

Abstract
Misregulation of hedgehog (Hh) signaling has been implicated in the pathogenesis of basal cell carcinoma (BCC) and medulloblastoma. Vismodegib, an orally bioavailable Hh signal pathway inhibitor targeting Smo, has been approved for the treatment of advanced BCC. However, acquired drug resistance to vismodegib induced by point mutation in Smo is emerging as a major problem to vismodegib treatment. In this study, we designed and synthesized a novel chimeric compound NL-103, which comprises structural elements of Hh pathway inhibitor vismodegib, and histone deacetylase (HDAC) inhibitor vorinostat. NL-103 simultaneously and significantly inhibited both HDACs and Hh pathway. Importantly, NL-103, as well as vorinostat, effectively overcame vismodegib resistance induced by Smoothened point mutations. Moreover, NL-103 and vorinostat, but not vismodegib, significantly downregulated the expression of Gli2 which plays an important role in Hh pathway. These results indicate that HDAC inhibitory activity is essential for NL-103 to overcome vismodegib resistance and that dual inhibition of HDAC and Hh signaling pathway may be a rational strategy for overcoming vismodegib resistance. Our findings suggest that NL-103 may be a promising compound for clinical development as a more effective Hh pathway inhibitor.
Keywords: Drug resistance, hedgehog pathway inhibitor, histone deacetylase inhibitor, NL-103, smoothened mutation
Introduction
The hedgehog (Hh) signaling pathway is conserved from Drosophila to humans and plays critical roles in cell differentiation during embryogenesis (Ingham and McMahon 2001). In mammals, the binding of Hh ligand to its 12-transmembrane protein receptor Patched-1 (Ptch-1) relieves Ptch1-mediated inhibition of Smoothened (Smo), a seven-pass transmembrane protein with homology to G-protein-coupled receptors (GPCRs). Through a series of poorly understood events, activated Smo productively interacts with its downstream targets and promotes the accumulation of full-length Gli transcription factors that act as transcription activators of Hh target genes. Many studies have demonstrated that Hh signaling is delicately coordinated by the primary cilium, a microtubule-based organelle that projects from the surface of certain mammalian cells (Goetz and Anderson 2010). In the absence of Hh, Ptch-1 localizes to the primary cilium of mammalian cells; its ciliary enrichment is abrogated after engagement with Hh ligand (Rohatgi et al. 2007). Conversely, Smo accumulates on the primary cilium upon treatment with Hh or small-molecule Smo agonists (May et al. 2005; Rohatgi et al. 2007; Kovacs et al. 2008; Wang et al. 2009). Downstream of Smo are multi-protein complexes, which comprise Gli transcription factors and other components implicated in the Hh signaling pathway. Many of these complexes also concentrate in the primary cilium or its basal body upon Hh pathway activation (Haycraft et al. 2005; Tran et al. 2008; Kim et al. 2009).
Inappropriate activation of Hh pathway has been associated with basal cell carcinoma (BCC) and medulloblastoma (MB) (Gailani et al. 1996; Goodrich et al. 1997; Raffel et al. 1997; Xie et al. 1998). BCC is the most common skin cancer. It rarely metastasizes or kills. However, because it can cause significant destruction and disfigurement by invading surrounding tissues, it is still considered malignant. MB is a highly malignant primary brain tumor. It is the most common brain malignancy among children 0–4 years old. Victims of BCC or MB suffer from debilitating side effects of conventional chemotherapy, highlighting the need for more effective and less harmful targeted therapies. Fortunately, vismodegib (formerly GDC-0449; Genentech, South San Francisco, CA), an orally bioavailable Smo antagonist, has produced promising antitumor responses in clinical trials of patients with advanced BCC harboring mutations in Hh pathway. Thus far, vismodegib has been approved by U.S. Food and Drug Administration (FDA) for the treatment of advanced BCC. Furthermore, treatment of a MB patient with vismodegib resulted in rapid regression of his metastatic tumors. However, the favorable response of this patient to vismodegib was transient, as metastatic tumors soon recurred, and biopsy molecular profiling revealed resistance to vismodegib due to a mutation in Smo (Asp473 to His, Smo-DH) (Yauch et al. 2009). Additionally, a constitutively active form of Smo (Trp535 to Leu, Smo-M2) frequently occurs in patients with BCC, and its sensitivity to vismodegib still remains unknown (Xie et al. 1998).
Targeting alternative pathways is emerging as a promising therapeutic strategy for tumors with primary or acquired drug resistance. A previous study has demonstrated that certain histone deacetylase inhibitors (HDACi) are capable of effectively shutting down Hh pathway signaling through novel mechanisms (Canettieri et al. 2010). To investigate whether the simultaneous inhibition of Hh pathway and histone deacetylases (HDACs) can achieve synergistic effects and overcome vismodegib resistance conferred by Smo mutations, we designed and synthesized a chimeric compound NL-103, which comprises structural elements of vismodegib, and of the prototypical HDACi vorinostat (also known as SAHA). In this study, we demonstrate that NL-103 simultaneously inhibit both HDAC activities and Hh signaling. More importantly, NL-103 effectively overcomes vismodegib resistance conferred by Smo-M2 and Smo-DH mutants. These results indicate that dual inhibition of HDAC and Hh signaling pathway may be a rational strategy for overcoming vismodegib resistance. Thus, for the first time, we provide a rational and novel strategy for overcoming vismodegib resistance induced by Smo mutations.
Materials and Methods
Compounds
NL-103 was synthesized and provided by Professor Wei Wang (Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai, China). Vismodegib and vorinostat were purchased from Selleck Chemicals (Shanghai, China). SAG (N-Methyl-N’-(3-pyridinylbenzyl)-N’-(3-chlorobenzo[b]thiophene-2-carbonyl)-1,4-diamino-cyclohexane) was purchased from EMD Millipore Chemicals (Darmstadt, Germany). BODIPY (boron-dipyrromethene)-cyclopamine was purchased from Toronto Research Chemicals (Ontario, Canada). 4′,6-diamidino-2-phenylindoledihydrochloride (DAPI) was purchased from Sigma-Aldrich (St. Louis, MO). All above reagents were dissolved in dimethyl sulfoxide (DMSO).
