

Abstract
At present, effective anticancer therapy remains one of the most challenging tasks facing the scientific community. A major limitation to most conventional low-molecular weight anticancer chemotherapeutics is their unfavourable uptake by healthy tissue, fast metabolism and lack of tumour cell selectivity. One way to solve this problem is the application of hybrid nanoparticles containing widely known therapeutic substances. This study was performed with the aim of investigating the potential of use hydroxyethyl starch (HES) as a high-molecular weight carrier for anticancer drug (methotrexate, MTX). HES-MTX conjugates were characterized in terms of MTX content, hydrodynamic size, zeta potential, and drug release kinetics. In vitro biological characteristics were determined using different cancer cell lines. The antitumor effect in vivo was tested in NOD/SCID mice subcutaneously inoculated with MV-4-11 human leukaemia cells and CDF1 mice intraperitoneally inoculated with P388 murine leukaemia cells. The in vivo experiments revealed the considerably higher antitumor efficacy of HES-MTX conjugates in comparison to unconjugated drug. The results presented in this article demonstrate that the application of HES as an anticancer drug carrier can improve the treatment efficacy and have significant implications for the future design and implementation of drug-carrier conjugates. The study should help create new opportunities in the design of HES-based innovative drug-carrier conjugates.
Keywords: Antitumor activity, drug delivery systems, hydroxyethyl starch, methotrexate, polymer–drug conjugate
Introduction
There were 14.1 million new cancer cases and approximately 8.2 million cancer-related deaths worldwide in 2012 (Bray et al. 2013; Ferlay et al. 2013). One of the important treatment options for patients with cancer is chemotherapy; however, usage of low-molecular weight anticancer drugs has certain disadvantages, including unfavourable uptake by healthy tissue, fast metabolism, and suboptimal accumulation in the tumour (Allen and Cullis 2004). Moreover, their clinical use is often limited by dose-dependent toxicity. Often, a low-molecular weight anticancer drug administered to an organism does not reach the tumour environment, but is distributed throughout the body, resulting in a variety of toxic effects. The goal is to deliver an anticancer agent at a dose high enough to achieve cytotoxicity within the tumour tissues without increasing toxicity to other vital organs. The delivery of drugs to the affected tissues is still a major hurdle in the treatment of various diseases, especially cancerous ones. The continued development of drug delivery system technologies is vital for future breakthroughs in medicine (Haag and Kratz 2006). Macromolecular drug-carrier conjugates offer a promising approach to achieving these goals (Duncan 2006). This kind of approach enables an improvement in the pharmacokinetic properties and bio-distribution of both standard and innovative drugs (Kopecek 2013). The bio-distribution of conjugates is, to a significant degree, determined by the properties of the carrier, for example, molecular weight, charge, conformation, hydrophobicity, and immunogenicity, and can be realized in several ways. One of the primary ways is via the “enhanced permeability and retention (EPR) effect” (Matsumura and Maeda 1986; Takakura and Hashida 1996). The EPR effect has been characterized as the ability of long-circulating macromolecules to extravasate through the abnormally leaky vasculature in tissues with deregulated neovascularization and inadequate lymphatic drainage, such as in tumours (Maeda 2010). Polymer-drug conjugates can alter pharmacokinetics and result in improved bio-distribution characteristics through the EPR effect leading to drug accumulation (Maeda et al. 2009). However, elimination of a carrier needs to happen in order to prevent the occurrence of side effects associated with polymer accumulation over time (Iyer et al. 2006).
Hydroxyethyl starch (HES) is an amylopectin-based modified polymer used as colloidal plasma volume expanders. The substrate for obtaining HES is one of the most abundant polysaccharides in nature – starch – and this is readily available from waxy maize or potato. Amylopectin is structurally similar to glycogen (a branched glucose storage polymer in humans) and this is one of the reasons why HES lacks immunogenicity (Brecher et al. 1997). For the HES polymer, no unfavourable accumulation has been observed in the liver or spleen (Hoffmann et al. 2013). This is an advantage over most synthetic and natural polymers or modified nanoparticles (Schadlich et al. 2012).
Nanoconjugates are a new class of therapeutic compounds, which may lead to new therapeutic quality due to combination with drugs already in clinical use. A methotrexate (MTX) was chosen as a model anticancer drug. This is one of the oldest antifolate drugs widely used in the treatment of cancer, rheumatoid arthritis, and other diseases (Visentin et al. 2012).
