

Abstract
COA-Cl (2Cl-C.OXT-A) is a recently developed adenosine-like nucleic acid analog that promotes angiogenesis via the mitogen-activated protein (MAP) kinases ERK1/2. Endothelial S1P1 receptor plays indispensable roles in developmental angiogenesis. In this study, we examined the functions of S1P1 in COA-Cl-induced angiogenic responses. Antagonists for S1P1, W146, and VPC23019, substantially but still partly inhibited the effects of COA-Cl with regard to ERK1/2 activation and tube formation in cultured human umbilical vein endothelial cells (HUVEC). Antagonists for adenosine A1 receptor and purinergic P2Y1 receptor were without effect. Genetic knockdown of S1P1 with siRNA, but not that of S1P3, attenuated COA-Cl-elicited ERK1/2 responses. The signaling properties of COA-Cl showed significant similarities to those of sphingosine 1-phosphate, an endogenous S1P1 ligand, in that both induced responses sensitive to pertussis toxin (Gα i/o inhibitor), 1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid tetrakis (acetoxymethyl ester) (BAPTA-AM), (calcium chelator), and PP2 (c-Src tyrosine kinase inhibitor). COA-Cl elevated intracellular Ca2+ concentration and induced tyrosine phosphorylation of p130Cas, a substrate of c-Src, in HUVEC. COA-Cl displaced [3H]S1P in a radioligand-binding competition assay in chem-1 cells overexpressing S1P1. However, COA-Cl activated ERK1/2 in CHO-K1 cells that lack functional S1P1 receptor, suggesting the presence of additional yet-to-be-defined COA-Cl target in these cells. The results thus suggest the major contribution of S1P1 in the angiogenic effects of COA-Cl. However, other mechanism such as that seen in CHO-K1 cells may also be partly involved. Collectively, these findings may lead to refinement of the design of this nucleic acid analog and ultimately to development of small molecule-based therapeutic angiogenesis.
Keywords: Angiogenesis, endothelial cells, nucleic acid, receptors, S1P1 receptors, signal transduction, sphingosine 1-phosphate
Introduction
Angiogenesis is defined as the formation of new blood vessels from preexisting ones. Disruption of blood flow can lead to tissue ischemia and body dysfunctions, and can have lethal consequences. Ischemic disorders represent the leading causes of mortality and morbidity in developed countries, and the number of people afflicted with ischemic disorders is increasing worldwide. Thus, development of a novel intervention to promote therapeutic angiogenesis is needed to help patients with various disease conditions. Several pro-angiogenic therapies are currently available, although their usage remains limited. For example, the polypeptide basic fibroblast growth factor (bFGF) is approved for the treatment of severe skin ulcers (Uchi et al. 2009). However, being a bacteria-derived recombinant protein, bFGF is not very stable at ambient conditions and is expensive. Other treatment options include gene transfer (Cho et al. 2008) and bone marrow cell transplantation (Silvestre 2012), which require specialized facilities and techniques.
We recently developed a novel adenosine-like nucleic acid analog 6-amino-2-chloro-9-[trans-trans-2,3-bis(hydroxymethyl) cyclobuthyl] purine (COA-Cl; previously abbreviated as 2Cl-C.OXT-A; Fig. 1A). COA-Cl was found to exert robust angiogenic potentials in multiple experimental models, including cultured human umbilical vein endothelial cells (HUVEC) in vitro as well as chicken chorioallantoic membrane and rabbit cornea in vivo (Tsukamoto et al. 2010). Importantly, the magnitude of COA-Cl-induced angiogenesis is comparable to that with vascular endothelial growth factor (VEGF), the best-characterized angiogenic polypeptide growth factor. COA-Cl activates the mitogen-activated protein (MAP) kinases ERK1/2 in HUVEC. Conversely, pharmacological inhibition of the ERK1/2 pathway abrogates angiogenic responses elicited by COA-Cl (Tsukamoto et al. 2010). COA-Cl does not appear to utilize VEGF receptors, and therefore, molecular mechanisms by which stimulation with COA-Cl activates intracellular signaling molecules remain to be elucidated. At present, relatively high concentrations are required for COA-Cl to exert its angiogenic effects. Therefore, we anticipate that the identification of “COA-Cl receptors” will lead to refinement of the drug design process of this compound and ultimately to the development of a clinically proficient proangiogenic chemical agent.
Figure 1.
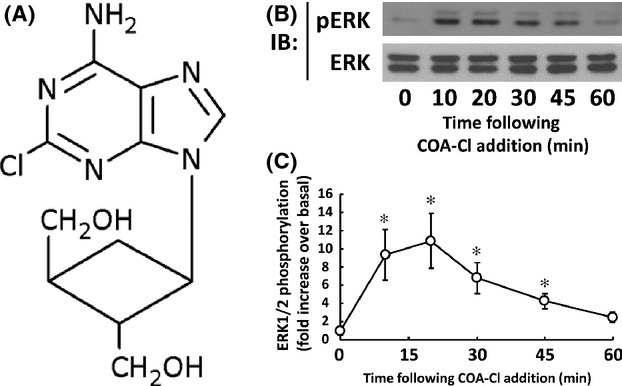
Structure of COA-Cl and time course assay of ERK1/2 phosphorylation induced by COA-Cl in HUVEC. (A) Structure of COA-Cl. (B) Results of immunoblot analyses (IB) using HUVEC lysates. Cells were treated with 100 μmol/L of COA-Cl for the times indicated. They were then lysed and equal amounts of the resulting cellular proteins were size-fractionated by SDS-PAGE. Proteins were electro-blotted to a nitrocellulose membrane and were probed with antibody directed to phosphorylated (activated) form of MAP kinases ERK1/2 (upper image). The membrane was stripped and was then reprobed with antibody directed to total ERK1/2 (lower image). The data shown are representative of four independent experiments that yielded equivalent results. (C) Results of densitometric analyses from pooled data, plotting the fold increase of the degree of ERK1/2 phosphorylation at the indicated time point, relative to the signals obtained in the absence of COA-Cl. *P < 0.05 versus control cells.
COA-Cl is a small molecule that partially resembles adenosine; therefore, we hypothesized that it may bind to G protein-coupled receptors (GPCR) rather than to receptor tyrosine kinases (RTK). Among numerous endothelial GPCR, S1P1 is well known for its ability to modulate angiogenesis (Blaho and Hla 2011). The endogenous ligand for S1P1 is a serum-borne lysophospholipid sphingosine 1-phosphate (S1P) that is produced by the enzyme sphingosine kinase in various cell types including vascular endothelial cells (Venkataraman et al. 2008). Studies in cell type-specific gene knockout mice and additional models have shown that S1P and S1P1 pathway plays an essential role during developmental angiogenesis in an endothelial cell-autonomous manner (Allende et al. 2003; Gaengel et al. 2012). Although the structure of COA-Cl is dissimilar to that of S1P, its functional similarity to S1P in the regulation of angiogenic responses prompted us to examine whether this adenosine-like agent modulates S1P1, thus helping vascular endothelial cells respond to extracellular stimulation by COA-Cl. In the present study, we provide evidence that COA-Cl induces angiogenic responses in cultured human vascular endothelial cells in a manner sensitive to the inhibition of S1P1 receptor.
Materials and Methods
Reagents
Rabbit monoclonal antibodies directed to S1P1, S1P3, and GAPDH and rabbit polyclonal antibody directed to cyclophilin-B were from Abcam (Cambridge, MA). Mouse monoclonal antibodies specific for phosphotyrosine and for p130Cas were from BD Biosciences (San Jose, CA). Rabbit polyclonal anti-S1P2 antibody was from Alomone (Jerusalem, Israel). Other antibodies were commercially obtained as described (Tsukamoto et al. 2010; Igarashi et al. 2013).
COA-Cl was synthesized as described previously (Tsukamoto et al. 2010). PD-98059 and the Raf Kinase Inhibitor IV (Raf K-I) were purchased from Calbiochem. 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrakis (acetoxymethyl ester) (BAPTA-AM) and fura-2 acetyoxymethyl ester (fura-2-AM) were from Dojindo (Kumamoto, Japan). All other materials were obtained from Sigma (St. Louis, MO) unless otherwise stated.
Cell culture and drug treatment
HUVEC were obtained from Kurabo (Osaka, Japan) (Tsukamoto et al. 2010; Igarashi et al. 2013). A heterologous stable expression cell line of rat S1P1 receptors derived from CHO-K1 cells was previously established (CHO-EDG1 cells, [Okamoto et al. 1998]). Cells were serum-starved overnight prior to assays unless otherwise stated. Commercially obtained reagents were resolved and stored as instructed by the suppliers. The final concentration of the organic solvents did not exceed 0.1% in any experiment.
Immunoblot analyses and immunoprecipitation
Immunoblot analysis and immunoprecipitation were performed as described (Igarashi et al. 2007, 2009).
Transfection with small interfering RNA
HUVEC were transiently transfected with small interfering RNA (siRNA) specific for human S1P1 (Hs_EDG1_1 from Qiagen (Valencia, CA) or S4447 from Ambion (Carlsbad, CA), both 40 nmol/L) or for S1P3 (Hs_EDG3_6 from Qiagen, 20 nmol/L), or an equal concentration of AllStars Negative Control siRNA (Qiagen), using OptiMEM and LipofecTAMINE RNAiMAX (Life Technologies, Carlsbad, CA) as instructed. Five hours after transfection, the cells were recovered in a growth medium.