HDAC enzyme inhibition assay
HDAC inhibitors were assessed using the HDAC Fluorometric Assay/Drug Discovery Kit-BML-AK500 (Biomol, Plymouth Meeting, PA). Test compounds were dissolved in DMSO. HDACs were immunoprecipitated from HeLa nuclear extract as recommended. Fluorescence was measured on a WALLAC Victor 2 plate reader (Perkin-Elmer Life & Analytical Science, Wellesley, MA) and reported as relative fluorescence units. Each assay was carried out according to the manufacturer’s instructions.
Luciferase reporter assay
Shh-N conditioned medium was prepared as previously described (Chen et al. 2002b). Cells were cultured, transfected, and the Hh pathway responsive reporter cell lines were constructed according to methods provided in supplementary data. Monoclonal NIH3T3 (ATCC, Manassas, VA) cells stably transfected with reporter gene were separately plated at 1 × 105 cells per well of 24-well plates. After cells reached confluence in about 2 days, they were shifted to serum-free medium containing 20% Shh-N conditioned medium or 20% HEK293 conditioned medium treatments. After 36 h, cells were lysed and luciferase activity was assayed on a Synergy-H4 Multi-Mode Microplate Reader (Bio-Tek Instruments, Winooski, VT) according to the protocol in Molecular Cloning (Sambrook and Russell 2001). Dozens of monoclonal cell lines were analyzed, and a cell line with optimal sensitivity to Shh-N stimulation and normal morphology was designated as NIH3T3-12Gli cell line.
Smo ciliary translocation assay
NIH3T3 cells were separately transfected with pEYFP-Smo-WT or pEYFP-Smo-M2 (EYFP, enhanced yellow fluorescence protein). Cells were split into new 96-well plates at an appropriate dilution and maintained in the selective medium containing 500 μg/mL G418 (geneticin; Amresco, Solon, OH) until colonies formed. Monoclonal colonies were picked, expanded, and analyzed according to a standard protocol provided in supplementary data. Each clone was examined for the translocation behavior of the Smo-WT-EYFP or Smo-M2-EYFP in response to Shh-N and/or SAG treatments. Clones that showed low level expression and expected translocation behaviors were selected for further study.
Quantitative real-time polymerase chain reaction
Total RNA was extracted with Trizol reagent (Sangon, Shanghai, China) and was quantitated (SPECTRA max190; Molecular Devices, Sunnyvale, CA). Complementary DNA was synthesized using PrimeScript® Reverse Transcription reagent Kit (Takara, Dalian, China) according to the manufacturer’s instructions. Real-time polymerase chain reaction (RT PCR) was performed by using SYBR® Premix Ex Taq™ (Takara) on the StepOnePlus real-time PCR system (Applied Biosystems, Foster City, CA). Quantitative real-time PCR was performed using primers against mouse Ptch-1, Smo, Gli-1, Gli-2, Gli-3, and glyceraldehyde-3-phosphate dehydrogenase (GAPDH). These primers were purchased from Sangon and their sequences were provided in supplementary data. The relative expression was calculated using the 2(-ΔΔCt) Method: the ΔCt was obtained by subtracting the Ct value of the reference gene GAPDH from the Ct of the gene of interest.
Data analysis
Data were presented as the means ± SD and were plotted using GraphPad Prism Ver. 5 (GraphPad Software, San Diego, CA). Concentrations required for 50% inhibition (IC50s) were calculated using a sigmoidal dose response curve fitting algorithm. Statistical comparisons between groups were performed using Student’s t-test. P < 0.05 was considered statistically significant.
Results
NL-103 simultaneously inhibits HDAC activities and Hh signaling
We designed and synthesized a dual-targeted inhibitor, NL-103, which integrates HDACs and Smo inhibitory functional groups into one single small molecule (molecular weight, 544.4; Fig. 1). We introduced hydroxamic acid with a flexible side chain onto the amino group of the pyridine backbone of a putative Smo inhibitor (Fig. 1). We anticipated that this engineered molecule would fit well into the substrate-binding pocket of HDACs and disrupt enzyme activities, yet still retain its ability to function as a Smo antagonist (Cai et al. 2010).
Figure 1.
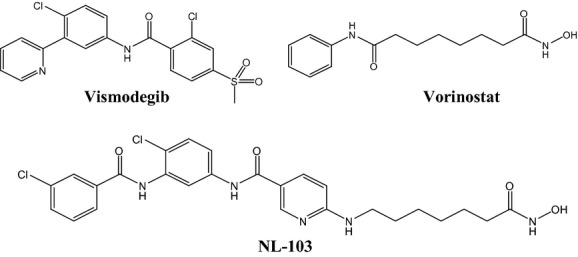
Schematic representation of the design and chemical structure of NL-103. To create NL-103, we introduced hydroxamic acid (functional group for HDAC inhibition) with a flexible side chain onto the pyridine backbone of a putative Smo inhibitor.
To confirm whether NL-103 can function as a HDACi, we examined its effects on both enzyme activities in vitro and substrate acetylation in cells. NL-103 inhibited enzyme activities of both class-I and class-II HDACs with high potency in vitro (21.3, 57.0, 71.4 and 680 nmol/L, respectively, for class-I HDAC1/2/3 and class-II HDAC6; Table 1). In NIH3T3 cells, 6 h treatment of NL-103 or vorinostat effectively and dose dependently increased the acetylation of total histones, as well as the acetylation of α-tubulin, a nonhistone substrate of HDACs (Fig. 2A). Moreover, NL-103 increased the acetylation of these proteins to a less extent than vorinostat, suggesting that NL-103 is a less potent HDACi (Fig. 2A).
Table 1.
