

Abstract
Mast cells are known to have a detrimental impact on a variety of pathological conditions. There is therefore an urgent need of developing strategies that limit their harmful effects. The aim of this study was to accomplish this by developing a means of inducing mast cell apoptosis. The strategy was to identify novel compounds that induce mast cell apoptosis by permeabilization of their secretory lysosomes (granules). As a candidate, we assessed mefloquine, an anti-malarial drug that has been proposed to have lysosome-permeabilizing activity. Mefloquine was added to mast cells and administered in vivo, followed by assessment of the extent and mechanisms of mast cell death. Mefloquine was cytotoxic to murine and human mast cells. Mefloquine induced apoptotic cell death of wild-type mast cells whereas cells lacking the granule compounds serglycin proteoglycan or tryptase were shown to undergo necrotic cell death, the latter finding indicating a role of the mast cell granules in mefloquine-induced cell death. In support of this, mefloquine was shown to cause compromised granule integrity and to induce leakage of granule components into the cytosol. Mefloquine-induced cell death was refractory to caspase inhibitors but was completely abrogated by reactive oxygen species inhibition. These findings identify mefloquine as a novel anti-mast cell agent, which induces mast cell death through a granule-mediated pathway. Mefloquine may thus become useful in therapy aiming at limiting harmful effects of mast cells.
Keywords: Apoptosis; granules; mast cells; mefloquine, mMCP-6; reactive oxygen species; serglycin; tryptase
Introduction
Mast cells are tissue-resident hematopoietic cells of the immune system (Metcalfe et al. 1997; Galli et al. 2005; Gurish and Austen 2012). A striking feature of mature mast cells is their remarkably high content of cytoplasmic secretory granules, which are filled with a plethora of bioactive compounds such as histamine, serotonin, serglycin proteoglycans, lysosomal hydrolases, cytokines, and a number of different mast cell-specific proteases (Wernersson and Pejler 2014). When mast cells are activated appropriately, they respond by degranulation, thereby releasing their contained secretory granule compounds. In addition, mast cell activation leads to de novo synthesis and release of numerous additional compounds such as prostaglandins, leukotrienes as well as different cytokines and chemokines (Galli et al. 2008). Altogether, mast cell activation can thus result in a massive release of a large variety of bioactive compounds, which collectively can produce a strong inflammatory response. Accordingly, mast cells have been implicated as detrimental players in numerous inflammatory disease settings, most notably in allergic conditions but also in diverse other pathologies including cancer, atherosclerosis, and inflammatory skin disorders (Kalesnikoff and Galli 2008).
Based on the known harmful impact of mast cells on a variety of pathological settings, there is currently a large need for identifying novel strategies for targeting mast cells or their products (Reber and Frossard 2014). Currently, various histamine receptor antagonists are being exploited as therapeutics and there is also the possibility to interfere with mast cell degranulation by using mast cell “stabilizers” such as chromoglycate or ketitofen (Reber and Frossard 2014). However, since mast cell activation is associated with release of pro-inflammatory compounds both from preformed stores in granules and from nongranule sources (from de novo synthesis), mere blockade of degranulation might not be sufficient to prevent mast cell-driven pathology. Moreover, even though agents blocking effects of individual mast cell-derived compounds can have beneficial effects (e.g., histamine receptor antagonists), it is likely that the overall effect of mast cells on any given pathological setting is a result of combined effects of several different proinflammatory compounds secreted by mast cells. Therefore, efficient blockade of harmful mast cell-mediated events may require a global intervention with mast cells, which potentially could be achieved through induction of selective mast cell death (Karra et al. 2009). Ideally, mast cell death should occur by apoptosis rather than necrosis, since apoptosis occurs without inflammatory side effects whereas necrosis is associated with substantial release of proinflammatory alarmins (Edinger and Thompson 2004; Elmore 2007; Taylor et al. 2008).
Various approaches have previously been used to induce mast cell apoptosis (Karra et al. 2009). However, since most proapoptotic pathways are ubiquitous among different cell types, it has so far not been possible to identify a cell death pathway that selectively targets mast cells. To approach this problem, we have hypothesized that mast cells potentially may be targeted by regimens that cause permeabilization of their secretory granules. The rationale behind this approach is that secretory granule permeabilization will lead to the release of granule-localized compounds including proteases into the cytosol where they potentially may induce apoptosis by, for example, proteolytic activation of latent proapoptotic factors or degradation of anti-apoptotic factors (Boya and Kroemer 2008; Turk and Turk 2009). Moreover, since mast cells are exceptionally rich in lysosome-like granules, we have hypothesized that mast cells may be more sensitive to such agents than are other cell types. Indeed, as a proof-of-principle for this scenario, we showed that the lysosomotropic agents l-leucyl-l-leucine methyl ester and siramesine induce mast cell death through a granule-mediated pathway (Melo et al. 2011b; Spirkoski et al. 2012). However, the mechanism of cell death in response to these agents was not fully elucidated and, moreover, neither siramesine nor l-leucyl-l-leucine methyl ester are approved for use in humans. The aim of this investigation was therefore to search for novel mast cell granule-permeabilizing agents that are compatible with use in humans and to identify the mechanism by which granule-permeabilizing agents cause mast cell death. We show that mefloquine, an approved anti-malarial drug that previously has been suggested to have lysosomotropic activity (Ginsburg 1990; Glaumann et al. 1992), has mast cell granule-permeabilizing activity and induces apoptotic death of mast cells. Moreover, we show that mefloquine-induced cell death is dependent on reactive oxygen species (ROS) production.
Materials and Methods
Reagents
Mefloquine, α-tocopherol and N-acetyl cysteine (NAC) were purchased from Sigma-Aldrich (St. Louis, MO). Mefloquine was prepared as a stock solution of 50 mmol/L in dimethyl sulfoxide, and was subsequently diluted in phosphate-buffered saline (PBS) for the further experiments. Acridine orange (AO), E64d, Z-DEVD-FMK, Z-VAD-FMK, Pefabloc SC, and Pepstatin A were from Sigma-Aldrich (Steinheim, Germany). Polyclonal antibodies to mouse mast cell protease 6 (mMCP-6) and carboxypeptidase A3 (CPA3) were as described (Rönnberg and Pejler 2012). Fluorescently labeled donkey anti-rabbit antibodies were purchased from Li-cor (Lincoln, NE).
Mice
Wild-type (WT) mice and mice lacking serglycin (Åbrink et al. 2004) or the tryptase mMCP-6 (Shin et al. 2008) were on C57BL/6J genetic background. All animal experiments were approved by the local ethical committee (Uppsala, Sweden).
