

Abstract
It has previously been reported that KU60019, as a highly effective radiosensitizer, inhibits the DNA damage response and blocks radiation-induced phosphorylation of key ataxia telangiectasia mutated targets in human glioma cells. The present study investigated whether KU60019 affects cell physiological activities and strengthens the efficacy of doxorubicin-induced DNA damage. It was demonstrated that the compound suppressed the proliferation of MCF-7 cells and significantly increased chemosensitization. In addition, KU60019 (without doxorubicin) inhibited MCF-7 cell motility and invasion, potentially by acting on the phosphorylated-Akt and E-cadherin signaling pathways. Although the majority of MCF-7 cells were arrested at the G1/S phase following treatment with KU60019, the combination of the two compounds did not result in such a marked effect on the cell cycle. In conclusion, KU60019 is a potent chemosensitizer in combination with doxorubicin, therefore, it may provide a promising strategy for non-invasive breast cancer.
Keywords: chemosensitivity, ataxia telangiectasia mutated, KU60019, p-p53 (Ser15), doxorubicin
Introduction
Despite advances in detection and therapeutic strategies, breast cancer remains the most common type of malignancy in females worldwide (1). MCF-7 (estrogen receptor (+), progesterone receptor (+), human epidermal growth factor receptor 2 (−) and wild-type p53) is one of the most representative non-invasive breast cancer cells in vitro. Doxorubicin, as an anthracycline antibiotic, possesses the ability to intercalate into DNA and block DNA topoisomerase II activity resulting in DNA double-strand breaks (2). Due to its high cytotoxic action towards tumor cells, doxorubicin-based chemotherapy remains a central component of current treatments for non-invasive breast cancer. Furthermore, doxorubicin is used as an adjunct to surgery in the majority of cases (3).
The ataxia telangiectasia mutated (ATM) protein is considered to directly or indirectly regulate the repair of double-strand breaks via cell cycle checkpoint control, and inhibition or absence of ATM increases radiosensitivity, indicating that it is critical for the cellular response to DNA damage (4,5). In a previous study, KU60019 was identified as a potent and specific ATP competitive inhibitor of ATM with regards to the inhibition of other members of the phosphoinositide 3-kinase family. KU60019 was shown to increase the sensitivity of breast cancer cells to ionizing radiation (IR), alter their cell cycle profile and inhibit the phosphorylation of a panel of ATM targets (6).
The current study was designed as a preclinical evaluation of the ATM inhibitor, KU60019, to investigate whether the compound affects cell physiological activities and strengthens the efficacy of doxorubicin-induced DNA damage.
Materials and methods
KU60019
The highly specific and potent ATM inhibitor, KU60019, was purchased from Selleck Chemicals (Houston, TX, USA). KU60019 was dissolved in 100% dimethyl sulfoxide (DMSO) and stored at −20°C.
Cell culture
The MCF-7 human breast cancer cell line was obtained from the American Type Culture Collection (Rockville, MD, USA). The cells were cultured in Dulbecco’s modified Eagle’s medium (Gibco-BRL, Grand Island, NY, USA) and supplemented with 10% fetal bovine serum (Gibco-BRL) in a humidified atmosphere of 5% CO2 at 37°C.
Cell survival analysis
Cells were seeded into 96-well plates (6×103/well) and following attachment for 12 h, the cells were exposed to 0.25 mg/l doxorubicin (Zhejiang Hisun Pharmaceutical Co., Ltd., Taizhou, China) and/or 3 μM KU60019 (Selleck Chemicals) for 24 h. The Cell Counting kit-8 (CCK-8) assay (Dojindo, Kunamoto, Japan) was used to determine relative cell growth, according to the manufacturer’s instructions. The data shown are representative of three independent experiments.