Inhibition of HDAC enzyme activities by NL-103 and reference compound vorinostat (n = 1–2; n = 2, IC50 in mean ± SD)
| IC50 (nmol/L) | ||||
|---|---|---|---|---|
| Compound | HDAC1 | HDAC2 | HDAC3 | HDAC6 |
| NL-103 | 21.3 ± 1.0 | 57.0 ± 12.6 | 71.4 ± 2.9 | 680 |
| Vorinostat | 16.3 | 22.9 | 114.7 | 104.6 ± 50.1 |
For HDAC activity assay, HeLa cell nuclear extracts were used as a source of HDAC enzymes.
Figure 2.

NL-103 increases the acetylation levels of α-tubulin and total histones, and effectively inhibits Hh signaling by targeting Smo. (A) NIH3T3 cells were treated with varying concentrations of NL-103 or vorinostat. After 6 h, cells were harvested and the acetylation levels of α-tubulin and total histones were measured using immunoblotting. (B) Vismodegib, NL-103 and vorinostat dose-dependently inhibit Shh-N-induced firefly luciferase expression in NIH3T3-12Gli cells. Seeded cells were divided into two groups. One group was cultured in HEK293 control medium and the other was maintained in Shh-N conditioned medium. Both groups were treated with various concentrations of each compound. For cells treated with the same compound at the same concentration, reporter activity in Shh-N was normalized to that in control medium. Empirically, this calculation method can effectively filter out the nonspecific effects of compound on reporter activity. Data are the average of three independent experiments ± SD. (C) GDC-0449 and NL-103 compete for the binding of BODIPY-cyclopamine to Smo-WT-expressing HEK293T cells, but not SAHA. Nonspecific binding as defined by BODIPY-cyclopamine levels of cells treated with mock transfection. (D) Flow cytometric quantitation of specific BODIPY-cyclopamine binding to Smo-expressing cells was used to determine the affinities of Smo antagonists through binding competitions.
We next investigated the effects of NL-103 on Hh signaling. Using the NIH3T3-12Gli reporter cell line, we showed that Hh signaling induced by Shh-N conditioned medium was potently inhibited by NL-103 and vismodegib in a dose-dependent manner (IC50s: 19.7 and 27.0 nmol/L, respectively, for vismodegib and NL-103; Fig. 2B). Consistent with previous findings (Canettieri et al. 2010), vorinostat also dose-dependently depressed Hh signaling (IC50s: 669.7 nmol/L; Fig. 2B), despite to a less extent than vismodegib or NL-103. To determine whether NL-103 targets Smo, we introduced BODIPY-cyclopamine, a fluorescent derivative of cyclopamine, which specifically binds to Smo-expressing cells (Chen et al. 2002a). As shown in Figure 2C and D, the association of BODIPY-cyclopamine with Smo-expressing HEK293T cells was effectively and dose-dependently inhibited by NL-103 (KD: 79.6 nmol/L) and vismodegib (KD: 44.9 nmol/L), but not vorinostat, suggesting that NL-103 can directly bind to Smo as vismodegib does and Smo is the direct cellular target of NL-103. Taken together, these results suggest that NL-103 displays activity against Hh signaling by targeting Smo.
NL-103 inhibits the translocation of Smo to primary cilium as vismodegib
Recently, several groups reported that diverse antagonists differentially affect the trafficking and localization of Smo to the primary cilium (Rohatgi et al. 2009; Wang et al. 2009; Wilson et al. 2009). To test the effects of vismodegib, NL-103 and vorinostat on Smo trafficking, we incubated monoclonal NIH3T3 cells stably expressing exogenous Smo-WT-EYFP fusion protein with these compounds in the absence or presence of stimulation, and determined whether exogenous Smo colocalized with acetylated tubulin, a primary cilium marker. Cells treated with vehicle (0.1% DMSO) displayed Smo accumulation in very few cilia (Fig. 3A). Treatment with either Shh-N or Smo agonist SAG still could not result in obvious translocation of Smo to cilia (Fig. 3B and C). However, stimulation with both Shh-N and SAG induced robust Smo accumulation in many cilia (Fig. 3D). Considering that Shh-N and SAG target Ptch-1 and Smo, respectively, these findings suggest that the abrogation of inhibitory effects of Ptch-1 upon Smo and an active conformation induced by SAG are both necessary for efficient Smo translocation. Expectedly, both NL-103 and vismodegib effectively suppressed Smo localization induced by the presence of Shh-N and SAG, but not vorinostat (Fig. 3E–H). At least two monoclonal cell lines were applied and similar results were obtained.
Figure 3.
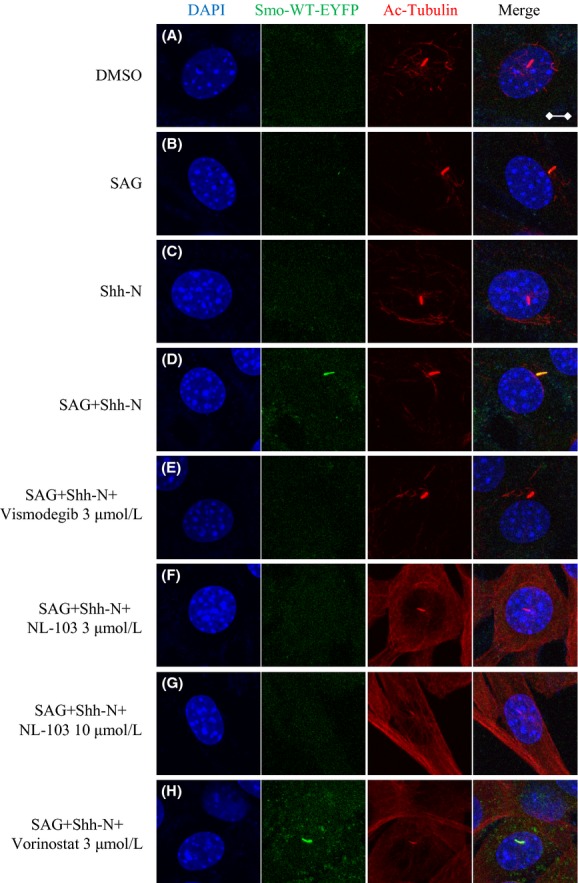
NL-103 prevents ciliary localization of Smo induced by Hh pathway agonists. (A–G) Monoclonal cells expressing Smo-WT-EYFP were treated with indicated compounds or conditions (DMSO, 0.1%; Shh-N, 20%; SAG, 100 nmol/L). Smo-WT-EYFP (green) was visualized by direct detection of EYFP. Primary cilia (red) were marked with acetylated-α-tubulin antibody by immunofluorescence. Nuclei (blue) were visualized by DAPI staining. Arrows point to cilia with weak (white) cilia staining. Representative images are provided (Scale bars: 5 μm). At least two monoclonal cell lines were applied and similar results were obtained.