There have already been studies of HES as a drug carrier, including conjugates with bioactive compounds such as deferoxamine (Mousa et al. 1992) and HES-based hydrogels for the controlled release of biomacromolecules (Wohl-Bruhn et al. 2012): however, to our knowledge, this is the first application of antifolate covalently conjugated to HES in experimental anticancer treatment.
This study was performed with the aim of investigating the potential for the use of HES as a high-molecular weight carrier for MTX and physicochemical, biological in vitro and in vivo characterization of the obtained conjugates.
Materials and Methods
Materials
MTX was purchased from EBEWE Pharma, Unterach, Austria and HES 130/0.4 (Voluven®) from Fresenius Kabi, Bad Homburg, Germany. Human plasma was a gift from the Military Blood Centre, Wroclaw, Poland. Inorganic salts were kindly provided by POCH, Gliwice, Poland. All other chemicals were purchased reagent grade from Sigma-Aldrich, St. Louis, MO and used without further purification. High-purity water was generated using a Direct-Q® apparatus (Millipore, Billerica, MA).
Conjugate preparation (HES-MTX)
HES 130/0.4 (Voluven®) was used in the reaction without further purification. Activation of the MTX carboxyl groups for covalent coupling with HES was performed according to well-established techniques (Nevozhay et al. 2006). The HES solution (20 mL, 1.2 g) was cooled to 4°C in an ice bath and pH was adjusted to 10.5 using 1 mol L−1 Na2CO3. Thereafter, activated MTX (300 mg, 0.66 mmol in 5 mL anhydrous dimethylformamide) was added drop wise to this solution at a rate of 0.5 mL min−1. During the conjugation reaction, pH was maintained at 10.5. After adding the total amount of activated MTX, the pH was immediately adjusted to 7.0 (using 1 mol L−1 HCl) and the obtained conjugate was dialyzed against ultrapure water for 3 h at a flow rate of 30 mL min−1 to remove free MTX (Pellicon® XL with Ultracel-10 PLCGC membrane, Millipore).
In this study, the MTX concentration in HES-MTX conjugate was based on the total contents of the MTX in the preparations.
Analytical procedures
The analysis of MTX in preparations was based on absorption spectrophotometry in 0.1 mol L−1 sodium bicarbonate at 372 nm with an absorption coefficient of 8571 L mol−1 cm−1 (total MTX content). All the spectrophotometric measurements were conducted on a Specord® 250 (AnalyticJena, Jena, Germany) spectrophotometer equipped with 1.0 cm quartz cells (Hellma Analytics, Müllheim, Germany). The analysis of unbound MTX in preparations was based on size exclusion chromatography according to published protocols (Ciekot et al. 2012). Size exclusion chromatography profiles were determined using an Ultimate 3000 HPLC system (Dionex, Sunnyvale, CA) equipped with a DAD detector connected to a Superdex® 30 (GE Healthcare, Little Chalfont, U.K.) Column (34 μm, 4.6 × 150 mm). Sodium bicarbonate solution (0.1 mol L−1; pH 8.30) was used as the mobile phase at a flow rate of 0.4 mL min−1, UV detection at λ = 220, 302 and 372 nm.
The total glucose contents were determined using the phenol-sulphuric acid method (Masuko et al. 2005).
The stability of HES-MTX
The stability of HES-MTX conjugate was investigated at 37°C in (a) phosphate buffered saline (PBS) pH 7.20 ± 0.05 and (b) human plasma pH 7.24 ± 0.05. Conjugate was dissolved in their final solutions to a final concentration of 0.8 mmol L−1 (MTX-equiv). At selected time intervals, each reaction solution was diluted with 0.1 mol L−1 NaHCO3 to a final concentration of 0.2 mmol L−1 and then analysed for unbound MTX via size exclusion chromatography with detection at a wavelength of 302 or 372 nm for inorganic buffer and human plasma, respectively. The observed hydrolysis half-time was calculated for MTX using the pseudo-first-order kinetics equation.
