

Abstract
Despite recent advances in therapy, chronic lymphocytic leukaemia (CLL) remains incurable and new treatment strategies are therefore urgently required. Inhibitor of apoptosis proteins (IAPs) are over-expressed in CLL, suggesting both a role in disease pathogenesis and the potential for therapeutic targeting. To explore these questions, we evaluated the effects on primary CLL cells of AZD5582, a novel potent and selective inhibitor of IAPs. AZD5582 at nanomolar concentrations induced extensive degradation of cIAP-1 and cIAP-2, but minimally of X chromosome-linked IAP (XIAP). However, these effects of AZD5582 produced little or no direct cytotoxicity, nor did they sensitize CLL cells to p53-dependent killing by fludarabine or p53-independent killing by dexamethasone. In contrast, AZD5582 significantly enhanced apoptosis induced by the death receptor (DR) agonist tumour necrosis factor-related apoptosis-inducing ligand (TRAIL). Importantly, killing by TRAIL plus AZD5582 was independent of adverse prognostic features including TP53 deletion which is strongly associated with chemoresistance in CLL. Coculture experiments involving transfected mouse fibroblasts expressing human CD40L (CD154) to mimic the effect of T cells at sites of tissue involvement showed that CD40 stimulation almost completely prevented the killing of CLL cells by TRAIL plus AZD5582 despite up-regulating TRAIL receptors 1 and 2. In conclusion, our findings confirm the rate-limiting, upstream involvement of IAPs in the extrinsic but not intrinsic apoptotic pathway of CLL cells and suggest that drug combinations that simultaneously activate DRs and inhibit IAPs may have therapeutic potential in patients with CLL who have failed T-cell-depleting chemotherapy.
Keywords: Apoptosis, CD40-stimulation, CLL, IAPs, TRAIL
Introduction
Chronic lymphocytic leukaemia (CLL) is the most common form of adult leukaemia in Western countries. Despite recent therapeutic advances involving purine analogues and monoclonal antibodies (Hallek et al. 2010), the disease remains incurable, with most patients requiring repeated treatment episodes and developing progressive drug resistance (Zenz et al. 2010). Developing new treatments for patients who have exhausted conventional ones is therefore a major priority.
Increased expression of inhibitor of apoptosis proteins (IAPs) has been implicated in the pathogenesis of many human cancers including leukaemia/lymphoma (Tamm et al. 2000; Kitada et al. 2002), and targeting IAPs has thus become an attractive strategy for the development of novel therapy (Schimmer and Dalili 2005; Fulda 2007). IAPs are endogenous inhibitors of caspases, a family of cysteine proteases that act as death effector molecules to bring about the biochemical and morphological changes characteristic of apoptosis (Salvesen and Duckett 2002). The mammalian IAP family consists of eight members including cIAP-1, cIAP-2 and X chromosome-linked IAP (XIAP). All IAPs contain at least one BIR (baculovirus inhibitor of apoptosis repeat) domain, which is made up of approximately 70 amino acids that are essential for protein–protein interaction (Srinivasula and Ashwell 2008; Mace et al. 2010). In addition, some IAPs also contain CARD (caspase activation and recruitment domain) and RING (really interesting new gene) domains. The RING domain can function as an E3 ligase and mediate the addition of ubiquitin to target proteins (Vaux and Silke 2005). For example, XIAP contains three BIR domains and a C-terminal RING domain (Eckelman et al. 2006). The BIR2 and BIR3 domains of XIAP mediate inhibition of caspase-3/-7 and caspase-9, respectively, whereas its RING domain is responsible for the ubiquitination and degradation of substrates such as caspases and SMAC (second mitochondrial activator of caspases) (Eckelman et al. 2006; Mace et al. 2010). More recently, accumulating evidence indicates that IAPs are also critically involved in regulating innate and adaptive immunity through modulation of signal transduction pathways involving NF-κB signalling, cytokine production and lymphocyte survival (Gyrd-Hansen and Meier 2010; Beug et al. 2012).
The role of IAPs in CLL is incompletely understood. Early studies have reported increased levels of cIAP-1, cIAP-2 and XIAP transcripts (Munzert et al. 2002; de Graaf et al. 2005), and up-regulation of the cIAP-1 transcript was associated with resistance to apoptosis following ex-vivo exposure of CLL cells to ionizing irradiation (Vallat et al. 2003). Increased expression of IAPs in CLL has also been reported at the protein level (Kitada et al. 2000; Schliep et al. 2004; Silva et al. 2006). A recent study has shown that resistance to apoptosis resulting from constitutive activation of Notch signalling in CLL cells is accompanied by increased NF-κB activity and expression of cIAP-2 and XIAP (Rosati et al. 2009). However, how Notch signalling might regulate NF-κB activity and IAP expression in CLL cells remain unclear.
Several previous studies have investigated the therapeutic potential of targeting IAPs in CLL. Two main approaches have been used to lower IAP activity: antisense oligonucleotides or small interfering RNA (siRNA) and small molecule inhibitors (Schimmer 2004; Fulda 2007). The latter are also known as SMAC mimetics as they mimic the dual action of SMAC in relieving IAP-mediated suppression of caspases and inducing auto-ubiquitination of IAPs by selectively binding to the corresponding BIR domains (Li et al. 2004; Schimmer et al. 2004). Polyphenylurea-based small molecule inhibitors selectively targeting the BIR2 domain of XIAP have been shown to reduce the viability of un-stimulated CLL cells (Schimmer et al. 2004) and sensitize CD40-activated CLL cells to Fas-induced apoptosis (Kater et al. 2005). More recent studies have shown that siRNA targeting of XIAP or small molecule inhibitors targeting the BIR3 domain of XIAP can sensitize primary CLL cells to TRAIL-induced apoptosis, albeit not in all cases (Loeder et al. 2009; Frenzel et al. 2011).
Although these studies provide some insight into the therapeutic potential of IAP inhibition in CLL, most of them did not take into account the modulating effect of the leukaemic micro-environment. Consequently, the true in-vivo potential of IAP inhibition as a potential therapeutic strategy for CLL remains unclear. Following on from these considerations, we sought to further explore the therapeutic potential of IAP inhibition in CLL, taking into account the microenvironment. We found that AZD5582, a highly potent and selective IAP inhibitor (Hennessy et al. 2013), sensitized CLL cells to killing by TRAIL but not fludarabine or dexamethasone under standard conditions. However, the cytotoxicity of AZD5582 plus TRAIL was almost completely abrogated when CLL cells were cocultured with transfected fibroblasts expressing CD40L to mimic CD40 stimulation by normal T cells at sites of tissue involvement.
Materials and Methods
Chemicals, antibodies and other reagents
The novel IAP inhibitor AZD5582 was provided by AstraZeneca (London, UK). Fludarabine and dexamethasone were obtained from Sigma-Aldrich (Gillingham, UK). Soluble recombinant human TRAIL was purchased from Enzo Life Sciences (Exeter, UK). Mouse monoclonal antibodies against XIAP (clone 48/hILP/XIAP) and cIAP-2 (clone F30-2285,) were obtained from BD Biosciences (Oxford, UK). Goat antibody to cIAP-1 was from R&D Systems (Minneapolis, MN). Rabbit antibodies against cFLIP, Bcl-2 and Bcl-xL were from Cell Signalling Technology (New England Biolabs, Herts, UK). Rabbit antibody to Mcl-1 was from Santa Cruz Biotechnology (Insight Biotechnology, Middlesex, UK). Mouse monoclonal antibody to β-actin (clone AC-74) was from Sigma-Aldrich. Other chemicals, unless otherwise stated, were also obtained from Sigma-Aldrich.
CLL samples
Peripheral blood samples from CLL patients were obtained with informed consent. The clinical details of the patients used for this study are given in Table 1. CLL cells were isolated by centrifugation of blood over Lymphoprep (Axis-Shield PoC AS, Oslo, Norway) and stored in liquid nitrogen prior to use. After thawing, the cells were cultured in RPMI 1640 medium supplemented with 10% heat-inactivated foetal bovine serum, 2 mmol/L L-glutamine, 100 U/mL penicillin and 100 μg/mL streptomycin (Life Sciences, Paisley, UK).
Table 1.
