

Abstract
The members of lethal-7 (Let-7) microRNA (miRNA) family are involved in regulation of cell differentiation and reprogramming of somatic cells into induced pluripotent stem cells. However, their function in the heart is not known. In this study, we examined the effect of inhibiting the function of Let-7c miRNA on the progression of postinfarction left ventricular (LV) remodeling in mice. Myocardial infarction was induced with permanent ligation of left anterior descending coronary artery with a 4-week follow-up period. Let-7c miRNA was inhibited with a specific antagomir administered intravenously. The inhibition of Let-7c miRNA downregulated the levels of mature Let-7c miRNA and its other closely related members of Let-7 family in the heart and resulted in increased expression of pluripotency-associated genes Oct4 and Sox2 in cardiac fibroblasts in vitro and in adult mouse heart in vivo. Importantly, Let-7c inhibitor prevented the deterioration of cardiac function postinfarction, as demonstrated by preserved LV ejection fraction and elevated cardiac output. Improvement in cardiac function by Let-7c inhibitor postinfarction was associated with decreased apoptosis, reduced fibrosis, and reduction in the number of discoidin domain receptor 2–positive fibroblasts, while the number of c-kit+ cardiac stem cells and Ki-67+ proliferating cells remained unaltered. In conclusion, inhibition of Let-7 miRNA may be beneficial for the prevention of postinfarction LV remodeling and progression of heart failure.
Keywords: Apoptosis, fibroblast, heart failure, Let-7c, microRNA, remodeling
Introduction
Heart failure is one of the most common causes of cardiovascular morbidity and mortality, and its prevalence is rapidly increasing as the mean age of the population advances (McMurray et al. 2012). As a result, novel therapeutic strategies are necessary. The key pathophysiological process that ultimately leads to heart failure is left ventricular (LV) remodeling (Sutton and Sharpe 2000; Shah and Mann 2011), caused by disorders that chronically increase workload, such as myocardial infarction (MI) or pressure overload due to hypertension (Jessup and Brozena 2003; McMurray and Pfeffer 2005). Myocardial remodeling is a complex process characterized by structural, functional, electrophysiological, cellular, and molecular events (Shah and Mann 2011; Burchfield et al. 2013). At the cellular level, myocyte loss, hypertrophy, fibrosis, inflammation, and the limited endogenous regenerative capacity of cardiac myocytes contribute to the adverse myocardial remodeling (Burchfield et al. 2013). Because these events are highly interrelated and multilayered molecular pathways are involved in LV remodeling (Lips et al. 2003; Heineke and Molkentin 2006), targeting a single molecule or process may not be sufficient to prevent progression into heart failure.
MicroRNAs (miRNAs) are small single-stranded, noncoding RNAs that bind to target mRNA and inhibit translation or promote degradation of the transcript, thereby suppressing protein expression (Bartel 2004). Any given miRNA has the potential to target multiple mRNAs. The involvement of miRNAs in various physiological and pathophysiological processes in the cardiovascular system includes cardiac development, heart failure, atherosclerosis, and arrhythmias (Quiat and Olson 2013; Condorelli et al. 2014; Santulli et al. 2014). Several miRNAs have been linked to myocyte hypertrophy, apoptosis, fibrosis, stem cell function, and regeneration (van Rooij and Olson 2012; Seeger et al. 2013), prompting the possibility for miRNAs as novel therapeutic targets for LV remodeling. One attractive candidate for this purpose is lethal-7 (Let-7), which was initially discovered as an essential developmental gene in Caenorhabditis elegans and later, as one of the first miRNAs (Pasquinelli et al. 2000; Reinhart et al. 2000). In embryonic stem (ES) cells, miRNAs of Let-7 family play essential role as suppressors of stem cell pluripotency and somatic cell reprogramming (Roush and Slack 2008; Peter 2009). Let-7 miRNAs are also expressed in the heart (Chen et al. 2007; Landgraf et al. 2007), but their role in myocardial remodeling and heart failure is unknown.
In this study, we provide the first evidence for the beneficial effect of inhibition of Let-7 miRNA on cardiac function. The inhibition of Let-7 increased expression of pluripotency genes Sox2 and Oct4 in cardiac fibroblasts in vitro and in the adult mouse heart in vivo. In experimental MI model, inhibition of Let-7 miRNA postinfarction maintained LV systolic function in vivo. This improvement of cardiac function was associated with attenuation of fibrosis and apoptosis, and alterations in fibroblast subpopulations. Together, these data suggest that inhibition of Let-7 miRNA is a promising therapy for postinfarction LV remodeling.
Materials and Methods
Animals
All experimental protocols were approved by the Animal Use and Care Committee of the University of Oulu and the Provincial Government of Western Finland Department of Social Affairs and Health (ESAVI-2010-03931/Ym-23). The investigations conform to the Guiding Principles for Research Involving Animals. The results were reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al. 2010; McGrath et al. 2010). Total number of 492 male C57BL/6JOlaHsd mice (Harlan, Horst, the Netherlands) were used for this study. The mice were maintained in plastic cages at a constant 21°C temperature with a 12 h light–dark cycle and had free access to food (Teklad Global Rodent diet, Harlan, Indianapolis, IN) and water.
MI in mice
MI model developed by Gao et al. (2010) was used. Mice were anesthetized with 2% isoflurane inhalation and pericardial incision was performed under thoracotomy. Left anterior descending (LAD) coronary artery was permanently ligated. Carprofen (5 mg/kg, Rimadyl) and buprenorphine (0.05–0.1 mg/kg, Vetergesic) were administered as perioperative analgesia and mice were hydrated with 0.5-mL glucose (50 mg/mL) solution subcutaneously. Postoperative analgesia (carprofen once per day, buprenorphine twice per day) was administered for 3 days.
Inhibition of Let-7c
To study the effects of Let-7c inhibition on cardiac function and myocardial structure, the single-stranded custom Let-7c antagomir was synthesized using locked-nucleic-acid-modified (LNA) technology (Exiqon, Vedbaek, Denmark). The sequence (UGAGGUAGUAGGUUGUAUGGUU) was obtained from miRBase (http://www.mirbase.org/). Lyophilized Let-7c inhibitor (sequence 5′-ACCATACAACCTACT) was resuspended into phosphate-buffered saline (PBS) as 10 mg/mL and stored at −20°C until use. A custom scramble-sequence LNA inhibitor provided by Exiqon (sequence 5′-ACGTCTATACGCCCA) was used as a negative control. Immediately after LAD ligation, Let-7c inhibitor was administered intravenously via tail vein at the dose of 20 mg/kg according to earlier studies demonstrating that single intravenous doses of antagomirs ranging between 3 and 80 mg/kg in vivo cause permanent downregulation of miRNAs in target tissue for several weeks (Frost and van Rooij 2010; Montgomery and van Rooij 2011).
Echocardiography
Cardiac function was analyzed by transthoracic echocardiography using Vevo 2100 high-frequency, high-resolution ultrasound system with 40 MHz linear transducer (MS-550S; Visual Sonics, Toronto, Ontario, Canada) as described (Kerkelä et al. 2013). Mice were sedated with isoflurane inhalation anesthesia. LV morphology and systolic function were evaluated by two-dimensional M-mode recording. To analyze diastolic function, transmitral flow velocity was measured through pulsed-wave Doppler targeted by apical four-chamber view.