Apoptosis assay
The MCF-7 cells were incubated with 0.25 mg/l doxorubicin and/or 3 μM KU60019 for 24 h. In total, 1×106 cells were later collected and washed twice with ice-cold phosphate-buffered saline (PBS). Cells were dual-stained using fluorescein isothiocyanate Annexin V apoptosis detection kit I (BD Biosciences, Franklin Lakes, NJ, USA), according to the manufacturer’s instructions. The stained cells were immediately analyzed using a flow cytometer (BD Accuri™ C6; BD Biosciences).
Cell cycle analysis
The cells were collected, washed twice with PBS supplemented with 1 ml ethanol (75%) and maintained at −20°C overnight. The cells were resuspended with PBS supplemented with propidium iodide and RNase A, incubated for 30 min at 37°C and analyzed using flow cytometry.
In vitro migration and invasion assays
For the migration assays, the blank cells or chemo survival fractions (MCF-7 cells) were detached and aliquots of 2×105 cells/ml were plated onto the inserts of the 8-μM pore size Transwell chambers (Corning Inc., Corning, NY, USA). For Transwell invasion assays, 1×106 cells were plated onto inserts containing a polycarbonate membrane with a thin layer of BD Matrigel matrix (BD Biosciences). Migration and invasion were detected 24 h later by counting the number of cells that had migrated or invaded through the membrane.
Western blot analysis
Cells were extracted and prepared in modified radio immunoprecipitation assay buffer (Beyotime, Jiangsu, China). Proteins were separated by 10% SDS-PAGE, transferred to polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA, USA) and incubated with primary antibodies against human BCL-2 (1:500; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), phosphorylated (p)-p53 (Ser15; 1:1,000; Cell Signaling Technology, Inc., Danvers, MA, USA), p53 (1:500; Bioworld Technology, Inc., St. Louis Park, MN, USA) and phosphorylated (p)-Akt (Ser473), ATM and E-cadherin (all 1:1,000; Cell Signaling Technology, Inc.) overnight at 4°C. The horseradish peroxidase-conjugated secondary antibody was purchased from DakoCytomation (Glostrup, Denmark). Immunoreactive bands were visualized by chemiluminescence with pierce enhanced chemiluminescence detection reagent (Millipore, Billerica, MA, USA) and β-actin (1:4,000; Bioworld Technology, Inc.) served as an internal loading control. Data shown are representative of individual experiments that were repeated at least three times.
Statistical analysis
The results presented are the average of at least three experiments each performed in triplicate with standard errors. Statistical analyses were performed by analysis of variance, followed by unpaired one- or two-tailed t-tests, using the SPSS 20.0 statistical package (IBM, Armonk, NY, USA). P<0.05 was considered to indicate a statistically significant difference.
Results
Effect of KU60019 on cell viability in MCF-7 cells
Results of the dose-dependent study are shown in Fig. 1A and indicated that KU60019 has an antiproliferative effect on MCF-7 cells, as assessed by CCK-8 assay. Treatment with KU60019 for 24 h significantly suppressed the proliferation of MCF-7 at concentrations of >6 μM (P<0.05, data not shown).
Figure 1.
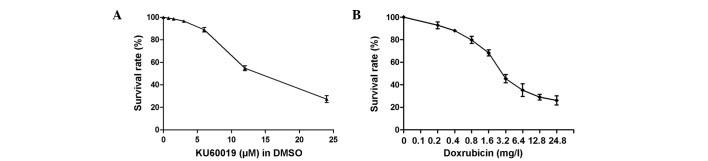
Survival rates of MCF-7 cells treated with different concentrations of (A) KU60019 and (B) doxorubicin for 24 h were tested using a Cell Counting Kit-8 assay. DMSO, dimethyl sulfoxide.