Reverse transcription-polymerase chain reaction analyses
RNA isolation and conventional reverse transcription-polymerase chain reaction (RT-PCR) assay were performed as described (Igarashi et al. 2007). Primer sequences of oligonucleotides used in these assays are described in “Supporting Information.”
Calcium measurement
Intracellular Ca2+ transients in the presence of COA-Cl were examined in HUVEC loaded with 2 μmol/L fura-2-AM for 30 min at 37°C and maintained in Tyrode's solution under continuous flow using a microperfusion system. Fura-2-loaded cells were alternately excited at 340 and 380 nm using a Lambda DG-4 Ultra High Speed Wavelength Switcher (Sutter Instruments, Novato, CA) coupled to an inverted IX71 microscope with a UApo 20×/0.75 objective lens (Olympus, Tokyo, Japan). Fura-2 fluorescent signals were recorded (ORCA-Flash 2.8; Hamamatsu Photonics, Hamamatsu, Japan) and analyzed by a ratiometric fluorescence method using MetaFluor software (version 7.7.5.0; Molecular Devices, Sunnyvale, CA). The fura-2 ratio at each time point following COA-Cl addition was normalized using the basal value and expressed as normalized amplitude. The difference between the peak fluorescence signal (10–30 sec after COA-Cl) and the baseline (▵F = F–F0) was divided by the baseline value to yield ▵F/F0 in each recording session.
Tube formation assay
Tube formation activity of cultured endothelial cells was assessed using a commercially available coculture system of HUVEC and normal human fibroblasts as described previously (Tsukamoto et al. 2010).
Receptor ligand-binding competition assay
S1P1 receptor ligand-binding competition assays were performed between [3H]S1P and test compounds using commercially available membrane preparations derived from chem-1 cells overexpressing human S1P1, essentially as described (Sanada et al. 2004). Briefly, the membrane was packaged in 50 mmol/L Tris-HCl (pH 7.4), 10% glycerol, and 1% bovine serum albumin and was stored at −80°C. Membrane, test compounds, and sphingosine, d-erythro-[3-3H]-1-phosphate ([3H]S1P, purchased from Muromachi Yakuhin (Tokyo, Japan), equivalent to ∼1 nmol/L) in assay buffer (50 mmol/L Tris-HCl, pH 7.4) were added to a 96-well MultiScreen HTS FC filter plate (MSFCN6B; Merck Millipore Japan, Tokyo, Japan) that had been treated with 0.3% polyethyleneimine (Nacalai Tesque, Kyoto, Japan). Binding was performed for 60 min at room temperature and was terminated by collecting the membranes using a MultiScreen HTS vacuum manifold (MSVMHTS00; Merck Millipore Japan). After drying the filter plates for 30 min, they were replaced in scintillation vials and measured using a liquid scintillation counter. Specific binding was calculated by subtracting the radioactivity that remained in the presence of 1000-fold excess of unlabeled S1P; typically, it was ∼50% of the total binding. The pKi values represent the −log of the inhibitory binding constant (Ki).
A1 receptor ligand-binding competition assays were similarly performed between [2,8-3H]adenosine and COA-Cl using commercially available membrane preparations derived from the chem-1 cells overexpressing human adenosine A1 receptor (Merck Millipore). Specific binding was typically ∼76% of total binding.
These experimental protocols had been approved by the Kagawa University RI Experiment Committee.
Other methods
All experiments were performed at least three times. Statistical differences were analyzed by analysis of variance (ANOVA) followed by Scheffe's F–test or by Student's t–test where appropriate using Statcel3 software (OMS, Saitama, Japan), except for the receptor ligand-binding assay in which the obtained values were analyzed with GraphPad Prism software (GraphPad Software, La Jolla, CA). All data are expressed as mean ± S.E.M. A P < 0.05 was considered statistically significant.
Results
COA-Cl is a novel nucleic acid analog that structurally resembles adenosine ( Fig. 1A; Mw = 283.71). We first examined the effects of COA-Cl on the MAP kinases ERK1/2 in time course and dose-response studies using an antibody directed to phosphorylated (activated) forms of ERK1/2. Immunoblot assays indicate that COA-Cl induced the phosphorylation (activation) of ERK1/2 in a time- and dose-dependent manner in HUVEC (Fig. 1B, C, 2A and B). The MAP kinases ERK1/2 are regulated by an upstream MAP kinase kinase MEK and a MAP kinase kinase kinase Raf. Therefore, we tested the effects of specific inhibitors of MEK and Raf, PD98059 and Raf K-I, respectively, on COA-Cl-induced responses. As depicted in Figure 2C and D, both inhibitors abolished COA-Cl-induced responses of the downstream protein kinases. Together, these results indicate that COA-Cl activates a classical MAP kinase cascade comprising Raf-MEK-ERK. COA-Cl elicits strong angiogenic activity that appears to be even more robust than VEGF (Tsukamoto et al. 2010), yet it is a nucleic acid-like small molecule and not a polypeptide. Hence, it is plausible that COA-Cl may bind to GPCR rather than to RTK for exerting its angiogenic effects. Among the many GPCR agonists found in endothelial cells, S1P represents a well-characterized ligand for S1P1 that is indispensable for normal developmental angiogenesis (Allende et al. 2003). We, therefore, hypothesized that extracellularly added COA-Cl mediates intracellular signaling in HUVEC by way of S1P1. To test this theory, we performed pharmacological and genetic loss-of-function approaches for S1P1. We first utilized two pharmacological agents, W146, a selective antagonist for S1P1 (Gaengel et al. 2012), and VPC23019, a dual antagonist for S1P1/S1P3 (Oo et al. 2007). Our results indicated that both W146 and VPC23019 attenuated COA-Cl-induced ERK activation by 77.2 ± 17.9% and 62.5 ± 11.9%, respectively (Fig. 3). They also declined ERK1/2 activation by S1P (Fig. 3). In immunoblot assays, we detected significant expression of S1P1 and S1P3, but not of S1P2 (Fig. 4A), which is in agreement with the results of an earlier report (Yoon et al. 2008). We transiently transfected siRNA oligonucleotides specifically designed for human S1P1 or S1P3, obtained from Qiagen, into HUVEC. Figure 4B shows that transfection with S1P1-specific siRNA led to a decrease in S1P1 protein levels to 34.2% ± 1.2% of the negative control cells, and S1P3-specific siRNA reduced S1P3 protein levels to 48.0% ± 3.6% (P < 0.01 for both). Levels of the nontarget S1P receptor subtype remained unchanged in both conditions (S1P3: 121.8% ± 11.4% in S1P1 siRNA cells and S1P1: 85.7% ± 4.6% in S1P3 siRNA cells; n.s. versus respective negative control cells), along with several other tested proteins. Although we detected mRNA encoding S1P2 in RT-PCR assays along with those for S1P1 and S1P3, siRNA specific for S1P1 did not alter its expression level (Fig. 4C). Figure 5 demonstrates that knockdown of S1P1 protein led to significant decreases in COA-Cl-induced ERK1/2 phosphorylation, as well as in S1P-induced responses. siRNA specific for S1P1 decreased the degree of COA-Cl-induced ERK1/2 phosphorylation by 73.0 ± 21.3% compared to the control oligonucleotide (Fig. 5A). Although the degree of basal ERK1/2 phosphorylation in S1P1 siRNA-treated cells was lower than that observed in the control cells, the difference was not statistically significant (Fig. 5A). In contrast, knockdown of S1P3 protein did not perturb ERK1/2 phosphorylation processes elicited by COA-Cl or S1P. Another siRNA directed to human S1P1, obtained from Ambion, similarly led to the knockdown of S1P1 protein expression and attenuation of ERK1/2 phosphorylation responses to COA-Cl (data not shown). Because numerous GPCR other than S1P1 are also capable of activating ERK1/2, we examined whether or not several additional endothelial GPCR antagonists attenuate ERK1/2 responses induced by COA-Cl. Figure 6 demonstrates that ERK1/2 phosphorylation induced by COA-Cl was insensitive to the following agents: KW-3902, an antagonist of adenosine A1 receptor (Muller and Jacobson 2011); MRS2179, an antagonist of purinergic P2Y1 receptor (Shen and DiCorleto 2008); and ginkgolide B, an antagonist of platelet-activating factor (PAF) receptor (PAF-R) (Singh et al. 2013). Moreover, each antagonist abrogated the effects of its target receptor agonist (Fig. 6). Taken together, these pharmacological and genetic loss-of-function studies demonstrate that S1P1 receptor plays a significant role in mediating stimulation with COA-Cl to phosphorylate/activate ERK1/2 in HUVEC.
Figure 2.