Stroke volume was evaluated from echocardiographic data by calculating the difference between LV volume in diastole and systole, determined from LV internal dimensions (LVID), respectively. The formula is the following: stroke volume = LV end diastolic volume ((7/(2,4 + LVID;d) × LVID;d3)) – LV end systolic volume ((7/(2,4 + LVID;s) × LVID;s3)).
Cell culture
Adult cardiac fibroblasts were isolated from 8-week-old C57BL6 mice as previously described (Kerkelä et al. 2013). Briefly, mouse was anesthetized with isoflurane, heart was excised rapidly and cannulated through the aorta. Fibroblasts were isolated after retrograde perfusion with HEPES-buffered Tyrode’s solution supplemented with 0.1% collagenase type II (Worthington, Lakewood, NJ) and 2,3-butandione-monoxime. Fibroblasts were resuspended into DMEM/F12 (Sigma, St. Louis, MO) supplemented with 10% fetal bovine serum (FBS) (Invitrogen, Life Technologies, Carlsbad, CA), 2 mmol/L l-glutamine and penicillin–streptomycin (Sigma). The medium was changed the following day and every 2 days thereafter. Let-7c inhibitor (100 nmol/L) or equal concentration/volume of control (PBS or scramble) was applied into the culture the following day after isolation. After 7 days of treatment, cells were washed once with PBS and frozen at −70°C.
RNA isolation and quantitative real-time PCR
Mice were euthanized with CO2 and sacrificed by decapitation. Heart, kidney, and liver were excised, frozen in liquid nitrogen, and stored at −70°C. RNA was isolated with Trizol (Invitrogen). To analyze the levels of Let-7 expression, cDNA was synthesized from 0.1 μg of total RNA with miScript RT kit (Qiagen, Hilden, Germany). miRNA levels were measured by real-time quantitative (qPCR) analysis with Let-7 (for the sequences, see Table S1) and reference gene RNU6B miScript primer assays and miScript SYBR green PCR kit (Qiagen). To analyze other genes (for the sequences, see Table S2), cDNA was synthesized from 1 μg of RNA with Transcriptor First Strand cDNA synthesis kit (Roche Applied Science, Penzberg, Germany). The expression levels were evaluated on an ABI Prism 7700 Sequence Detection System (Applied Biosystems, Life Technologies, Carlsbad, CA) as previously described (Tenhunen et al. 2006).
Western blotting
To extract the cytoplasmic proteins, the LV free wall tissue was broken and reduced to a powder in liquid nitrogen (Tenhunen et al. 2006). The thawed LV powder or cardiac fibroblasts were lysed with 20 mmol/L Tris-HCl, 150 mmol/L NaCl, 1 mmol/L ethylenediaminetetraacetic acid, 1 mmol/L ethyleneglycoltetraacetic acid, 1% Triton-X100, 2.5 mmol/L sodium pyrophosphate, 1 mmo/L β-glycerophosphate, and 1 mmol/L dithiothreitol (pH 7.5) supplemented with protease and phosphatase inhibitor cocktails (Sigma). The lysate was centrifuged at 12,500 rpm for 20 min at 4°C and the supernatant was transferred to a new tube as the total protein extract. The protein concentrations were determined with colorimetric protein assay (Bio-Rad Laboratories, Hercules, CA). Protein samples (40 μg) were denatured at 97°C for 5 min and loaded on sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), run with 200 V and transferred to 0.45 μm Optitran BA-S 85 nitrocellulose membrane (Whatman, GE Healthcare Life Sciences, Uppsala, Sweden) with 100 V for 2 h. The membranes were incubated for 1 h at RT in blocking buffer (Odyssey, LI-COR, Lincoln, NE) diluted into tris-buffered saline (TBS) (50 mmol/L Tris, 200 mmol/L NaCl, pH 7.4). Membranes were then incubated overnight with primary antibody Sox2 (0.5 μg/mL; Pierce Antibodies, Thermo Scientific, Rockford, IL); extracellular signal-regulated kinase (ERK) or p38 mitogen-activated protein kinase (MAPK) (0.1 μg/mL Cell Signaling, Danvers, MA); or GAPDH (0.01 μg/mL, Millipore, Billerica, MA) in blocking buffer, washed with TBS-0.05% Tween-20 and incubated with secondary antibody (goat-anti-rabbit Alexa Fluor 680; Molecular Probes, Life Technologies, Carlsbad, CA; goat-anti-mouse IRDye 800; Rockland Immunochemicals, Gilbertsville, PA) 1 h at RT. Antibody binding was detected by Odyssey Infrared Imaging System (LI-COR) and quantified using the public domain NIH Image program (developed at the U.S. National Institutes of Health, Bethesda, MD).
Histology and image analysis
Tissue sections from LV were fixed in phosphate-buffered 10% formaline (pH 7.0) and embedded in paraffin (Serpi et al. 2011). Five-μm thick transversal sections were cut at the level of the papillary muscles. Sections were stained with Masson’s trichrome to evaluate the fibrosis and cardiomyocyte cross-sectional area. Fibrosis and cardiomyocyte area were measured by the Nikon (Melville, NY) NIS-Elements BR 2.30 program from five representative high-power fields from the infarcted myocardium. Infarction scar was measured as a percentage from the LV circumference. Cardiomyocytes were also stained with 5 μg/mL of wheat germ agglutinin Alexa Fluor 488-conjugate (Molecular Probes), as described (Condorelli et al. 2002; Santulli et al. 2012). The ApopTag in situ apoptosis detection kit (S7100; Chemicon, Millipore, Billerica, MA) for 3′-end labeling of apoptotic DNA by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) method was used to analyze apoptosis (Rysä et al. 2010). Primary antibody for c-kit (sc-168; Santa Cruz Biotechnology, Dallas, TX) and Ki-67 (M7248; Dako, Glostrup, Denmark) with EnVision Detection System (Dako) for secondary detection was used to determine the number of cardiac stem cells and cell proliferation, respectively. Fibroblasts were double stained by primary antibodies for discoidin domain receptor 2 (DDR2, sc-8989; Santa Cruz Biotechnology) and octamer-binding transcription factor 4 (Oct4, sc-5279; Santa Cruz Biotechnology). Number of Oct4+/DDR2+ double-positive cells were analyzed by immunofluorescence microscopy. CY2-conjugated anti-mouse IgG (800-656-7625; Rockland) and Alexa Fluor 568 goat anti-rabbit IgG (A11036; Invitrogen) were used for secondary detection. Fibroblast activation protein (FAP, ab28244; Abcam, Cambridge, UK) was used to further evaluate fibroblast subpopulations. Antibody for CD31 (sc-1506R; Santa Cruz Biotechnology) was used to stain endothelial cells in the LV. The number and cross-sectional area of capillaries was calculated from three representative high-power fields (40× objective) with NIS-Elements BR 2.30 program from each section choosing corresponding fields from epicardial and endocardial side of the LV at the level of the papillary muscles as described (Rysä et al. 2010).
Statistical analysis
Results are expressed as the mean ± SEM. Mann–Whitney U test or Student’s t-test was used for comparisons between two groups. Statistical significance between multiple groups was evaluated by one-way analysis of variance (ANOVA) followed by a least significant difference (LSD) post hoc test. For histological data, nonparametric Mann–Whitney U test was used. P < 0.05 was considered statistically significant.