KU60019 is a potent chemosensitizer in combination with doxorubicin
A gradual decrease in cell survival was identified in response to increased concentrations of doxorubicin (Fig. 1B). The IC50 values of doxorubicin and KU60019 were 2.5 mg/l and 12.4 μM, respectively. The response to a fixed concentration of each compound was subsequently investigated (Fig. 2). When the apoptosis rate of MCF-7 cells was measured, it appeared that the cells were much more sensitive to the combination of the two compounds rather than KU60019 or doxorubicin alone. At a concentration of 0.25 mg/l, doxorubicin induced only 9.6±0.83% of cell apoptosis (Fig. 2C). Similarly, no marked difference was identified between the cells that were treated with KU60019 at a concentration of 3 μM and the control groups (Fig. 2A, B and D). However, combining the two inhibitors significantly increased the chemosensitization of MCF-7 cells, which indicated an advanced role of doxorubicin-based chemotherapy in non-invasive breast cancer (Fig. 2E and F).
Figure 2.

Apoptotic rate of MCF-7 cells induced by different compounds. (A,B and D) No marked difference was identified among the cells treated with KU60019 at a concentration of 3 μM and the control groups. (C) At a concentration of 0.25 mg/l, Dox induced 9.6±0.83% cell apoptosis. (E) Combining the two inhibitors significantly increased the chemosensitization of MCF-7 cells. (F) Apoptotic rate of MCF-7 cells induced by different compounds. *P<0.05 vs. the blank control. DMSO, dimethyl sulfoxide; Dox, doxorubicin.
Blocking the cell cycle may be a key mechanism of KU60019 at a concentration of 3 μM
It was of interest in the present study whether KU60019 induced alteration of the cell cycle in MCF-7 cells. The results presented in Table I demonstrate that, compared with the control groups, the vast majority of MCF-7 cells were arrested at G1/S phase (62.2±2.9%) following treatment with 3 μM KU60019. In the combination group, the two compounds caused G1/G2 phase arrest (G1/S, 41.8±3.1%; and G2/M, 35.5±2.4%).
Table I.
Cell cycle distribution in MCF-7 cells.
| Phase | Blank, % | DMSO, % | Dox, % | KU60019, % | Dox and KU60019, % |
|---|---|---|---|---|---|
| G0G1 | 44.3±0.96 | 43.8±2.12 | 48.4±2.07a | 69.2±0.93a | 54.8±1.08a |
| S | 34.0±0.33 | 34.2±0.54 | 21.1±0.76 | 8.7±0.22 | 13.5±0.93 |
| G2/M | 21.9±1.07 | 21.7±1.82 | 30.4±1.03a | 21.6±0.94 | 30.1±1.02a |
P<0.05 vs. blank control. Data are presented as the mean ± standard deviation.
DMSO, dimethyl sulfoxide; Dox, doxorubicin.
KU60019 affects the ability of migration and invasion in the MCF-7 cell line in vitro
It was hypothesized that KU60019 may restrict MCF-7 cell migration and invasion. Thus, migration and invasion assays of MCF-7 cells were performed and established in vitro conditions (Fig. 3A). Fewer KU60019-treated cells crossed the membrane compared with control group cells (KU60019 group [142.2±3.4] vs. blank [274±7.3] and DMSO [263±5.1] groups; P<0.01, Fig. 3B). Compared with the control groups, following treatment with KU60019, the number of MCF-7 cells that invaded the Matrigel-coated filter was significantly lower (KU60019 group [7.5±0.6] vs. blank [19.0±0.7] and DMSO [20.1±0.9] groups; P<0.01, Fig. 3C). In addition to its negative effects on migration and invasion, KU60019 weakened these abilities when it was combined with doxorubicin at a concentration of 0.25 mg/l. For the migration assay, the cell numbers were; doxorubicin group (117±6.2) vs. combination group (60±4.4) as shown in Fig. 3B (P<0.01). For the invasion assay, the cell numbers were; doxorubicin group (9±0.8) vs. combination group (3.5±0.7) as shown in Fig. 3C (P<0.01).
Figure 3.

Effect of KU60019 and/or Dox on the migration and invasion ability of MCF-7 cells. (A) Images of representative crystal violet-stained membranes are shown from the migration and invasion assays. (B) Quantitative evaluation of cell migration ability (chambers without Matrigel); the migrating cells were counted in six predetermined fields. (C) Quantitative evaluation of cell invasion ability (chambers with Matrigel); the invading cells were counted in six predetermined fields. *P<0.05 vs. blank control. DMSO, dimethyl sulfoxide; Dox, doxorubicin.