Intriguingly, our Smo translocation assays revealed that the acetylation of primary cilium was reduced by NL-103 or vorinostat treatment. To determine whether this reduction in ciliary acetylation is due to shrinkage or loss of the axoneme, we further investigated the level of detyrosinated α-tubulin, another maker of primary cilium. Our results showed that NL-103 and vorinostat downregulated the acetylation but not the detyrosination of cilia (Fig. S1), indicating that the axoneme of primary cilium still exists.
NL-103 is capable of effectively overcoming the drug resistance conferred by point mutations of Smo
Thus far, two clinically relevant Smo mutants have been identified. Smo-M2 is a constitutively active mutant (Xie et al. 1998), and Smo-DH is a vismodegib-resistant mutant with an amino acid substitution that disrupts the ability of vismodegib to bind Smo (Yauch et al. 2009). To further study the functional consequences of these mutations, we transfected NIH3T3-12Gli cells with retroviral expression vectors encoding Smo-WT-EYFP, Smo-M2-EYFP or Smo-DH-EYFP fusion proteins, and then cells stably transfected with exogenous Smo were selected by puromycin treatment. These fusion Smo proteins were expressed at similar levels as determined with fluorescence microscope. Consistent with previous findings (Yauch et al. 2009), Smo-M2 transfection induced Hh pathway activity to levels substantially higher than that seen with Smo-WT and Smo-DH, two of which were comparable, regardless of the presence or absence of Shh-N (Fig. 4A), further suggesting that Smo-M2 has inherent oncogenic potential and Smo-DH has similar activity as Smo-WT.
Figure 4.

Effects of vismodegib, NL-103 or vorinostat on Hh signaling mediated by exogenous human wildtype Smo and two clinically relevant Smo mutants. (A) Luciferase reporter activity after transfection of Smo variants in the absence (blank bars) or presence (gray bars) of Shh-N. (B–D) Luciferase reporter activity in NIH3T3-12Gli cells expressing Smo-WT (B), Smo-M2 (C), or Smo-DH (D) after treatment with various doses of vismodegib (circles), NL-103 (triangles) or vorinostat (squares) in the absence (blank) or presence (black) of Shh-N. Reporter activity is normalized to DMSO-treated cultures. Data are the average of three independent experiments ± SD. (E) Flow cytometric determination of specific BODIPY-cyclopamine binding to HEK293T cells expressing Smo-DH, and the competitions of vismodegib and NL-103 for the binding of BODIPY-cyclopamine to Smo mutants.
Next, we investigated the effects of vismodegib, NL-103 and vorinostat on Hh signaling mediated by these Smo mutants. In the absence of Shh-N, vismodegib and NL-103 slightly but dose-dependently inhibited reporter activity of NIH3T3-12Gli cells expressing Smo-WT-EYFP and that was not observed in parental NIH3T3-12Gli cells (Fig. 4B), suggesting that exogenously overexpressed Smo partially activates Hh signaling. In the presence of Shh-N, although vismodegib and NL-103 inhibited reporter activity, respectively, at IC50s of 19.7 and 27.0 nmol/L in NIH3T3-12Gli cells (Fig. 2B), weaker inhibitions (IC50s of 114.3 and 134.9 nmol/L for vismodegib and NL-103, respectively) were observed in Smo-WT-EYFP–transfected cells (Fig. 4B), indicating that overexpression of exogenous Smo causes resistance to Smo antagonists to some degrees. Both NL-103 and vismodegib significantly inhibited Hh signaling mediated by Smo-M2 in the absence of Shh-N with nearly the same activities, however, their activities were weakened in the presence of Shh-N (Fig. 4C), indicating that Smo-M2 is still subjected to the negative regulation of Ptch-1, and it becomes resistant to certain Smo antagonists when the inhibitory effect of Ptch-1 upon Smo is relieved by Shh-N binding to Ptch-1. Intriguingly, we also observed that the activity of NL-103 against Smo-M2 was much stronger than that of vismodegib in the presence of Shh-N, for NL-103 fully blocked Hh pathway at 3 μmol/L, but vismodegib did not even at 10 μmol/L (Fig. 4C), suggesting that NL-103 is able to overcome vismodegib resistance induced by Smo-M2 to some extent.
Consistent with previous findings (Yauch et al. 2009), although vismodegib almost maximally blocked reporter activity at 1 μmol/L in NIH3T3-12Gli cells expressing Smo-WT (Fig. 4B), no inhibition was observed in Smo-DH–transfected cells (Fig. 4D), regardless of Shh-N. In contrast, in the absence or presence of Shh-N, NL-103 effectively and dose-dependently inhibited the reporter activity in Smo-DH–transfected cells (Fig. 4D), indicating that NL-103 can efficiently overcome vismodegib resistance induced by Smo-DH. Interestingly, vorinostat always maximally inhibited Hh pathway at about 2 μmol/L, regardless of Smo variants (Fig. 4B–D), further suggesting that vorinostat may inhibit Hh pathway downstream of Smo.