Results
Cardiac function, fibrosis, and LV Let-7c mRNA expression postinfarction
To study the effects of Let-7c inhibitor on infarcted myocardium, we first analyzed the effects of MI on cardiac function and LV fibrosis in mice. Cardiac function was analyzed 7 days after LAD ligation by transthoracic echocardiography. A significant decrease in LV ejection fraction (P < 0.001, Fig. 1A) and fractional shortening (P < 0.001, Fig. 1B; for other echocardiography parameters, see Table S3) was observed. When compared to sham-operated mice, MI resulted in 3.6-fold increase in the fibrotic area in the LV (P < 0.01, Fig. 1C).
Figure 1.
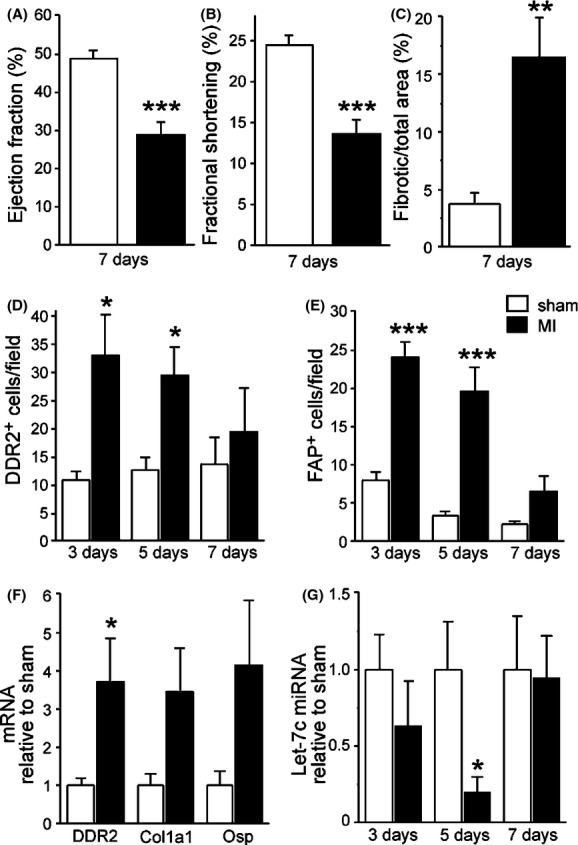
Effects of myocardial infarction (MI) induced by ligation of left anterior descending (LAD) artery in mice. (A) Left ventricular (LV) ejection fraction and (B) fractional shortening decreased significantly postinfarction. (C) MI increased the fibrotic area in the LV. (D) Number of DDR2+ and (E) FAP+ cells were significantly elevated at 3 and 5 days postinfarction. (F) MI resulted an increase in DDR2 mRNA levels at 4 weeks postinfarction. (G) Let-7c miRNA levels were significantly decreased in LV 5 days after MI. Results are mean ± SEM. (A–C) n = 8–11, (D, E, G) n = 3–6, (F) n = 6–9. White column: sham-operated; black column: MI. Data were analyzed with Student’s t-test. *P < 0.05, **P < 0.01, and ***P < 0.001 versus sham. DDR2, discoidin domain receptor 2; FAP, fibroblast activation protein; Let-7c, lethal-7c.
We next evaluated the alterations in the fibroblast subpopulations postinfarction. Fibroblasts can be divided into subpopulations based on the morphology and expression of cell surface markers (Zeisberg and Kalluri 2010). Of these markers, DDR2 has been used to identify cardiac fibroblasts (Goldsmith et al. 2004; Camelliti et al. 2005). LV fibrosis was accompanied by increase in the number of DDR2+ cells as observed 3 and 5 days after MI (Fig. 1D). Because DDR2 is not specific to cardiac fibroblasts but is also expressed in cells of lymphocytic lineages (Zeisberg and Kalluri 2010), FAP was used to further evaluate fibroblast subpopulations. Also the amount of FAP+ cells increased both at 3 and 5 days after MI (Fig. 1E). The absolute number of FAP+ cells was lower than that of DDR2+ cells at day 3 postinfarction (Fig. 1D and E) suggesting a more important role for DDR2+ fibroblast population in the progression of cardiac fibrosis. Furthermore, an increase in the mRNA levels of DDR2 was observed (Fig. 1F). Interestingly, levels of Let-7c miRNA were 70% lower 5 days after MI (P < 0.05, Fig. 1G).
Efficacy of Let-7c miRNA inhibitor
To evaluate the effects of Let-7c inhibition on myocardial remodeling, we injected Let-7c inhibitor, control nonsense sequence (scramble), or PBS intravenously via tail vein. When compared to PBS control group 4 weeks after injections and LAD ligation, a 50% decrease in LV levels of Let-7c miRNA was noted (P < 0.05, Fig. 2A). This decrease was 63% in comparison to scramble group (P < 0.001, Fig. 2B). We also injected Let-7c inhibitor into normal mice without MI and observed a 42% decrease in LV Let-7c miRNA levels after 2 weeks (P = 0.12, Fig. 2C). Moreover, Let-7c inhibitor resulted in a 98% decrease in the levels of Let-7c miRNA both in the liver and kidney (P < 0.05, Fig. 2D), organs responsible for the elimination of antagomirs.
Figure 2.

Efficacy of Let-7c inhibitor in vivo and in vitro. Let-7c inhibitor decreased significantly the levels of Let-7c miRNA in the infarcted myocardium compared to PBS-treated (A) or scramble-treated (B) mice 4 weeks after i.v. injection. (C) There was also a tendency for Let-7c miRNA to decrease in normal mice 2 weeks after administration of Let-7c inhibitor. (D) Let-7c inhibition reduced Let-7c miRNA levels in the kidney and liver in mice 4 weeks postinfarction. (E) Let-7c inhibitor decreased the expression of closely related members of Let-7 family. (F) Let-7c is expressed in cardiomyocytes and fibroblasts to the similar extent as analyzed from adult mouse heart immediately after ex vivo fractionation of cell types. Inhibition of Let-7c increased Sox2 (G) protein and (H) mRNA levels in isolated cardiac mouse fibroblasts in vitro. (I) Let-7c inhibitor resulted in elevation of LV Oct4 and Sox2 mRNA levels in normal mice 2 weeks after administration of Let-7c inhibitor. Results are mean ± SEM. (A, B, E) n = 7–8, (C, F, I) n = 4–5, (D) n = 4–6, (G) n = 3, (H) n = 8–9. White column: control; black column: Let-7c inhibitor. Data were analyzed with Student’s t-test. *P < 0.05 and ***P < 0.001 versus control. Let-7c, lethal-7c; miRNA, microRNA; PBS, phosphate-buffered saline; LV, left ventricular; Oct4, octamer-binding transcription factor 4.