Cell culture
Mouse bone marrow-derived mast cells (BMMCs) from either WT, mMCP-6−/− or serglycin−/− mice were prepared and cultured as described (Rönnberg and Pejler 2012). Peritoneal cell-derived mast cells (PCMCs) were generated according to the method described (Malbec et al. 2007). Human mast cell line-1 (HMC-1), a kind gift from the Mayo Foundation for Medical Education and Research, was cultured under conditions described previously (Melo et al. 2011a). Human cord blood-derived mast cells (CBMCs) were obtained from normal deliveries at Uppsala University hospital. Mononuclear cells were separated by centrifugation on Ficoll-Paque. CD34+ mononuclear cells were enriched using the MACS column separation and were seeded in StemPro-34 serum-free medium (Gibco/Invitrogen, Carlsbad, CA) supplemented with StemPro-Nutrient Supplement (Gibco/Invitrogen), recombinant human IL-6 (10 ng/mL) and stem cell factor (100 ng/mL), 10% fetal bovine serum (FBS), 50 μg/mL streptomycin sulfate, 60 μg/mL penicillin G, 2 mmol/L l-glutamine. Recombinant human IL-3 (1 ng/mL) was added only during the 1st week. Cells were kept at 37°C in 5% CO2-balanced air and the medium was replaced once a week. After 4 weeks, mast cells were identified by enzyme-histochemical staining for tryptase. The human breast cancer cell line, MDA-MB-231, was kindly provided by Ludwig Institute for Cancer Research (Uppsala, Sweden). The cells were propagated in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS, 50 μg/mL streptomycin sulfate, and 60 μg/mL penicillin G. Human Embryonic Kidney 293 cells (HEK-293) were cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated FBS, 2 mmol/L l-glutamine, 60 μg/mL penicillin G, and 50 μg/mL streptomycin. Primary human dermal fibroblast neonatal cells (HDFn) were cultured in Dulbecco’s modified Eagle’s medium containing 5% heat-inactivated FBS, 2 mmol/L l-glutamine, 60 μg/mL penicillin G, and 50 μg/mL streptomycin. The U-937 cell line isolated from histiocytic lymphoma was cultured in RPMI 1640 – GlutaMAX™-1 supplemented with 5% heat-inactivated FBS, 60 μg/mL penicillin G, and 50 μg/mL streptomycin.
Cell viability assessment
The cytotoxicity of mefloquine was examined by the CellTiter-Blue® Reagent cell viability assay (Promega-Invitrogen, Carlsbad, CA) as described previously (Spirkoski et al. 2012). To test the effect of different protease inhibitors on cell death, BMMCs were preincubated with a panel of inhibitors including E-64d (20 μmol/L), Z-DEVD-FMK (20 μmol/L), Z-VAD-FMK (20 μmol/L), Pepstatin A (50 μmol/L), and Pefabloc SC (0.1 mmol/L), 30 min prior to mefloquine treatment. After 24 h, cell death was assessed. Moreover, to test the effect of different antioxidants on cell death, BMMCs were preincubated with α-tocopherol (0.4 mmol/L) or NAC (8 mmol/L), 2 h prior to mefloquine treatment. After 8 h, cell death was assessed.
Staining of acidic compartments
BMMCs (0.5 × 106 cells/mL) were either treated with 10 μmol/L of mefloquine for 24 h or left untreated. Thereafter, cells were incubated with AO (5 μg/mL) for 15 min at 37°C and then rinsed 3 times with PBS supplemented with bovine serum albumin (BSA; 100 μg/mL). Finally, triplicates of 100 μL of cell suspension were transferred to a 96-well plate and fluorescence intensity was determined with excitation at 485 nm and emission at 650 nm using a microplate reader (Infinite M200–TECAN, Männedorf, Switzerland).
Cytosolic extract preparation
Cytosolic extracts were prepared using digitonin extraction as described (Melo et al. 2011b).
Cysteine cathepsin-like activity measurement
Cysteine cathepsin-like activity was measured using chromogenic peptide substrate Z-Phe-Arg-AMC (Bachem, Bubendorf, Switzerland) (Melo et al. 2011b).
Western blot analysis
Western blot analysis was carried out as described previously (Rönnberg and Pejler 2012), using 500-fold dilutions of the antibodies towards mMCP-6 and CPA3, respectively. Membranes were incubated overnight with primary antibody; incubation with secondary antibody was performed for 1 hour.
Measurement of ROS
BMMCs (0.5 × 106 cells) were preincubated with or without NAC (8 mmol/L) in a 24-well plate for 2 h. Next, cells were either left untreated or treated with 20 μmol/L of mefloquine. After 30 min of incubation, cells were washed with PBS and incubated with 5 μmol/L CM-H2DCFDA (Life Technologies, Carlsbad, CA) in PBS at 37°C in dark for 30 min. Finally, cells were washed and resuspended in PBS followed by analysis with a FACScan® flow cytometer (BD Biosciences, San Jose, CA). Data analysis was performed using the FlowJo software (TreeStar Inc., Ashland, OR).
In vivo effect of mefloquine
Eight- to 10-week-old female mice (C57BL/6J) were treated with intraperitoneal injections of PBS (control group) or 10 or 30 mg−1 kg−1 day−1 of mefloquine for 4 consecutive days. One day after the last injection, mice were killed using isoflurane and peritoneal lavage fluid, ear skin, trachea and tongues were collected. The treatment with mefloquine did not induce weight loss in comparison with nontreated animals, indicating that mefloquine did not adversely affect animal welfare. Total peritoneal cells were counted and cytospin slides were prepared. Collected tissues were paraffin-embedded and sectioned. Cytospin slides were stained with May-Grünwald/Giemsa, whereas the tissue sections were stained with toluidine blue. Peritoneal cells were stained with antibodies against surface markers and were subjected to flow cytometric analysis.
Flow cytometry and monoclonal antibodies
Apoptotic and necrotic cell death was assessed by Annexin V/propidium iodide (PI) staining using an Annexin V-FITC apoptosis detection kit (BD Biosciences) according to the manufacturer’s instructions. Stained cells were analyzed using a FACScan® flow cytometer, and data analysis was performed using the FlowJo software. For in vivo experiments, peritoneal cell suspensions were preincubated with anti-CD16/CD32 mAb (2.4G2) and then stained with monoclonal antibodies against: Gr-1 (RB6-8C5), CD3 (17A2), CD4 (GK1.5), CD8b (eBioH35-17.2), CD19 (ebio1D3), B220 (RA3-6B2), c-kit (2B8), CD11c (N418), F4/80 (BM8), CD45 (30-F11), and Siglec-F (E50-2440). Antibodies were from either BD Biosciences or eBioscience. Stained cells were analyzed using a LSR II flow cytometer (BD Biosciences), and data analysis was performed using the FlowJo software.
Confocal microscopy
BMMCs (0.5 × 106 cells) were incubated with or without 10 or 20 μmol/L of mefloquine. After 3 h of incubation at 37°C, 1 μL of LysoTracker stock solution (1 mmol/L, Life Technologies, Carlsbad, CA) was added followed by 1 h incubation at 37°C in dark. After washing, cells were resuspended in DMEM media and aliquots of 100 μL were dropped into round areas (prepared using liquid-repellent slide marker pen) on microscopic glasses, followed by confocal microscopy analysis as described (Garcia-Faroldi et al. 2013).