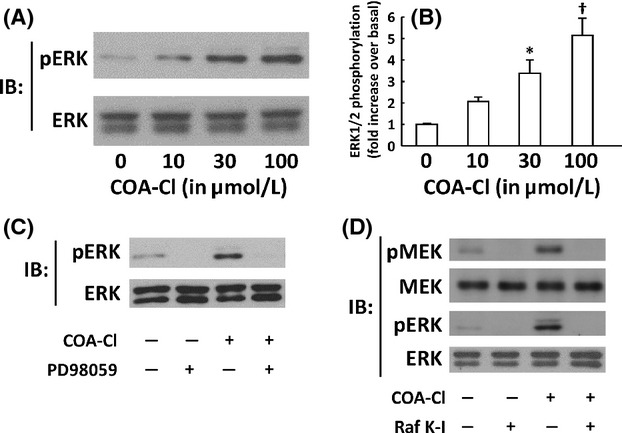
Characterization of ERK1/2 responses to COA-Cl in HUVEC. Shown are the results of immunoblot (IB) assays using lysates derived from HUVEC. (A) Results of dose-response assay of COA-Cl. HUVEC were treated with increasing concentrations of COA-Cl or vehicle for 15 min at the indicated concentrations, and were subjected to IB for phospho- and total-ERK1/2 as above (n = 4). (B) Results of pooled data. *P < 0.05 and †P < 0.01 versus vehicle-treated cells. (C and D) Results of IB analyses in which HUVEC were treated with COA-Cl either in the presence and absence of pretreatment with inhibitors of MAP kinase pathways. (C) Effects of PD98059, an inhibitor of a MAP kinase kinase MEK. HUVEC were treated with 20 μmol/L of PD98059 for 30 min, followed by 100 μmol/L of COA-Cl for 15 min. They were then subjected to IB assay as above. Shown are the representative of three independent experiments, which yielded equivalent data. (D) HUVEC were pretreated with Raf K-I (10 μmol/L for 30 min), an inhibitor of a MAP kinase kinase kinase Raf, prior to COA-Cl. Proteins were probed with antibodies directed to phosphorylated forms of a MAP kinase kinase MEK as well as MAP kinases ERK1/2. They were then reprobed with antibodies directed to total forms of MEK as well as ERK1/2, as indicated (n = 3).
Figure 3.
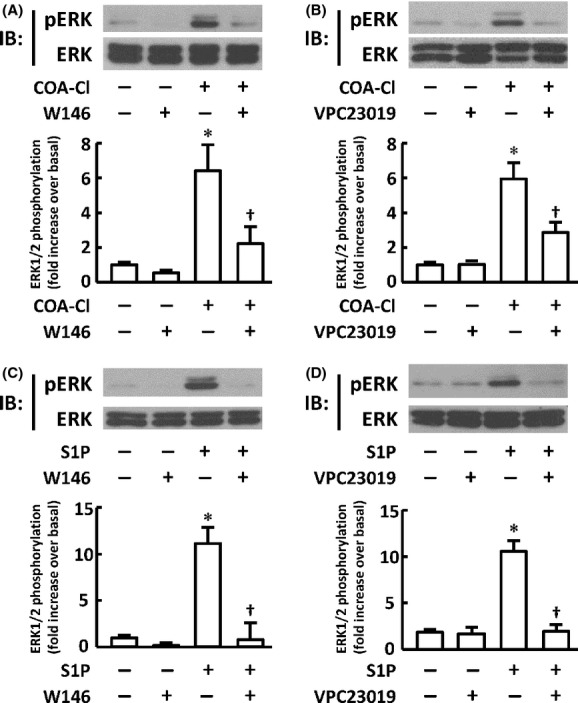
S1P receptor antagonists attenuate phosphorylation responses of ERK1/2 in HUVEC induced by COA-Cl and S1P. The figure shows the results of immunoblot (IB) analyses in which HUVEC were treated with COA-Cl or S1P either in the presence or absence of pretreatment with antagonists specific for S1P receptors. (A) Effects of the S1P1 antagonist W146. HUVEC were treated with 10 μmol/L of W146 for 30 min, followed by 100 μmol/L of COA-Cl for 15 min. They were then subjected to IB analyses for phospho- and total-ERK1/2. The upper half of the panel shows the representative results of four independent experiments, which yielded equivalent data. The results are summarized in the lower half graphs. (B) VPC23019, a dual antagonist for S1P1/S1P3 (5 μmol/L for 15 min), was used instead of W146 (n = 6). (C and D) Cells were treated with 1 μmol/L of S1P for 5 min instead of COA-Cl (n = 4), respectively. *P < 0.05 versus vehicle alone. †P < 0.05 versus cells not treated with W146 or VPC23019.
Figure 4.
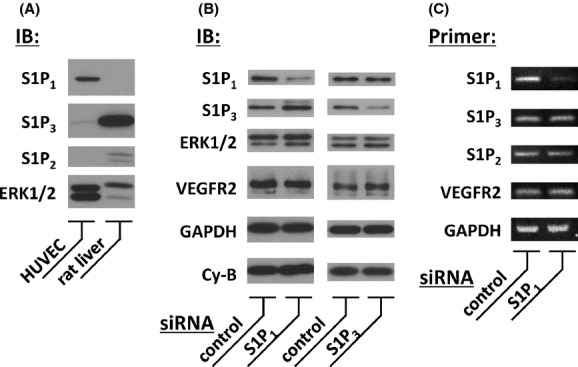
Immunoblot assay and RT-PCR analysis of HUVEC for S1P receptor subtypes and effects of siRNA. (A) Results of immunoblot (IB) analyses using lysates derived from HUVEC and tissue homogenates derived from healthy male rats. Equal quantities of cellular proteins were separated and probed using antibodies directed to S1P receptor subtypes S1P1–3 or to MAP kinases ERK1/2, as indicated. HUVEC expressed abundant S1P1 and detectable S1P3 immunoreactive signals but did not express S1P2 (n = 3). (B) Effective knockdown of S1P receptor proteins in HUVEC transiently transfected with siRNA directed to S1P1 or S1P3. In the left half of the figure, the cells were transfected with siRNA specific for S1P1 (Qiagen Hs_EDG1_1, 40 nmol/L) or negative control siRNA. Forty-eight hours after transfection, the cells were harvested and subjected to a series of IB assays using antibodies directed to S1P1, S1P3, ERK1/2, VEGFR2, GAPDH and cyclophilin-B (Cy-B). In the right half, the cells were transfected with siRNA specific for S1P3 (Qiagen Hs_EDG3_6, 20 nmol/L) or control siRNA. For both halves, six independent cultures were tested that yielded similar results. siRNA directed to S1P1 or S1P3 leads to significant and specific knockdown of their target protein. (C) Results of RT-PCR assays. HUVEC were transiently transfected with Qiagen siRNA directed to human S1P1 receptor or with negative control. They were then subjected to RNA isolation and RT-PCR assays using primer pairs as indicated. Note that these cells express detectable levels of S1P receptor subtypes S1P1−3 along with VEGFR2 and GAPDH. Densitometric analysis revealed that the expression level of S1P1 transcript relative to GAPDH in cells treated with siRNA specific to S1P1 decreased to 24.5% ± 5.5% of the control, while those of S1P3, S1P2, and VEGFR2 remained unchanged (n = 3).
Figure 5.
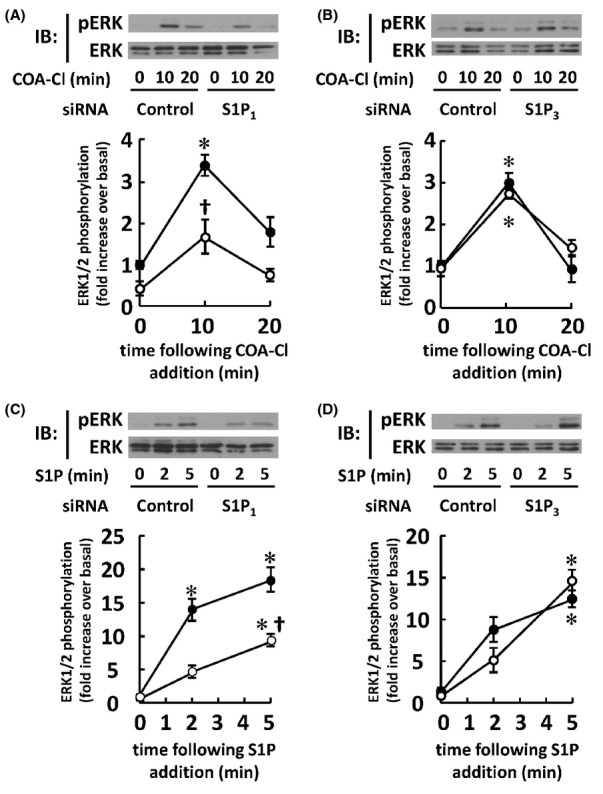
Effects of siRNA directed to S1P receptors on HUVEC stimulated with COA-Cl or S1P. The results of immunoblot (IB) analyses using lysates derived from HUVEC treated with COA-Cl or S1P that had been transfected with siRNA directed to S1P receptor subtypes are shown. (A) HUVEC transfected with siRNA specific for S1P1 or control oligonucleotides were treated with COA-Cl (100 μmol/L, upper half) for the times indicated, followed by IB analyses for phosphorylated and total forms of ERK1/2. (B) Transfection was performed using siRNA specific for S1P3. (C and D) Cells were treated with S1P (100 nmol/L) instead of COA-Cl (n = 4) for each subset. *P < 0.05 versus vehicle alone. †P < 0.05 versus control siRNA.
Figure 6.