The sequences of other members of Let-7 family closely resemble to that of Let-7c (Table S1) differing one to few nucleotides from Let-7c sequence. Therefore, we performed qPCR to analyze the effect of Let-7c inhibitor treatment on other members of the Let-7 family. As shown in Figure 2E, Let-7c antagomir also significantly decreased miRNA levels of other closely related members of Let-7c family, namely -7b, -7d, and -7f. Overall, the expression of the members of Let-7 family whose sequences differ from Let-7c more prominently, that is, -7g and -7i were not reduced by Let-7c inhibitor and the expression of -7i even increased with the treatment of Let-7c inhibitor (Fig. 2E), probably due to compensatory mechanism(s). Despite the fact that Let-7c inhibitor affected other members of the Let-7 family, the highest reduction was seen in Let-7c miRNA levels over other miRNAs of Let-7 family both in heart (Fig. 2E) and in liver (data not shown). To study the Let-7c expression in cardiac cell types, we fractionated cardiomyocytes and fibroblasts from adult mouse hearts as described previously (Kerkelä et al. 2013). As shown in Figure 2F, the Let-7c miRNA expression was similar in cardiomyocytes and fibroblasts when analyzed from fractionated cells.
When the effects of loss of Let-7c function were analyzed in cell culture using cardiac fibroblasts, an activation of induced pluripotent stem (iPS) cell–related genes was observed. Sox2 protein levels were elevated in response to Let-7c miRNA inhibition (P < 0.05, Fig. 2G), and also mRNA levels of Oct4 (6.6-fold) and Sox2 (10.9-fold) increased in fibroblasts (P < 0.05, Fig. 2H). Comparable changes were observed in normal adult mouse heart in vivo: mRNA levels of Oct4 (5.0-fold) and Sox (1.8-fold) increased 2 weeks after Let-7c inhibitor injection (Fig. 2I). On the other hand, the levels of cMyc and Kruppel-like factor 4 (Klf4) mRNA remained unaltered by Let-7c inhibition (Fig. 2I). To further characterize the selectivity of Let-7c inhibitor in vivo, we studied the expression of miRNA-125b, a negative regulator of the p53 tumor suppressor pathway (Le et al. 2009), and miRNA-126, a regulator of angiogenesis (Liu et al. 2009), and did not detect any changes in their expression in the heart, liver, or kidney (data not shown).
Inhibition of Let-7c improves cardiac function after MI
Cardiac function was evaluated by echocardiography 3 days and 4 weeks after MI. Interestingly, inhibition of Let-7c miRNA prevented the decrease in LV ejection fraction postinfarction (Fig. 3A and B). When the change in LV ejection fraction in scramble and Let-7c inhibitor groups was analyzed, a statistically significant difference was noted (P < 0.05, Fig. 3C). Accordingly, Let-7c inhibitor increased stroke volume by 30% (P = 0.08, Fig. 3D) and cardiac output by 20% (P < 0.05, Fig. 3E) at 4 weeks after MI. Let-7c inhibitor did not affect LV posterior wall thickness (Fig. 3F and G). Also, no alterations in diastolic function in response to inhibition of Let-7c were observed (Table S4).
Figure 3.

Let-7c inhibition prevented the deterioration of systolic function after MI. (A) Representative images from echocardiography M-mode view. (B, C) Let-7c inhibitor maintained LV ejection fraction postinfarction. (D) There was a tendency for stroke volume to increase in response to Let-7c inhibitor. (E) Let-7c inhibitor enhanced cardiac output 4 weeks after MI. (F, G) Let-7c inhibitor had no effect on the LV posterior wall thickness in systole (LVPW;s) or diastole (LVPW;d). Results are mean ± SEM. (B–G) n = 7–8. Data were analyzed with ANOVA. *P < 0.05 versus scramble. Let-7c, lethal-7c; MI, myocardial infarction; LV, left ventricular.
Let-7c inhibition reduces cardiac fibrosis postinfarction
Inhibition of Let-7c miRNA decreased the level of fibrosis in the LV after MI. Percentage of fibrotic area in the noninfarcted myocardium was 64% lower in Let-7c inhibitor-treated mice (P < 0.05, Fig. 4A). There was also a tendency for a smaller infarction scar size, as measured from the LV circumference (Fig. 4B). Interestingly, the reduction in fibrosis triggered by Let-7c inhibitor was accompanied by a lower number of DDR2+ cells as analyzed by immunofluorescence microscopy (Fig. 4C). There was also a tendency for the number of Oct+ and Oct+/DDR2+ double-positive cells to increase (Fig. 4C and D), however, these changes were not statistically significant. On the other hand, Let-7c inhibitor did not significantly alter LV expression of genes involved in the induction of iPS cells (Fig. 5A) or fibrosis (Fig. 5B) measured from the MI border zone. Moreover, the LV levels of p38, ERK, and phospho-ERK proteins remained unchanged by Let-7c inhibition (data not shown).
Figure 4.

Let-7c inhibition reduced fibrosis in the LV. (A) Let-7c inhibitor significantly decreased the fibrotic area in the LV 4 weeks after MI and (B) also a nonsignificant reduction in the size of infarction scar was observed. (C) The number of DDR2+ cells decreased in response to Let-7c inhibition, and there was a trend for the number of Oct+ and Oct+DDR2+ cells to increase postinfarction. (D) A representative image of Oct+DDR2+ cells. Results are mean ± SEM. (A–C) n = 5–8. White column: scramble; black column: Let-7c inhibitor. Scale bar, 50 μm (A), 1 mm (B), 10 μm (D). Data were analyzed with Mann–Whitney U test. *P < 0.05 versus scramble. Let-7c, lethal-7c; LV, left ventricular; MI, myocardial infarction; DDR2, discoidin domain receptor 2; Oct4, octamer-binding transcription factor 4.
Figure 5.
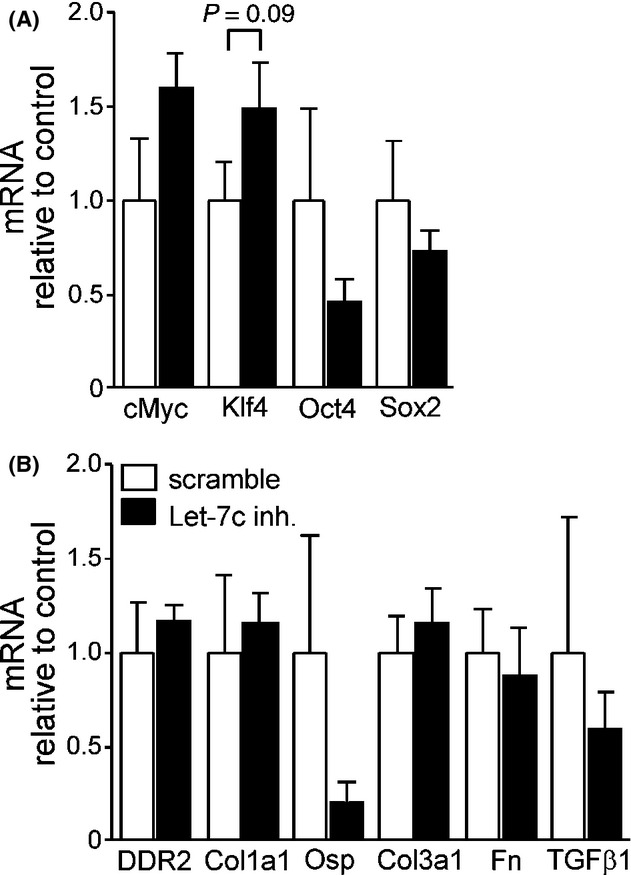
Effects of Let-7c inhibition on LV gene expression after MI. (A) There were no significant changes in pluripotency-associated genes with Let-7c inhibitor treatment when analyzed 4 weeks postinfarction. (B) The activation of genes involved in the regulation of fibrosis remained unaltered in response to Let-7c inhibition. Results are mean ± SEM (n = 7–8/group). White column: scramble; black column: Let-7c inhibitor. Data were analyzed with Student’s t-test. Let-7c, lethal-7c; LV, left ventricular; MI, myocardial infarction.