Hydrodynamic parameters
Hydrodynamic parameters of HES and HES-MTX conjugate were characterized by dynamic light scattering (DLS) technique to obtain their hydrodynamic size, polydispersity information, and zeta potential. The sample solution was illuminated by a 633 nm laser, and the light intensity scattered at an angle of 173° was measured. At least six consecutive measurements were carried out for each sample. The experimental results consist of the overall average size, size distributions by intensity, and the overall polydispersity index (PDI). All samples were measured at 25°C using a Zetasizer Nano ZS (Malvern Instruments, Worcestershire, U.K.) in a 12-μL quartz cuvette (size measurement) and folded capillary cells (zeta potential). The HES and HES-MTX conjugate concentrations were 5.5 mmol L−1 (glucose-equiv). DLS data were analysed using DTS 6.10 software (Malvern Instruments). The intensity particle size distributions were obtained by using the General Purpose algorithm included in the DTS software.
Cell lines
P388 (murine leukaemia), and MV-4-11 (human Myelomonocytic Leukaemia, ATCC® Number: CRL-9591™, Lot Number: 58352230, date of purchase: 30 March 2012) were obtained from the American Type Culture Collection (Manassas, VA) and either were maintained in culture or were frozen at the Cell Culture Collection of the Institute of Immunology and Experimental Therapy, Wrocław, Poland.
In vitro assays
The antiproliferative tests were performed according to our standard procedure (Opolski et al. 1999). The cells were placed in 96-well plates at a density of 0.5 × 104 cells per well in 100 μL of culture medium. Twenty-four hours later, various concentrations of the tested compounds were added to the cells and incubated for 72 h. The cytotoxic effect was measured using an MTT assay (MTT: 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide). The results were calculated as the IC50 value (inhibitory concentration 50%) calculated for each experiment separately and presented as the mean value ± SD. Each experiment was repeated three to seven times.
Experimental animals
CDF1 (C57Bl/6 × DBA2, female, male) 10–16-week-old mice were supplied from the Medical University (Wroclaw, Poland). NOD/SCID (male) 8–12-week-old mice were supplied from the Children’s University Hospital (Krakow, Poland). Animals were maintained under standard laboratory conditions. All experiments were performed according to Interdisciplinary Principles and Guidelines for the Use of Animals in Research, Testing and Education issued by the New York Academy of Sciences Ad Hoc Committee on Animal Research and were approved by the First Local Committee for Experiments with the Use of Laboratory Animals, Wroclaw, Poland.
In vivo studies on P388 murine leukaemia
Passages of P388 leukaemia in mice were carried out according to NIH/NCI standard screening protocols for in vivo assessment (Khleif and Curt 1993). For the experiments, CDF1 mice were intraperitoneally inoculated with 1 × 106 P388 cells per mouse in 0.2 mL saline and then randomly divided into three groups receiving MTX (number of mice in group n = 7), HES-MTX conjugate (n = 8), both at a dose of 80 μmol kg−1 of body weight MTX-equiv, or saline (control group, n = 8) intraperitoneally (i.p), once, on day 1 after cell inoculation. Body weight was measured daily. The increase in life span (ILS) of treated mice was calculated from the following formula: ILS [%] = (LST/LSC) × 100–100%, where LST is the mean life span of treated mice, LSC is the mean life span of untreated mice.
In vivo studies on MV-4-11 human leukaemia
NOD/SCID mice were subcutaneously inoculated in the right flank region with 8 × 106 of MV-4-11 cells suspended in 0.2 mL Hanks solution (ChL, IIET). When the tumours reached a mean volume of 180–200 mm3 (Day 6), mice were randomized into three groups (n = 7) that intravenously (i.v.) received MTX or HES-MTX in a single dose of 40 μmol kg−1 (MTX-equiv) of body weight, or saline (10 mL kg−1) – control animals. On day 22, the experiment was terminated. Tumours and body weight were measured three times a week. Tumour volume (mm3) was calculated using the formula: a2 × b/2 (a = shorter diameter, b = longer diameter, in mm). Tumour growth inhibition (TGI) was calculated using the formula: TGI% = (VT/VC) × 100–100%, (VT - mean tumour volume of treated mice, VC - that of the untreated control animals).
Statistical analysis
Statistical analysis was performed using STATISTICA version 7.1 (StatSoft, Tulsa, OK). The data were analysed with Kruskal–Wallis analysis of variance; or, the Peto & Peto modification of the Gehan-Wilcoxon test for survival analysis was used. P-values less than 0.05 were considered significant.