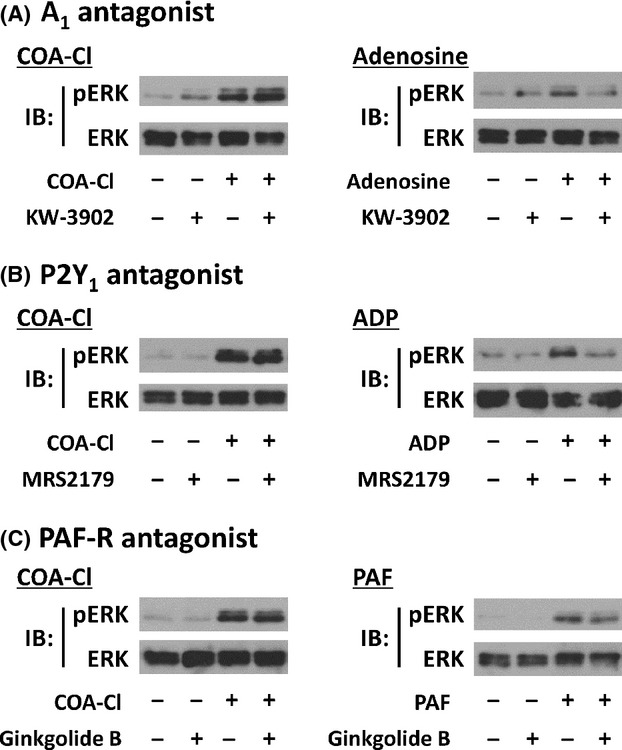
Effects of various endothelial GPCR antagonists for COA-Cl-induced ERK1/2 phosphorylation responses. The figure shows the results of immunoblot (IB) analyses, in which HUVEC were treated with COA-Cl or with various endothelial agonists/antagonists. Panel A shows the effects of KW-3902, an antagonist for adenosine A1 receptor. The cells were treated with KW-3902 (20 μmol/L for 30 min), followed by COA-Cl (100 μmol/L for 15 min, left half) or adenosine (10 μmol/L for 10 min, right half). Panel B shows the effects of MRS2179, an antagonist for purinergic P2Y1 receptor. The cells were treated with MRS2179 (100 μmol/L for 30 min), followed by COA-Cl (left half) or ADP (1 μmol/L for 15 min, right half). (C) Effects of ginkgolide B, an antagonist for PAF-R. The cells were treated with ginkgolide B (1 μmol/L for 30 min), followed by COA-Cl (left half) or PAF (5 μmol/L for 5 min, right half). HUVEC lysates were probed with antibodies directed to phospho- or total-ERK1/2 as described above. In each set of experiments, the receptor antagonist was capable of effectively attenuating responses elicited by its cognate agonist, without affecting responses elicited by COA-Cl (n = 3) for each subset.
We then sought to explore the roles of intervening signaling machineries activated in response to S1P1 stimulation by its bona fide agonist S1P in the context of COA-Cl responses. We focused on the following three groups of molecules: PTx-sensitive G-proteins, intracellular calcium, and c-Src tyrosine kinases. The S1P1 receptor subtype, unlike S1P2 or S1P3, is principally coupled to PTx-sensitive G protein components, presumably Gαi/o and Gαi/o-associated-Gβγ subunits (Ancellin and Hla 1999; Igarashi and Michel 2001; Igarashi et al. 2001). We therefore tested whether or not the toxin alters ERK1/2 responses to COA-Cl. PTx abrogated ERK1/2 phosphorylation induced by COA-Cl (Fig. 7A). We also observed inhibitory effects of COA-Cl on forskolin-stimulated cAMP production in HUVEC (Fig. S1A). S1P stimulation in HUVEC leads to increases in cytosolic Ca2+ concentration (Muraki and Imaizumi 2001). Thus, we investigated the involvement of Ca2+ in COA-Cl-activation of ERK1/2. The results demonstrated that the intracellular calcium chelator BAPTA-AM diminished the effects of COA-Cl on ERK1/2 (Fig. 7B). COA-Cl markedly increased cytosolic Ca2+ concentrations in HUVEC, determined using fura-2 as an indicator (Fig. 7C). This increase in Ca2+ was completely blocked by BAPTA-AM, PTx, or W146 (Fig. 7C and D). c-Src tyrosine kinase is known to promote both physiological and pathological angiogenesis (Ishizawar and Parsons 2004). S1P has been reported to activate c-Src in at least some vascular endothelial cells (Gonzalez et al. 2006). We, therefore, explored whether or not COA-Cl modulates the c-Src tyrosine kinase pathways in HUVEC. We assessed c-Src tyrosine kinase activity by immunoprecipitation of p130Cas, a well-known c-Src substrate, followed by an immunoblot assay directed to phospho-tyrosine. Figure 8A shows that COA-Cl led to a marked increase in tyrosine phosphorylation of p130Cas in a manner sensitive to PP2, an inhibitor of c-Src family tyrosine kinases. PP2 abrogated the phosphorylation of ERK1/2 by COA-Cl (Fig. 8B). Collectively, these pharmacological experiments indicate that the signaling profile of COA-Cl in HUVEC exhibits many similarities with previously established signaling patterns of the S1P–S1P1 receptor pathway, which involves PTx-sensitive G-proteins, intracellular calcium, and c-Src tyrosine kinases. We examined the roles of the S1P-producing enzyme sphingosine kinase (Venkataraman et al. 2008). Pharmacological inhibition of sphingosine kinase by the sphingosine derivative dimethylsphingosine (DMS) did not affect COA-Cl responses to ERK1/2 in HUVEC (Fig. S1B). ERK response to COA-Cl was attenuated by pretreatment with COA-Cl itself or S1P (Fig. S1C). We confirmed that W146, an antagonist of S1P1, did not interfere with MAP kinase activation by VEGF or adenosine in HUVEC (data not shown).
Figure 7.
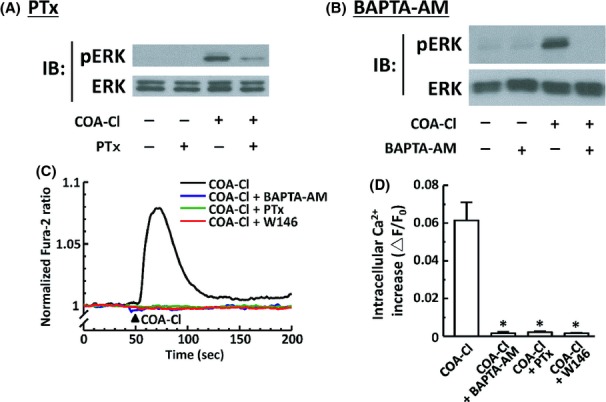
Roles of PTx-sensitive G-proteins and intracellular Ca2+ in COA-Cl-induced ERK1/2 phosphorylation responses in HUVEC. (A) Results of immunoblot (IB) analyses in which HUVEC were treated with COA-Cl either in the presence or absence of pretreatment with PTx. HUVEC were treated with 50 ng mL−1 of PTx overnight, followed by 100 μmol/L of COA-Cl for 15 min. They were then subjected to IB analyses for phospho- and total-ERK1/2, as described above. (B) BAPTA-AM, a chelator of intracellular calcium (20 μmol/L for 30 min), was used instead of PTx. (A and B) Representative results of three independent experiments, which yielded equivalent data. (C and D) Results of intracellular Ca2+ transients assays in HUVEC loaded with fura-2-AM. (C) Representative trace of 1 mmol/L COA-C1-induced intracellular Ca2+ increase; some cells had been treated with various inhibitors as described above prior to COA-Cl treatment. (D) Summarizes the results derived from pooled records obtained from 8−28 cells in each group. Fura-2 fluorescent signals were recorded and analyzed as described in the main text. Changes in the fura-2 ratio that corresponded to peak increases in intracellular Ca2+ concentrations are shown as ▵F/F0. *P < 0.01 versus COA-Cl alone.
Figure 8.
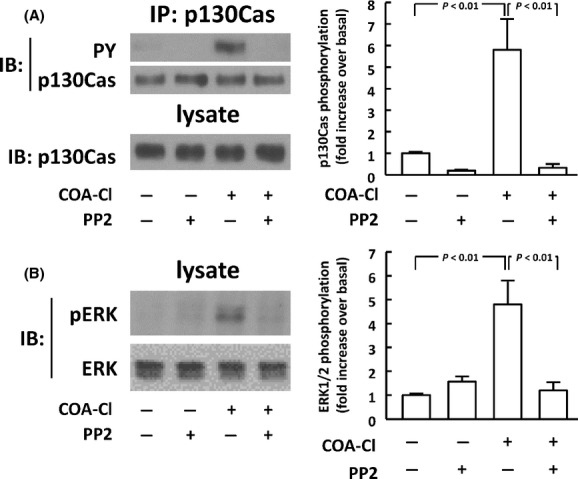
Roles of c-Src tyrosine kinases in COA-Cl-elicited ERK1/2 responses in HUVEC. (A) Results of immunoprecipitation (IP) assay of p130Cas, a c-Src tyrosine kinase substrate. HUVEC were treated with 100 μmol/L of COA-Cl for 15 min; some cells had been treated with the c-Src inhibitor PP2 (30 min at 10 μmol/L). An aliquot of cell lysate was set aside, and the rest was subjected to IP using antibody specific for p130Cas. Immunoprecipitated proteins were probed with antiphosphotyrosine antibody and were reprobed with anti-p130Cas antibody. Pre-IP cell lysates were also subjected to IB analyses for p130Cas. (B) HUVEC were subjected to IB analyses for phospho- and total-ERK1/2 as described above (n = 4) in each panel.
