luciferase reagents that provided robust signal and was linear in the range of our assay (Figure 6). Our method is adapted from a protocol generously provided by Ron Johnson at the NIH and a previously described non-commercial dual-luciferase assay20. The firefly assay buffer lyses cells and provides ATP and luciferin, the substrates of firefly luciferase. This assay yields a glow reaction with a half-life of approximately 1 hr (Figure 7). The Renilla assay buffer quenches the firefly signal and provides appropriate substrate (coelenterazine). Note that the Renilla assay buffer itself will not lyse cells and must be used in combination with either Triton lysis buffer or firefly assay buffer. We found that previously described Renilla assay buffers readily precipitated at RT, thus we eliminated sodium sulfate and decreased the amount of sodium pyrophosphate by 60%. Our assay also includes PTC124, a potent inhibitor of firefly luciferase 21 that contributes additional quenching in the assay. Quenching activity is also provided by EDTA, NaCl, and pyrophosphate in the Renilla buffer. With these new formulations, approximately 99.5% of firefly activity is quenched by the Renilla buffer. The intensity and reproducibility of both firefly and Renilla luciferase signals were slightly improved by mixing after the addition of the buffers (Steps 4.6 and 4.10).
In our experience, the complete alpha-synuclein promoter in pGL4.10 is uniquely difficult to amplify and clone, perhaps because of GC-rich regions and complex secondary structure in intron 2. We were most successful with STBL4 competent cells, which are optimized for the cloning of unstable inserts. Even following these precautions, we frequently recovered a mutant in which E. coli genomic DNA was inserted into the 3' end of the construct. This mutant creates bands of approximately 5.8 kb and 4 kb when digested with BamHI (versus 5.8 kb and 2.8 kb in the correct clone) and appears as larger colonies on selective plates. Careful growth of competent cells at 30 °C and preventing cultures from reaching saturation decreases, but does not eliminate, the likelihood of recovering the mutant. We recommend verifying the purity of all DNA preparations by restriction digest and gel electrophoresis prior to transfection.
This screening protocol is easily adapted to other promoters, and must be optimized accordingly, using as positive and negative controls genes already known to regulate the target promoter. Several considerations apply to the cloning and transfection of the reporter plasmids. When cloning the target promoter into the firefly luciferase reporter plasmid, the start site of the luciferase should approximate the translational start site in the target gene. Dual-luciferase screens typically place the Renilla plasmid under the control of a minimal promoter, such as the HSV-tk promoter, to minimize effects of the plasmid backbone. However, we found that one of our positive controls induced this promoter, thus we opted for the CMV promoter. The relative amount of each reporter plasmid should be determined empirically and is influenced by the constitutive activity of each promoter. Ideal baseline (transfected, but uninduced) luciferase counts should be at least five- to ten-fold over background from untransfected cells, to allow detection of library genes that repress or that cause toxicity, which will result in lower signal. Prior to screening, verify that expected luciferase counts fall within the linear range of each assay, which may vary between luminometers. (Note that our Renilla counts typically fall at the upper end of the linear range in our assay.) We found that increasing the ratio of library DNA to firefly reporter plasmid resulted in higher inductions, thus we used the minimal amount of this reporter.
The transfection protocol must be optimized as well. We chose 293T cells for their relative ease of transfection, the efficiency of which increased 50-fold with the addition of a centrifugation step (Step 3.5). The dopaminergic line SY5Y is over 100-fold less transfectable in our hands, and, as discussed above, may not be optimal for performing overexpression screening. Our optimal assay time was empirically determined to be 48 hr, thus our cells were plated at a low initial density, necessitating the use of Optifect, which is designed to transfect cells at 10-70% confluency. Other cell lines may require different starting densities and different transfection reagents. We chose to change the cell media after transfection to add antibiotics (which could cause toxicity if present during transfection). Although this decreases the likelihood of bacterial contamination, it increases cost in materials (especially robot pipet tips), thus the necessity of this step should be evaluated when adapting this protocol to other systems. The luciferase assays may be used with minimal optimization, but before performing this assay in high-throughput with a different cell type, verify that cells are appropriately lysed by the Triton lysis buffer. Note that Triton X-100 causes coelenterazine autofluorescence. The Renilla signal in our assay was strong enough to trivialize this effect, but when optimizing this assay for other cell types, limit Triton X-100 volumes to the minimal amount required for cell lysis.
We have presented a rapid and relatively inexpensive screening method for identifying novel transcriptional regulators of alpha-synuclein using cells transiently transfected with a dual-luciferase reporter system. This protocol is easily modifiable to target other genes of interest and may also be adapted for any application requiring high-throughput transfections, such as lentivirus production22.
Disclosures
This work was supported by royalties obtained by licensing BacMamreagents (US patent 5,731,182).