Results
Activation of the carboxyl groups of MTX initially formed carbodiimide adducts producing the activated intermediate, presumably the cyclic MTX-anhydride, which reacts directly with the hydroxyl groups of HES glycosyl units. In this study, commercially available HES (Voluven®) was used as a potential, macromolecular carrier for MTX. HES-MTX conjugate was obtained and purified from an excess of the free drug. Eventually, the conjugate containing a 52 × 10−3 number of covalently bound MTX residues per anhydroglucose unit was obtained (Table S1).
The results of in vitro stability experiments indicate that a specific base-catalysis was involved in the hydrolysis of the ester conjugate, which is usually observed in weak alkaline solution. The stability of the conjugates (half-life time) in plasma reaches 65.5 ± 5 h and is almost twice as short as the half-life time in phosphate buffer at an identical pH (Table S2).
The hydrodynamic size distribution profile, as shown in Figure 1, represents a typical batch of HES polymers with a mean hydrodynamic diameter of 15.2 ± 6.2 nm and a narrow size distribution (PDI = 0.17). It may be noted from the data presented that both the hydrodynamic diameter of HES-MTX conjugates and their polydispersity is characterized by higher values when compared to the initial (unmodified) polymer. Figure S1 shows that the surfaces of HES-MTX conjugates have a negative charge of about −27.7 ± 8.13 mV, while those of unmodified HES are electrically neutral. Changes in the electrical properties of these molecules may be of key significance with respect to their biological properties. Zeta potential can influence polymer/conjugate stability in suspension through electrostatic repulsion between molecules. It can also determine conjugate interaction with the cell membrane in vivo.
Figure 1.
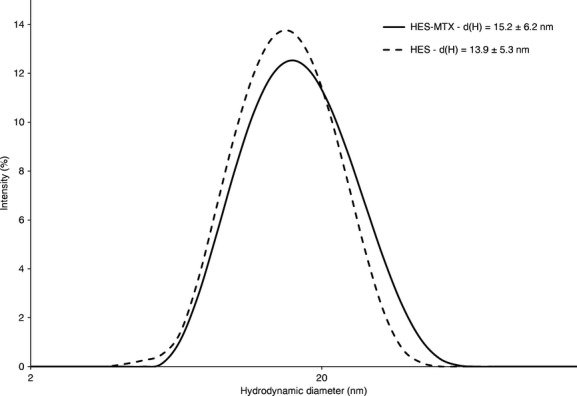
Characterization of HES 130/0.4 and HES-MTX conjugates using the dynamic light scattering technique. Size distributions are shown according to intensity. d(H) – mean hydrodynamic diameter ± size distribution width (nm). See methods section for details.
The antiproliferative activity assay was performed on human (MV4-11) and murine (P388) leukaemia cell lines. As shown in Table 1, the HES-MTX conjugate revealed an approximately 10-fold weaker cytotoxic activity in vitro than MTX alone. HES itself did not reveal any cytotoxic activity in vitro.
Table 1.
The in vitro antiproliferative activity of HES–MTX conjugates in comparison with free MTX
| Cell line, IC50 ± SD (nmol L−1)1 | ||
|---|---|---|
| Compound | MV4-11 | P388 |
| HES-MTX | 106 ± 27 | 196 ± 32 |
| MTX | 10.4 ± 3.1 | 15.1 ± 1.1 |
The compounds were tested in concentrations ranging from 2 to 2000 nmol L−1 for HES-MTX (MTX-equiv) and 0.2 to 200 nmol L−1 for MTX.
In vivo experiments (murine leukaemia P388 model) were performed to investigate the effect of controlled, sustained release of the active drug from the macromolecular conjugate on treatment effectiveness in comparison to unbound MTX. The results demonstrate that the survival time of leukaemia-bearing mice treated with HES-MTX conjugate was indeed longer than that of untreated animals (ILS = 55%, P < 0.05) and of mice treated with unbound MTX (ILS = 38%, P < 0.05) (Fig. 2).
Figure 2.