Clinical characteristics of the CLL samples used
| Sample no. | Gender | Age | Stage (Binet) | WBC (109/L) | IGHV status | Chromosomal abnormalities | Prior therapy* |
|---|---|---|---|---|---|---|---|
| 2096 | M | 65 | A | 60 | M | 17p- & 13q- | N |
| 2103 | M | 56 | A | 125 | UM | 17p- & 13q- | N |
| 2156 | F | 66 | C | 183 | UM | +12 | N |
| 2263 | M | 65 | B | 366 | UM | 11q- & 13q- | Y |
| 2441 | M | 44 | A | 153 | UM | Normal | N |
| 2474 | M | 55 | B | 108 | UM | 11q- & 13q- | Y |
| 2808 | F | 74 | nd | 76 | nd | nd | nd |
| 2866 | M | 72 | A | 229 | UM | normal | N |
| 2911 | M | 72 | A | 223 | UM | normal | N |
| 2968 | M | 80 | A | 87 | M | Normal | N |
| 3035 | M | 61 | C | 282 | UM | Normal | N |
| 3155 | M | 70 | nd | 236 | nd | 17p- | Y |
| 3159 | M | 44 | B | 153 | UM | Normal | Y |
| 3174 | M | 77 | C | 246 | UM | 13q- | Y |
IGHV status refers to somatic mutation in IGHV gene of CLL cells as compared with the gene sequence of germ-line using 2% as a cut-off. nd, not done/unknown.
Prior therapy consisted of the therapeutic administration of various combinations of steroid, chlorambucil, fludarabine or fludarabine plus cyclophosphamide.
Coculture of CLL cells with CD154-transfected cells
Stably transfected mouse fibroblasts expressing human CD154 (CD40L) and empty-plasmid transfected control fibroblasts (both kindly provided by Prof. Gerald Cohen at University of Leicester, UK) were maintained as described (Vogler et al. 2009). For CD40 stimulation, CLL cells were seeded on an adherent monolayer of fibroblasts expressing CD154 or control fibroblasts at a ratio of 10:1 and cultured at 37°C for an appropriate period of time. To determine the effect of CD40 ligation on killing induced by fludarabine, dexamethasone or the IAP inhibitor, CLL cells were cocultured for 48h with the CD154-expressing or control fibroblasts in the presence or absence of the above drugs before harvesting. To determine the effect of IAP inhibition on CD40-stimulated CLL cells to TRAIL-induced apoptosis, CLL cells were cocultured with the CD154-expressing or control fibroblasts for 48h in the presence or absence of AZD5582. The CLL cells were then gently removed from the respective adherent monolayers, resuspended in fresh culture medium and co-incubated under standard conditions with different concentrations of recombinant human TRAIL.
Flow cytometric analysis of cell death
CLL cells were incubated in multiwell plates at a density of 4 × 106 cells/mL in the presence or absence of indicated reagents. At the end of the treatment period, drug-induced killing was measured using a flow cytometry method employing dual staining with FITC labelled annexin-V and propidium iodide as described (Zhuang et al. 2010). Cell death was defined as phosphatidylserine exposure on the cell surface detected by annexin-V staining. Aliquots of cells from the same experiment were analysed by Western blotting for protein expression.
Western blotting analysis
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and immunoblotting were performed essentially as described (Zhuang et al. 2010). Briefly, cellular proteins were separated on an SDS-polyacrylamide gel and transferred to Immobilon-P polyvinilidene difluoride membranes (Millipore Corporation, Bedford, MA), which were probed with the appropriate primary antibodies. Immunoreactivity was detected with the relevant HRP-labelled secondary antibodies (Santa Cruz Biotechnology), which in turn were visualized on an Image Reader LAS-1000 (Fujifilm, Tokyo, Japan) using an enhanced chemiluminescence kit (GE Healthcare Life Sciences, Buckinghamshire, UK). For quantification of the signals, the images were further analysed on the same instrument using 2D Densitometry Aida Image Analyzer software (Fujifilm).
Flow cytometric analysis of TRAIL receptors in CLL cells
Detection of cell surface expression of TRAIL receptors by flow cytometry was performed essentially as described (Harper and MacFarlane 2008). Briefly, at the end of the incubation period, 1 × 106 cells were collected by centrifugation and resuspended in 1 mL of ice-cold fresh medium with all subsequent steps performed with the cells kept on ice or at 4°C. 250 μL of cell suspension was aliquoted into each of four individual flow cytometry tubes and centrifuged again. The cells were then resuspended in 100 μL of phosphate-buffered saline (PBS) containing 0.3% bovine serum albumin (BSA). 5 μL of phycoerythrin (PE)-conjugated anti-TRAIL-R1, anti-TRAIL-R2 or isotype-matched control antibody (all obtained from eBiosciences, San Diego, CA) was added to the appropriate tubes. Cells in the remaining fourth tube with no added antibody were used as a negative control. Cells were incubated on ice for 1h in the dark and then washed three times with PBS before analysis by flow cytometry using excitation and emission wavelengths of 488 and 575 nm, respectively.
Results
AZD5582 is a potent inhibitor of IAPs in CLL cells
The characterization of the small molecule SMAC mimetic, AZD5582, has recently been described and biochemical studies showed that the molecule bound potently to the BIR3 domains of cIAP1, cIAP2 and XIAP (Hennessy et al. 2013). Our initial dose-response and time-course experiments indicated that incubation of CLL cells or breast cancer cells with AZD5582 at a concentration of 0.1 nmol/L resulted in a significant loss of cIAP-2 as early as 4h after treatment (Fig. S1). Exposure of 14 individual CLL samples to AZD5582 at 1 nmol/L for 48 h produced almost complete degradation of cIAP-1 and cIAP-2 in all samples studied, but with more variable loss of XIAP (Fig. 1A). Pooled analysis of densitometric signals corresponding to individual IAPs showed an average reduction in levels of XIAP, cIAP-1 and cIAP-2 of approximately 50%, 90% and 80%, respectively (Fig. 1B). These observations therefore confirmed that AZD5582 is highly potent at targeting IAPs for degradation in CLL cells and identified 1 nmol/L as a drug concentration that achieves near-maximal inhibition.
Figure 1.

AZD5582 is a potent inhibitor of IAP proteins in CLL cells, but is not directly cytotoxic. (A) Primary CLL cells were incubated with 1 nmol/L AZD5582 for 48 h and then examined for levels of IAP proteins by Western blotting. β-actin was used as a loading control for densitometric analysis. The numbers under the lanes refer to the ratio of the respective IAP protein relative to β-actin, normalised to that of untreated cells in each sample. (B) Pooled quantitative analysis of the effect of AZD5582 on levels of XIAP, cIAP-1 and cIAP-2 in CLL cells as described in (A). In this and subsequent statistical analyses, a two-tailed t-test was performed to determine the statistical significance of the difference between the two groups of data (mean ± SD). (C) CLL cells were incubated with AZD5582 at the indicated concentrations for 24 and 48 h. Cell death was measured by flow cytometry following dual staining with FITC-conjugated annexin V and PI. The percentage of drug-induced cell death was calculated as: 100 × [(% cell death of drug-treated cells − % cell death of untreated cells)/(100 − % cell death of untreated cells)]. Each data point in the graph represents the mean ± SD of independent experiments using primary CLL cells from at least three patients.
AZD5582 is not cytotoxic to CLL cells cultured under standard conditions
Having established the biological activity of AZD5582 in CLL cells, we next performed dose-response and time-course experiments to determine whether the IAP inhibitor could kill CLL cells cultured under standard conditions. As shown in Figure 1C, incubation of CLL cells with 1 nmol/L AZD5582 for 24 and 48 h did not significantly reduce their viability. Even when applied at a 1000-fold higher concentration, AZD5582 induced only a modest amount of killing at both 24 and 48 h time points (Fig. 1C). These results indicate that AZD5582 is not cytotoxic to CLL cells at concentrations that induce significant IAP degradation.
AZD5582 does not sensitize CLL cells to killing by fludarabine or dexamethasone
We next investigated the possibility that AZD5582, although not cytotoxic on its own, may sensitize CLL cells to killing by clinically relevant drugs that activate the intrinsic death pathway by p53-dependent (fludarabine) and p53-independent (dexamethasone) mechanisms (Melarangi et al. 2012). First, CLL cells were co-incubated for 48 h with either AZD5583 (1 nmol/L) and/or fludarabine at different concentrations. As shown in Figure 2A, AZD5582 did not sensitize CLL cells to killing by fludarabine in any of the cases examined despite inducing degradation of IAPs in these cells (Fig. 2A, inset). To further test the possibility that higher concentrations of AZD5582 might be needed for the sensitization effect to occur, the experiment was repeated using AZD5582 at 10 and 100 nmol/L. However, no sensitization was observed (Fig. 2B) despite extensive degradation of IAPs (Fig. 2C). Although one of the CLL samples (#2968) showed some sensitivity to AZD5582 treatment at 100 nmol/L, this did not translate into clear sensitization to fludarabine (Fig. 2B). We also investigated the ability of AZD5582 to sensitize CLL cells to p53-independent killing by dexamethasone. As shown in Figure 2D, no such sensitization was observed. Taken together, these results indicate that AZD5582 at the concentration that induced significant degradation of IAPs does not sensitize CLL cells to drugs that activate the intrinsic death pathway.