Apoptosis, angiogenesis, cell proliferation, and c-kit+ cardiac stem cells
In addition to fibrosis, apoptotic cell death, angiogenesis, cell proliferation, and cardiac stem cell recruitment play key roles in the regulation of myocardial remodeling (Quintavalle et al. 2011; Santulli et al. 2012; Burchfield et al. 2013). Therefore, we performed immunohistochemical staining procedures to evaluate morphological changes in the heart in response to administration of Let-7c inhibitor with MI. A significantly reduced number of TUNEL+ apoptotic cells in the border zone of infarction was observed by Let-7c inhibition (P < 0.05, Fig. 6A). Double immunofluorescence staining of TUNEL+ cells with cardiomyocyte marker α-actinin or fibroblast marker DDR2 showed that the apoptotic cells were mainly fibroblasts (Fig. 6B). Instead, myocyte cross-sectional area (Fig. 6C), the number of c-kit+ cardiac stem cells (Fig. 6D) and Ki-67+ proliferating cells (Fig. 6E) remained unaltered in the border zone by Let-7c inhibitor postinfarction. In addition, Let-7c inhibitor did not affect capillary density or area as assessed from LV (Fig. 6F).
Figure 6.

Morphological changes in the LV analyzed by immunohistochemistry. (A) The number of TUNEL+ apoptotic cells decreased statistically significantly in response to Let-7c inhibition 4 weeks after MI. (B) Apoptotic cells were noncardiomyocytes and DDR2+ fibroblasts. (C) Let-7c inhibitor did not affect cardiomyocyte hypertrophy as assessed with cardiomyocyte cross-sectional area after Masson’s trichrome stain and when observed with wheat germ agglutinin stain. The number of c-kit+ stem cells (D) and Ki-67+ proliferative cells (E) remained unaltered. (F) Let-7c inhibitor had no effect on angiogenesis. Results are mean ± SEM. (A–F), n = 6–8/group. White column: scramble; black column: Let-7c inhibitor. Scale bar, 20 μm (A, C–F) and 10 μm (B). Data were analyzed by Mann–Whitney U test. *P < 0.05 versus scramble. LV, left ventricular; TUNEL, transferase-mediated dUTP nick end labeling; MI, myocardial infarction; DDR2, discoidin domain receptor 2; Let-7c, lethal-7c.
Discussion and Conclusions
Despite substantial efforts in the recent years, the detrimental postinfarction remodeling with concomitant fibrosis, hypertrophy, cell death, and limited regenerative capacity remains an unsolved challenge (Burchfield et al. 2013). The finding that functions of miRNAs are intensified under pathophysiological conditions and that miRNAs influence the progression of LV remodeling has opened new therapeutic possibilities (van Rooij and Olson 2012; Kumarswamy and Thum 2013; Seeger et al. 2013). Depending on the abundance of a miRNA and its targets, as well as the physiological state of a cell, a miRNA can act as a fine-tuner of gene expression or as an on/off switch (van Rooij and Olson 2012). MiRNAs are known to regulate cardiac hypertrophy, apoptosis, endothelial cell function, and vessel growth as well as fibrosis (Kumarswamy and Thum 2013; Quiat and Olson 2013; Seeger et al. 2013; Condorelli et al. 2014; Santulli et al. 2014). In this study, we observed preserved systolic function with reduction in fibrosis and apoptosis postinfarction in mice in response to Let-7c inhibitor suggesting that inhibition of Let-7 miRNA may be a novel therapeutic tool for LV remodeling.
MiRNAs are involved in diverse biological functions, including development, life span, cell proliferation, differentiation, signaling pathways, apoptosis, and metabolism (Sun and Lai 2013). The first two known miRNAs, Lin-4 and Let-7, were discovered in the nematode C. elegans and controlling the timing of stem-cell division and differentiation (Pasquinelli et al. 2000; Reinhart et al. 2000). Let-7 and its family members are highly conserved across species in sequence and function, and play a significant role in the regulation of cell reprogramming (Roush and Slack 2008; Peter 2009). In ES cell differentiation, Let-7 miRNA family members are augmented to suppress pluripotency factors (Melton et al. 2010). In this study, we targeted a Let-7 family member, Let-7c, with a miRNA inhibitor. The Let-7c inhibitor was designed based on the mature sequence of mouse Let-7c. With this approach, the expression of Let-7c miRNA in the target tissue (the heart) as well as in the tissues responsible for elimination of antagomirs (liver and kidney) was downregulated. The inhibition of Let-7c was functional, as reflected by increased expression of specific pluripotency genes (Oct4, Sox2) in cardiac fibroblasts in vitro and in the adult mice heart in vivo. However, although Let-7c inhibitor had the highest influence on the expression of Let-7c, also other closely related members of the Let-7 family were downregulated by the inhibitor. Thus, effects of Let-7c antagomir used in this study may result from inhibition of other members of the Let-7 family alongside with the inhibition of Let-7c.
A hallmark feature of ventricular remodeling is deposition of excessive extracellular matrix and fibrosis (Dobaczewski et al. 2010; Burchfield et al. 2013). In cardiac pathologies, fibroblasts proliferate and secrete various growth factors, collagen, and fibronectin (Camelliti et al. 2005; Krenning et al. 2010; Zeisberg and Kalluri 2010). Fibroblasts can be divided into subpopulations based on the morphology and expression of cell surface markers, such as DDR2 and FAP (Zeisberg and Kalluri 2010). DDR2+ fibroblasts are involved in cell migration and differentiation (Camelliti et al. 2005; Krenning et al. 2010). To date, no therapeutic strategy has been developed to specifically target fibrosis and fibroblast subpopulations in the heart. In this study, parallel with the maintained systolic function, we detected significantly decreased fibrosis, measured by the amount of interstitial collagen deposits, in mice treated with Let-7c inhibitor postinfarction. This was associated with a lower number of DDR2+ fibroblasts and a tendency for a smaller infarction scar size after MI. Hence, the decreased fibrotic response may contribute to the improved cardiac function in mice postinfarction by the Let-7 miRNA inhibition. Interestingly, it has been reported recently that in cultured human proximal tubular epithelial cells, lipoxin-mediated upregulation of Let-7c suppresses transforming growth factor β1 (TGFβ1)–induced fibrosis (Brennan et al. 2013) and that in pancreatic cancer cells both collagen and TGFβ1 can attenuate Let-7c expression (Dagni-Garimella et al. 2011). However, in this study, the expression of TGFβ1 in heart was not affected by inhibition of Let-7c. Taken together, these studies suggest distinct cell- and tissue-specific interplay and regulatory circuits between profibrotic factors and Let-7c miRNA.