Statistical analysis
All calculations were done using the Microsoft Office Excel 2010 software with QI Macros SPC Software (KnowWare International Inc., Denver, CO) for Excel. Statistical differences between groups were assessed using unpaired Student t-test or one-way analysis of variance (ANOVA) followed by Fisher’s least significant difference (LSD) post hoc test (two-sided P-value of less than 0.05 was considered significant). The data are presented as mean ± SEM. All graphs were prepared using GraphPad Prism 4.0 (GraphPad software Inc., San Diego, CA). The results shown are from individual experiments, representative of at least 2 experiments.
Results
Mefloquine is cytotoxic for mast cells
Mefloquine is an approved and widely spread drug used for the treatment of malaria. Earlier investigations have suggested that its mechanism of action may involve permeabilization of the parasite’s lysosomes, that is, the drug has lysosomotropic activity (Ginsburg 1990). Since mast cells are exceptionally rich in lysosome-like secretory granules (“secretory lysosomes”), we reasoned that mefloquine might be cytotoxic for mast cells by inducing permeabilization of their lysosome-like secretory granules. To evaluate this possibility, we first added mefloquine at various concentrations to mouse BMMCs and assessed cell viability after 24 h. As seen in Figure 1A, mefloquine induced cell death of BMMCs with an IC50 value of ∼9 μmol/L (Table 1). At a concentration of 10 μmol/L, time course experiments revealed ∼80% cell death after 24 h (Fig. 1B), and an increasing extent of cell death after prolonged incubation (Fig. 1B). BMMCs are commonly used for studies of mast cell function, but are considerably less granulated than fully mature mast cells found in vivo. Since our hypothesis was that mefloquine-induced cell death might occur via a granule-mediated pathway, we reasoned that fully mature mast cells, that is, with a higher density of granules, could be more sensitive to mefloquine than are BMMCs. To address this possibility, we incubated mefloquine with fully mature PCMCs (Malbec et al. 2007) and monitored cytotoxicity. Indeed, as shown in Figure 1A and in Table 1, PCMCs were markedly more sensitive to mefloquine than were BMMCs, with an IC50 value of ∼3 μmol/L. Mefloquine was also cytotoxic for a HMC-1 and for CBMCs, although with lower efficiency than for BMMCs and PCMCs. However, it should be noted that both HMC-1 and CBMCs display a low extent of granule maturation, which possibly could explain their lower sensitivity to mefloquine. Mefloquine has previously been shown to have cytotoxic activity on MDA-MB-231 breast cancer cells (Sharma et al. 2012) but, as seen in Table 1, the drug was considerably less efficient on MDA-MB-231 cells than on mast cells. Moreover, human fibroblasts (HDFn), HEK-293 cells and U-937 cells were markedly less sensitive to mefloquine than were the fully mature mast cells (Table 1). Together, these data indicate that mast cells are highly sensitivity to mefloquine.
Figure 1.
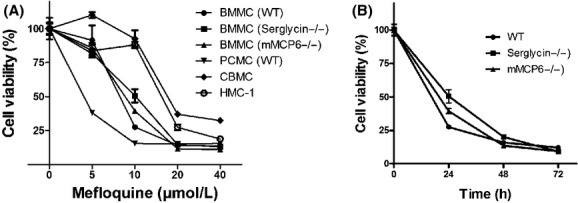
Mefloquine has cytotoxic activity on mast cells. (A) Concentration response of the effect of mefloquine on murine bone marrow-derived mast cells (BMMCs) (WT, serglycin−/− and mMCP-6−/−), peritoneal cell-derived mast cells (PCMCs), human cord blood-derived mast cells (CBMCs) and human mast cell line-1 (HMC-1). Cell viability was measured after 24 h of treatment. (B) Time course of mefloquine-induced cell death. BMMCs (WT, serglycin−/− or mMCP-6−/−) were treated with 10 μmol/L mefloquine and cell viability was determined at the indicated time points. Results are expressed as % cell survival (mean ± SEM, n = 3) in comparison with nontreated control cells.
Table 1.
IC50 values for the cytotoxic effect of mefloquine on different cell types
| Cell type1 | IC50 (μmol/L) |
|---|---|
| BMMC (WT) | 8.8 ± 1.9 |
| BMMC (Serglycin−/−) | 8.4 ± 0.9 |
| BMMC (mMCP6−/−) | 7.4 ± 0.6 |
| PCMC | 3.0 ± 0.4 |
| HMC-1 | 14.2 ± 0.6 |
| CBMC | 16.9 ± 0.3 |
| U-937 | 12.2 ± 0.7 |
| HEK-293 | 19.7 ± 0.2 |
| MDA-MB-231 | 40.1 ± 2.5 |
| HDFn | 18.2 ± 1.26 |
Data are displayed as mean ± SEM.
BMMC, bone marrow-derived mast cells; HMC-1, human mast cell line 1; CBMC, cord blood-derived mast cells; PCMC, peritoneal cell-derived mast cells; U-937, macrophage-like cell line; HEK-293, human embryonic kidney 293 cells; MDA-MB-231, human breast cancer cell line; HDFn, human dermal fibroblast neonatal cells.
Mefloquine induces apoptotic cell death in mast cells but necrosis in mast cells lacking a serglycin-tryptase axis
To address the mechanism of cell death in response to mefloquine, we stained mast cells with Annexin V and PI. As shown in Figure 2, mefloquine treatment resulted in the appearance of cell populations that were either Annexin V+/PI− (apoptotic) or double Annexin V/PI-positive (necrotic). However, the single Annexin V-positive population dominated over the double positive cells, indicating that mefloquine induces predominantly apoptotic cell death in mast cells. Previous studies have suggested that serglycin, a proteoglycan that is abundant in mast cell granules, and tryptase, the latter a serine protease that is stored in complex with serglycin, can have an impact on the mechanism of cell death in mast cells (Melo et al. 2012). To assess the role of these compounds in mefloquine-induced cell death, we incubated WT, serglycin−/− and tryptase (mMCP-6)−/− mast cells with mefloquine followed by Annexin V/PI staining. These experiments showed that WT, serglycin−/− and tryptase−/− mast cells showed approximately equal sensitivity to mefloquine-induced cell death (Table 1, Figs. 1A and B, 2). However, the mechanism of cell death differed profoundly, with WT cells predominantly dying by an apoptotic mechanism whereas cells lacking either serglycin or tryptase died to a larger extent by necrosis (Fig. 2).
Figure 2.