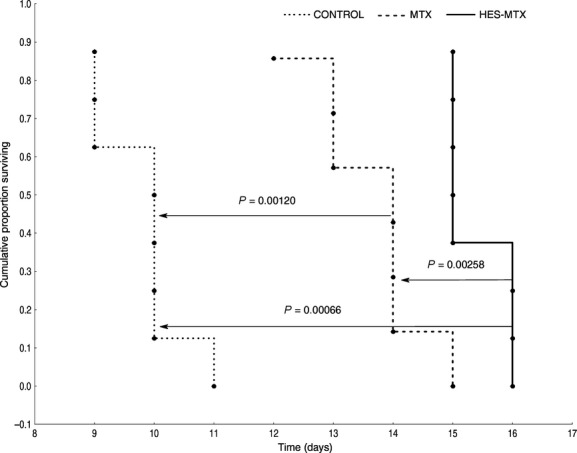
Survival data of leukaemia-bearing mice in control group (untreated, n = 8), and mice treated with either free MTX (80 μmol kg−1, n = 7) or HES-MTX conjugate (80 μmol kg−1 MTX-equiv, n = 8). The increase in life span over the control group (ILS) was 38% for the group treated with free MTX and 55% for the group treated with HES-MTX conjugate.
No acute toxicity was observed and none of the mice died due to the therapy in any of the experimental groups. Changes in body weight suggested that, overall, no toxicity of either unbound MTX or HES-MTX conjugate was observed. The weight gain was comparable in mice from the all groups as illustrated in Figure S2. A similar decrease in body weight was observed in all groups of mice during the first few days after compound administration.
In the second in vivo experiment the MV-4-11 human leukaemia cell line was chosen for the xenograft study because of its ability to form tumours when the cells are implanted subcutaneously (Shankar et al. 2007). The examined HES-MTX conjugate revealed a definitely increased effectiveness in the inhibition of MV-4-11 tumour growth in relation to free MTX. The HES-MTX conjugate reduced the volume of MV4-11 tumours to a significant degree (P < 0.05) when compared to the control group, starting from day 8 and up to day 22. Moreover, the tumour growth in the group that received the HES-MTX preparation was significantly retarded in comparison with the MTX-treated group. Significant differences were noted on days 11–22 (Table S3).
Analysis of the TGI parameter on day 22 of the experiment showed that the highest inhibition in tumour growth was 89.7% in the HES-MTX group. However, the TGI value for free MTX on day 22 was only 18.9% (Fig. 3.)
Figure 3.
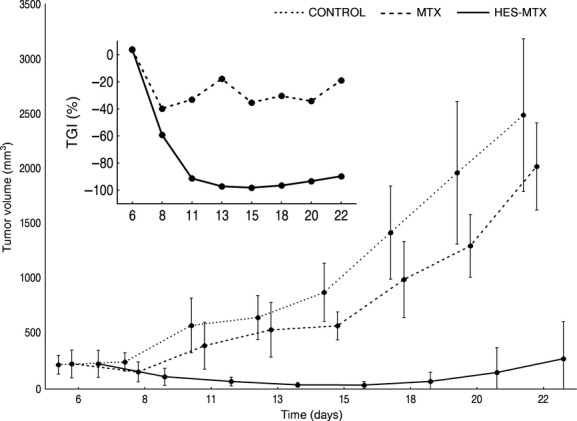
Tumour growth kinetics and tumour growth inhibition parameters (inset) of MV-4-11 bearing NOD/SCID mice. The mice were treated either with HES-MTX conjugate or with MTX alone. The control group received saline. Number of mice in each group – n = 7. Data are presented as mean tumour volumes (mm3) ± standard deviation.
Moreover, the HES-MTX conjugate positively influenced the kinetics of the growth of experimental tumours in mice causing, in individual cases, their temporary regression. Simultaneously, the effectiveness of MTX administered alone decreased during the course of the experiment; the TGI value was reduced from 32% on day 11 to 18.9% on day 22 of the experiment. No mice died during the therapy in any of the experimental groups and the body weight changes suggested no overall toxicity. Body weight changes were comparable between the groups, as is illustrated in Figure S3.
Discussion
In this article, the innovative idea of combining two medical substances currently widely used in medicine, and thus endowing them with a new therapeutic quality, was achieved. High doses of HES have been used in clinics for many years (volume replacement therapy) and are known to be safe, biocompatible and well tolerated (Westphal et al. 2009). However, in specific cases in critically ill adult patients, HES can lead to the risk of side effects (Perner et al. 2012), so the quantitative difference in using HES either as a plasma volume expander (infusion of comparatively high doses) or in perspective tumour therapy (using HES-based conjugates) needs to be taken into account (van der Linden et al. 2013).
We have described the physicochemical and biological characteristics of a novel conjugate, prepared from MTX and HES via esterification of the HES hydroxyl groups. The linker between the drug and the polymer was glutamic acid – an integral part of the MTX molecule. There is, however, a lack of additional linking substances. Since esterification is an equilibrium reaction, and a by-product of this reaction is water, reconstruction of the substrates of the reaction occurs during the drug release from the conjugate (hydrolysis of esters), that is, free MTX and HES are obtained.