Figure 2.
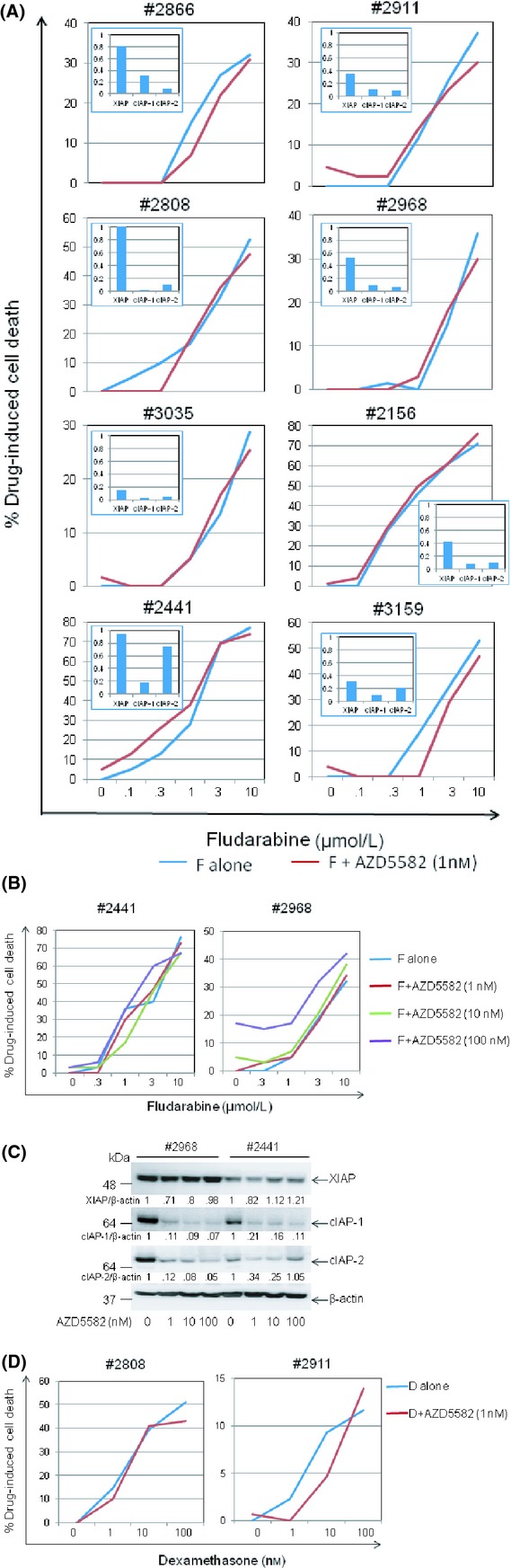
AZD5582 does not sensitise CLL cells to killing by fludarabine or dexamethasone. (A) Available CLL samples from among the cases shown in Figure 1A were incubated for 48 h with 1 nmol/L AZD5582 and/or fludarabine (F) at the indicated concentrations. Cell death was measured as in Figure 1C. (B) CLL cells were incubated for 48 h with 1, 10 or 100 nmol/L AZD5582 and/or fludarabine (F) at the indicated concentrations and cell death measured as above. (C) CLL cells were incubated for 48 h with AZD5582 at the indicated concentrations before analysing for levels of IAP proteins by Western blotting as in Figure 1A. The numbers under the lanes refer to the ratio of the respective IAP protein relative to β-actin, normalised to that of untreated cells in each sample. (D) CLL cells were incubated for 48 h with either 1 nmol/L AZD5582 and/or dexamethasone (D) at the indicated concentrations and cell death measured as above.
AZD5582 sensitizes CLL cells to killing by TRAIL
Previous studies have shown that certain IAP inhibitors can sensitize CLL cells to apoptosis induced by agents that activate the extrinsic death pathway via cell-surface death receptors (DR) such as CD95 and tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) (Kater et al. 2005; Loeder et al. 2009). We therefore sought to establish whether AZD5582 exhibited a similar effect. To address this question, CLL cells were incubated with AZD5582 and/or human recombinant TRAIL which engages DR4 (TRAIL-R1) and DR5 (TRAIL-R2). As shown in Figure 3A, treatment with TRAIL alone produced minor increases in cytotoxicity over baseline apoptosis detected in untreated cells, particularly at the higher concentration of 500 ng/mL. However, the addition of AZD5582 to TRAIL produced significantly more cytotoxicity compared to that induced by TRAIL alone. Additional experiments using CLL samples with known cytogenetic defects including 17p or 11q deletion showed that AZD5582 was still able to increase TRAIL-induced apoptosis in these cells regardless of the presence of these adverse features (Fig. 3B). Further analysis showed that there was no correlation between the AZD5582-induced reduction in levels of individual IAPs and the amount of killing observed (Fig. S2). This suggests that IAPs function collectively rather than individually to oppose activation of the extrinsic death pathway.
Figure 3.
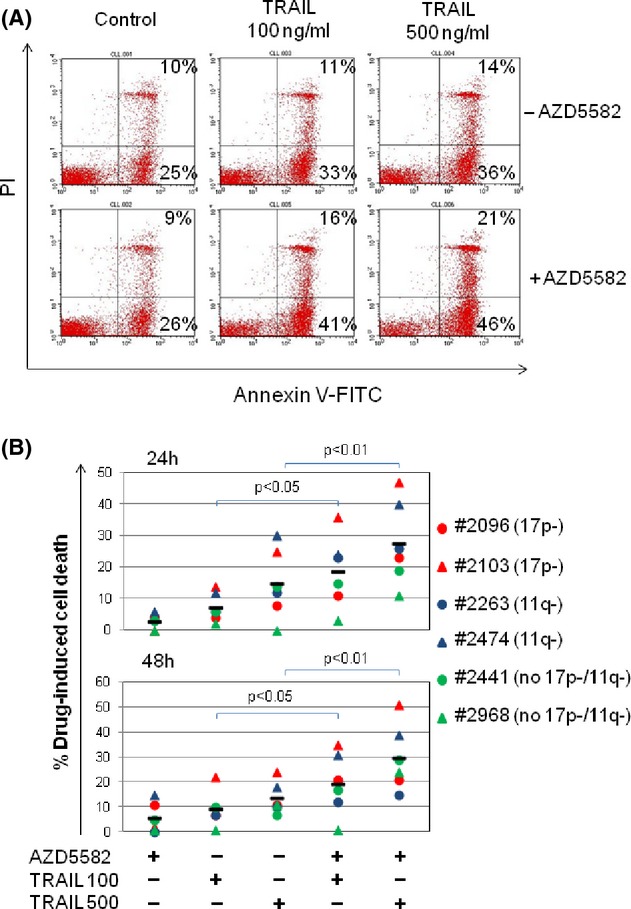
AZD5582 sensitises CLL cells to killing by TRAIL. (A) CLL cells were incubated with 1 nmol/L AZD5582 and/or TRAIL at the indicated concentrations and cell death was measured as in Figure 1C. A representative series of flow cytometry dot-plots is shown. Live cells are annexin/PI dim, early apoptotic cells are annexin bright/PI dim and late apoptotic/necrotic cells are annexin/PI bright. (B) CLL samples with or without 17p or 11q deletions were incubated for 24 and 48 h with 1 nmol/L AZD5582 and/or TRAIL at the indicated concentrations (100 or 500 ng/mL) and cell death measured as described above.
CD40 stimulation protects CLL cells from spontaneous apoptosis and killing by fludarabine and dexamethasone independently of IAPs
It is widely accepted that the survival of CLL cells in vivo is strongly influenced by interactions with the leukaemic microenvironment and that one of the most important interactions involves stimulation of CD40 on CLL cells by CD154 on T cells, which initiates a potent signalling cascade that activates NF-κB (Furman et al. 2000; Granziero et al. 2001). Since IAPs are established transcriptional targets of NF-κB (Chu et al. 1997; Stehlik et al. 1998; Wang et al. 1998), we sought to establish to what extent these proteins contribute to the cytoprotective effect of CD40 stimulation and whether AZD5582 can block the protective effect of CD40 stimulation. To this end, CLL cells were cocultured with transfected mouse fibroblasts expressing human CD154, an established in-vitro model that mimics CD40 stimulation by T cells in the tissue microenvironment. Empty-plasmid transfected fibroblasts were used as a control. In agreement with previous reports (Kitada et al. 1999; Kater et al. 2004), CD40 stimulation protected CLL cells from spontaneous apoptosis and killing induced by either fludarabine or dexamethasone (Fig. S3A). As expected (Kater et al. 2004; Vogler et al. 2009), this protective effect was associated with increased levels of anti-apoptotic Bcl-2 family proteins such as Bcl-xL and Mcl-1 (Fig. S3B) and IAP proteins (Fig. 4A, compare lane 7 to lane 1). Importantly, AZD5582 retained its ability to degrade IAP proteins in CD40-stimulated CLL cells (Fig. 4A, lanes 8, 11 and 12). However, the inhibitor had little or no effect on the viability of these cells, nor did it sensitize them to killing by fludarabine or dexamethasone (Fig. 4B). This suggests that the protective effect of CD40 stimulation is not mediated by IAPs.