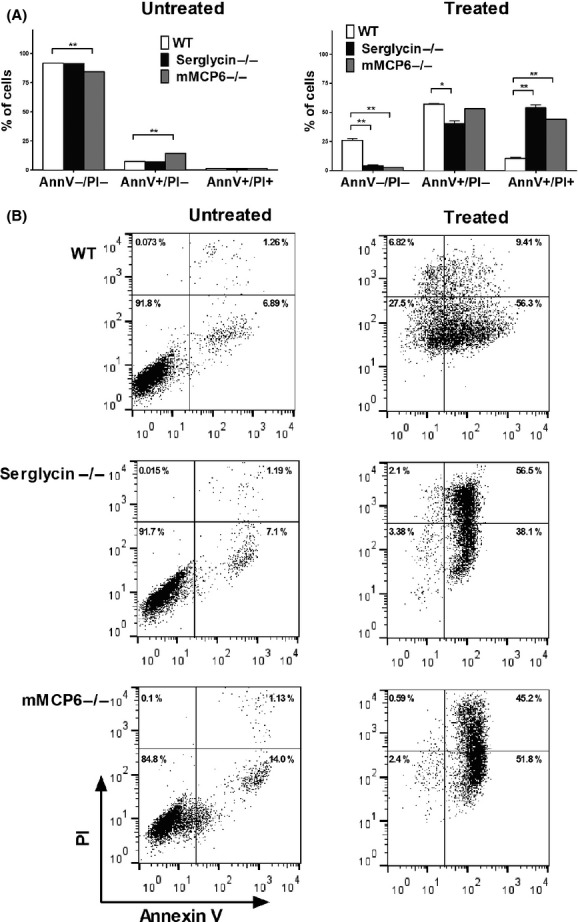
Mefloquine induces apoptotic mast cell death. Samples of 0.5 × 106 bone marrow-derived mast cells (BMMCs) (WT, serglycin−/− and mMCP-6−/−) were cultured in the presence or absence of 20 μmol/L of mefloquine. (A) After 8 h incubation, cells were collected and cell death was assessed by Annexin V/PI staining and flow cytometry analysis. The Annexin V−/PI− population corresponds to viable cells whereas Annexin V+/PI− and Annexin V+/PI+ populations represent apoptotic and necrotic cells, respectively. (B) Representative dot plots showing Annexin V/PI staining of non- and mefloquine-treated WT, serglycin−/− and mMCP-6−/− mast cells. Data represent means of duplicates ± SEM. **P < 0.01; *P < 0.05. AnnV, Annexin V.
Mefloquine-induced mast cell death is caspase independent
To further clarify the cell death mechanism in response to mefloquine, we investigated the effect of caspase inhibition. However, caspase inhibition did not prevent cell death in response to mefloquine (Fig. 3). Moreover, broad-spectrum inhibitors of either cysteine proteases (E64d) or aspartic acid proteases (Pepstatin A) had no significant effect on cell death. Similarly, a broad-spectrum inhibitor of serine proteases (Pefabloc SC) did not prevent cell death in response to mefloquine. However, in agreement with the marked effect of tryptase (a serine protease) in promoting apoptotic vs. necrotic cell death (see Fig. 2), serine protease inhibition caused a marked deviation from apoptotic to necrotic cell death (Fig. 3).
Figure 3.
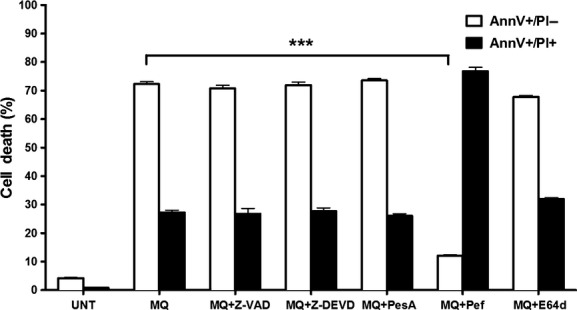
Mefloquine-induced cell death in bone marrow-derived mast cells (BMMCs) is caspase-independent. BMMCs (0.5 × 106 cells) were preincubated with or without a panel of inhibitors 30 min before addition of mefloquine. After 8 h incubation with 20 μmol/L of mefloquine, cell viability assessment was performed. Data are displayed as mean ± SEM (n = 3). ***P < 0.0001; UNT, untreated; MQ, mefloquine; PesA, pepstatin A; Pef, Pefabloc SC; MQ, mefloquine. Inhibitors were used at the following concentrations: E-64d (20 μmol/L), Z-DEVD-FMK (20 μmol/L), Z-VAD-FMK (20 μmol/L), Pepstatin A (50 μmol/L) and Pefabloc SC (0.1 mmol/L).
Mefloquine causes granule permeabilization in mast cells
Previous studies have suggested that mefloquine may have lysosomotropic activity on Plasmodium (Ginsburg 1990), and we therefore reasoned that mefloquine could have an analogous effect on mast cells, that is, to cause permeabilization of their lysosome-like secretory granules. To assess this possibility, we first incubated untreated and mefloquine-treated mast cells with AO. AO is a dye that localizes to acidic compartments (such as secretory granules) and produces strong fluorescence when acidic compartments are intact, but loses fluorescence upon compromised integrity of acidic compartments. As depicted in Figure 4A, incubation of mast cells with mefloquine resulted in rapid loss of AO fluorescence, in agreement with lost integrity of secretory granules. Further, staining of cells with LysoTracker, a dye that preferentially localizes to lysosome-like organelles, produced the expected granular staining in untreated cells (Fig. 4C). However, LysoTracker staining was abrogated upon incubation of mast cells with mefloquine, that is, in agreement with compromised secretory granule integrity (Fig. 4C). In contrast, mefloquine treatment did not induce any detectable reduction in Nonyl-AO fluorescence, indicating that mitochondrial damage in response to mefloquine was minimal (data not shown). To verify that mefloquine caused secretory granule disruption, we prepared cytosolic extracts from untreated and mefloquine-treated cells and assayed them for activity towards Z-Phe-Arg-AMC, a substrate that is commonly used to detect lysosomal cysteine cathepsin activity (Ivanova et al. 2008). As mast cell granules are rich in cysteine cathepsins (Wernersson and Pejler 2014), we thus expected that mefloquine, if acting by granule permeabilization, would cause release of cysteine cathepsin activity into the cytosolic compartment. Indeed, mefloquine caused a rapid and biphasic release of Z-Phe-Arg-AMC-cleaving activity into the cytosol (Fig. 4B). In further agreement with a granule-permeabilizing effect, mefloquine treatment caused the release of granule-specific proteases (tryptase [mMCP-6] and carboxypeptidase A3 [CPA3]) into the cytosol (Fig. 4D). Similar to the release of Z-Phe-Arg-AMC-cleaving activity, the release of tryptase and CPA3 into the cytosolic compartment occurred in a biphasic manner. Importantly, the loss of AO fluorescence and LysoTracker staining, as well as appearance of granular proteases in the cytosol, was seen at considerably earlier time points than profound loss of cell viability (see Fig. 1B). Together, these data indicate that mefloquine causes disruption of mast cell secretory granules, thereby causing the release of granule-localized compounds into the cytosol.