We then performed a series of tube formation assay as a surrogate indicator of COA-Cl-induced angiogenic responses in HUVEC. In this system, HUVEC were cocultured with normal human skin fibroblasts for 10 days prior to staining with an anti-CD31 antibody, followed by quantification of the degrees of endothelial tube formation (Tsukamoto et al. 2010). W146 and VPC23019 attenuated COA-Cl-induced tube formation by 65.0 ± 5.6% and by 80.3 ± 10.0%, respectively (Fig. 9A). PP2, a c-Src tyrosine kinase inhibitor, also decreased the degree of COA-Cl-elicited tube formation by 46.5 ± 9.3% (Fig. 9B). These results demonstrate that S1P1 plays a major role in mediating the tube formation activity of HUVEC in response to COA-Cl in a manner sensitive to a c-Src inhibitor. We compared the effects of COA-Cl with those of S1P and VEGF in the identical cellular preparations. The results indicate that ERK1/2 phosphorylation by COA-Cl is comparable to that by S1P (Fig. S2A and B). Both VEGF and S1P induced tube formation in HUVEC with statistical significance. However, the degree of tube formation induced by COA-Cl appeared to be even higher than that induced by these classic angiogenic agents (Fig. S2C and D). S1P stimulated marked phosphorylation of the kinase Akt, whereas COA-Cl was without effect (Fig. S2A).
Figure 9.
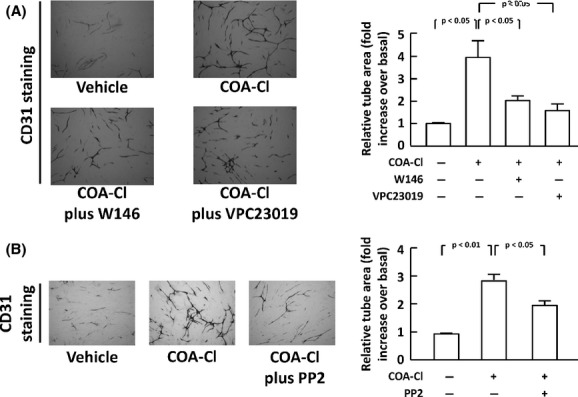
Promotion of tube formation activity by COA-Cl in HUVEC and inhibitory effects of S1P1 antagonists and a Src tyrosine kinase. The results of tube formation analyses are shown. (A) HUVEC were cocultured with normal human fibroblasts for 10 days in the presence or absence of COA-Cl (100 μmol/L), with/without W146 (20 μmol/L) or VPC23019 (5 μmol/L), as indicated. Endothelial tube formation was then identified as CD31-positive signals in microphotographs, which were subsequently quantified in the graph. The right half of the figure summarizes the results derived from four independent assays, representative images of which are shown in the left half. (B) Certain cells were treated with PP2 (2.5 μmol/L). In both panels, scale bars correspond to 500 micrometer (n = 4).
We sought to examine whether or not COA-Cl is able to bind with the S1P1 receptor. Using membrane preparations derived from chem-1 cells overexpressing S1P1, we performed a competition assay between COA-Cl and [3H]S1P. We employed this technique because radio-labeled COA-Cl has not yet become commercially available. The results demonstrated that COA-Cl attenuated binding of [3H]S1P to S1P1-expressing membrane preparations in a dose-dependent manner, and unlabeled S1P displaces [3H]S1P whereas adenosine did not (Fig. 10). COA-Cl and S1P displaced [3H]S1P at pKi values of 4.71 ± 0.18 and 7.60 ± 0.08, respectively. We noted that COA-Cl competed with radio-labeled adenosine in terms of A1 receptor binding at high concentrations (Fig. 11).
Figure 10.
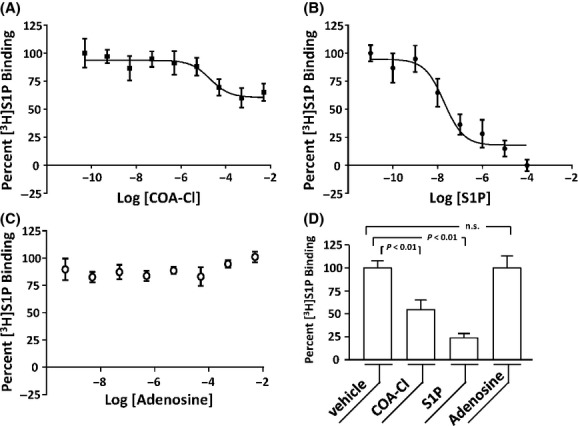
Displacement of [3H]S1P by COA-Cl in receptor ligand-binding competition assay using membrane preparation derived from S1P1-overexpressing chem-1 cells. The figure shows the results of receptor ligand-binding competition assay in which [3H]S1P was bound to membrane preparation derived from chem-1 cells overexpressing S1P1. Increasing concentrations of COA-Cl, unlabeled S1P, or unlabeled adenosine were included along with [3H]S1P (A−C, respectively). In each panel, 100% and 0% correspond to the [3H]S1P binding values obtained in the presence of the vehicle and excess S1P, respectively (n = 6−9). (D) Degrees of [3H]S1P binding obtained in the presence of the vehicle, COA-Cl (500 μmol/L), S1P (10 μmol/L) and adenosine (2.5 mmol/L). COA-Cl displaced [3H]S1P but adenosine did not.
Figure 11.
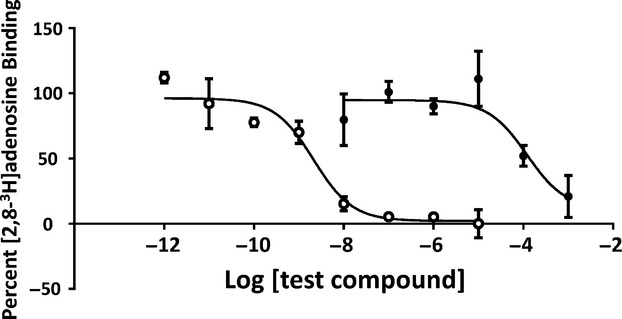
Displacement of [2,8-3H]adenosine by COA-Cl in receptor ligand-binding competition assay using membrane preparation derived from A1-overexpressing chem-1 cells. Shown are the results of receptor ligand-binding competition assay, in which [2,8-3H]adenosine was bound to membrane preparation derived from chem-1 cells that overexpress adenosine A1 receptor. Increasing concentrations of COA-Cl (closed circles) and adenosine (open circles) were included along with [2,8-3H]adenosine. 100% and 0% correspond to the [2,8-3H]adenosine binding values obtained in the presence of vehicle and excess adenosine (n = 6).
We tested whether or not overexpression of S1P1 was sufficient to promote responses to COA-Cl. Consequently, we exploited CHO-EDG1 cells that stably express heterologous rat S1P1 (Okamoto et al. 1998). CHO-EDG1 cells, but not their parental CHO-K1 cells, showed marked ERK1/2 responses to S1P. However, COA-Cl-induced ERK1/2 phosphorylation in a dose-dependent manner in both CHO-K1 and CHO-EDG1 cells to similar degrees (Fig. 12). These results indicate that heterologous overexpression of S1P1 is not sufficient to promote effects of COA-Cl.
Figure 12.
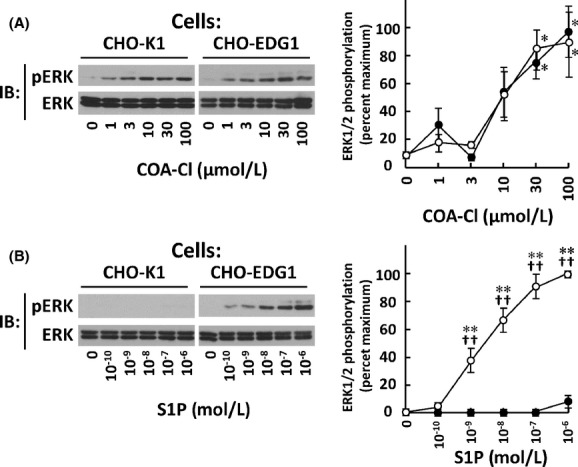
Characterization of ERK1/2 responses to COA-Cl in CHO-EDG1 cells. Shown are the results of protein immunoblot (IB) assay of cell lysates derived from CHO-EDG1 cells and their parental CHO-K1 cells. They were treated with increasing concentrations of COA-Cl (A) and S1P (B). They were then subjected to IB with antibodies directed to phosphorylated and total forms of ERK, as indicated. Left side shows representative images and right side shows graphic summaries, where closed and open circles represent values derived from CHO-K1 and CHO-EDG1 cells, respectively (n = 4). *P < 0.05 and **P < 0.01 versus vehicle-treated cells. ††P < 0.01 versus CHO-K1 cells.
Discussion
COA-Cl is an adenosine-like lead compound that exerts strong angiogenic activities in multiple experimental models. In the present study, we attempted to elucidate the mechanism by which this molecule modulates endothelial cell functions and observed that S1P1 plays a major role in converting extracellular stimulation by COA-Cl into intracellular signaling and ultimately to tube formation in HUVEC. COA-Cl promoted the phosphorylation/activation of the MAP kinases ERK1/2, key protein kinases that promote angiogenesis, and tube formation activity, a hallmark of angiogenic responses of HUVEC, which is in agreement with the results of our earlier report (Tsukamoto et al. 2010). COA-Cl-modulated ERK1/2 phosphorylation and tube formation in a manner similar to S1P, a naturally occurring ligand for S1P1 (Rikitake et al. 2002; Igarashi et al. 2007; Biswas et al. 2013). Thus, these results identify COA-Cl as a novel and potent angiogenic agent. The magnitude of the COA-Cl response was comparable to that of S1P in ERK phosphorylation assays, but appeared even higher in tube formation assays. This difference may reflect the fact that ERK phosphorylation assays were performed in mono-cultures of HUVEC for a short time period, whereas tube formation assays were performed in a coculture of HUVEC and fibroblasts for 10 days.