Figure 4.
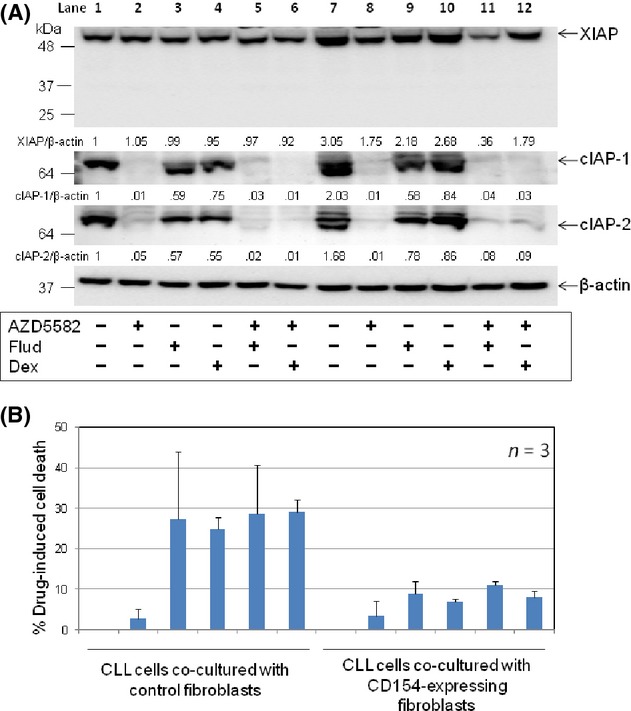
CD40 stimulation protects CLL cells from killing by fludarabine and dexamethasone independently of IAPs. (A) CLL cells were cultured for 48 h on a monolayer of parental or CD154-expressing fibroblasts in the presence of AZD5582 (100 nmol/L) and/or fludarabine (Flud) (10 μmol/L) or dexamethasone (Dex) (100 nmol/L). CLL cells were then analysed for levels of IAPs by Western blotting and densitometry as in Figure 1A. A representative set of blots from three CLL samples examined is shown. The numbers under the lanes refer to the ratio of the respective IAP proteins relative to β-actin, which are normalized to that of untreated cells cocultured with control fibroblasts in each sample. (B) CLL cells were cocultured with control or CD154-transfected fibroblasts for 48 h in the presence of AZD5582 (100 nmol/L) and/or fludarabine (Flud) (10 μmol/L) or dexamethasone (Dex) (100 nmol/L) and analysed for apoptosis as described in Figure 1C.
CD40 stimulation protects CLL cells from killing by TRAIL plus AZD5582
Stimulation of CLL cells with membrane-bound CD154 has been shown to increase the expression of DRs including CD95 (Chu et al. 2002; Kater et al. 2005) and TRAIL-R2 (DR5) (Dicker et al. 2005) and also to enhance apoptosis induced by membrane-bound TRAIL (Dicker et al. 2005). We therefore postulated that CD40 stimulation might sensitize CLL cells to killing by TRAIL plus AZD5582. We first sought to confirm that CD40 stimulation up-regulates surface expression of TRAIL receptors 1 and 2 (DR4 and DR5, respectively). As shown in Figure 5A, unstimulated CLL cells expressed low but detectable levels of DR4 and DR5 (upper panel). Expression of both DRs was increased when CLL cells were cocultured with control fibroblasts and further increased by CD40 stimulation (Fig. 5A, lower panels, left). Treatment with AZD5582 did not significantly alter the expression of DR4 and DR5 in CLL cells cocultured with the control or CD154-expressing fibroblasts (Fig. 5A, lower panels, right). Further analysis showed that the up-regulation of DR4 and DR5 following CD40 stimulation was statistically significant (Fig. 5B). Together, these observations indicate that CD40 stimulation increases the surface expression of TRAIL receptors 1 and 2 via IAP-independent mechanisms.
Figure 5.
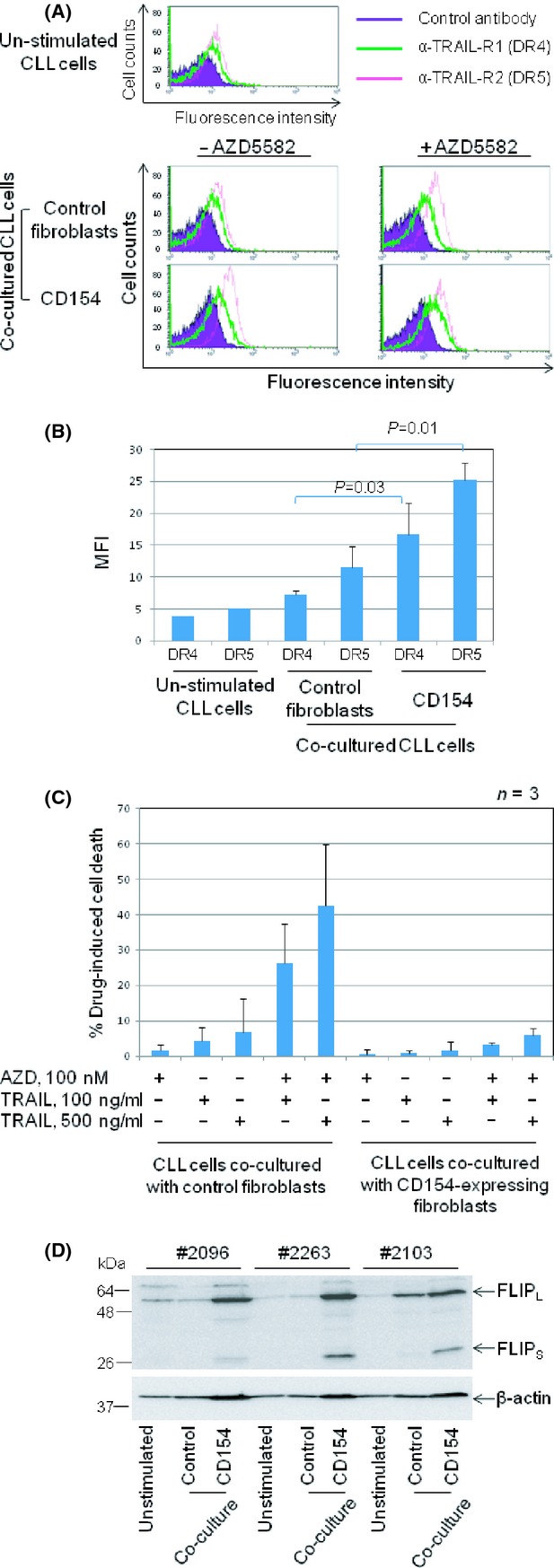
CD40 stimulation protects CLL cells from killing by TRAIL plus AZD5582. (A) CLL samples (n = 4) were cultured for 48 h on control or CD154-expressing fibroblasts in the presence or absence of AZD5582 (100 nmol/L) and then examined for expression of TRAIL receptor 1 (DR4) and 2 (DR5) by flow cytometry. Unstimulated CLL samples (n = 2) were also examined for basal levels of DR4 and DR5 (upper panel). Representative histograms are shown. (B) Quantification and statistical analysis of the data illustrated in A showing the effect of CD40 stimulation on TRAIL receptors 1 (DR4) and 2 (DR5). (C) CLL cells were cultured for 48 h on the respective fibroblasts monolayers in the presence or absence of AZD5582 (100 nmol/L). They were then removed from the cocultures, respectively, and incubated under standard conditions with or without TRAIL at the indicated concentrations for a further 24 h before being analysed for viability. (D) Unstimulated and cocultured CLL cells on control or CD154-expressing fibroblasts for 48 h as described in (C) were examined for level of cFLIP expression by Western blotting. β-actin was used as a loading control.
We next investigated the effect of CD40 stimulation on the killing of CLL cells by TRAIL plus AZD5582. As shown in Figure 5C, TRAIL or AZD5582 alone produced little or no killing of CLL cells cocultured with control fibroblasts. However, the combination of TRAIL plus AZD5582 produced extensive killing, indicating a sensitization effect similar to that observed in CLL cells cultured under standard conditions (Fig. 3). In contrast, no such killing was observed in CLL cells cocultured with CD154-expressing fibroblasts (Fig. 5C). This observation indicates that CD40 stimulation strongly inhibits the killing of CLL cells via the extrinsic death pathway despite the increased surface expression of TRAIL receptors.