Next, we addressed how these mutations affect the ability of different compounds to bind Smo. Consistent with previous observations (Wang et al. 2012), BODIPY-cyclopamine specifically bound to Smo-WT rather than Smo-M2 (data not shown), suggesting that Smo-M2 may be resistant to Smo antagonist derived from cyclopamine. Similarly, the ability of BODIPY-cyclopamine binding to Smo-DH was severely compromised compared with that to Smo-WT, but not completely destroyed (Fig. 4E). Moreover, the association of BODIPY-cyclopamine with Smo-DH was effectively inhibited by NL-103 (Fig. 4E), suggesting that NL-103 is still capable of binding to Smo-DH. Thus, the ability of NL-103 to suppress Hh signaling in the context of the Smo-DH may be partially associated with its ability to bind this mutant.
To elucidate the potential mechanisms underlying the special responses of Smo-M2 to targeted compounds, we established a monoclonal NIH3T3 cell line expressing low level of human Smo-M2-EYFP, and then investigated the effects of vismodegib, NL-103 and vorinostat on the translocation of Smo-M2 to primary cilium. We showed that the presence of Smo-M2 in primary cilia was observable even in the absence of stimulators of Hh pathway (Fig. 5A), and treatment with either Shh-N or SAG strongly promoted the translocation of Smo-M2 to cilia (Fig. 5B and C), indicating that Smo-M2 can translocate to primary cilium more easily than Smo-WT. Vismodegib did not inhibit the accumulation of Smo-M2 in cilia induced by Hh pathway stimulators, further demonstrating that Smo-M2 is resistant to vismodegib when Ptch-1 is engaged by Shh-N (Fig. 5D and E). Additionally, NL-103 also showed no effects on Smo-M2 enrichment and eliminated the acetylation of primary cilium (Fig. 5F). At least two monoclonal cell lines were applied and similar results were obtained.
Figure 5.
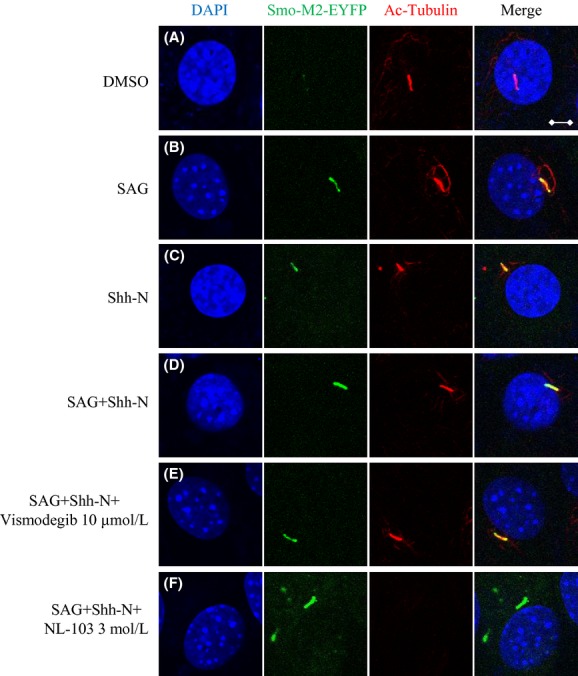
Effects of Hh pathway stimulators or inhibitors on ciliary localization of Smo-M2. (A–F) Monoclonal cells expressing Smo-M2-EYFP were treated with indicated compounds or conditions (DMSO, 0.1%; Shh-N, 20%; SAG, 100 nmol/L). Smo-M2-EYFP (green), primary cilia (red), and nuclei (blue) were visualized as before. Representative images are provided (Scale bars: 5 μm). At least two monoclonal cell lines were applied and similar results were obtained.
NL-103 differentially modulates the expression of Gli transcription factors
Our prior results indicate that vorinostat probably blocks Hh pathway though its inhibitory activity against HDACs, and we confer that NL-103 has similar effects on Hh signaling. To further reveal the potential mechanisms underlying the negative effects of HDAC inhibitors on Hh pathway, we examined the effects of NL-103 and vorinostat on the transcription of critical components, such as Ptch-1, Gli-1, Gli-2, and Gli-3. Our real-time PCR results showed that the transcription of Gli-1 increased robustly after Shh-N treatment (Fig. 6A), which is consistent with the fact that Gli-1 is a downstream target gene of Hh pathway. Vismodegib, NL-103 and vorinostat all potently inhibited the transcription induced by Shh-N (Fig. 6A). Unexpectedly, vorinostat did not downregulate the expression of Gli-1 to background levels (Fig. 6A), which was inconsistent with the prior observation that vorinostat at 3 μmol/L almost completely blocked the reporter activity. Additionally, the Gli-1 transcription was substantially enhanced by NL-103 treatment at 10 μmol/L compared with that at 3 μmol/L (Fig. 6A). Thus, these results seemed to be contradictory and it was unclear how HDAC inhibition affected Gli-1 expression. To address this, we investigated the effects of these compounds on the Gli-1 mRNA levels in the absence of Shh-N. We showed that NL-103 and vorinostat significantly increased the transcription of Gli-1 even when Hh pathway is in off-state (Fig. 6B), clearly indicating that HDAC inhibition can stimulate the expression of Gli-1.
Figure 6.

Effects of vismodegib, NL-103 or vorinostat on the transcription of Gli-1 and Gli-2. (A, B) Gli-1 mRNA levels after treatment with various doses of compounds in the presence (A) or absence of Shh-N (B). (C, D) Gli-2 mRNA levels after treatment with various doses of compounds in the presence (C) or absence of Shh-N (D). NIH3T3-12Gli cells were maintained in either HEK293 control medium (blank bars) or Shh-N conditioned medium (gray bars). Cells were treated with DMSO control, vismodegib, NL-103 or vorinostat in triplicate. After 24 h, samples were collected and total RNA was extracted. Levels of mRNA were assessed by quantitative real-time PCR. Data indicate mean ± SD (n = 3). *P < 0.05, **P < 0.01, ***P < 0.001; Student’s t-test.