Various kinases are involved in the doxorubicin/KU6- 0019-dependent signaling pathway
The expression levels of proteins regulated by doxorubicin and/or KU60019 were examined. Compared with the control groups, following treatment with doxorubicin and/or KU60019, the accumulation of wild-type p53 and E-cadherin was observed, whereas the p-Akt (Ser473) level decreased. The results are presented as the ratio of Δp-p53 (Ser15)/Δp53. It was evident that this ratio was always >1.0 (mean ± standard deviation, 1.9±0.3), which indicated that treatment with doxorubicin, while leading to a rise of total p53, induced a much greater rise in the proportion of p-p53 at Ser15. It was evident that the key intracellular targets of the ATM kinase and doxorubicin-induced p53 phosphorylation (Ser15) were inhibited or almost completely abrogated in the presence of low micromolar concentrations of KU60019. Additionally, it was identified that the anti-apoptotic protein, Bcl-2, was only marginally inhibited by KU60019 alone, whereas it was significantly suppressed by the combination of the two compounds (Fig. 4).
Figure 4.

Western blot analysis in MCF-7 cell lines. *P<0.05 vs. blank control. ATM, ataxia telangiectasia mutated; DMSO, dimethyl sulfoxide; Dox, doxorubicin.
Discussion
Surgery and chemoradiation significantly hinder the progression of breast cancer, however, more effective treatments for doxorubicin-based therapy remain a crucial requirement. Doxorubicin-induced DNA damage response (DDR) involves numerous highly conserved checkpoint pathways that are activated by genotoxic stress. Checkpoint activation triggers a cascade of events, which ultimately lead to cell cycle arrest or apoptosis. One of key components of this response is ATM kinase, which typically functions as a sensor of DNA damage (7).
The robust DNA repair capacity of cancer cells results in a resistance to therapies, such as IR and cytotoxic drugs, which are intended to cause lethal DNA damage (8). Specific improved ATM kinase inhibitors have been developed in the context of the classic role of ATM in DNA repair, with the rationale that inhibition of DNA repair is likely to increase the efficacy of radiation or chemical therapy (6). A previous study indicated that KU60019, a highly effective radiosensitizer, inhibits DDR and blocks radiation-induced phosphorylation of key ATM targets in human glioma cells (9). The present study demonstrated that the ATM kinase-specific inhibitor, KU60019, effectively inhibits migration and invasion as well as chemosensitization in the non-invasive MCF-7 human breast cancer cell line.
An upregulation of the level of total p53 and p-p53 (Ser15) that was concurrent with an increased expression of ATM, was observed in the cells that were treated with doxorubicin. Previous studies showed that phosphorylation of p53 at Ser15, in response to DNA double-strand breaks, was by the ATM protein kinase (10). Furthermore, other N-terminal sites may be phosphorylated by kinases that require the prior phosphorylation of p53 at Ser15 (11). As hypothesized in the present study, blocking of doxorubicin-induced ATM phosphorylation by KU60019 markedly reduced the level of p-p53 (Ser15). This indicated that the loss of ATM and p-p53 (Ser15) may marginally inhibit doxorubicin-induced DNA repair. However, similar to the alteration observed in the apoptosis rate, exposure to KU60019 alone did not suppress the expression of the anti-apoptotic protein, BCL-2, and combining the two compounds induced an increased expression of BCL-2. In certain cases, KU60019 and doxorubicin may synergize in apoptosis. Thus, the combined use of the compounds (by inhibiting the ATM-mediated DNA repair pathway and enhancing apoptosis) was anticipated to produce a larger repair deficit and a corresponding greater chemosensitization compared with each compound alone.