Two chemically distinct S1P1 antagonists, W146 (Gaengel et al. 2012) and VPC23019 (Oo et al. 2007), inhibited both ERK1/2 phosphorylation and tube formation. Although the structure of COA-Cl resembles the structure of nucleic acids, the adenosine A1 receptor antagonist KW-3992 (Muller and Jacobson 2011), and the purinergic P2Y1 receptor antagonist MRS2179 (Shen and DiCorleto 2008) failed to influence ERK1/2 responses to COA-Cl. PAF is a potent lipid inflammatory mediator (Bernatchez et al. 2003), similar to S1P in terms of being a lipid-agonist of an endothelial GPCR. However, ginkgolide B, an antagonist of PAF-R (Singh et al. 2013) was also without effect. These antagonists KW-3992, MRS2179, and ginkgolide B effectively attenuate ERK1/2 responses to their cognate receptor agonists under identical treatment conditions. In contrast, ERK1/2 phosphorylation responses evoked by COA-Cl were subjected to homo- and cross-desensitization by pretreatment with COA-Cl itself or S1P, indicating that COA-Cl and S1P share a receptor-binding site in HUVEC. W146 did not attenuate ERK phosphorylation elicited by VEGF or adenosine. Following these pharmacological studies, we performed a subtype-specific genetic knockdown experiment of S1P receptor by means of siRNA transfection. Immunoblot analysis of HUVEC lysates indicates that, among three major S1P receptor subtypes, these cells expressed detectable levels of S1P1 and S1P3, but not S1P2, which is in accordance with the results of a previous study (Yoon et al. 2008). In genetic knockdown experiments with siRNA combined with phospho-western assays, downregulation of S1P1, but not that of S1P3, significantly decreased the magnitude of ERK1/2 activation evoked by COA-Cl or by S1P. Two distinct siRNA oligonucleotides specific for S1P1 led to similar knockdown of S1P1 protein expression and attenuation of ERK1/2 phosphorylation induced by COA-Cl. Although we detected mRNA encoding S1P2 in RT-PCR assays, siRNA specific for S1P1 did not alter its expression level. Thus, these pharmacological and genetic loss-of-function studies collectively serve to demonstrate that the S1P1 receptor subtype plays a significant role in mediating signals induced by COA-Cl to MAP kinases and to tube formation. In the present study, tube formation assays were not performed in cells transfected with siRNA targeting S1P1. This is because the effects of introduced siRNAs were expected to disappear after 10-days in cultures, which is required for the tube formation assay. Moreover, it has been reported that endothelial S1P1null mice are embryonic lethal (Allende et al. 2003). Thus, genetic inhibition studies for S1P1 remain to be performed in relation with COA-Cl-induced tube formation. In addition, both W146 and VPC23019 partially attenuated ERK1/2 responses elicited by COA-Cl, whereas they completely inhibited responses to S1P. This may be consistent with our observation that COA-Cl binds to S1P1 with much more moderate affinity than that observed with the bona fide agonist S1P, and suggest that COA-Cl has additional target molecules besides S1P1.
S1P1 represents one of the best-characterized endothelial GPCR that modulates angiogenic processes. Its cognate ligand S1P is a serum-borne lysophospholipid. The S1P−S1P1 pathway plays an indispensable role in proper vascular development in utero in an endothelial cell-autonomous manner by regulating a wide variety of signaling molecules (Allende et al. 2003; Blaho and Hla 2011; Gaengel et al. 2012). After identifying COA-Cl as an activator of S1P1, we sought to explore the similarities and differences between intracellular signaling patterns evoked by COA-Cl with S1P. We primarily focused on the MAP kinases ERK1/2 as a readout of COA-Cl responses in this series of experiments, because of the following reasons: (1) ERK1/2 have been established as a key player of angiogenesis (Takahashi et al. 1999); (2) PD98059, an inhibitor of the MAP kinase kinase MEK, abrogates tube formation responses induced by COA-Cl (Tsukamoto et al. 2010); and (3) COA-Cl does not appear to elicit detectable levels of activation of several other well-known protein kinases implicated in angiogenic responses in endothelial cells, including Akt (examined in the present study). Our results demonstrate that the following signaling molecules act as major players of the COA-Cl−S1P1 pathway and are common to the S1P−S1P1 pathway: PTx-sensitive G-proteins; intracellular calcium; and c-Src tyrosine kinase. S1P1 preferentially couples to Gαi/o proteins that are sensitive to PTx (Ancellin and Hla 1999) and attenuates adenylate cyclase activity (Kon et al. 1999; Means et al. 2008; Means and Brown 2009;). S1P2 and S1P3 subtypes are more promiscuously coupled with various G protein subunits, yet they do not inhibit adenylate cyclase. Thus, the sensitivity of COA-Cl responses to PTx and the inhibition of cAMP production by COA-Cl in the presence of forskolin further support the notion that S1P1 plays a crucial role in mediating HUVEC signals elicited by COA-Cl. Intracellular Ca2+ acts as a key second messenger of receptor signal transduction in vascular endothelial cells as well as in other excitable cells, and an earlier study clearly revealed that activation of S1P1 leads to augmentation of intracellular Ca2+ concentration in HUVEC (Muraki and Imaizumi 2001). Our study with fura-2-AM demonstrated that COA-Cl actually increases intracellular Ca2+ concentration. Thus, intracellular Ca2+ plays a critical function in mediating COA-Cl-induced responses in HUVEC. c-Src tyrosine kinase plays an indispensable role in developmental as well as pathophysiological angiogenesis (Ishizawar and Parsons 2004). Endothelial S1P1 activates c-Src tyrosine kinase activity under certain conditions (Gonzalez et al. 2006). In our study, treatment of HUVEC with COA-Cl leads to robust tyrosine phosphorylation of p130Cas, a known substrate for c-Src (Ishizawar and Parsons 2004). In addition, inhibition of c-Src by PP2 attenuated the effects of COA-Cl in terms of both ERK1/2 activation and tube formation. Taken together, these pharmacological experiments reveal marked similarities between HUVEC signaling profiles in response to COA-Cl and the profile in response to S1P, the bona fide S1P1 agonist. Although COA-Cl did not appear to activate Akt in the present study, the S1P−S1P1 system utilizes numerous endothelial signaling pathways besides ERK1/2 to promote angiogenesis (Blaho and Hla 2011). The roles of these S1P−S1P1 effector pathways in the context of COA-Cl-induced angiogenic responses remain to be elucidated.
After demonstrating that S1P1 mediates pharmacological responses of HUVEC to COA-Cl, we sought to determine whether COA-Cl is capable of binding with S1P1 in vitro. At present radio-labeled COA-Cl is not commercially available; thus, we used [3H]S1P as an indicator of the degrees of receptor binding in order to examine whether or not COA-Cl displaces radioactive S1P from cellular membrane preparations with overexpressed S1P1. Our data clearly indicate that COA-Cl competes with [3H]S1P for binding to S1P1 in a dose-dependent manner. Competitive binding took place at relatively high concentrations; however, this finding is not surprising given that the natural ligand (S1P) would exert a much higher affinity for S1P1 than a chemically distinct agent such as COA-Cl. Unlabeled S1P displaced [3H]S1P, whereas adenosine did not, both molecules serving as positive and negative controls in this assay, respectively. We observed relatively high nonspecific binding of [3H]S1P (∼50%) in S1P receptor radioligand-binding assays, and similar values were reported by other investigators (Okamoto et al. 1998; Murata et al. 2000; Yonesu et al. 2009). It should be noted that even at the highest concentration tested, the disruption of [3H]S1P by COA-Cl for S1P1 binding was incomplete; the reason for this lack of complete binding is not fully understood. Concentrations of COA-Cl more than 10 mmol/L were not tested due to the limitation of solubility. Receptor-binding competition assay using membrane preparations bearing S1P2−5 remains to be tested.