As the major finding of this study, we observed decreased number of apoptotic cells in myocardium after Let-7c inhibition, likely playing an important role in preserving systolic function postinfarction. Let-7 family of miRNAs is known to be involved in the regulation of cell apoptosis (Roush and Slack 2008; Peter 2009). In cardiovascular system, for example, Let-7c overexpression enhances apoptosis in endothelial cells, whereas inhibition of Let-7c partly alleviates apoptotic cell death mediated by oxidized-LDL (Qin et al. 2012). In that study, apoptosis was related to inhibition of Bcl-xL, an antiapoptotic member of the Bcl-2 family, and a direct target of Let-7c in endothelial cells. Apoptosis has also been linked to the ability of Let-7 to target caspase-3, Bcl-2, cyclin-dependent kinase 5, and Fas proteins (Barh et al. 2010). Interestingly, the expression of Let-7a and Let-7b decreases significantly following exposure to agents that induce cellular stress, such as ionizing radiation (Saleh et al. 2011). Therefore, it is tempting to speculate that the decrease in Let-7c miRNA levels observed in this study is caused by MI-induced LV wall cell stress. Importantly, because inhibition of Let-7c improved cardiac function, our results suggest that the downregulation of Let-7c after MI is beneficial, yet a short-term compensatory mechanism. Interestingly, the expression of Let-7c miRNA has also been shown to be downregulated in patients with atrial fibrillation (Cooley et al. 2012), and thus Let-7c miRNA may serve as a potential drug target in the prevention and treatment of atrial fibrillation (Santulli et al. 2014). Recently, the functional role of the reverse atrial remodeling in preventing atrial fibrillation has been demonstrated also in the clinical setting (D’Ascia et al. 2011), and thus, further studies are warranted to dissect the possible effect of Let-7 inhibition on atrial remodeling.
One of the key functions of Let-7 is to promote differentiation of cells (Roush and Slack 2008; Peter 2009; Heinrich and Dimmeler 2012). In C. elegans, this role was shown by the requirement of Let-7 in halting the stem-cell-like divisions of the seam cells and their consequent adoption of a fully differentiated state (Reinhart et al. 2000). In more complex organisms, Let-7 expression dramatically increases as cells become more differentiated (Roush and Slack 2008; Peter 2009). Recently, the stem-cell factor Lin28 has been shown to be a major contributor to the posttranscriptional regulation of mammalian Let-7 (Thornton and Gregory 2012; Shyh-Chang and Daley 2013). Lin28 is highly expressed in undifferentiated ES cells and represents part of a pluripotency network in these cells with key ES cell transcription factors, such as Oct4, Sox2, and Nanog (Marson et al. 2008). Reprogramming of differentiated cells to iPS cells can be achieved by the introduction of pluripotency transcription factors into somatic cells (Yamanaka and Blau 2010). One of the first studies reporting the derivation of human iPS cells utilized ectopic expression of Oct4, Sox2, Nanog, and Lin28 as reprogramming factors (Yu et al. 2007). This observation, together with the report that Let-7 inhibition enhances reprogramming of mouse fibroblasts to iPS cells (Melton et al. 2010), shows that Lin28/Let-7 axis manipulates cellular pluripotency. In line with these studies, we provide evidence that the inhibition of Let-7 miRNA increases expression of pluripotency genes Sox2 and Oct4 in cardiac fibroblasts in vitro and in normal mice heart in vivo, and that modulation of Let-7 miRNAs can be used to alter the phenotype of cardiac fibroblasts and concomitantly affect cardiac function.
The fact that any given miRNA has the potential to target multiple mRNAs as well as a given mRNA is likely to be targeted by several miRNAs (Dorn 2012) makes the interpretation of the experimental results challenging. As described above, in this study the inhibition of Let-7 miRNA increased expression of pluripotency genes Sox2 and Oct4 in cardiac fibroblasts in vitro and in normal mice heart in vivo. However, postinfarction alterations in gene expression were not significant. The reasons for distinct changes remain to be established, but may be due to the masking effect of myocardial tissue damage and associated stress response (Dorn 2012). The ability of miRNAs to impact on multiple effectors within a biological pathway, on the other hand, facilitates their putative use as therapeutic targets (van Rooij and Olson 2012; Seeger et al. 2013). In this study, the inhibition of one family member of Let-7 miRNAs led to altered expression of several target genes in normal mice, and likely several others – known or unknown – we did not evaluate in this study. Moreover, because of a very high degree of homology between members of Let-7 family, Let-7c antagomir used in this study resulted in inhibition of other members of the Let-7 family alongside with the inhibition of Let-7c. Thus, the results obtained with one antagomir should be interpreted with caution. Nevertheless, the fact that we observed altered recovery of myocardium after ischemic insult confirms that the modulation of miRNAs can be a powerful tool.
Despite optimal treatment with existing therapeutics, the rates of morbidity and mortality are still high in patients with heart failure (Jessup and Brozena 2003; McMurray and Pfeffer 2005), and novel therapeutic strategies are necessary. In this study, we observed decreased fibrosis and apoptosis, and subsequently maintained systolic function postinfarction in response to Let-7c inhibitor. Thus, inhibition of Let-7 miRNA’s function may be a novel therapeutic approach for detrimental postinfarction LV remodeling. One concern of this approach, arising from the understanding of Let-7 regulation, is the dual role of Let-7 in being both protective and causal in different types of disease. Like reported here with MI model, Let-7 inhibition has also protective effect in mice with diet-induced obesity (Frost and Olson 2011). However, Let-7 is a tumor suppressor raising the likelihood that persistent Let-7 repression may lead to hyperplastic disorders and, in certain cases, cancer (Barh et al. 2010; Boyerinas et al. 2010). Thus, long-term experiments are required to establish the safety of Let-7 inhibition.
Acknowledgments
We thank Kirsi Salo, Kati Lampinen, Marja Arbelius, and Sirpa Rutanen for their expert technical assistance. This work was supported by the Academy of Finland (Center of Excellence to H. R., 251736 to R. S.), Sigrid Juselius Foundation (to H. R.), Finnish Foundation for Cardiovascular Research (to H. R. and R. S.), Finnish Cultural Foundation (to R. S.), the National Technology Foundation TEKES (to H. R.), and Biocenter Finland (to H. R. and R. S.).
Glossary
- LSD
least significant difference
- ANOVA
analysis of variance
- TUNEL
transferase-mediated dUTP nick end labeling
- MAPK
mitogen-activated protein kinase
- ERK
extracellular signal-regulated kinase
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- FBS
fetal bovine serum
- LVID
LV internal dimensions
- PBS
phosphate-buffered saline
- LNA
locked nucleic acid
- Col1a1
collagen 1α1
- Col3a1
collagen 3α1
- DDR2
discoidin domain receptor 2
- ES
embryonic stem
- FAP
fibroblast activation protein
- FS
fractional shortening
- iPS
induced pluripotent stem
- Klf4
Kruppel-like factor 4
- LAD
left anterior descending
- Let-7
lethal-7
- LV
left ventricle
- MI
myocardial infarction
- miRNA
microRNA
- Oct4
octamer-binding transcription factor 4
- Osp
osteopontin
- TGFβ1
transforming growth factor β1
Disclosures
None declared.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. Probes used for miRNA quantitative PCR analysis. Let-7c sequence is shown in blue and nucleotide differences compared to Let-7c are indicated with red color.
Table S2. Fluorogenic probe, forward and reverse primer sequences used for quantitative PCR analysis. Fluorogenic probe sequence is indicated as 5′-[FAM…TAMRA]), forward and reverse primers as (5′-…).