HES-MTX conjugate formation does not significantly alter the hydrodynamic diameter and polydispersity of HES polymers, but it does affect the electrokinetic properties. Conjugate size tones with those required for preferred tumour accumulation as a result of the EPR effect (Maeda 2010). Due to their hydrodynamic size range, HES-MTX conjugates are able to avoid renal clearance (Noguchi et al. 1998). The surfaces of HES-MTX conjugates have a negative charge similar to those on the vascular endothelial surface. This fact may result in a longer half-life in plasma and consequently increased tumour accumulation of HES-MTX conjugate via the EPR effect (Campbell et al. 2002). For HES polymer, tumour-specific accumulation properties have recently been reported in the literature (Hoffmann et al. 2013). However, it is noteworthy that even a slight covalent modification of the macromolecular carrier can cause a great change in physicochemical and biological properties, including accumulation and elimination in vivo. Moreover, the scope of action is not solely limited to taking advantage of the EPR effect. The therapeutic activity of conjugates in an organism is never a result of acting through one given pathway. In fact, the observed effect is the result of a range of physiological and physicochemical factors and overlapping interactions. Thus, when designing drugcarrier systems, attention should be paid to all factors that may affect the realization of the assumed effect.
Our stability studies suggest that MTX may be cleaved from the polymer both via chemical and enzymatic hydrolysis (e.g., by esterases). An additional mechanism of MTX release from a conjugate is the enzymatic hydrolysis of an HES carrier by amylases (Schaeffer et al. 1986). Conjugate subjected to partial enzymatic degradation may exhibit favourable properties. Enzymatic HES hydrolysis may also be caused by the fact that no toxicity was observed as a result of HES-MTX in in vivo examinations. They are subject, as in the case of application of HES as plasma volume expanders, to gradual degradation and removal from an organism. Furthermore, in contrast to most synthetic polymers, degradation of the HES and HES-based conjugates leads to obtaining only glucose derivatives, without there being in the body any exogenous degradation products of the polymer. The release of MTX from the conjugate may be carried out via different pathways; and finally, it was observed that the effect of the overlapping of all these processes is the impressive therapeutic efficacy of the HES-MTX conjugate.
The results of the in vitro antiproliferative study of HES-MTX conjugate are consistent with our previous evidence that the poor performance of conjugates in vitro does not necessarily predict diminished activity in vivo (Goszczynski et al. 2013). The absence of the enzymatic apparatus of an organism in this type of experiment means that MTX released mainly as a result of chemical hydrolysis is responsible for the antiproliferative effect. A similar correlation can be observed in P388 in vivo experiments. The study showed that the survival rate of leukaemia-bearing mice treated with HES-MTX conjugate was significantly higher than that of untreated animals and of mice treated with unbound MTX. The in vivo experiment performed on the MV-4-11 leukaemia model revealed the high antitumor efficacy of HES-MTX conjugate when injected intravenously. HES-MTX revealed significant activity in inhibiting the growth of MV-4-11 tumours beginning from day 11 until the last day of the experiment (22 day).
This experiment clearly demonstrated that the HES-MTX conjugate is more effective than MTX alone, when administered intravenously. Moreover, our data indirectly confirm the ability of HES-MTX to accumulate in the tumour micro-environment.
Conclusion
The in vivo study indicated that the HES-MTX conjugates exhibit high levels of efficacy for the treatment of experimental tumours and have potential clinical applications. The proposed approach and results we obtained open new avenues to increase the effectiveness of many well-known substances. The study helps create new opportunities in the design of HES-based innovative drug-carrier conjugates.
Acknowledgments
This project was supported by the National Science Centre, Poland (N N302 098434).
Glossary
- ATCC
American type culture collection
- DLS
dynamic light scattering
- EPR
enhanced permeability and retention
- HES
hydroxyethyl starch
- ILS
increase in life span
- MTX
methotrexate
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
- PBS
phosphate buffered saline
- PDI
polydispersity index
- TGI
tumour growth inhibition
Disclosures
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. Physicochemical characterization of HES and HES-MTX conjugates.
Table S2. The stability of HES-MTX conjugate.
Table S3. In vivo studies on MV-4-11 human leukaemia – statistical significance between experimental groups.