We next considered the possible mechanism responsible for this blockade of the extrinsic death pathway induced by CD40 stimulation. We focused on FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein (cFLIP) since it is a known transcriptional target of NF-κB that binds to components of the extrinsic death pathway to prevent formation of the death-inducing signalling complex (DISC) (Yu and Shi 2008). We speculated that the resistance of CD40-stimulated CLL cells to killing by TRAIL plus AZD5582 might be mediated by up-regulation of cFLIP. To investigate this possibility, levels of cFLIP protein were examined by Western blotting in CLL cells cultured under standard conditions and in contact with control or CD154-expressing fibroblasts. In keeping with our hypothesis, both long and short isoforms of cFLIP were greatly increased following CD40 stimulation (Fig. 5D).
Discussion and Conclusions
This study was performed to improve our understanding of how IAPs regulate CLL-cell survival and at the same time explore the therapeutic potential of a novel SMAC mimetic inhibitor (AZD5582). In the first part of the study involving primary CLL cells cultured under standard conditions, we showed that AZD5582 was highly potent in inducing the degradation of IAPs. However, it did not induce apoptosis or sensitize CLL cells to killing by clinically relevant drugs that activate the intrinsic death pathway. We used fludarabine and dexamethasone as activators of the intrinsic death pathway as both drugs are in current clinical use in CLL and act via distinct upstream death signalling mechanisms. Fludarabine is a purine nucleoside analogue that has been used in the treatment of CLL since early 1990s; its cytotoxic effects are known to involve DNA damage and p53-dependent apoptosis (Pettitt et al. 1999; Rosenwald et al. 2004). In contrast, dexamethasone in common with other glucocorticoids induces p53-independent apoptosis by binding to the glucocorticoid receptor and regulating gene expression (Pettitt et al. 1999; Melarangi et al. 2012). Both glucocorticoids and DNA-damaging agents induce apoptosis via the mitochondrial (intrinsic) death pathway since their cytotoxic effects require the presence of the BH3-only proteins BIM and/or PUMA (Erlacher et al. 2005; Happo et al. 2010) which are essential initiators of this pathway (Huang and Strasser 2000; Willis and Adams 2005; Giam et al. 2008).
In contrast with its effect on killing induced by fludarabine or dexamethasone, AZD5582 did sensitize CLL cells to apoptosis induced by TRAIL, which activates the extrinsic death pathway. Importantly, this sensitizing effect was observed in CLL samples displaying chromosomal defects associated with chemotherapy resistance. These findings suggest that IAPs play an important modulatory role in the extrinsic but not intrinsic death pathway of CLL cells and indicate the therapeutic potential of combining an IAP inhibitor such as AZD5582 with a DR agonist.
In the second part of the study, primary CLL cells were cocultured with fibroblasts expressing CD154 to mimic CD40 stimulation by nonmalignant T cells at sites of tissue involvement. Compared to CLL cells cocultured with control fibroblasts, CD40-stimulated CLL cells expressed increased levels of IAPs, underwent less spontaneous apoptosis and were more resistant to killing induced by fludarabine and dexamethasone. AZD5582 induced extensive degradation of IAPs under these conditions and completely abrogated the up-regulation of IAPs induced by CD40 stimulation. However, the inhibitor had little or no effect on spontaneous apoptosis or drug-induced killing. These observations confirm that CD40 stimulation protects CLL cells from apoptosis induced via the intrinsic death pathway and indicate that this protective effect is not mediated by IAPs or overcome by IAP inhibition. Finally, we showed that CD40 stimulation also protected CLL cells from killing by TRAIL plus AZD5582 despite up-regulating TRAIL receptors 1 and 2. This observation indicates that the IAP-independent protective effect of CD40 stimulation extends to the extrinsic death pathway and over-rides the sensitizing effect of IAP inhibition on this pathway. The prominent up-regulation of cFLIP observed in CD40-stimulated CLL cells provides a possible explanation for this functional blockade of the extrinsic death pathway, although other mechanisms cannot be excluded. In particular, a recent report suggested that the apoptosis of CLL cells induced by SMAC mimetics could be inhibited by CD40 stimulation via a mechanism involving aberrant upstream NF-κB signalling and failure of ripoptosome formation upon IAP degradation (Maas et al. 2013).
Our pharmacodynamic studies confirmed that AZD5582 is a rapidly acting and highly potent inhibitor of IAPs which produces near-maximal degradation of IAPs in intact primary CLL cells at a concentration of only 1 nmol/L. Although SMAC mimetics were originally designed to target XIAP (Gyrd-Hansen and Meier 2010), we found that AZD5582 preferentially degrades cIAP-1 and cIAP-2. Thus, within the overall cohort of samples studied, XIAP, cIAP-1 and cIAP-2 levels were reduced by approximately 50%, 90% and 80%, respectively, following 48 h incubation with 1 nmol/L AZD5582. However, it is noteworthy that the inhibitory effects of AZD5582 varied considerably between individual cases (Fig. 2A). For example, in case #2808, 1 nM AZD5582 induced near complete degradation of cIAP-1 and cIAP-2 but had virtually no effect on XIAP. In contrast, the same concentration of inhibitor induced the selective loss of cIAP-1 in case #2441 and extensive degradation of all 3 IAPs in case #3035. The resistance of XIAP to AZD5582-induced degradation in some cases cannot be explained by dose–response considerations since increasing the concentration of AZD5582 from 1 nmol/L to 100 nmol/L did not result in any additional degradation of XIAP (Fig. 2C).
The functional effect of AZD5582 in sensitizing CLL cells to TRAIL-induced killing did not seem to depend on which IAPs were preferentially degraded (Supplementary Fig. 2), suggesting that IAPs function collectively rather than individually to oppose activation of the extrinsic death pathway. However, the overall level of IAP degradation did not always correspond with the amount of killing observed. For example, case #2968 was resistant to killing by TRAIL (100 ng/mL) plus AZD5582 (1 nmol/L) (Fig. 3B) despite the significant degradation of all 3 IAPs induced by AZD5582 in this case (Fig. 2A). This observation suggests that sensitivity to TRAIL-induced killing is, at least in some cases, modulated by factors other than IAP levels. Although it was beyond the scope of the present study to identify these factors, from a practical perspective, these considerations present major challengers for the development of the pharmacodynamic biomarkers required to optimize AZD5582 drug dosing and scheduling as part of combination regimens.
Since IAPs are endogenous inhibitors of caspases, the failure of IAP inhibition to sensitize CLL cells to killing by fludarabine and dexamethasone supports a view of caspases as downstream effectors in the mitochondrial (intrinsic) death pathway and suggests that the availability of caspases to perform this role is not rate limiting. In contrast, our observation that IAP inhibition sensitizes CLL cells to killing by TRAIL suggests that caspases play a major upstream role in the DR-mediated (extrinsic) apoptosis pathway and that under these circumstances their availability is rate limiting.
Our findings concerning the effect of CD40 stimulation were of particular interest. First, our demonstration that coculture of CLL cells with CD154-expressing fibroblasts increased levels of Bcl-xL, IAPs and FLIP is in agreement with previous studies (Kitada et al. 1999; Furman et al. 2000; Granziero et al. 2001; Kater et al. 2004; Vogler et al. 2009) and strongly implicates NF-κB activation as a major signalling event induced by CD40 stimulation (Figs. 4A, 5D and S3B). Second, although the prosurvival effects of NF-κB have been attributed to the transactivation of numerous prosurvival genes including IAPs (Chu et al. 1997; Stehlik et al. 1998; Wang et al. 1998), our demonstration that AZD5582 failed to reverse the protective effects of CD40 stimulation appears to indicate that IAPs do not contribute in a significant way to the prosurvival effects of NF-κB activation in CLL cells.
However, the situation may be more complex in that recent studies have shown a role for cIAP-1 and cIAP-2 in regulating NF-κB function (Ghosh and Hayden 2008; Baud and Karin 2009; Gyrd-Hansen and Meier 2010). Thus, in addition to being involved in activating the canonical NF-κB signalling pathway, cIAP-1 and cIAP-2 also inhibit the noncanonical pathway of NF-κB signalling by targeting NF-κB inducing kinase (NIK) for degradation (Varfolomeev et al. 2007; Vince et al. 2007). This latter effect is mediated by the function of cIAP1 and cIAP2 as E3 ubiquitin ligases for NIK. Since ubiquitylation of NIK results in its proteasomal degradation, loss of IAP proteins leads to the stabilization of NIK, activation of the noncanonical NF-κB signalling pathway and protection from apoptosis. The importance of this paradoxical, alternative function of IAPs in B lymphoproliferative disorders is highlighted by the fact that the E3 ubiquitin ligase RING domain of the gene encoding cIAP-2 (BIRC3) is disrupted by somatic mutations which occur in about 10% of patients with CLL and splenic marginal zone lymphoma and by the t(11;18) translocation that is characteristic of extranodal marginal zone lymphoma (Rossi et al. 2011, 2012). Following on from these considerations, it is possible that any pro-apoptotic or sensitizing effects of IAP inhibition in CD40-stimulated CLL cells could have been masked by a dominant prosurvival effect resulting from paradoxical activation of the noncanonical NF-κB signalling pathway.