Acknowledgments
We thank John Darga of the MGH DNA Core for preparation of the DNA screening library. Christopher Chigas of Perkin Elmer provided invaluable support for our Wallac 1420 luminometer. Steven Ciacco and Martin Thomae of Agilent provided support for the Bravo robot. We thank Ron Johnson and Steve Titus at the NIH for generously providing their dual-glow luciferase assay protocol.
References
- Chiba-Falek O, Nussbaum RL. Effect of allelic variation at the NACP-Rep1 repeat upstream of the α-synuclein gene (SNCA) on transcription in a cell culture luciferase reporter system. Hum. Mol. Genet. 2001;10(26):3101–3109. doi: 10.1093/hmg/10.26.3101. [DOI] [PubMed] [Google Scholar]
- Chiba-Falek O, Touchman JW, Nussbaum RL. Functional analysis of intra-allelic variation at NACP-Rep1 in the alpha-synuclein gene. 2003;113(5):426–431. doi: 10.1007/s00439-003-1002-9. [DOI] [PubMed] [Google Scholar]
- Scherzer CR, et al. GATA transcription factors directly regulate the Parkinson's disease-linked gene α-synuclein. Proc. Natl. Acad. Sci. USA. 2008;105(31):10907–10912. doi: 10.1073/pnas.0802437105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M. α-Synuclein in Lewy Bodies. Nature. 1997;388(6645):839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- Pankratz N, et al. Meta-analysis of Parkinson's disease: identification of a novel locus, RIT2. Ann. Neurol. 2012;71(3):370–384. doi: 10.1002/ana.22687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satake W, et al. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson's disease. Nat. Genet. 2009;41(12):1303–1307. doi: 10.1038/ng.485. [DOI] [PubMed] [Google Scholar]
- Simón-Sánchez J, et al. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat. Genet. 2009;41(12):1308–1312. doi: 10.1038/ng.487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, et al. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science. 1997;276(5321):2045–2047. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- Ibáñez P, et al. Causal relationship between a-synuclein gene duplication and familial Parkinson's disease. Lancet. 2004;364(9440):1169–1171. doi: 10.1016/S0140-6736(04)17104-3. [DOI] [PubMed] [Google Scholar]
- Chartier-Harlin M-C, et al. a-Synuclein locus duplication as a cause of familial Parkinson's disease. Lancet. 2004;364(9440):1167–1169. doi: 10.1016/S0140-6736(04)17103-1. [DOI] [PubMed] [Google Scholar]
- Singleton AB, et al. a-Synuclein locus triplication causes Parkinson's disease. Science. 2003;302(5646) doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- Ahn T-B, et al. a-Synuclein gene duplication is present in sporadic Parkinson disease. Neurology. 2008;70(1):43–49. doi: 10.1212/01.wnl.0000271080.53272.c7. [DOI] [PubMed] [Google Scholar]
- Boyce FM, Bucher NL. Baculovirus-mediated gene transfer into mammalian cells. Proc. Nat. Acad. USA. 1996;93(6):2348–2352. doi: 10.1073/pnas.93.6.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montigny WJ, et al. Parameters influencing high-efficiency transfection of bacterial artificial chromosomes into cultured mammalian cells. Biotechniques. 2003;35:796–807. doi: 10.2144/03354rr02. [DOI] [PubMed] [Google Scholar]
- Gorman CM, Moffat LF, Howard BH. Recombinant genomes which express chloramphenicol acetyltransferase in mammalian cells. Mol. Cell. Biol. 1982;2(9):1044–1051. doi: 10.1128/mcb.2.9.1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JJ, Herskowitz I. Isolation of ORC6, a Component of the Yeast Origin Recognition Complex by a One-Hybrid System. Science. 1993;262:1870–1874. doi: 10.1126/science.8266075. [DOI] [PubMed] [Google Scholar]
- Dejardin J, Kingston RE. Purification of Proteins Associated with Specific Genomic Loci. Cell. 2009;136:175–186. doi: 10.1016/j.cell.2008.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, et al. A map of the cis-regulatory sequences in the mouseGenome. Nature. 2012;488:116–120. doi: 10.1038/nature11243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan F, Wood K. Bioluminescent assays for high-throughput screening. Assay Drug Dev. Technol. 2007;5(1):127–136. doi: 10.1089/adt.2006.053. [DOI] [PubMed] [Google Scholar]
- Hampf M, Gossen M. A protocol for combined Photinus and Renilla luciferase quantification compatible with protein assays. Anal. Biochem. 2006;356(1):94–99. doi: 10.1016/j.ab.2006.04.046. [DOI] [PubMed] [Google Scholar]
- Auld DS, Thorne N, Maguire WF, Inglese J. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc. Natl. Acad. Sci. USA. 2009;106(9):3585–3590. doi: 10.1073/pnas.0813345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearlberg J, et al. Screens using RNAi and cDNA expression as surrogates for genetics in mammalian tissue culture cells. Cold Spring Harb.Symp. Quant. Biol. 2005;70:449–459. doi: 10.1101/sqb.2005.70.047. [DOI] [PubMed] [Google Scholar]