In glioma cells, KU60019 inhibits the migration/motility and invasion of cells via deregulated receptor tyrosine kinase-mediated signaling (9). Similarly, the expression of activated Akt in fibrosarcoma or pancreatic cancer cells increases cell invasion through Matrigel (12–14), an effect that is recapitulated by an overexpression of Akt2 in breast and ovarian cancer cells (15). In addition, the expression of Akt promotes epithelial-mesenchymal transition, a process that is closely associated with tumor progression to invasive and metastatic carcinoma (16). Akt is activated by a dual regulatory mechanism whereby maximal activation requires additional phosphorylation at Ser473 (17,18). Furthermore, E-cadherin is a calcium-dependent cell adhesion molecule that mediates cell-cell adhesion and modulates cell migration and tumor invasiveness (19). Compelling evidence exists, which indicates that treating cells with KU60019 and/or doxorubicin results in a decrease of p-Akt Ser473 and the increased expression of E-cadherin, which is consistent with the diminished capacity of migration and invasion exhibited by MCF-7 cells.
Another focus of the present study was to understand the mechanism by which the combination of the two compounds affected cell cycle arrest. Upon genotoxic damage, p53 contributes to cell cycle arrest at the G1/S and/or G2/M checkpoints via various mechanisms (9). Treatment with KU60019 alone was found to markedly induce G1/S phase arrest and doxorubicin resulted in G1/S and G2/M phase arrest via activation of p53 (20,21). The cell cycle was halted at the transition from the G1/S to the G2/M phase that was induced by the combination of the two compounds, which may also be involved in ATM and the downstream signaling of ATM pathways. This indicated that the damage induced by the combination of the two compounds provoked p53- and ATM-mediated cell cycle arrest in MCF-7 cells (22).
In conclusion, the present study demonstrated that KU60019 is a specific ATM kinase inhibitor, which is capable of chemosensitizing MCF-7 cells when it is combined with doxorubicin. Chemosensitization may result from the ability of KU60019 to inhibit the phosphorylation of ATM and p53 (Ser15), alter cell cycle checkpoints, decrease DNA repair (via inhibiting the phosphorylation of p53; Ser15) and increase cell apoptosis. Additionally, the results indicated that KU60019 (without doxorubicin) inhibits MCF-7 cell motility and invasion, potentially by acting on the p-Akt and E-cadherin signaling pathways. Thus, KU60019 may be developed into a highly effective, cancer therapeutic agent, acting as a chemosensitizer and curtailing tumor dispersal. Furthermore, accelerated pharmaceutical development of KU60019 is critical to expand the available treatment strategies for non-invasive breast cancer.
Acknowledgements
The current study was supported by a grant from the National Natural Science Foundation of China (grant no. 81272470).
References
- 1.Jeng KS, Sheen IS, Jeng WJ, et al. High expression of Sonic Hedgehog signaling pathway genes indicates a risk of recurrence of breast carcinoma. Onco Targets Ther. 2013;7:79–86. doi: 10.2147/OTT.S54702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Binaschi M, Capranico G, Dal Bo L, Zunino F. Relationship between lethal effects and topoisomerase II-mediated double-stranded DNA breaks produced by anthracyclines with different sequence specificity. Mol Pharmacol. 1997;51:1053–1059. doi: 10.1124/mol.51.6.1053. [DOI] [PubMed] [Google Scholar]
- 3.Violet JA, Harmer C. Breast cancer: improving outcome following adjuvant radiotherapy. Br J Radiol. 2004;77:811–820. doi: 10.1259/bjr/44576710. [DOI] [PubMed] [Google Scholar]
- 4.Valerie K, Povirk LF. Regulation and mechanisms of mammalian double-strand break repair. Oncogene. 2003;22:5792–5812. doi: 10.1038/sj.onc.1206679. [DOI] [PubMed] [Google Scholar]