Our demonstration that AZD5582 sensitizes CLL cells to TRAIL-induced killing under standard conditions is in agreement with previous reports (Loeder et al. 2009; Frenzel et al. 2011) and indicates the therapeutic potential of combining SMAC mimetics with a DR agonist. However, the potent inhibition of AZD5582/TRAIL-induced killing by CD40 stimulation suggests that this drug combination is unlikely to be effective at sites of tissue involvement where significant numbers of T cells are present. It is therefore important to consider the implications of our findings in the context of what is known about T cells in CLL and how they are affected by anti-CLL therapy. In patients with previously untreated CLL, T cells are abundant in the bone marrow and lymph nodes (Ghia et al. 2002; Patten et al. 2008), suggesting that IAP inhibitor/DR agonist combination therapy is unlikely to be effective at these anatomical sites. However, several drugs currently employed in the treatment of CLL, most notably fludarabine and alemtuzumab, induce profound T-cell depletion as an unintended effect. In fact, the discrepancy between the established clinical effectiveness of fludarabine in CLL and its failure to kill CD40-stimulated CLL cells in vitro suggests that the drug’s T-cell depleting properties may contribute to its therapeutic efficacy in vivo (Consoli et al. 1998; Beyer et al. 2005). The possibility therefore arises that IAP inhibitor/DR antagonist combinations should display clinical efficacy in those patients whose tissues have been inadvertently depleted of T cells by prior therapy with fludarabine and/or alemtuzumab.
Our study did not specifically address the question of whether IAP inhibition by AZD5582 could induce the apoptosis of CLL cells that were proliferating. However, the likely answer to this question can be inferred from three considerations. First, CLL cells can be induced to proliferate by coculture with CD40L-expressing fibroblasts (Hamilton et al. 2012). Second, CD40 stimulation strongly promotes the survival of cultured CLL cells via activation NF-κB (Furman et al. 2000) and up-regulation of NF-κB target genes including Bcl-xL and c-FLIP (Kitada et al. 1999; Kater et al. 2004; Vogler et al. 2009). Third, in our study AZD5582 had little or no effect on the survival of CLL cells stimulated via CD40.
It is interesting to note that the role of IAPs in cell survival and proliferation - and consequently the effect of IAP inhibition - appears to vary with cell type. Thus, whereas inhibition of IAPs by AZD5582 reduced the viability of the human breast cancer cell line MDA-MB-231 and significantly reduced tumour growth in an MDA-MB-231 xenograft model, 166 of 200 cancer cell lines representing a variety of tumour types were insensitive to the antiproliferative effects of AZD5582 (Hennessy et al. 2013). This observation illustrates the difficulty of making generalizations regarding the therapeutic potential of IAP inhibitors in cancer and instead justifies careful preclinical evaluation in specific malignancies as a prerequisite for clinical studies.
In conclusion, our study has explored the therapeutic potential of IAP inhibition in CLL and, in doing so, the role of IAPs in maintaining the survival of CLL cells under different conditions. Our findings support a rate-limiting, upstream role for IAPs (and by implication caspases) in the extrinsic death pathway and a non-rate-limiting, downstream role in the intrinsic death pathway. Furthermore, IAPs do not appear to mediate the cytoprotective effect of CD40 stimulation which blocks apoptosis induced via both the intrinsic and extrinsic death pathways. From a clinical perspective, our data suggest that IAP inhibitor/DR agonist combinations may have therapeutic potential in CLL but only in situations of T-cell depletion such as that induced by prior treatment with fludarabine or alemtuzumab.
Acknowledgments
This project was funded in part by AstraZeneca.
Glossary
- BIR
baculovirus inhibitor of apoptosis repeat
- BSA
bovine serum albumin
- CARD
caspase activation and recruitment domain
- CLL
chronic lymphocytic leukaemia
- DISC
death-inducing signalling complex
- DR
death receptor
- FLIP
FLICE (FADD-like IL-1β-converting enzyme)-inhibitory protein
- IAPs
inhibitor of apoptosis proteins
- NIK
NF-κB inducing kinase
- PBS
phosphate-buffered saline
- PE
phycoerythrin
- RING
really interesting new gene
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- siRNA
small interfering RNA
- SMAC
second mitochondrial activator of caspases
- TRAIL
tumour necrosis factor-related apoptosis-inducing ligand
- XIAP
X chromosome-linked IAP
Disclosures
NL is an employee of AstraZeneca. ARP and JZ have received research funding from AstraZeneca. KL, GJ and MO declare no conflict of interest.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Figure S1. AZD5582 is potent in inducing degradation of IAP proteins. Primary CLL cells (#3144) were incubated for 4 h with AZD5582 at the indicated concentrations and then examined for expression of cIAP-2 by Western blotting. β-actin was used as control for protein loading. The human breast cancer cell line MDA-MB-231 was used as a positive control as it has been shown that IAPs were extensively degraded by SMAC mimetic IAP inhibitor AZD5582 (Hennessy et al., 2013).
Figure S2. Lack of correlation between levels of individual IAPs after AZD5582 treatment and sensitivity to TRAIL-induced apoptosis. Primary CLL cells were co-incubated for 48 h with 1 nmol/L AZD5582 and recombinant human TRAIL (500 ng/mL) and cell death measured by flow cytometry. Pearson’s correlation analysis was performed to determine the statistical significance of the correlation between the AZD5582-induced reduction in levels of XIAP, cIAP-1 and cIAP-2 and sensitivity to TRAIL-induced killing among the six cases examined.
Figure S3. CD40 stimulation protects CLL cells from spontaneous apoptosis and killing induced by fludarabine and dexamethasone. (A) CLL cells were cultured for 48 h under standard conditions or on parental or CD154-expressing fibroblasts in the absence or presence of fludarabine (10 μmol/L) or dexamethasone (Dex) (100 nmol/L). CLL cells were then harvested for analysis of viability by flow cytmetry. (B) CLL cells cultured for 48 h under the same conditions as in (A) were also examined for the expression of Bcl-xL, Mcl-1 and Bcl-2 proteins by Western blotting.