Intriguingly, NL-103 and vorinostat, but not vismodegib, significantly and dose-dependently suppressed the transcription of Gli-2, no matter whether Hh pathway was activated (Fig. 6C and D). Similar results were observed in NIH3T3 parental cell line (data not shown). Additionally, vismodegib, NL-103 and vorinostat all effectively suppressed the expression of Ptch-1 induced by Shh-N in NIH3T3-12Gli cell line (data not shown), further confirming that all three drugs have inhibitory effects on Hh pathway. None of three compounds substantially changed the expression of Gli-3 (data not shown).
Discussion
Vismodegib has revolutionized the treatment of BCC since it received FDA approval in 2012, and has been shown to be a promising therapeutic agent for metastatic MB associated with aberrant Hh signaling (Rudin et al. 2009). However, acquired drug resistance to vismodegib is challenging this targeted therapy. Here, we found that NL-103, a rationally designed dual-targeted inhibitor of Smo and HDACs, is capable of effectively suppressing Hh signaling.
The primary cilium has proved to be indispensable for normal and efficient Hh signaling in mammalian systems (Gerdes et al. 2009; Goetz and Anderson 2010). Recent studies have suggested that tubulin posttranslational modifications (PTMs) could be creating a “tubulin code” and some molecular motors use them like road signs to recruit protein complexes to specific cellular locations (Hammond et al. 2008). Our immunoblotting results showed that the HDAC inhibitor activity of NL-103 increases the total amount of acetylated α-tubulin, whereas our immunostaining data revealed that it decreases the tubulin acetylation of cilia. Considering that primary cilium is pivotal scaffold for components of Hh pathway, reduction in acetylation of primary cilium induced by HDACi is likely to have implications for Hh signaling.
Smo-M2 can fully activate Hh pathway independently of Shh-N. Interestingly, although Shh-N is not able to further boost the activation of Hh pathway, it confers Smo-M2 drug resistance to Smo antagonists. These findings prompt us to further assume that relief of inhibitory effects of Ptch-1 upon Smo-M2 by Shh-N binding further facilitates the conformational equilibrium from a drug-sensitive inactive form to the drug-resistant active form, and that results in loss of pharmacological inhibition on Smo-M2. This study suggests that tumors harboring Smo-M2 mutants may become drug resistant to Smo antagonists through Hh overexpression, downregulation or inactivating mutations of Ptch-1. Although Smo-M2 has been characterized as a constitutively active mutant, little is known of the molecular mechanism leading to its activation. Our observations indicate that Smo-M2 is still subjected to the negative regulation by Ptch-1, but it is more prone to accumulate in primary cilium than Smo-WT. We can reasonably infer that Smo-M2 is predisposed to assume an active conformation that is more suitable for ciliary translocation.
Smo-DH has similar signaling potency to Smo-WT (Yauch et al. 2009). Moreover, our findings consistently confirmed that the inability of vismodegib to suppress Smo-DH-mediated signaling is because of its inability to bind Smo-DH. Therefore, the rationale for targeting Hh pathway in tumors which are driven by dysregulated Hh signaling continues to be applicable and highlights the need to either identify second-generation Smo inhibitors capable of overcoming acquired resistance, or identify inhibitors targeting downstream signaling molecules. Thus, new compounds capable of binding this mutant are promising candidates for second-generation Smo antagonists. NL-103 specifically binds to Smo-DH and effectively inhibits Smo-DH-mediated signaling in dose-dependent manner, suggesting that NL-103 may be a promising second-generation Smo antagonist for clinical development. We also note that another group reported that certain compounds with similar structural elements to NL-103 are also capable of overcoming drug resistance to vismodegib (Dijkgraaf et al. 2011). Additionally, BODIPY-cyclopamine cannot bind to Smo-DH as effectively as Smo-WT, indicating that Smo-DH may be partially resistant to cyclopamine derivatives. These findings have direct implications for clinical development and applications of cyclopamine-derived Smo antagonists such as saridegib (formerly IPI-926) (Tremblay et al. 2009; Lee et al. 2012).
Pharmacological inhibition of HDACs induces the accumulation of hyper-acetylated nucleosome core histones, leading to transcriptional activation of some genes, and repression of some other genes (Bolden et al. 2006). There are three transcriptional mediators of Hh pathway activation: Gli-1, Gli-2, and Gli-3. Gli-1 and Gli-2 are transcriptional activators, whereas Gli3 functions primarily as a transcriptional repressor (Jacob and Briscoe 2003). Intriguingly, Gli-1mutant mice lack any developmental defects while Gli-2 rather than Gli-1 seems to mediate the initial response of cells to Shh (Park et al. 2000; Bai and Joyner 2001; Bai et al. 2002). In this study, we showed that HDAC inhibition significantly downregulates the expression of Gli-2. Therefore, we speculate that HDAC inhibitors, such as NL-103 and vorinostat, block Hh signaling through downregulating Gli-2 expression.
Although relatively rare, Gli-2 amplifications have been observed in human MB (Northcott et al. 2009). And the oncogenic potential of Gli-2 has been firmly established in mouse models of MB or BCC (Grachtchouk et al. 2000; Han et al. 2009). Moreover, a previous study suggested that Gli2 amplification may be a potential mechanism responsible for vismodegib resistance downstream of Smo, and Smo antagonists are of limited use if resistance occurs downstream of the molecular target (Dijkgraaf et al. 2011). Given that NL-103 can effectively and dose-dependently decrease Gli-2 levels through inhibition of HDACs, it may have therapeutic potentials for tumors associated with Gli-2 amplification, and may reasonably overcome the drug resistance mediated by Gli-2 overexpression. In addition, suppressor-of-fused (Sufu) is a negative regulator of Hh pathway downstream of Smo and upstream of Gli-2 and its mutations have been associated with MB and other tumors (Taylor et al. 2002; Lee et al. 2007). In this case, NL-103 may be also therapeutically advantageous over conventional Smo antagonists. Collectively, our findings suggest that future clinical applications of NL-103 may improve response rate, overcome drug resistance to some Smo antagonists, and prevent or delay the onset of acquired resistance in patients.