Figure S1. Zeta potential distribution of HES-MTX conjugate.
Figure S2. Dynamics of body weight (murine leukaemia P388 model).
Figure S3. Dynamics of body weight (human leukaemia MV-4-11 model).
References
- Allen TM, Cullis PR. Drug delivery systems: entering the mainstream. Science. 2004;303:1818–1822. doi: 10.1126/science.1095833. [DOI] [PubMed] [Google Scholar]
- Bray F, Ren JS, Masuyer E, Ferlay J. Global estimates of cancer prevalence for 27 sites in the adult population in 2008. Int J Cancer. 2013;132:1133–1145. doi: 10.1002/ijc.27711. [DOI] [PubMed] [Google Scholar]
- Brecher ME, Owen HG, Bandarenko N. Alternatives to albumin: starch replacement for plasma exchange. J Clin Apher. 1997;12:146–153. doi: 10.1002/(sici)1098-1101(1997)12:3<146::aid-jca8>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Campbell RB, Fukumura D, Brown EB, Mazzola LM, Izumi Y, Jain RK, et al. Cationic charge determines the distribution of liposomes between the vascular and extravascular compartments of tumors. Cancer Res. 2002;62:6831–6836. [PubMed] [Google Scholar]
- Ciekot J, Goszczynski T, Boratynski J. Methods for methotrexate determination in macromolecular conjugates drug carrier. Acta Pol Pharm. 2012;69:1342–1346. [PubMed] [Google Scholar]
- Duncan R. Polymer conjugates as anticancer nanomedicines. Nat Rev Cancer. 2006;6:688–701. doi: 10.1038/nrc1958. [DOI] [PubMed] [Google Scholar]
- Ferlay J, Soerjomataram I, Ervik M, Dikshit R, Eser S, Mathers C, et al. GLOBOCAN 2012 v1.0, cancer incidence and mortality worldwide:. IARC CancerBase No. 11 [Internet] Lyon, France: International Agency for Research on Cancer; 2013. Available at http://globocan.iarc.fr (accessed 30 December 2013) [Google Scholar]
- Goszczynski T, Nevozhay D, Wietrzyk J, Omar MS, Boratynski J. The antileukemic activity of modified fibrinogen-methotrexate conjugate. Biochim Biophys Acta. 2013;1830:2526–2530. doi: 10.1016/j.bbagen.2012.11.005. [DOI] [PubMed] [Google Scholar]
- Haag R, Kratz F. Polymer therapeutics: concepts and applications. Angew Chem Int Ed Engl. 2006;45:1198–1215. doi: 10.1002/anie.200502113. [DOI] [PubMed] [Google Scholar]
- Hoffmann S, Caysa H, Kuntsche J, Kreideweiss P, Leimert A, Mueller T, et al. Carbohydrate plasma expanders for passive tumor targeting: in vitro and in vivo studies. Carbohydr Polym. 2013;95:404–413. doi: 10.1016/j.carbpol.2013.03.033. [DOI] [PubMed] [Google Scholar]
- Iyer AK, Khaled G, Fang J, Maeda H. Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today. 2006;11:812–818. doi: 10.1016/j.drudis.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Khleif SN, Curt GA. Animal models in drug development. In: Bast RC, Gansler TS, Holland JF, Frei E, editors. Cancer medicine. London: Lea & Febiger; 1993. pp. 653–666. [Google Scholar]
- Kopecek J. Polymer-drug conjugates: origins, progress to date and future directions. Adv Drug Deliv Rev. 2013;65:49–59. doi: 10.1016/j.addr.2012.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Linden P, James M, Mythen M, Weiskopf RB. Safety of modern starches used during surgery. Anesth Analg. 2013;116:35–48. doi: 10.1213/ANE.0b013e31827175da. [DOI] [PubMed] [Google Scholar]
- Maeda H. Tumor-selective delivery of macromolecular drugs via the EPR effect: background and future prospects. Bioconjug Chem. 2010;21:797–802. doi: 10.1021/bc100070g. [DOI] [PubMed] [Google Scholar]
- Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71:409–419. doi: 10.1016/j.ejpb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- Masuko T, Minami A, Iwasaki N, Majima T, Nishimura S, Lee YC. Carbohydrate analysis by a phenol-sulfuric acid method in microplate format. Anal Biochem. 2005;339:69–72. doi: 10.1016/j.ab.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Matsumura Y, Maeda H. A new concept for macromolecular therapeutics in cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the antitumor agent smancs. Cancer Res. 1986;46:6387–6392. [PubMed] [Google Scholar]