References
- Baud V, Karin M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat Rev Drug Discov. 2009;8:33–40. doi: 10.1038/nrd2781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beug ST, Cheung HH, Lacasse EC, Korneluk RG. Modulation of immune signalling by inhibitors of apoptosis. Trends Immunol. 2012;33:535–545. doi: 10.1016/j.it.2012.06.004. [DOI] [PubMed] [Google Scholar]
- Beyer M, Kochanek M, Darabi K, Popov A, Jensen M, Endi E, et al. Reduced frequencies and suppressive function of CD4+CD25hi regulatory T cells in patients with chronic lymphocytic leukemia after therapy with fludarabine. Blood. 2005;106:2018–2025. doi: 10.1182/blood-2005-02-0642. [DOI] [PubMed] [Google Scholar]
- Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW. Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-IAP2 is under NF-kappaB control. Proc Natl Acad Sci USA. 1997;94:10057–10062. doi: 10.1073/pnas.94.19.10057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu P, Deforce D, Pedersen IM, Kim Y, Kitada S, Reed JC, et al. Latent sensitivity to Fas-mediated apoptosis after CD40 ligation may explain activity of CD154 gene therapy in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99:3854–3859. doi: 10.1073/pnas.022604399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consoli U, El-Tounsi I, Sandoval A, Snell V, Kleine HD, Brown W, et al. Differential induction of apoptosis by fludarabine monophosphate in leukemic B and normal T cells in chronic lymphocytic leukemia. Blood. 1998;91:1742–1748. [PubMed] [Google Scholar]
- Dicker F, Kater AP, Fukuda T, Kipps TJ. Fas-ligand (CD178) and TRAIL synergistically induce apoptosis of CD40-activated chronic lymphocytic leukemia B cells. Blood. 2005;105:3193–3198. doi: 10.1182/blood-2003-10-3684. [DOI] [PubMed] [Google Scholar]
- Eckelman BP, Salvesen GS, Scott FL. Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep. 2006;7:988–994. doi: 10.1038/sj.embor.7400795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlacher M, Michalak EM, Kelly PN, Labi V, Niederegger H, Coultas L, et al. BH3-only proteins Puma and Bim are rate-limiting for gamma-radiation- and glucocorticoid-induced apoptosis of lymphoid cells in vivo. Blood. 2005;106:4131–4138. doi: 10.1182/blood-2005-04-1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenzel LP, Patz M, Pallasch CP, Brinker R, Claasen J, Schulz A, et al. Novel X-linked inhibitor of apoptosis inhibiting compound as sensitizer for TRAIL-mediated apoptosis in chronic lymphocytic leukaemia with poor prognosis. Br J Haematol. 2011;152:191–200. doi: 10.1111/j.1365-2141.2010.08426.x. [DOI] [PubMed] [Google Scholar]
- Fulda S. Inhibitor of apoptosis proteins as targets for anticancer therapy. Expert Rev Anticancer Ther. 2007;7:1255–1264. doi: 10.1586/14737140.7.9.1255. [DOI] [PubMed] [Google Scholar]
- Furman RR, Asgary Z, Mascarenhas JO, Liou HC, Schattner EJ. Modulation of NF-kappa B activity and apoptosis in chronic lymphocytic leukemia B cells. J Immunol. 2000;164:2200–2206. doi: 10.4049/jimmunol.164.4.2200. [DOI] [PubMed] [Google Scholar]
- Ghia P, Strola G, Granziero L, Geuna M, Guida G, Sallusto F, et al. Chronic lymphocytic leukemia B cells are endowed with the capacity to attract CD4+, CD40L+ T cells by producing CCL22. Eur J Immunol. 2002;32:1403–1413. doi: 10.1002/1521-4141(200205)32:5<1403::AID-IMMU1403>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Ghosh S, Hayden MS. New regulators of NF-kappaB in inflammation. Nature Rev Immunol. 2008;8:837–848. doi: 10.1038/nri2423. [DOI] [PubMed] [Google Scholar]
- Giam M, Huang DC, Bouillet P. BH3-only proteins and their roles in programmed cell death. Oncogene. 2008;27(Suppl. 1):S128–S136. doi: 10.1038/onc.2009.50. [DOI] [PubMed] [Google Scholar]
- de Graaf AO, van Krieken JH, Tonnissen E, Wissink W, van de Locht L, Overes I, et al. Expression of CIAP-1, CIAP-2 and survivin discriminates different types of lymphoid malignancies. Br J Haematol. 2005;100:852–859. doi: 10.1111/j.1365-2141.2005.05690.x. [DOI] [PubMed] [Google Scholar]
- Granziero L, Ghia P, Circosta P, Gottardi D, Strola G, Geuna M, et al. Survivin is expressed on CD40 stimulation and interfaces proliferation and apoptosis in B-cell chronic lymphocytic leukemia. Blood. 2001;97:2777–2783. doi: 10.1182/blood.v97.9.2777. [DOI] [PubMed] [Google Scholar]
- Gyrd-Hansen M, Meier P. IAPs: from caspase inhibitors to modulators of NF-kappaB, inflammation and cancer. Nat Rev Cancer. 2010;10:561–574. doi: 10.1038/nrc2889. [DOI] [PubMed] [Google Scholar]
- Hallek M, Fischer K, Fingerle-Rowson G, Fink AM, Busch R, Mayer J, et al. Addition of rituximab to fludarabine and cyclophosphamide in patients with chronic lymphocytic leukaemia: a randomised, open-label, phase 3 trial. Lancet. 2010;376:1164–1174. doi: 10.1016/S0140-6736(10)61381-5. [DOI] [PubMed] [Google Scholar]
- Hamilton E, Pearce L, Morgan L, Robinson S, Wane V, Brennan P, et al. Mimicking the tumour microenvironment: three different co-culture systems induce a similar phenotype but distinct proliferative signals in primary chronic lymphocytic leukaemia cells. Br J Haematol. 2012;158:589–599. doi: 10.1111/j.1365-2141.2012.09191.x. [DOI] [PubMed] [Google Scholar]
- Happo L, Cragg MS, Phipson B, Haga JM, Jansen ES, Herold MJ, et al. Maximal killing of lymphoma cells by DNA damage-inducing therapy requires not only the p53 targets Puma and Noxa, but also Bim. Blood. 2010;116:5256–5267. doi: 10.1182/blood-2010-04-280818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper N, MacFarlane M. Recombinant TRAIL and TRAIL receptor analysis. Methods Enzymol. 2008;446:293–313. doi: 10.1016/S0076-6879(08)01618-2. [DOI] [PubMed] [Google Scholar]
- Hennessy EJ, Adam A, Aquila BM, Castriotta LM, Cook D, Hattersley M, et al. Discovery of a novel class of dimeric Smac mimetics as potent IAP antagonists resulting in a clinical candidate for the treatment of cancer (AZD5582) J Med Chem. 2013;56:9897–9919. doi: 10.1021/jm401075x. [DOI] [PubMed] [Google Scholar]
- Huang DC, Strasser A. BH3-Only proteins-essential initiators of apoptotic cell death. Cell. 2000;103:839–842. doi: 10.1016/s0092-8674(00)00187-2. [DOI] [PubMed] [Google Scholar]
- Kater AP, Evers LM, Remmerswaal EB, Jaspers A, Oosterwijk MF, van Lier RA, et al. CD40 stimulation of B-cell chronic lymphocytic leukaemia cells enhances the anti-apoptotic profile, but also Bid expression and cells remain susceptible to autologous cytotoxic T-lymphocyte attack. Br J Haematol. 2004;127:404–415. doi: 10.1111/j.1365-2141.2004.05225.x. [DOI] [PubMed] [Google Scholar]
- Kater AP, Dicker F, Mangiola M, Welsh K, Houghten R, Ostresh J, et al. Inhibitors of XIAP sensitize CD40-activated chronic lymphocytic leukemia cells to CD95-mediated apoptosis. Blood. 2005;106:1742–1748. doi: 10.1182/blood-2005-02-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitada S, Zapata JM, Andreeff M, Reed JC. Bryostatin and CD40-ligand enhance apoptosis resistance and induce expression of cell survival genes in B-cell chronic lymphocytic leukaemia. Br J Haematol. 1999;106:995–1004. doi: 10.1046/j.1365-2141.1999.01642.x. [DOI] [PubMed] [Google Scholar]
- Kitada S, Zapata JM, Andreeff M, Reed JC. Protein kinase inhibitors flavopiridol and 7-hydroxy-staurosporine down-regulate antiapoptosis proteins in B-cell chronic lymphocytic leukemia. Blood. 2000;96:393–397. [PubMed] [Google Scholar]
- Kitada S, Pedersen IM, Schimmer AD, Reed JC. Dysregulation of apoptosis genes in hematopoietic malignancies. Oncogene. 2002;21:3459–3474. doi: 10.1038/sj.onc.1205327. [DOI] [PubMed] [Google Scholar]
- Li L, Thomas RM, Suzuki H, De Brabander JK, Wang X, Harran PG. A small molecule Smac mimic potentiates TRAIL- and TNFalpha-mediated cell death. Science. 2004;305:1471–1474. doi: 10.1126/science.1098231. [DOI] [PubMed] [Google Scholar]
- Loeder S, Zenz T, Schnaiter A, Mertens D, Winkler D, Döhner H, et al. A novel paradigm to trigger apoptosis in chronic lymphocytic leukemia. Cancer Res. 2009;69:8977–8986. doi: 10.1158/0008-5472.CAN-09-2604. [DOI] [PubMed] [Google Scholar]
- Maas C, Tromp JM, van Laar J, Thijssen R, Elias JA, Malara A, et al. CLL cells are resistant to smac mimetics because of an inability to form a ripoptosome complex. Cell Death Dis. 2013;4:e782. doi: 10.1038/cddis.2013.305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mace PD, Shirley S, Day CL. Assembling the building blocks: structure and function of inhibitor of apoptosis proteins. Cell Death Differ. 2010;17:46–53. doi: 10.1038/cdd.2009.45. [DOI] [PubMed] [Google Scholar]