It is noteworthy that COA-Cl is more similar to adenosine than to S1P with regard to its structure, yet it mimics S1P with respect to its receptor signaling, tube-forming activity, and S1P1 binding. However, our data do not exclude the possibility that vascular endothelial cells additionally express ≥1 unidentified sites that are bound and modulated by COA-Cl. First, we noted that COA-Cl competed with radio-labeled adenosine in adenosine A1 receptor-binding assays, suggesting that binding of COA-Cl to GPCR is not limited to S1P1. Antagonism of adenosine A1 receptor by KW-3992 did not affect COA-Cl-induced responses in HUVEC in this study; thus, functional consequences of A1 receptor binding by COA-Cl remains unknown at this stage. Plausibly, we have observed that COA-Cl exerts protective effects against oxygen−glucose deprivation in primary cortical neurons in a manner sensitive to suramin (Okabe et al. 2013). Suramin is a nonspecific antagonist for purinergic receptors; it may also perturb numerous signaling molecules, including certain G-proteins (Voogd et al. 1993). Second, we noted that CHO-K1 cells were capable of responding to COA-Cl without expression of S1P1. Because these cells did not respond to S1P unless they were heterologously overexpressed with S1P1 receptor (CHO-EDG-1 cells), these cells clearly express yet unidentified COA-Cl targets. Heterologous overexpression of S1P1 was insufficient for these cells to acquire hyper-responsiveness to COA-Cl. Although it is beyond the scope of the present study, it would be interesting to explore the identity of target molecules of COA-Cl in CHO-K1 cells. In addition, the sphingosine kinase inhibitor DMS did not affect the activation of ERK1/2 by COA-Cl, indicating that it is unlikely that COA-Cl stimulates sphingosine kinase to release S1P and modulates S1P1 receptor in an autocrine manner. Collectively, although S1P1 in HUVEC appears to play a significant functional role in mediating angiogenic actions of COA-Cl, both endothelial and nonendothelial cell types may express yet unidentified binding sites for COA-Cl. Based on these considerations, we propose that a deeper understanding of the COA-Cl-elicited signaling responses will lead to the identifications of novel regulatory pathways of angiogenic responses in vascular cells.
Several polypeptide growth factors are readily available that can induce therapeutic angiogenesis. For example, bFGF is clinically used to treat skin ulcers (Uchi et al. 2009). However, many of these growth factors are recombinant human proteins that are expensive and not very stable. In contrast, COA-Cl is a chemically stable molecule that is inexpensively synthesized with a Mw of 283.71, yet it exhibits strong angiogenic activity in multiple models (Tsukamoto et al. 2010). Other clinical protocols for therapeutic angiogenesis require specialized facilities and/or techniques (Cho et al. 2008; Silvestre 2012). Although high concentrations of COA-Cl are required to induce tube formation and ERK1/2 phosphorylation response at this stage, S1P1 was still identified as a target of COA-Cl. Therefore, there are merits in developing of additional lead compounds based on this adenosine analog for clinical application. Clearly, much remains to be elucidated for us to attain this ultimate goal; however, the present discovery of the linkage between COA-Cl and S1P1 represents a significant advance in this field.
In conclusion, the present study identified S1P1 receptor as a molecule that couples extracellular stimulation by COA-Cl, a novel adenosine-like nucleic acid analog, to intracellular signaling molecules and tube formation in cultured human vascular endothelial cells. COA-Cl competed with S1P in vitro in terms of S1P1 binding capacity, although it may also bind with A1 adenosine receptor and may activate yet unidentified sites in CHO-K1 cells. Further exploration of the pharmacological effects of COA-Cl and its potential derivatives may lead to a deeper understanding of angiogenesis, and ultimately to drug-based angiogenic therapy using inexpensive and stable small molecules.
Acknowledgments
We thank Daisuke Nakano (Kagawa University) for kindly providing rat liver tissue sections. This study was supported in part by Grants-in-Aid for Scientific Research (C) to J. I. (25461132) and Y. Kubota (24591654) from the Ministry of Education, Culture, Sports, Science and Technology of Japan, a Grant-in-Aid to J. I. from the SENSHIN Medical Research Foundation (Osaka, Japan), and a Grant-in-Aid to J. I. from the “JUTENKA” Research Programs from Kagawa University Faculty of Medicine. The authors thank Enago (http://www.enago.jp) for their English language review.
Glossary
- [3H]S1P
sphingosine d-erythro-[3-3H]-1-phosphate
- BAPTA-AM
1,2-bis(2-aminophenoxy)ethane-N,N,N',N'-tetraacetic acid tetrakis (acetoxymethyl ester)
- bFGF
basic fibroblast growth factor
- COA-Cl
6-amino-2-chloro-9-[trans-trans-2,3-bis(hydroxymethyl) cyclobuthyl] purine
- DMS
dimethylsphingosine
- Fura-2-AM
fura-2 acetoxymethyl ester
- GPCR
G protein-coupled receptors
- HUVEC
human umbilical vein endothelial cells
- MAP
mitogen-activated protein
- PAF
platelet-activating factor
- PAF-R
PAF receptor
- Raf K-I
Raf Kinase Inhibitor IV
- RTK
receptor tyrosine kinases
- RT-PCR
reverse transcription-polymerase chain reaction
- S1P
sphingosine 1-phosphate
- siRNA
small interfering RNA
- VEGF
vascular endothelial growth factor
Disclosures
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. Pharmacological characterization of HUVEC responses to COA-Cl. (A) Results of cAMP assay. HUVEC were treated for 30 min with forskolin (25 μmol/L) or W146 (10 μmol/L), followed by COA-Cl (100 μmol/L for 10 min), as indicated. They were then subjected to cAMP determination (n = 3). (B) Results of immunoblot (IB) analyses, in which HUVEC were treated with DMS (10 μmol/L for 30 min), an inhibitor of sphingosine kinase, followed by COA-Cl (100 μmol/L for 10 min). Cells were harvested and were subjected to IB using antibodies directed to phospho- and total-ERK1/2, as above (n = 3). (C) Results of receptor desensitization assay. Some cells had been pretreated (“Pre-Tx”) with COA-Cl (100 μmol/L), S1P (100 nmol/L), or vehicle for 60 min prior to COA-Cl (100 μmol/L for 10 min) (n = 3).
Figure S2. Comparison of COA-Cl actions with S1P in HUVEC. (A) Results of immunoblot assays using the cell lysates derived from HUVEC treated with S1P (1 μmol/L) or with COA-Cl (100 μmol/L) for the times indicated, performed in an identical culture batch. They were subjected to IB for phospho- and total-ERK1/2 as above. Blots were also reprobed with antibodies directed to phospho- and total-Akt, as well. The degrees of ERK1/2 phosphorylation were quantified and are summarized in a graph shown in the (B) (n = 4). (C and D) Results of tube formation assays, in which cells were treated with vehicle, VEGF (10 ng/mL), S1P (1 μmol/L), or COA-Cl (100 μmol/L), performed in the identical cellular preparations (n = 5).
References
- Allende ML, Yamashita T, Proia RL. G-protein-coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood. 2003;102:3665–3667. doi: 10.1182/blood-2003-02-0460. [DOI] [PubMed] [Google Scholar]
- Ancellin N, Hla T. Differential pharmacological properties and signal transduction of the sphingosine 1-phosphate receptors EDG-1, EDG-3, and EDG-5. J Biol Chem. 1999;274:18997–19002. doi: 10.1074/jbc.274.27.18997. [DOI] [PubMed] [Google Scholar]
- Bernatchez PN, Tremblay F, Rollin S, Neagoe PE, Sirois MG. Sphingosine 1-phosphate effect on endothelial cell PAF synthesis: role in cellular migration. J Cell Biochem. 2003;90:719–731. doi: 10.1002/jcb.10686. [DOI] [PubMed] [Google Scholar]
- Biswas K, Yoshioka K, Asanuma K, Okamoto Y, Takuwa N, Sasaki T, et al. Essential role of class II phosphatidylinositol-3-kinase-C2alpha in sphingosine 1-phosphate receptor-1-mediated signaling and migration in endothelial cells. J Biol Chem. 2013;288:2325–2339. doi: 10.1074/jbc.M112.409656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaho VA, Hla T. Regulation of mammalian physiology, development, and disease by the sphingosine 1-phosphate and lysophosphatidic acid receptors. Chem Rev. 2011;111:6299–6320. doi: 10.1021/cr200273u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KR, Choi JS, Hahn W, Kim DS, Park JS, Lee DS, et al. Therapeutic angiogenesis using naked DNA expressing two isoforms of the hepatocyte growth factor in a porcine acute myocardial infarction model. Eur J Cardiothorac Surg. 2008;34:857–863. doi: 10.1016/j.ejcts.2008.05.045. [DOI] [PubMed] [Google Scholar]
- Gaengel K, Niaudet C, Hagikura K, Lavina B, Muhl L, Hofmann JJ, et al. The sphingosine-1-phosphate receptor S1PR1 restricts sprouting angiogenesis by regulating the interplay between VE-cadherin and VEGFR2. Dev Cell. 2012;23:587–599. doi: 10.1016/j.devcel.2012.08.005. [DOI] [PubMed] [Google Scholar]