Figure 4.
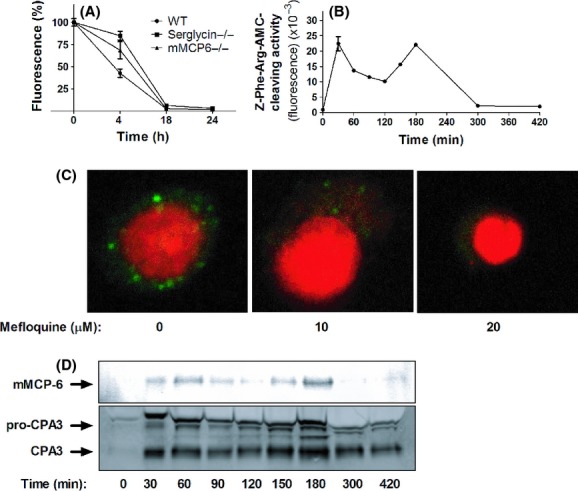
Mefloquine causes secretory granule permeabilization in mast cells. (A) Bone marrow-derived mast cells (BMMCs) (WT, serglycin−/− or mMCP-6−/−; 0.5 × 106 cells) were cultured in the absence or presence of 10 μmol/L mefloquine for the time periods indicated, followed by staining with AO. Results are expressed as % fluorescence in comparison with nontreated control cells (mean ± SEM, n = 3). (B) Z-Phe-Arg-AMC-cleaving activity present in the cytosol after treatment of BMMCs (106 cells) with 10 μmol/L mefloquine. After addition of mefloquine, cytosolic extracts were prepared at the time points indicated and were analyzed for Z-Phe-Arg-AMC-cleaving activity. (C) BMMCs (106 cells) were treated with the indicated concentrations of mefloquine. After 3 h of incubation, cells were stained with LysoTracker (green) to visualize acidic compartments and nuclei were visualized by DAPI staining (Red). (C) BMMCs (106 cells) were treated with 10 μmol/L mefloquine. At the time points indicated, cytosolic extracts were prepared and subjected to western blot analysis for presence of granule proteases (mMCP-6 and CPA3).
Mefloquine-induced mast cell death depends on ROS generation
The results depicted above indicated that mefloquine-induced mast cell death occurs independently of caspases and a range of other proteases. To search for alternative mechanisms in the execution of cell death, we assessed the contribution of ROS. Incubation of mast cells with mefloquine caused a marked increase in staining with a fluorescent ROS probe 30 min after addition of mefloquine, indicating that ROS are generated during mefloquine-induced mast cell death (Fig. 5A and B). When assessing ROS levels at 1 h after mefloquine addition, ROS levels were similar (data not shown). To assess the contribution of ROS in the actual execution of cell death, we treated mast cells with mefloquine in the presence of either of the ROS scavengers α-tocopherol or NAC. As seen in Figure 5B, α-tocopherol was without effect whereas NAC had a profound inhibitory activity on mefloquine-induced cell death. In fact, in the presence of NAC, cell viability in mefloquine-treated cells was indistinguishable from that of nonmefloquine-treated control cells (NAC-treated only). In further agreement with a role of ROS production in mefloquine-induced mast cell death, NAC was shown to inhibit the elevation of ROS levels in response to mefloquine (Fig. 5A). Together, these data indicate that ROS production is a major cause of mast cell death in response to mefloquine.
Figure 5.
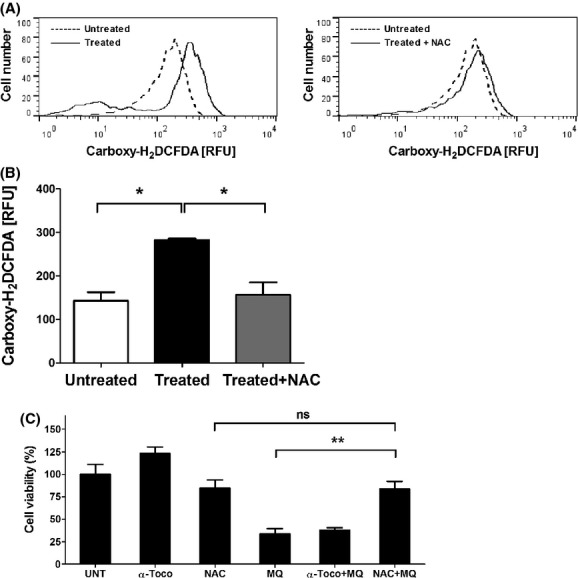
Mefloquine-induced mast cell death is dependent on ROS production. (A) Histogram showing ROS generation (measured with the fluorescent ROS probe Carboxy-H2DCFDA) after 30 min incubation of BMMCs (0.5 × 106 cells) with 20 μmol/L of mefloquine in the presence or absence of N-acetyl cysteine (NAC). Dashed lines represent untreated cells whereas solid lines represent mefloquine-treated cells in the absence (left panel) or presence of NAC (right panel). (B) Quantification of the reactive oxygen species (ROS) production after 30 min incubation of BMMCs (0.5 × 106 cells) with 20 μmol/L of mefloquine in the presence or absence of NAC. Data represent the geometric mean of carboxy-H2DCFDA fluorescence intensity for each sample. (C) The effect of the antioxidants α-tocopherol or NAC on cell viability. BMMCs (0.5 × 106 cells) were preincubated with or without α-tocopherol (0.4 mmol/L) or NAC (8 mmol/L). After 2 h incubation, cells were either left untreated or treated with 20 μmol/L of mefloquine for 8 h. The experiments were performed in triplicate; the bars represent mean ± SEM. **P < 0.01. UNT, untreated; α-Toco, α-tocopherol; MQ, mefloquine.
Mefloquine causes mast cell death in vivo
In order to assess whether mefloquine can induce mast cell death in vivo, we injected mice intraperitoneally with mefloquine at either 10 or 30 mg−1 kg−1 day−1 for 4 days, and then quantified the numbers of peritoneal mast cells and other peritoneal cell populations. In agreement with the in vitro analysis, mefloquine induced a reduction in the peritoneal mast cell population as judged both by flow cytometry analysis (Fig. 6A) and staining of cytospin slides with May-Grünwald/Giemsa (Fig. 6B). Mefloquine additionally caused a moderate but significant reduction in the total lymphocyte population. In contrast, no significant effect on the populations of peritoneal macrophages or neutrophils was seen (Fig. 6A), although there was a trend towards a reduction in the neutrophil populations. To assess if mefloquine has systemic effects on mast cell populations distant from the site of administration, we stained sections from ear skin, tongue and trachea of non- and mefloquine-treated mice for mast cells. However, mefloquine treatment did not reduce the mast cell numbers in any of these tissues (shown for ear skin; Fig. 6C); the latter notion was also confirmed by quantification of the data showing no statistically significant difference in mast cell counts of ear tissue from non- versus mefloquine-treated animals (not shown). Hence, mefloquine has local cytotoxic activity on mast cells in vivo.