Table S3. Effects of myocardial infarction on cardiac function 7 days after MI. Results are mean ± SEM (n = 8–11). Data were analyzed with Student’s t-test.
Table S4. Effects of inhibition of Let-7c on cardiac function 4 weeks after MI. Results are mean ± SEM (n = 7–8). Data were analyzed with ANOVA.
References
- Barh D, Malhotra R, Ravi B, Sindhurani P. MicroRNA let-7: an emerging next-generation cancer therapeutic. Curr Oncol. 2010;17:70–80. doi: 10.3747/co.v17i1.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Boyerinas B, Park SM, Hau A, Murmann AE, Peter ME. The role of let-7 in cell differentiation and cancer. Endocr Relat Cancer. 2010;17:F19–F36. doi: 10.1677/ERC-09-0184. [DOI] [PubMed] [Google Scholar]
- Brennan EP, Nolan KA, Börgeson E, Gough OS, McEvoy CM, Docherty NG, et al. Lipoxins attenuate renal fibrosis by inducing let-7c and suppressing TGFβR1. J Am Soc Nephrol. 2013;24:627–637. doi: 10.1681/ASN.2012060550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burchfield JS, Xie M, Hill JA. Pathological ventricular remodelling. Part 1 of 2. Circulation. 2013;128:388–400. doi: 10.1161/CIRCULATIONAHA.113.001878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camelliti P, Borg TK, Kohl P. Structural and functional characterization of cardiac fibroblasts. Cardiovasc Res. 2005;65:40–51. doi: 10.1016/j.cardiores.2004.08.020. [DOI] [PubMed] [Google Scholar]
- Chen C, Ridzon D, Lee CT, Blake J, Sun Y, Strauss WM. Defining embryonic stem cell identity using differentiation-related microRNAs and their potential targets. Mamm Genome. 2007;18:316–327. doi: 10.1007/s00335-007-9032-6. [DOI] [PubMed] [Google Scholar]
- Condorelli G, Drusco A, Stassi G, Bellacosa A, Roncarati R, Iaccarino G, et al. Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc Natl. Acad Sci USA. 2002;99:12333–12338. doi: 10.1073/pnas.172376399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condorelli G, Latronico MV, Cavarretta E. microRNAs in cardiovascular diseases: current knowledge and the road ahead. J Am Coll Cardiol. 2014;63:2177–2187. doi: 10.1016/j.jacc.2014.01.050. [DOI] [PubMed] [Google Scholar]
- Cooley N, Cowley MJ, Lin RCY, Marasco S, Wong C, Kaye DM, et al. Influence of atrial fibrillation on microRNA expression profiles in left and right atria from patients with valvular heart disease. Physiol Genomics. 2012;44:211–219. doi: 10.1152/physiolgenomics.00111.2011. [DOI] [PubMed] [Google Scholar]
- Dagni-Garimella S, Strouch MJ, Grippo PJ, Bentrem DJ, Munshi HG. Collagen regulation of let-7 in pancreatic cancer involves TGF-1β-mediated membrane type 1-matrix metalloproteinase expression. Oncogene. 2011;30:1002–1008. doi: 10.1038/onc.2010.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ascia SL, D’Ascia C, Marino V, Lombardi A, Santulli R, Chiariello M, et al. Cardiac resynchronisation therapy response predicts occurrence of atrial fibrillation in non-ischaemic dilated cardiomyopathy. Int J Clin Pract. 2011;11:1149–1155. doi: 10.1111/j.1742-1241.2011.02732.x. [DOI] [PubMed] [Google Scholar]
- Dobaczewski M, Gonzalez-Quesada C, Frangogiannis NG. The extracellular matrix as a modulator of the inflammatory and reparative response following myocardial infarction. J Mol Cell Cardiol. 2010;48:504–511. doi: 10.1016/j.yjmcc.2009.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorn GW., II Decoding the cardiac message. The 2011 Thomas W. Smith Memorial Lecture. Circ Res. 2012;110:755–763. doi: 10.1161/CIRCRESAHA.111.256768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost RJ, Olson EN. Control of glucose homeostasis and insulin sensitivity by the Let-7 family of microRNAs. Proc Natl. Acad Sci USA. 2011;108:21075–21080. doi: 10.1073/pnas.1118922109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost RJ, van Rooij E. miRNAs as therapeutic targets in ischemic heart disease. J Cardiovasc Transl Res. 2010;3:280–289. doi: 10.1007/s12265-010-9173-y. [DOI] [PubMed] [Google Scholar]
- Gao E, Lei YH, Huang ZM, Zuo L, Boucher M, Fan Q, et al. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res. 2010;107:1445–1453. doi: 10.1161/CIRCRESAHA.110.223925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith EC, Hoffman A, Morales MO, Potts JD, Price RL, McFadden A, et al. Organization of fibroblasts in the heart. Dev Dyn. 2004;230:787–794. doi: 10.1002/dvdy.20095. [DOI] [PubMed] [Google Scholar]
- Heineke J, Molkentin J. Regulation of cardiac hypertrophy by intracellular signalling pathways. Mol Cell Biol. 2006;7:581–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- Heinrich E-M, Dimmeler S. MicroRNAs and stem cells. Control of pluripotency, reprogramming, and lineage commitment. Circ Res. 2012;110:1014–1022. doi: 10.1161/CIRCRESAHA.111.243394. [DOI] [PubMed] [Google Scholar]
- Jessup M, Brozena S. Heart failure. N Engl J Med. 2003;348:2007–2018. doi: 10.1056/NEJMra021498. [DOI] [PubMed] [Google Scholar]
- Kerkelä R, Karsikas S, Szabo Z, Serpi R, Magga J, Gao E, et al. Activation of hypoxia response in endothelial cells contributes to ischemic cardioprotection. Mol Cell Biol. 2013;16:3321–3329. doi: 10.1128/MCB.00432-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenning G, Zeisberg EM, Kalluri R. The origin of fibroblasts and mechanism of cardiac fibrosis. J Cell Physiol. 2010;225:631–637. doi: 10.1002/jcp.22322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumarswamy T, Thum T. Non-coding RNAs in cardiac remodelling and heart failure. Circ Res. 2013;113:676–689. doi: 10.1161/CIRCRESAHA.113.300226. [DOI] [PubMed] [Google Scholar]
- Landgraf P, Rusu M, Sheridan R, Sewer A, Iovino N, Aravin A, et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401–1414. doi: 10.1016/j.cell.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le MTN, Teh K, Shyh-Chang N, Xie H, Zhou B, Korzh V, et al. MicroRNA-125b is a novel negative regulator of p53. Genes Dev. 2009;23:862–876. doi: 10.1101/gad.1767609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lips D, deWindt LJ, van Kraaij D, Doevendans PA. Molecular determinants of myocardial hypertrophy and failure: alternative pathways for beneficial and maladaptive hypertrophy. Eur Heart J. 2003;24:883–896. doi: 10.1016/s0195-668x(02)00829-1. [DOI] [PubMed] [Google Scholar]
- Liu B, Peng XC, Zheng XL, Wang J, Qin YW. MiR-126 restoration down-regulate VEGF and inhibit the growth of lung cancer cell lines in vitro and in vivo. Lung Cancer. 2009;66:169–175. doi: 10.1016/j.lungcan.2009.01.010. [DOI] [PubMed] [Google Scholar]