- Melarangi T, Zhuang J, Lin K, Rockliffe N, Bosanquet AG, Oates M, et al. Glucocorticoid resistance in chronic lymphocytic leukaemia is associated with a failure of upregulated Bim/Bcl-2 complexes to activate Bax and Bak. Cell Death Dis. 2012;3:e372. doi: 10.1038/cddis.2012.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munzert G, Kirchner D, Stobbe H, Bergmann L, Schmid RM, Döhner H, et al. Tumour necrosis factor receptor-associated factor 1 gene overexpression in B-cell chronic lymphocytic leukemia: analysis of NF-κB/Rel–regulated inhibitors of apoptosis. Blood. 2002;100:3749–3756. doi: 10.1182/blood.V100.10.3749. [DOI] [PubMed] [Google Scholar]
- Patten PE, Buggins AG, Richards J, Wotherspoon A, Salisbury J, Mufti GJ, et al. CD38 expression in chronic lymphocytic leukemia is regulated by the tumor microenvironment. Blood. 2008;111:5173–5181. doi: 10.1182/blood-2007-08-108605. [DOI] [PubMed] [Google Scholar]
- Pettitt AR, Sherrington PD, Cawley JC. The effect of p53 dysfunction on purine analogue cytotoxicity in chronic lymphocytic leukaemia. Br J Haematol. 1999;106:1049–1051. doi: 10.1046/j.1365-2141.1999.01649.x. [DOI] [PubMed] [Google Scholar]
- Rosati E, Sabatini R, Rampino G, Tabilio A, Di Ianni M, Fettucciari K, et al. Constitutively activated Notch signaling is involved in survival and apoptosis resistance of B-CLL cells. Blood. 2009;113:856–865. doi: 10.1182/blood-2008-02-139725. [DOI] [PubMed] [Google Scholar]
- Rosenwald A, Chuang EY, Davis RE, Wiestner A, Alizadeh AA, Arthur DC, et al. Fludarabine treatment of patients with chronic lymphocytic leukemia induces a p53-dependent gene expression response. Blood. 2004;104:1428–1434. doi: 10.1182/blood-2003-09-3236. [DOI] [PubMed] [Google Scholar]
- Rossi D, Deaglio S, Dominguez-Sola D, Rasi S, Vaisitti T, Agostinelli C, et al. Alteration of BIRC3 and multiple other NF-κB pathway genes in splenic marginal zone lymphoma. Blood. 2011;118:4930–4934. doi: 10.1182/blood-2011-06-359166. [DOI] [PubMed] [Google Scholar]
- Rossi D, Fangazio M, Rasi S, Vaisitti T, Monti S, Cresta S, et al. Disruption of BIRC3 associates with fludarabine chemorefractoriness in TP53 wild-type chronic lymphocytic leukemia. Blood. 2012;119:2854–2862. doi: 10.1182/blood-2011-12-395673. [DOI] [PubMed] [Google Scholar]
- Salvesen GS, Duckett CS. IAP proteins: blocking the road to death’s door. Nat Rev Mol Cell Biol. 2002;3:401–410. doi: 10.1038/nrm830. [DOI] [PubMed] [Google Scholar]
- Schimmer AD. Inhibitor of apoptosis proteins: translating basic knowledge into clinical practice. Cancer Res. 2004;64:7183–7190. doi: 10.1158/0008-5472.CAN-04-1918. [DOI] [PubMed] [Google Scholar]
- Schimmer AD, Dalili S. Targeting the IAP family of caspase inhibitors as an emerging therapeutic strategy. Hematology Am Soc Hematol Educ Program. 2005;2005:215–219. doi: 10.1182/asheducation-2005.1.215. [DOI] [PubMed] [Google Scholar]
- Schimmer AD, Welsh K, Pinilla C, Wang Z, Krajewska M, Bonneau MJ, et al. Small-molecule antagonists of apoptosis suppressor XIAP exhibit broad antitumor activity. Cancer Cell. 2004;5:25–35. doi: 10.1016/s1535-6108(03)00332-5. [DOI] [PubMed] [Google Scholar]
- Schliep S, Deckerb T, Schnellerb F, Wagnera H, Hacker G. Functional evaluation of the role of inhibitor of apoptosis proteins in chronic lymphocytic leukemia. Exp Hematol. 2004;32:556–562. doi: 10.1016/j.exphem.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Silva KL, Vasconcellos DV, de Paula Castro ED, Coelho AM, Linden R, Maia RC. Apoptotic effect of fludarabine is independent of expression of IAPs in B-cell chronic lymphocytic leukemia. Apoptosis. 2006;11:277–285. doi: 10.1007/s10495-006-3560-5. [DOI] [PubMed] [Google Scholar]
- Srinivasula SM, Ashwell JD. IAPs: what’s in a name? Mol Cell. 2008;30:123–135. doi: 10.1016/j.molcel.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stehlik C, de Martin R, Kumabashiri I, Schmid JA, Binder BR, Lipp J. Nuclear factor (NF)-kappaB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J Exp Med. 1998;188:211–216. doi: 10.1084/jem.188.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamm I, Kornblau SM, Segall H, Krajewski S, Welsh K, Kitada S, et al. Expression and prognostic significance of IAP-family genes in human cancers and myeloid leukemias. Clin Cancer Res. 2000;6:1796–1803. [PubMed] [Google Scholar]
- Vallat L, Magdelenat H, Merle-Beral H, Masdehors P, Potocki de Montalk G, Davi F, et al. The resistance of B-CLL cells to DNA damage-induced apoptosis defined by DNA microarrays. Blood. 2003;101:4598–4606. doi: 10.1182/blood-2002-06-1743. [DOI] [PubMed] [Google Scholar]
- Varfolomeev E, Blankenship JW, Wayson SM, Fedorova AV, Kayagaki N, Garg P, et al. IAP antagonists induce autoubiquitination of c-IAPs, NF-kappaB activation, and TNFalpha-dependent apoptosis. Cell. 2007;131:669–681. doi: 10.1016/j.cell.2007.10.030. [DOI] [PubMed] [Google Scholar]
- Vaux DL, Silke J. IAPs–the ubiquitin connection. Cell Death Differ. 2005;12:1205–1207. doi: 10.1038/sj.cdd.4401696. [DOI] [PubMed] [Google Scholar]
- Vince JE, Wong WW, Khan N, Feltham R, Chau D, Ahmed AU, et al. IAP antagonists target cIAP1 to induce TNFalpha-dependent apoptosis. Cell. 2007;131:682–693. doi: 10.1016/j.cell.2007.10.037. [DOI] [PubMed] [Google Scholar]
- Vogler M, Butterworth M, Majid A, Walewska RJ, Sun XM, Dyer MJ, et al. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood. 2009;113:4403–4413. doi: 10.1182/blood-2008-08-173310. [DOI] [PubMed] [Google Scholar]
- Wang CY, Mayo MW, Korneluk RG, Goeddel DV, Baldwin AS., Jr NF-kappaB antiapoptosis: induction of TRAF1 and TRAF2 and c-IAP1 and c-IAP2 to suppress caspase-8 activation. Science. 1998;281:1680–1683. doi: 10.1126/science.281.5383.1680. [DOI] [PubMed] [Google Scholar]
- Willis SN, Adams JM. Life in the balance: how BH3-only proteins induce apoptosis. Curr Opin Cell Biol. 2005;17:617–625. doi: 10.1016/j.ceb.2005.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu JW, Shi Y. FLIP and the death effector domain family. Oncogene. 2008;27:6216–6227. doi: 10.1038/onc.2008.299. [DOI] [PubMed] [Google Scholar]
- Zenz T, Mertens D, Küppers R, Döhner H, Stilgenbauer S. From pathogenesis to treatment of chronic lymphocytic leukaemia. Nat Rev Cancer. 2010;10:37–50. doi: 10.1038/nrc2764. [DOI] [PubMed] [Google Scholar]
- Zhuang J, Hawkins SF, Glenn MA, Lin K, Johnson GG, Carter A, et al. Akt is activated in chronic lymphocytic leukemia cells and delivers a pro-survival signal: the therapeutic potential of Akt inhibition. Haematologica. 2010;95:110–118. doi: 10.3324/haematol.2009.010272. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. AZD5582 is potent in inducing degradation of IAP proteins. Primary CLL cells (#3144) were incubated for 4 h with AZD5582 at the indicated concentrations and then examined for expression of cIAP-2 by Western blotting. β-actin was used as control for protein loading. The human breast cancer cell line MDA-MB-231 was used as a positive control as it has been shown that IAPs were extensively degraded by SMAC mimetic IAP inhibitor AZD5582 (Hennessy et al., 2013).
Figure S2. Lack of correlation between levels of individual IAPs after AZD5582 treatment and sensitivity to TRAIL-induced apoptosis. Primary CLL cells were co-incubated for 48 h with 1 nmol/L AZD5582 and recombinant human TRAIL (500 ng/mL) and cell death measured by flow cytometry. Pearson’s correlation analysis was performed to determine the statistical significance of the correlation between the AZD5582-induced reduction in levels of XIAP, cIAP-1 and cIAP-2 and sensitivity to TRAIL-induced killing among the six cases examined.
Figure S3. CD40 stimulation protects CLL cells from spontaneous apoptosis and killing induced by fludarabine and dexamethasone. (A) CLL cells were cultured for 48 h under standard conditions or on parental or CD154-expressing fibroblasts in the absence or presence of fludarabine (10 μmol/L) or dexamethasone (Dex) (100 nmol/L). CLL cells were then harvested for analysis of viability by flow cytmetry. (B) CLL cells cultured for 48 h under the same conditions as in (A) were also examined for the expression of Bcl-xL, Mcl-1 and Bcl-2 proteins by Western blotting.