Nonhistone proteins such as transcription factors are also targets for acetylation with varying functional effects (Vigushin and Coombes 2004; Lane and Chabner 2009). A previous study has demonstrated that Gli-1 and Gli-2 can be acetylated and their HDAC-mediated deacetylation promotes transcriptional activation and sustains a positive auto-regulatory loop (Canettieri et al. 2010). Thus, according to this hypothesis, inhibition of HDACs by NL-103 should be able to increase the acetylation of Gli-1 and Gli-2, decrease their transcriptional activities, disrupt the positive regulatory loop, and to eventually block Hh signaling at the transcriptional level. Although inhibition of HDACs can increase the transcription of Gli-1, the enhanced expression of Gli-1 may not further activate the expression of target genes when its transcriptional activity is suppressed by acetylation.
Our data suggest that simultaneous inhibition of Smo and HDACs through NL-103 may achieve synergistic effects on Hh pathway suppression. NL-103 alters the posttranslational modification status of primary cilia, reduces Gli-2 transcription, and putatively increases the acetylation of downstream Gli transcription activators, suggesting that Smo inhibition could be enhanced by these indirect effects. These results provide further support for the therapeutic rationale to simultaneously target HDACs and Smo to boost antiproliferative effects on cancer cells relying on aberrant Hh pathway. Our findings further confirm the notion that multiple steps in Gli regulation are pharmacologically targetable (Hyman et al. 2009).
In brief, our results suggest that NL-103, a chimeric compound with its unique dual-targeted activities, can effectively inhibit Hh signaling via multiple mechanisms at several levels. Considering its potential to outperform current single-targeted therapies, it is an attractive candidate for future clinical development as an extra therapeutic means to improve the effectiveness of marketed Smo inhibitors.
Acknowledgments
This study was supported by research funding from the National Natural Science Foundation of China (nos. Y201181042 and 81273546) and from the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program,” China (nos. 2013ZX09102008 and 2013ZX09402102-001).
Glossary
- BCC
basal cell carcinoma
- DAPI
4′,6-diamidino-2-phenylindoledihydrochloride
- DMSO
dimethyl sulfoxide
- EYFP
enhanced yellow fluorescence
- EYFP
enhanced yellow fluorescence protein
- FDA
Food and Drug Administration
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- GPCR
G-protein-coupled receptors
- HDAC
histone deacetylase
- HDACi
histone deacetylase inhibitors
- Hh
hedgehog
- MB
medulloblastoma
- Ptch-1
Patched-1
- PTM
protein posttranslational modification
- Smo
Smoothened
- Sufu
suppressor-of-fused
Disclosure
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Data S1. Supplementary Materials and Methods.
Figure S1. NL-103 and vorinostat downregulates the acetylation but not the detyrosination levels of cilia. NIH3T3-12Gli cells were treated with indicated compounds or conditions. Primary cilia were marked with detyrosinated-tubulin antibody (also known as glu-tubulin) (green) and acetylated-α-tubulin antibody (red) simultaneously by immunofluorescence. Nuclei (blue) were visualized by DAPI staining. Representative images are provided (Scale bars: 5 μm).
References
- Bai CB, Joyner AL. Gli1 can rescue the in vivo function of Gli2. Development. 2001;128:5161–5172. doi: 10.1242/dev.128.24.5161. [DOI] [PubMed] [Google Scholar]
- Bai CB, Auerbach W, Lee JS, Stephen D, Joyner AL. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development. 2002;129:4753–4761. doi: 10.1242/dev.129.20.4753. [DOI] [PubMed] [Google Scholar]
- Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discovery. 2006;5:769–784. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- Cai X, Zhai H-X, Wang J, Forrester J, Qu H, Yin L, et al. Discovery of 7-(4-(3-ethynylphenylamino)-7-methoxyquinazolin-6-yloxy)-N-hydroxyheptanamide (CUDC-101) as a potent multi-acting HDAC, EGFR, and HER2 inhibitor for the treatment of cancer. J Med Chem. 2010;53:2000–2009. doi: 10.1021/jm901453q. [DOI] [PubMed] [Google Scholar]
- Canettieri G, Di Marcotullio L, Greco A, Coni S, Antonucci L, Infante P, et al. Histone deacetylase and Cullin3–RENKCTD11 ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat Cell Biol. 2010;12:132–142. doi: 10.1038/ncb2013. [DOI] [PubMed] [Google Scholar]
- Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002a;16:2743–2748. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci USA. 2002b;99:14071–14076. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkgraaf GJ, Alicke B, Weinmann L, Januario T, West K, Modrusan Z, et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011;71:435–444. doi: 10.1158/0008-5472.CAN-10-2876. [DOI] [PubMed] [Google Scholar]
- Gailani MR, Ståhle-Bäckdahl M, Leffell DJ, Glyn M, Zaphiropoulos PG, Undén AB, et al. The role of the human homologue of Drosophila patched in sporadic basal cell carcinomas. Nat Genet. 1996;14:78–81. doi: 10.1038/ng0996-78. [DOI] [PubMed] [Google Scholar]
- Gerdes JM, Davis EE, Katsanis N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 2009;137:32–45. doi: 10.1016/j.cell.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet. 2010;11:331–344. doi: 10.1038/nrg2774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrich LV, Milenković L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science. 1997;277:1109–1113. doi: 10.1126/science.277.5329.1109. [DOI] [PubMed] [Google Scholar]
- Grachtchouk M, Mo R, Yu S, Zhang X, Sasaki H, C-c Hui, et al. Basal cell carcinomas in mice overexpressing Gli2 in skin. Nat Genet. 2000;24:216–217. doi: 10.1038/73417. [DOI] [PubMed] [Google Scholar]