- Marson A, Levine SS, Cole MF, Frampton GM, Brambrink T, Johnstone S, et al. Connecting microRNA genes to the core transcriptional circuitry of embryonic stem cells. Cell. 2008;134:521–533. doi: 10.1016/j.cell.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurray JJ, Pfeffer MA. Heart failure. Lancet. 2005;365:1877–1889. doi: 10.1016/S0140-6736(05)66621-4. [DOI] [PubMed] [Google Scholar]
- McMurray JJ, Adamopoulos S, Anker SD, Auricchio A, Böhm M, Dickstein K, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: The Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2012 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association (HFA) of the ESC. Eur Heart J. 2012;33:1787–1847. doi: 10.1093/eurheartj/ehs104. [DOI] [PubMed] [Google Scholar]
- Melton C, Judson RL, Blelloch R. Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature. 2010;463:621–626. doi: 10.1038/nature08725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RL, van Rooij E. Therapeutic advances in MicroRNA targeting. J Cardiovasc Pharmacol. 2011;57:1–7. doi: 10.1097/FJC.0b013e3181f603d0. [DOI] [PubMed] [Google Scholar]
- Pasquinelli AE, Reinhart BJ, Slack F, Martindale MQ, Kuroda MI, Maller B, et al. Conservation of the sequence and temporal expression of let-7 heterochronic regulatory RNA. Nature. 2000;408:86–89. doi: 10.1038/35040556. [DOI] [PubMed] [Google Scholar]
- Peter ME. Let-7 and miR-200 microRNAs: guardians against pluripotency and cancer progression. Cell Cycle. 2009;8:843–852. doi: 10.4161/cc.8.6.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin B, Xiao B, Liang D, Li Y, Jiang T, Yang H. MicroRNA let-7c inhibits Bcl-xl expression and regulates ox-LDL-induced endothelial apoptosis. BMP Rep. 2012;45:464–469. doi: 10.5483/BMBRep.2012.45.8.033. [DOI] [PubMed] [Google Scholar]
- Quiat D, Olson EN. MicroRNAs in cardiovascular disease: from pathogenesis to prevention and treatment. J Clin Invest. 2013;123:11–18. doi: 10.1172/JCI62876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintavalle C, Garofalo M, Croce CM, Condorelli G. “ApoptomiRs” in vascular cells: their role in physiological and pathological angiogenesis. Vascul Pharmacol. 2011;55:87–91. doi: 10.1016/j.vph.2011.07.004. [DOI] [PubMed] [Google Scholar]
- Reinhart BJ, Slack FJ, Basson M, Pasquinelli AE, Bettinger JC, Rougvic AE, et al. The 21-nucleotide let-7 RNA regulates developmental timing in Caenorhabditis elegans. Nature. 2000;403:901–906. doi: 10.1038/35002607. [DOI] [PubMed] [Google Scholar]
- van Rooij E, Olson EN. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discov. 2012;11:860–872. doi: 10.1038/nrd3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roush S, Slack FJ. The let-7 family of microRNAs. Trends Cell Biol. 2008;18:505–516. doi: 10.1016/j.tcb.2008.07.007. [DOI] [PubMed] [Google Scholar]
- Rysä J, Tenhunen O, Serpi R, Soini Y, Nemer M, Leskinen H, et al. GATA-4 is an angiogenic survival factor of the infarcted heart. Circ Heart Fail. 2010;3:440–450. doi: 10.1161/CIRCHEARTFAILURE.109.889642. [DOI] [PubMed] [Google Scholar]
- Saleh AD, Savage JE, Cao L, Soule BP, Ly D, DeGraff W, et al. Cellular stress induced alterations in microRNA let-7a and let-7b expression are dependent on p53. PLoS One. 2011;6:e24429. doi: 10.1371/journal.pone.0024429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli G, Cipolletta E, Sorriento D, Del Giudice C, Anastasio A, Monaco S, et al. CaMK4 gene deletion induces hypertension. J Am Heart Assoc. 2012;4:e001081. doi: 10.1161/JAHA.112.001081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santulli G, Iaccarino G, De Luca N, Trimarco B, Condorelli G. Atrial fibrillation and microRNAs. Front Physiol. 2014;5:15. doi: 10.3389/fphys.2014.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeger FH, Zeiher AM, Dimmeler S. MicroRNAs in stem cell function and regenerative therapy of the heart. Arterioscler Thromb Vasc Biol. 2013;33:1739–1746. doi: 10.1161/ATVBAHA.113.300138. [DOI] [PubMed] [Google Scholar]
- Serpi R, Tolonen AM, Huusko J, Rysä J, Tenhunen O, Ylä-Herttuala S, et al. Vascular endothelial growth factor-B gene transfer prevents angiotensin II-induced diastolic dysfunction via proliferation and capillary dilatation in rats. Cardiovasc Res. 2011;89:204–213. doi: 10.1093/cvr/cvq267. [DOI] [PubMed] [Google Scholar]
- Shah AM, Mann DL. In search of new therapeutic targets and strategies for the heart failure: recent advances in basic research. Lancet. 2011;378:704–712. doi: 10.1016/S0140-6736(11)60894-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyh-Chang N, Daley GQ. Lin28: primal regulator of growth and metabolism in stem cells. Cell Stem Cell. 2013;12:395–406. doi: 10.1016/j.stem.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Lai EC. Adult-specific functions of animal microRNAs. Nat Rev Genet. 2013;14:535–548. doi: 10.1038/nrg3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutton MG, Sharpe N. Left ventricular remodelling after myocardial infarction: pathophysiology and therapy. Circulation. 2000;101:2981–2988. doi: 10.1161/01.cir.101.25.2981. [DOI] [PubMed] [Google Scholar]
- Tenhunen O, Soini Y, Ilves M, Rysä J, Tuukkanen J, Serpi R, et al. p38 kinase rescues failing myocardium after myocardial infarction; evidence for angiogenic and anti-apoptotic mechanisms. FASEB J. 2006;20:1907–1909. doi: 10.1096/fj.05-5618fje. [DOI] [PubMed] [Google Scholar]
- Thornton JE, Gregory RI. How does Lin28 Let-7 control development and disease? Trends Cell Biol. 2012;22:474–482. doi: 10.1016/j.tcb.2012.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka S, Blau HM. Nuclear reprogramming to a pluripotent state by three approaches. Nature. 2010;465:704–712. doi: 10.1038/nature09229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Zeisberg E, Kalluri R. Origins of cardiac fibroblasts. Circ Res. 2010;107:1304–1312. doi: 10.1161/CIRCRESAHA.110.231910. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Probes used for miRNA quantitative PCR analysis. Let-7c sequence is shown in blue and nucleotide differences compared to Let-7c are indicated with red color.
Table S2. Fluorogenic probe, forward and reverse primer sequences used for quantitative PCR analysis. Fluorogenic probe sequence is indicated as 5′-[FAM…TAMRA]), forward and reverse primers as (5′-…).
Table S3. Effects of myocardial infarction on cardiac function 7 days after MI. Results are mean ± SEM (n = 8–11). Data were analyzed with Student’s t-test.
Table S4. Effects of inhibition of Let-7c on cardiac function 4 weeks after MI. Results are mean ± SEM (n = 7–8). Data were analyzed with ANOVA.