- Gonzalez E, Kou R, Michel T. Rac1 modulates sphingosine 1-phosphate-mediated activation of phosphoinositide 3-kinase/Akt signaling pathways in vascular endothelial cells. J Biol Chem. 2006;281:3210–3216. doi: 10.1074/jbc.M510434200. [DOI] [PubMed] [Google Scholar]
- Igarashi J, Michel T. Sphingosine 1-phosphate and isoform-specific activation of phosphoinositide 3-kinase beta. Evidence for divergence and convergence of receptor-regulated endothelial nitric-oxide synthase signaling pathways. J Biol Chem. 2001;276:36281–36288. doi: 10.1074/jbc.M105628200. [DOI] [PubMed] [Google Scholar]
- Igarashi J, Bernier SG, Michel T. Sphingosine 1-phosphate and activation of endothelial nitric oxide synthase: differential regulation of Akt and MAP kinase pathways by EDG and bradykinin receptors in vascular endothelial cells. J Biol Chem. 2001;276:12420–12426. doi: 10.1074/jbc.M008375200. [DOI] [PubMed] [Google Scholar]
- Igarashi J, Miyoshi M, Hashimoto T, Kubota Y, Kosaka H. Hydrogen peroxide induces S1P1 receptors and sensitizes vascular endothelial cells to sphingosine 1-phosphate, a platelet-derived lipid mediator. Am J Physiol Cell Physiol. 2007;292:C740–C748. doi: 10.1152/ajpcell.00117.2006. [DOI] [PubMed] [Google Scholar]
- Igarashi J, Shoji K, Hashimoto T, Moriue T, Yoneda K, Takamura T, et al. Transforming growth factor-beta1 downregulates caveolin-1 expression and enhances sphingosine 1-phosphate signaling in cultured vascular endothelial cells. Am J Physiol Cell Physiol. 2009;297:C1263–C1274. doi: 10.1152/ajpcell.00109.2009. [DOI] [PubMed] [Google Scholar]
- Igarashi J, Hashimoto T, Shoji K, Yoneda K, Tsukamoto I, Moriue T, et al. Dexamethasone induces caveolin-1 in vascular endothelial cells: implications for attenuated responses to VEGF. Am J Physiol Cell Physiol. 2013;304:C790–C800. doi: 10.1152/ajpcell.00268.2012. [DOI] [PubMed] [Google Scholar]
- Ishizawar R, Parsons SJ. c-Src and cooperating partners in human cancer. Cancer Cell. 2004;6:209–214. doi: 10.1016/j.ccr.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Kon J, Sato K, Watanabe T, Tomura H, Kuwabara A, Kimura T, et al. Comparison of intrinsic activities of the putative sphingosine 1-phosphate receptor subtypes to regulate several signaling pathways in their cDNA-transfected Chinese hamster ovary cells. J Biol Chem. 1999;274:23940–23947. doi: 10.1074/jbc.274.34.23940. [DOI] [PubMed] [Google Scholar]
- Means CK, Brown JH. Sphingosine-1-phosphate receptor signalling in the heart. Cardiovasc Res. 2009;82:193–200. doi: 10.1093/cvr/cvp086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Means CK, Miyamoto S, Chun J, Brown JH. S1P1 receptor localization confers selectivity for Gi-mediated cAMP and contractile responses. J Biol Chem. 2008;283:11954–11963. doi: 10.1074/jbc.M707422200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller CE, Jacobson KA. Xanthines as adenosine receptor antagonists. Handb Exp Pharmacol. 2011;200:151–199. doi: 10.1007/978-3-642-13443-2_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muraki K, Imaizumi Y. A novel function of sphingosine 1-phosphate to activate a non-selective cation channel in human endothelial cells. J Physiol. 2001;537:431–441. doi: 10.1111/j.1469-7793.2001.00431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murata N, Sato K, Kon J, Tomura H, Okajima F. Quantitative measurement of sphingosine 1-phosphate by radioreceptor-binding assay. Anal Biochem. 2000;282:115–120. doi: 10.1006/abio.2000.4580. [DOI] [PubMed] [Google Scholar]
- Okabe N, Nakamura E, Himi N, Narita K, Tsukamoto I, Maruyama T, et al. Delayed administration of the nucleic acid analog 2Cl-C.OXT-A attenuates brain damage and enhances functional recovery after ischemic stroke. Brain Res. 2013;1506:115–131. doi: 10.1016/j.brainres.2013.02.009. [DOI] [PubMed] [Google Scholar]
- Okamoto H, Takuwa N, Gonda K, Okazaki H, Chang K, Yatomi Y, et al. EDG1 is a functional sphingosine-1-phosphate receptor that is linked via a Gi/o to multiple signaling pathways, including phospholipase C activation, Ca2+ mobilization, Ras-mitogen-activated protein kinase activation, and adenylate cyclase inhibition. J Biol Chem. 1998;273:27104–27110. doi: 10.1074/jbc.273.42.27104. [DOI] [PubMed] [Google Scholar]
- Oo ML, Thangada S, Wu MT, Liu CH, Macdonald TL, Lynch KR, et al. Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J Biol Chem. 2007;282:9082–9089. doi: 10.1074/jbc.M610318200. [DOI] [PubMed] [Google Scholar]
- Rikitake Y, Hirata K, Kawashima S, Ozaki M, Takahashi T, Ogawa W, et al. Involvement of endothelial nitric oxide in sphingosine-1-phosphate-induced angiogenesis. Arterioscler Thromb Vasc Biol. 2002;22:108–114. doi: 10.1161/hq0102.101843. [DOI] [PubMed] [Google Scholar]
- Sanada Y, Mizushima T, Kai Y, Nishimura J, Hagiya H, Kurata H, et al. Therapeutic effects of novel sphingosine-1-phosphate receptor agonist W-061 in murine DSS colitis. PLoS One. 2004;6:e23933. doi: 10.1371/journal.pone.0023933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, DiCorleto PE. ADP stimulates human endothelial cell migration via P2Y1 nucleotide receptor-mediated mitogen-activated protein kinase pathways. Circ Res. 2008;102:448–456. doi: 10.1161/CIRCRESAHA.107.165795. [DOI] [PubMed] [Google Scholar]
- Silvestre JS. Pro-angiogenic cell-based therapy for the treatment of ischemic cardiovascular diseases. Thromb Res. 2012;130(Suppl. 1):S90–S94. doi: 10.1016/j.thromres.2012.08.287. [DOI] [PubMed] [Google Scholar]
- Singh P, Singh IN, Mondal SC, Singh L, Garg VK. Platelet-activating factor (PAF)-antagonists of natural origin. Fitoterapia. 2013;84:180–201. doi: 10.1016/j.fitote.2012.11.002. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Ueno H, Shibuya M. VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene. 1999;18:2221–2230. doi: 10.1038/sj.onc.1202527. [DOI] [PubMed] [Google Scholar]
- Tsukamoto I, Sakakibara N, Maruyama T, Igarashi J, Kosaka H, Kubota Y, et al. A novel nucleic acid analogue shows strong angiogenic activity. Biochem Biophys Res Commun. 2010;399:699–704. doi: 10.1016/j.bbrc.2010.08.003. [DOI] [PubMed] [Google Scholar]
- Uchi H, Igarashi A, Urabe K, Koga T, Nakayama J, Kawamori R, et al. Clinical efficacy of basic fibroblast growth factor (bFGF) for diabetic ulcer. Eur J Dermatol. 2009;19:461–468. doi: 10.1684/ejd.2009.0750. [DOI] [PubMed] [Google Scholar]
- Venkataraman K, Lee YM, Michaud J, Thangada S, Ai Y, Bonkovsky HL, et al. Vascular endothelium as a contributor of plasma sphingosine 1-phosphate. Circ Res. 2008;102:669–676. doi: 10.1161/CIRCRESAHA.107.165845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voogd TE, Vansterkenburg EL, Wilting J, Janssen LH. Recent research on the biological activity of suramin. Pharmacol Rev. 1993;45:177–203. [PubMed] [Google Scholar]
- Yonesu K, Kawase Y, Inoue T, Takagi N, Tsuchida J, Takuwa Y, et al. Involvement of sphingosine-1-phosphate and S1P1 in angiogenesis: analyses using a new S1P1 antagonist of non-sphingosine-1-phosphate analog. Biochem Pharmacol. 2009;77:1011–1020. doi: 10.1016/j.bcp.2008.12.007. [DOI] [PubMed] [Google Scholar]
- Yoon CM, Hong BS, Moon HG, Lim S, Suh PG, Kim YK, et al. Sphingosine-1-phosphate promotes lymphangiogenesis by stimulating S1P1/Gi/PLC/Ca2+ signaling pathways. Blood. 2008;112:1129–1138. doi: 10.1182/blood-2007-11-125203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Pharmacological characterization of HUVEC responses to COA-Cl. (A) Results of cAMP assay. HUVEC were treated for 30 min with forskolin (25 μmol/L) or W146 (10 μmol/L), followed by COA-Cl (100 μmol/L for 10 min), as indicated. They were then subjected to cAMP determination (n = 3). (B) Results of immunoblot (IB) analyses, in which HUVEC were treated with DMS (10 μmol/L for 30 min), an inhibitor of sphingosine kinase, followed by COA-Cl (100 μmol/L for 10 min). Cells were harvested and were subjected to IB using antibodies directed to phospho- and total-ERK1/2, as above (n = 3). (C) Results of receptor desensitization assay. Some cells had been pretreated (“Pre-Tx”) with COA-Cl (100 μmol/L), S1P (100 nmol/L), or vehicle for 60 min prior to COA-Cl (100 μmol/L for 10 min) (n = 3).
Figure S2. Comparison of COA-Cl actions with S1P in HUVEC. (A) Results of immunoblot assays using the cell lysates derived from HUVEC treated with S1P (1 μmol/L) or with COA-Cl (100 μmol/L) for the times indicated, performed in an identical culture batch. They were subjected to IB for phospho- and total-ERK1/2 as above. Blots were also reprobed with antibodies directed to phospho- and total-Akt, as well. The degrees of ERK1/2 phosphorylation were quantified and are summarized in a graph shown in the (B) (n = 4). (C and D) Results of tube formation assays, in which cells were treated with vehicle, VEGF (10 ng/mL), S1P (1 μmol/L), or COA-Cl (100 μmol/L), performed in the identical cellular preparations (n = 5).