Figure 6.
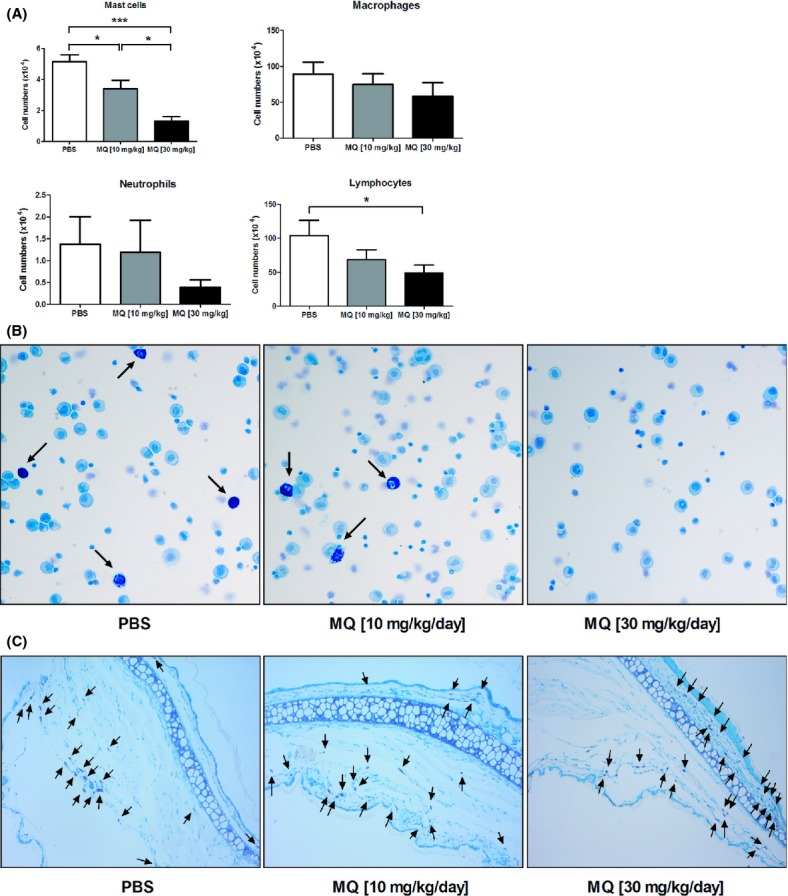
Mefloquine depletes mast cell populations in vivo. Mice (n = 6) were injected i.p. with PBS (control) or mefloquine (10 or 30 mg−1 kg−1 day−1, in PBS) for 4 consecutive days. After 4 days of treatment, mice were sacrificed and peritoneal lavage fluid and ear skin tissue were collected. (A) Quantification of peritoneal cell populations after staining with antibodies against surface markers: CD45+ c-kit+ (mast cells); CD45+ CD11c− F4/80+ (macrophages); CD45+ CD11c− F4/80− SiglecF− Gr-1high (neutrophils); CD45+ CD19+ B220+ CD3+ CD4+ CD8+ (lymphocytes). (B) Representative cytospin slides showing May-Grünwald/Giemsa staining of peritoneal cells obtained from mice either untreated or treated with mefloquine as indicated. Arrows indicate mast cells. (C) Representative ear skin tissue sections stained with toluidine blue. Tissue sections were obtained from mice treated with PBS or mefloquine as indicated. Arrows indicate mast cells. MQ, mefloquine.
Discussion and Conclusions
This study introduces mefloquine as a novel anti-mast cell agent showing efficiency both in vitro and in vivo. Moreover, we show that mefloquine is more cytotoxic towards mast cells than towards a number of other cell types. It should, however, be noted that the extent of selectivity is relatively modest when comparing the sensitivity of BMMCs with the other cell types tested (up to ∼4-fold). On the other hand, when assessing the sensitivity of fully mature mast cells (PCMCs) with those of the other cell types included in the study, the extent of selectivity was considerably higher (up to ∼10-fold). When administering mefloquine in vivo, we noted a significant reduction in the peritoneal mast cell populations, while macrophages were not affected. However, there was also a moderate reduction in the lymphocyte population at high doses of mefloquine and a trend towards a decrease in the peritoneal neutrophil population. Possibly, the latter observations could be in line with previous reports (Thong et al. 1979; Labro and Babin-Chevaye 1988).
Mefloquine is an established, approved drug for the treatment of malaria that has been used widely for many decades. This study thus introduces a novel use of this drug. The potential therapeutic implications for this new activity of mefloquine are manifold. Potentially, mefloquine may be useful as a therapeutic option in various pathologies where mast cells are implicated, including allergic asthma and various inflammatory skin diseases. It is important to emphasize that, while mefloquine had potent cytotoxic activity on mast cells at the site of administration, we did not see any systemic effect of the drug under the conditions employed. This suggests that mefloquine may be primarily useful for topical administration in order to ablate local mast cell populations. This could in fact be an advantage, since a systemic ablation of mast cells at multiple sites of the body could result in unwanted side effects such as impaired host defense towards various infectious agents whose clearance may be mast cell-dependent (Marshall 2004).
Having identified a cytotoxic effect of mefloquine on mast cells, a major focus of the present investigation was to investigate its mechanism of action, one important question being to determine if mefloquine-treated mast cells undergo apoptotic or necrotic cell death. As judged by Annexin V/PI staining, mefloquine-treated mast cells showed clear signs of apoptotic cell death. This was an important finding in the context of the potential clinical use of the drug, since apoptotic cell death occurs without an inflammatory component while necrotic cell death is associated with extensive release of harmful proinflammatory alarmins from dying cells (Edinger and Thompson 2004). In agreement with this notion, we did not see any signs of inflammation when assessing the effect of mefloquine in vivo. Interestingly, while WT cells predominantly underwent apoptotic cell death, mast cells lacking either serglycin or tryptase died to a large extent by necrosis in response to mefloquine. The latter is in agreement with previous findings showing that a serglycin-tryptase axis is an important regulator of apoptotic versus necrotic cell death in response to various other types of cell stress (Melo et al. 2011b, 2012; Spirkoski et al. 2012). Moreover, since both serglycin and tryptase are exclusively located to the mast cell granules, their extensive impact on cell death in response to mefloquine indicates that granules and their contained substances are important players in mefloquine-induced cell death. In line with this, we demonstrate that mefloquine induces mast cell granule permeabilization, as shown by compromised granule integrity and by the release of granule-contained proteases into the cytosol. An important observation was that the loss of granule integrity and release of granular proteases into the cytosol was observed at earlier time points than when profound loss of viability was seen. This argues against that the granule permeabilization is a secondary effect of cell death, and is instead compatible with granule permeabilization being an important step in the mechanism of mefloquine-induced cell death. Taken together, these data thus suggest that mefloquine induces mast cell death via a pathway involving secretory granule permeabilization. Interestingly, treatment of malaria patients with mefloquine has been associated with adverse effects such as urticaria and pruritis (Smith et al. 1999). A plausible explanation for such clinical conditions may be therefore be that mefloquine induces cell death of mast cells leading to release of mast cell mediators. On the other hand, since our data indicate mefloquine predominantly induces apoptotic cell death in mast cells, it is likely that most of the mast cell remnants are phagocytosed by macrophages as a part of the normal apoptotic process, thus minimizing release of mast cell mediators.