- 5.Lavin MF. Ataxia-telangiectasia: from a rare disorder to a paradigm for cell signalling and cancer. Nat Rev Mol Cell Biol. 2008;9:759–769. doi: 10.1038/nrm2514. [DOI] [PubMed] [Google Scholar]
- 6.Hickson I, Zhao Y, Richardson CJ, et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 7.Ramachandran S, Tran DD, Klebba-Faerber S, et al. An ataxia-telangiectasia-mutated (ATM) kinase mediated response to DNA damage down-regulates the mRNA-binding potential of THOC5. RNA. 2011;17:1957–1966. doi: 10.1261/rna.2820911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Helleday T, Petermann E, Lundin C, et al. DNA repair pathways as targets for cancer therapy. Nat Rev Cancer. 2008;8:193–204. doi: 10.1038/nrc2342. [DOI] [PubMed] [Google Scholar]
- 9.Golding SE, Rosenberg E, Valerie N, et al. Improved ATM kinase inhibitor KU-60019 radiosensitizes glioma cells, compromises insulin, AKT and ERK prosurvival signaling, and inhibits migration and invasion. Mol Cancer Ther. 2009;8:2894–2902. doi: 10.1158/1535-7163.MCT-09-0519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Saito S, Goodarzi AA, Higashimoto Y, et al. ATM mediates phosphorylation at multiple p53 sites, including Ser(46), in response to ionizing radiation. J Biol Chem. 2002;277:12491–12494. doi: 10.1074/jbc.C200093200. [DOI] [PubMed] [Google Scholar]
- 11.Saito S, Yamaguchi H, Higashimoto Y, et al. Phosphorylation site interdependence of human p53 post-translational modifications in response to stress. J Biol Chem. 2003;278:37536–37544. doi: 10.1074/jbc.M305135200. [DOI] [PubMed] [Google Scholar]
- 12.Park BK, Zeng X, Glazer RI. Akt1 induces extracellular matrix invasion and matrix metalloproteinase-2 activity in mouse mammary epithelial cells. Cancer Res. 2001;61:7647–7653. [PubMed] [Google Scholar]
- 13.Kim D, Kim S, Koh H, et al. Akt/PKB promotes cancer cell invasion via increased motility and metalloproteinase production. FASEB J. 2001;15:1953–1962. doi: 10.1096/fj.01-0198com. [DOI] [PubMed] [Google Scholar]
- 14.Tanno S, Tanno S, Mitsuuchi Y, et al. AKT activation up-regulates insulin-like growth factor I receptor expression and promotes invasiveness of human pancreatic cancer cells. Cancer Res. 2001;61:589–593. [PubMed] [Google Scholar]
- 15.Arboleda MJ, Lyons JF, Kabbinavar FF, et al. Overexpression of AKT2/protein kinase Bbeta leads to up-regulation of beta1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res. 2003;63:196–206. [PubMed] [Google Scholar]
- 16.Grille SJ, Bellacosa A, Upson J, et al. The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Res. 2003;63:2172–2178. [PubMed] [Google Scholar]
- 17.Andjelković M, Alessi DR, Meier R, et al. Role of translocation in the activation and function of protein kinase B. J Biol Chem. 1997;272:31515–31524. doi: 10.1074/jbc.272.50.31515. [DOI] [PubMed] [Google Scholar]
- 18.Bellacosa A, Chan TO, Ahmed NN, et al. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998;17:313–325. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- 19.Hazan RB, Phillips GR, Qiao RF, et al. Exogenous expression of N-cadherin in breast cancer cells induces cell migration, invasion, and metastasis. J Cell Biol. 2000;148:779–790. doi: 10.1083/jcb.148.4.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ciciarello M, Mangiacasale R, Casenghi M, et al. p53 displacement from centrosomes and p53-mediated G1 arrest following transient inhibition of the mitotic spindle. J Biol Chem. 2001;276:19205–19213. doi: 10.1074/jbc.M009528200. [DOI] [PubMed] [Google Scholar]
- 21.Zhang T, Tan Y, Zhao R, Liu Z. DNA damage induced by oridonin involves cell cycle arrest at G2/M phase in human MCF-7 cells. Contemp Oncol (Pozn) 2013;17:38–44. doi: 10.5114/wo.2013.33772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]