- Mousa SA, Ritger RC, Smith RD. Efficacy and safety of deferoxamine conjugated to hydroxyethyl starch. J Cardiovasc Pharmacol. 1992;19:425–429. doi: 10.1097/00005344-199203000-00019. [DOI] [PubMed] [Google Scholar]
- Nevozhay D, Budzynska R, Kanska U, Jagiello M, Omar MS, Boratynski J, et al. Antitumor properties and toxicity of dextran-methotrexate conjugates are dependent on the molecular weight of the carrier. Anticancer Res. 2006;26:1135–1143. [PubMed] [Google Scholar]
- Noguchi Y, Wu J, Duncan R, Strohalm J, Ulbrich K, Akaike T, et al. Early phase tumor accumulation of macromolecules: a great difference in clearance rate between tumor and normal tissues. Jpn J Cancer Res. 1998;89:307–314. doi: 10.1111/j.1349-7006.1998.tb00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opolski A, Wietrzyk J, Chrobak A, Marcinkowska E, Wojdat E, Kutner A, et al. Antiproliferative activity in vitro of side-chain analogues of calcitriol against various human normal and cancer cell lines. Anticancer Res. 1999;19:5217–5222. [PubMed] [Google Scholar]
- Perner A, Haase N, Guttormsen AB, Tenhunen J, Klemenzson G, Åneman A, et al. Hydroxyethyl starch 130/0.42 versus ringer’s acetate in severe sepsis. N Engl J Med. 2012;367:124–134. doi: 10.1056/NEJMoa1204242. [DOI] [PubMed] [Google Scholar]
- Schadlich A, Hoffmann S, Mueller T, Caysa H, Rose C, Gopferich A, et al. Accumulation of nanocarriers in the ovary: a neglected toxicity risk? J Control Release. 2012;160:105–112. doi: 10.1016/j.jconrel.2012.02.012. [DOI] [PubMed] [Google Scholar]
- Schaeffer RC, Renkiewicz RR, Chilton SM, Marsh D, Carlson RW. Preparation and high-performance size-exclusion chromatographic (Hpsec) analysis of fluorescein isothiocyanate-hydroxyethyl starch - macromolecular probes of the blood-lymph barrier. Microvas Res. 1986;32:230–243. doi: 10.1016/0026-2862(86)90057-9. [DOI] [PubMed] [Google Scholar]
- Shankar DB, Li JL, Tapang P, Mccall JO, Pease LJ, Dai Y, et al. ABT-869, a multitargeted receptor tyrosine kinase inhibitor: inhibition of FLT3 phosphorylation and signaling in acute myeloid leukemia. Blood. 2007;109:3400–3408. doi: 10.1182/blood-2006-06-029579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takakura Y, Hashida M. Macromolecular carrier systems for targeted drug delivery: pharmacokinetic considerations on biodistribution. Pharm Res. 1996;13:820–831. doi: 10.1023/a:1016084508097. [DOI] [PubMed] [Google Scholar]
- Visentin M, Zhao RB, Goldman ID. The Antifolates. Hematol Oncol Clin North Am. 2012;26:629–648. doi: 10.1016/j.hoc.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westphal M, James MF, Kozek-Langenecker S, Stocker R, Guidet B, Van Aken H. Hydroxyethyl starches: different products-different effects. Anesthesiology. 2009;111:187–202. doi: 10.1097/ALN.0b013e3181a7ec82. [DOI] [PubMed] [Google Scholar]
- Wohl-Bruhn S, Bertz A, Harling S, Menzel H, Bunjes H. Hydroxyethyl starch-based polymers for the controlled release of biomacromolecules from hydrogel microspheres. Eur J Pharm Biopharm. 2012;81:573–581. doi: 10.1016/j.ejpb.2012.04.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Physicochemical characterization of HES and HES-MTX conjugates.
Table S2. The stability of HES-MTX conjugate.
Table S3. In vivo studies on MV-4-11 human leukaemia – statistical significance between experimental groups.
Figure S1. Zeta potential distribution of HES-MTX conjugate.
Figure S2. Dynamics of body weight (murine leukaemia P388 model).
Figure S3. Dynamics of body weight (human leukaemia MV-4-11 model).