The exact mechanism responsible for the execution of cell death in response to mefloquine is intriguing. Since mefloquine-treated mast cells showed clear signs of apoptosis we considered it likely that caspase activation contributed to cell death. However, caspase inhibition did not preserve cellular viability, arguing against a role of caspases in the execution of cell death. An alternative scenario would be that the various proteases that are located in the granules, that is, cysteine cathepsins, aspartic acid proteases or serine proteases, may contribute to cell death, based on the known contribution of these types of proteases to certain types of cell death (Boya and Kroemer 2008; Turk and Turk 2009) and on our observed release of granule-localized proteases into the cytosol of mefloquine-treated mast cells. However, inhibition of these protease types failed to rescue mast cells from mefloquine-induced cell death, suggesting that granule-localized proteases do not contribute substantially to the execution of cell death. Notably though, although not rescuing cells from cell death, serine protease inhibition caused a dramatic shift in the mode of cell death from apoptosis to necrosis. The latter finding is clearly in line with the role of tryptase (a serine protease) in promoting apoptotic versus necrotic cell death, and suggests that tryptase inhibition was the main explanation for the impact of serine protease inhibition on mast cell death.
As an alternative mechanism of cell death in response to mefloquine we considered the possibility that ROS generation is involved, based on the known contribution of ROS generation to apoptosis in various settings (Mates et al. 2012; Sinha et al. 2013). Indeed, our data suggest that ROS generation has a key role in the induction of cell death in response to mefloquine. Intriguingly, the lipophilic ROS scavenger α-tocopherol did not rescue mast cells from mefloquine-induced cell death whereas essentially complete blockade of cell death was seen when administering the hydrophilic ROS scavenger NAC, a finding that is in agreement with a recent report showing that mefloquine-induced cell death of prostate cancer PC3 cells is inhibited by NAC (Yan et al. 2013). Although the implication of this observation is not entirely clear, it is notable that α-tocopherol is typically an effective inhibitor of mitochondrial ROS generation (Majima et al. 2011; Cesen et al. 2013; Quast et al. 2013). Hence, the lack of effect of α-tocopherol on ROS generation in response to mefloquine may suggest that the generation of cytotoxic ROS occurs in a compartment outside of the mitochondria and, consequently, that mitochondrial damage is not the prime cause of mast cell death in response to mefloquine. Interestingly, recent studies have indicated that ROS generation in response to certain types of agents can occur in compartments other than the mitochondria, including lysosomes (Kubota et al. 2010; Denamur et al. 2011). Hence, it is possible that the cytotoxic ROS that are generated in mefloquine-treated mast cells arise from a compartment outside of the mitochondria, for example from the secretory granules.
In summary, this study identifies the anti-malarial drug mefloquine as a novel anti-mast cell agent that induces mast cell death through a pathway involving granule permeabilization and ROS generation.
Acknowledgments
This work was supported by grants from the Swedish Research Council, the Swedish Cancer Foundation, and the Swedish Heart and Lung Foundation and Formas.
Glossary
- AO
acridine orange
- BMMC
bone marrow-derived mast cell
- FBS
fetal bovine serum
- mMCP-6
mouse mast cell protease 6
- PCMC
peritoneal cell-derived mast cell
- ROS
reactive oxygen species
Disclosures
None declared.
References
- Åbrink M, Grujic M, Pejler G. Serglycin is essential for maturation of mast cell secretory granule. J Biol Chem. 2004;279:40897–40905. doi: 10.1074/jbc.M405856200. [DOI] [PubMed] [Google Scholar]
- Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death. Oncogene. 2008;27:6434–6451. doi: 10.1038/onc.2008.310. [DOI] [PubMed] [Google Scholar]
- Cesen MH, Repnik U, Turk V, Turk B. Siramesine triggers cell death through destabilisation of mitochondria, but not lysosomes. Cell Death Dis. 2013;4:e818. doi: 10.1038/cddis.2013.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denamur S, Tyteca D, Marchand-Brynaert J, Van Bambeke F, Tulkens PM, Courtoy PJ, et al. Role of oxidative stress in lysosomal membrane permeabilization and apoptosis induced by gentamicin, an aminoglycoside antibiotic. Free Radic Biol Med. 2011;51:1656–1665. doi: 10.1016/j.freeradbiomed.2011.07.015. [DOI] [PubMed] [Google Scholar]
- Edinger AL, Thompson CB. Death by design: apoptosis, necrosis and autophagy. Curr Opin Cell Biol. 2004;16:663–669. doi: 10.1016/j.ceb.2004.09.011. [DOI] [PubMed] [Google Scholar]
- Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galli SJ, Kalesnikoff J, Grimbaldeston MA, Piliponsky AM, Williams CM, Tsai M. Mast cells as “tunable” effector and immunoregulatory cells: recent advances. Annu Rev Immunol. 2005;23:749–786. doi: 10.1146/annurev.immunol.21.120601.141025. [DOI] [PubMed] [Google Scholar]
- Galli SJ, Grimbaldeston M, Tsai M. Immunomodulatory mast cells: negative, as well as positive, regulators of immunity. Nat Rev. 2008;8:478–486. doi: 10.1038/nri2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Faroldi G, Melo FR, Ronnberg E, Grujic M, Pejler G. Active Caspase-3 Is Stored within Secretory Compartments of Viable Mast Cells. J Immunol. 2013;191:1445–1452. doi: 10.4049/jimmunol.1300216. [DOI] [PubMed] [Google Scholar]
- Ginsburg H. Antimalarial drugs: is the lysosomotropic hypothesis still valid? Parasitol Today. 1990;6:334–337. doi: 10.1016/0169-4758(90)90178-7. [DOI] [PubMed] [Google Scholar]
- Glaumann H, Motakefi AM, Jansson H. Intracellular distribution and effect of the antimalarial drug mefloquine on lysosomes of rat liver. Liver. 1992;12:183–190. doi: 10.1111/j.1600-0676.1992.tb01045.x. [DOI] [PubMed] [Google Scholar]