- Hammond JW, Cai D, Verhey KJ. Tubulin modifications and their cellular functions. Curr Opin Cell Biol. 2008;20:71–76. doi: 10.1016/j.ceb.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han Y-G, Kim HJ, Dlugosz AA, Ellison DW, Gilbertson RJ, Alvarez-Buylla A. Dual and opposing roles of primary cilia in medulloblastoma development. Nat Med. 2009;15:1062–1065. doi: 10.1038/nm.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman JM, Firestone AJ, Heine VM, Zhao Y, Ocasio CA, Han K, et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc Natl Acad Sci USA. 2009;106:14132–14137. doi: 10.1073/pnas.0907134106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham PW, McMahon AP. Hedgehog signaling in animal development: paradigms and principles. Genes Dev. 2001;15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- Jacob J, Briscoe J. Gli proteins and the control of spinal-cord patterning. EMBO Rep. 2003;4:761–765. doi: 10.1038/sj.embor.embor896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kato M, Beachy PA. Gli2 trafficking links Hedgehog-dependent activation of Smoothened in the primary cilium to transcriptional activation in the nucleus. Proc Natl Acad Sci USA. 2009;106:21666–21671. doi: 10.1073/pnas.0912180106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs JJ, Whalen EJ, Liu R, Xiao K, Kim J, Chen M, et al. {beta}-Arrestin-mediated localization of smoothened to the primary cilium. Science Signalling. 2008;320:1777. doi: 10.1126/science.1157983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AA, Chabner BA. Histone deacetylase inhibitors in cancer therapy. J Clin Oncol. 2009;27:5459–5468. doi: 10.1200/JCO.2009.22.1291. [DOI] [PubMed] [Google Scholar]
- Lee Y, Kawagoe R, Sasai K, Li Y, Russell H, Curran T, et al. Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene. 2007;26:6442–6447. doi: 10.1038/sj.onc.1210467. [DOI] [PubMed] [Google Scholar]
- Lee MJ, Hatton BA, Villavicencio EH, Khanna PC, Friedman SD, Ditzler S, et al. Hedgehog pathway inhibitor saridegib (IPI-926) increases lifespan in a mouse medulloblastoma model. Proc Natl Acad Sci USA. 2012;109:7859–7864. doi: 10.1073/pnas.1114718109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May S, Ashique A, Karlen M, Wang B, Shen Y, Zarbalis K, et al. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev Biol. 2005;287:378. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- Northcott PA, Nakahara Y, Wu X, Feuk L, Ellison DW, Croul S, et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat Genet. 2009;41:465–472. doi: 10.1038/ng.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, Bai C, Platt K, Matise M, Beeghly A, Hui C, et al. Mouse Gli1 mutants are viable but have defects in SHH signaling in combination with a Gli2 mutation. Development. 2000;127:1593–1605. doi: 10.1242/dev.127.8.1593. [DOI] [PubMed] [Google Scholar]
- Raffel C, Jenkins RB, Frederick L, Hebrink D, Alderete B, Fults DW, et al. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997;57:842–845. [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Scott MP. Patched1 regulates hedgehog signaling at the primary cilium. Sci Signal. 2007;317:372. doi: 10.1126/science.1139740. [DOI] [PubMed] [Google Scholar]
- Rohatgi R, Milenkovic L, Corcoran RB, Scott MP. Hedgehog signal transduction by Smoothened: pharmacologic evidence for a 2-step activation process. Proc Natl Acad Sci USA. 2009;106:3196–3201. doi: 10.1073/pnas.0813373106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, et al. Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med. 2009;361:1173–1178. doi: 10.1056/NEJMoa0902903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Molecular cloning: a laboratory manual (3-volume set) New York: Cold Spring Harbor Laboratory Press; 2001. pp. 1357–1361. Chap. 17. [Google Scholar]
- Taylor MD, Liu L, Raffel C, C-C Hui, Mainprize TG, Zhang X, et al. Mutations in SUFU predispose to medulloblastoma. Nat Genet. 2002;31:306–310. doi: 10.1038/ng916. [DOI] [PubMed] [Google Scholar]
- Tran PV, Haycraft CJ, Besschetnova TY, Turbe-Doan A, Stottmann RW, Herron BJ, et al. THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat Genet. 2008;40:403–410. doi: 10.1038/ng.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay MR, Lescarbeau A, Grogan MJ, Tan E, Lin G, Austad BC, et al. Discovery of a potent and orally active hedgehog pathway antagonist (IPI-926) J Med Chem. 2009;52:4400–4418. doi: 10.1021/jm900305z. [DOI] [PubMed] [Google Scholar]
- Vigushin D, Coombes R. Targeted histone deacetylase inhibition for cancer therapy. Curr Cancer Drug Targets. 2004;4:205–218. doi: 10.2174/1568009043481560. [DOI] [PubMed] [Google Scholar]
- Wang Y, Zhou Z, Walsh CT, McMahon AP. Selective translocation of intracellular Smoothened to the primary cilium in response to Hedgehog pathway modulation. Proc Natl Acad Sci USA. 2009;106:2623–2628. doi: 10.1073/pnas.0812110106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Arvanites AC, Davidow L, Blanchard J, Lam K, Yoo JW, et al. Selective identification of hedgehog pathway antagonists by direct analysis of smoothened ciliary translocation. ACS Chem Biol. 2012;7:1040–1048. doi: 10.1021/cb300028a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CW, Chen M-H, Chuang P-T. Smoothened adopts multiple active and inactive conformations capable of trafficking to the primary cilium. PLoS ONE. 2009;4:e5182. doi: 10.1371/journal.pone.0005182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie J, Murone M, Luoh S-M, Ryan A, Gu Q, Zhang C, et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature. 1998;391:90–92. doi: 10.1038/34201. [DOI] [PubMed] [Google Scholar]
- Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Sci Signal. 2009;326:572. doi: 10.1126/science.1179386. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. Supplementary Materials and Methods.
Figure S1. NL-103 and vorinostat downregulates the acetylation but not the detyrosination levels of cilia. NIH3T3-12Gli cells were treated with indicated compounds or conditions. Primary cilia were marked with detyrosinated-tubulin antibody (also known as glu-tubulin) (green) and acetylated-α-tubulin antibody (red) simultaneously by immunofluorescence. Nuclei (blue) were visualized by DAPI staining. Representative images are provided (Scale bars: 5 μm).