- Gurish MF, Austen KF. Developmental origin and functional specialization of mast cell subsets. Immunity. 2012;37:25–33. doi: 10.1016/j.immuni.2012.07.003. [DOI] [PubMed] [Google Scholar]
- Ivanova S, Repnik U, Bojic L, Petelin A, Turk V, Turk B. Lysosomes in apoptosis. Methods Enzymol. 2008;442:183–199. doi: 10.1016/S0076-6879(08)01409-2. [DOI] [PubMed] [Google Scholar]
- Kalesnikoff J, Galli SJ. New developments in mast cell biology. Nat Immunol. 2008;9:1215–1223. doi: 10.1038/ni.f.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karra L, Berent-Maoz B, Ben-Zimra M, Levi-Schaffer F. Are we ready to downregulate mast cells? Curr Opin Immunol. 2009;21:708–714. doi: 10.1016/j.coi.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Kubota C, Torii S, Hou N, Saito N, Yoshimoto Y, Imai H, et al. Constitutive reactive oxygen species generation from autophagosome/lysosome in neuronal oxidative toxicity. J Biol Chem. 2010;285:667–674. doi: 10.1074/jbc.M109.053058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labro MT, Babin-Chevaye C. Effects of amodiaquine, chloroquine, and mefloquine on human polymorphonuclear neutrophil function in vitro. Antimicrob Agents Chemother. 1988;32:1124–1130. doi: 10.1128/aac.32.8.1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majima HJ, Indo HP, Suenaga S, Matsui H, Yen HC, Ozawa T. Mitochondria as possible pharmaceutical targets for the effects of vitamin E and its homologues in oxidative stress-related diseases. Curr Pharm Des. 2011;17:2190–2195. doi: 10.2174/138161211796957490. [DOI] [PubMed] [Google Scholar]
- Malbec O, Roget K, Schiffer C, Iannascoli B, Dumas AR, Arock M, et al. Peritoneal cell-derived mast cells: an in vitro model of mature serosal-type mouse mast cells. J Immunol. 2007;178:6465–6475. doi: 10.4049/jimmunol.178.10.6465. [DOI] [PubMed] [Google Scholar]
- Marshall JS. Mast-cell responses to pathogens. Nat Rev. 2004;4:787–799. doi: 10.1038/nri1460. [DOI] [PubMed] [Google Scholar]
- Mates JM, Segura JA, Alonso FJ, Marquez J. Oxidative stress in apoptosis and cancer: an update. Arch Toxicol. 2012;86:1649–1665. doi: 10.1007/s00204-012-0906-3. [DOI] [PubMed] [Google Scholar]
- Melo FR, Lundequist A, Calounova G, Wernersson S, Pejler G. Lysosomal membrane permeabilization induces cell death in human mast cells. Scand J Immunol. 2011a;74:354–362. doi: 10.1111/j.1365-3083.2011.02589.x. [DOI] [PubMed] [Google Scholar]
- Melo FR, Waern I, Rönnberg E, Åbrink M, Lee DM, Schlenner SM, et al. A role for serglycin proteoglycan in mast cell apoptosis induced by a secretory granule-mediated pathway. J Biol Chem. 2011b;286:5423–5433. doi: 10.1074/jbc.M110.176461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo FR, Grujic M, Spirkoski J, Calounova G, Pejler G. Serglycin proteoglycan promotes apoptotic versus necrotic cell death in mast cells. J Biol Chem. 2012;287:18142–18152. doi: 10.1074/jbc.M112.344796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalfe DD, Baram D, Mekori YA. Mast cells. Physiol Rev. 1997;77:1033–1079. doi: 10.1152/physrev.1997.77.4.1033. [DOI] [PubMed] [Google Scholar]
- Quast SA, Berger A, Eberle J. ROS-dependent phosphorylation of Bax by wortmannin sensitizes melanoma cells for TRAIL-induced apoptosis. Cell Death Dis. 2013;4:e839. doi: 10.1038/cddis.2013.344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reber LL, Frossard N. Targeting mast cells in inflammatory diseases. Pharmacol Ther. 2014;142:416–435. doi: 10.1016/j.pharmthera.2014.01.004. [DOI] [PubMed] [Google Scholar]
- Rönnberg E, Pejler G. Serglycin: the master of the mast cell. Methods Mol Biol. 2012;836:201–217. doi: 10.1007/978-1-61779-498-8_14. [DOI] [PubMed] [Google Scholar]
- Sharma N, Thomas S, Golden EB, Hofman FM, Chen TC, Petasis NA, et al. Inhibition of autophagy and induction of breast cancer cell death by mefloquine, an antimalarial agent. Cancer Lett. 2012;326:143–154. doi: 10.1016/j.canlet.2012.07.029. [DOI] [PubMed] [Google Scholar]
- Shin K, Watts GF, Oettgen HC, Friend DS, Pemberton AD, Gurish MF, et al. Mouse mast cell tryptase mMCP-6 is a critical link between adaptive and innate immunity in the chronic phase of Trichinella spiralis infection. J Immunol. 2008;180:4885–4891. doi: 10.4049/jimmunol.180.7.4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha K, Das J, Pal PB, Sil PC. Oxidative stress: the mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch Toxicol. 2013;87:1157–1180. doi: 10.1007/s00204-013-1034-4. [DOI] [PubMed] [Google Scholar]
- Smith HR, Croft AM, Black MM. Dermatological adverse effects with the antimalarial drug mefloquine: a review of 74 published case reports. Clin Exp Dermatol. 1999;24:249–254. doi: 10.1046/j.1365-2230.1999.00471.x. [DOI] [PubMed] [Google Scholar]
- Spirkoski J, Melo FR, Grujic M, Calounova G, Lundequist A, Wernersson S, et al. Mast cell apoptosis induced by siramesine, a sigma-2 receptor agonist. Biochem Pharmacol. 2012;84:1671–1680. doi: 10.1016/j.bcp.2012.09.028. [DOI] [PubMed] [Google Scholar]
- Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. 2008;9:231–241. doi: 10.1038/nrm2312. [DOI] [PubMed] [Google Scholar]
- Thong YH, Ferrante A, Rowan-Kelly B, O’Keefe DE. Effect of mefloquine on the immune response in mice. Trans R Soc Trop Med Hyg. 1979;73:388–390. doi: 10.1016/0035-9203(79)90160-3. [DOI] [PubMed] [Google Scholar]
- Turk B, Turk V. Lysosomes as “suicide bags” in cell death: myth or reality? J Biol Chem. 2009;284:21783–21787. doi: 10.1074/jbc.R109.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernersson S, Pejler G. Mast cell granules: armed for battle. Nat Rev Immunol. 2014;14:478–494. doi: 10.1038/nri3690. [DOI] [PubMed] [Google Scholar]
- Yan KH, Yao CJ, Hsiao CH, Lin KH, Lin YW, Wen YC, et al. Mefloquine exerts anticancer activity in prostate cancer cells via ROS-mediated modulation of Akt, ERK, JNK and AMPK signaling. Oncol Lett. 2013;5:1541–1545. doi: 10.3892/ol.2013.1211. [DOI] [PMC free article] [PubMed] [Google Scholar]