

Abstract
Glutamate carboxypeptidase II (GCP II) is a therapeutic target in neurologic disorders associated with excessive activation of glutamatergic systems. The potent, orally bioavailable GCP II inhibitor 2-(3-mercaptopropyl) pentanedioic acid (2-MPPA) is effective in preclinical models of diseases where excess glutamate release is implicated, including neuropathic pain, and was the first GCP II inhibitor to be administered to man. The relationships between dosing regimen, pharmacokinetics, and analgesia in a neuropathic pain model were examined in rats to aid development of clinical dosing. The efficacy of oral 2-MPPA in the chronic constrictive injury model was not simply related to plasma concentrations. Even though maximal concentrations were observed within 1 hour of dosing, the analgesic effect took at least 8 days of daily dosing to become significant. The delay was not due to tissue drug accumulation since inhibitory concentrations of the drug were achieved in the nerve within 1 hour of dosing. There was also no accumulation of drug in plasma or tissue after multiple daily dosing. Effects were dependent on reaching a threshold concentration since dividing the daily dose led to a loss of effect. The analgesic effect outlasted plasma exposure and was maintained for days even after daily dosing was halted. The delayed onset, dependence on threshold plasma concentration, and sustained effects after exposure support the hypothesis that an indirect, long-lived mechanism of action exists. Although these longer lasting secondary mechanisms are not yet identified, daily clinical dosing of a GCP II inhibitor seems justified.
Introduction
Glutamate is a major excitatory neurotransmitter in the central and peripheral nervous systems. Excessive activation of glutamatergic systems has been implicated in a number of neurologic disorders, including neuropathy and neuropathic pain (Carozzi et al., 2008a; Yogeeswari et al., 2009). The neuropeptide N-acetyl-aspartyl glutamate (NAAG) is abundant in both brain and peripheral nervous system, where it appears to serve as a neurotransmitter both as a dipeptide and as a precursor of glutamate. As a dipeptide, it appears to act through mGluR3 receptors to decrease glutamate release (Neale et al., 2011; Bařinka et al., 2012).
Glutamate carboxypeptidase II (GCP-II, also termed NAALADase and NAAG peptidase) is a 94-kDa membrane-bound zinc metalloenzyme that catalyzes the hydrolysis of neuropeptide NAAG to N-acetyl-aspartate and glutamate (Slusher et al., 1990). GCP-II acts to terminate the neurotransmitter activity of NAAG at mGluR3 receptors resulting both in increased synaptic glutamate release (Wroblewska et al., 1993; Coyle, 1997; Slusher et al., 1999) and liberation of glutamate from NAAG that can further activate various glutamate receptors. Inhibition of GCP-II therefore attenuates glutamatergic effects both by causing an increase in NAAG’s inhibition of glutamate release and simultaneously decreasing glutamate levels.
The first potent and selective GCP-II inhibitor identified was 2-(phosphonomethyl) pentanedioic acid (2-PMPA) (Jackson et al., 2001). 2-PMPA was shown to possess therapeutic efficacy in various preclinical models, including ischemia (Slusher et al., 1999), spinal cord injury (Long et al., 2005), neuropathic pain (Jackson et al., 2001; Wozniak et al., 2012), cocaine addiction (Xi et al., 2010a; Xi et al., 2010b), and schizophrenia (Olszewski et al., 2008; Zuo et al., 2012). Thus, GCP-II inhibition may provide broad therapeutic utility in neurologic diseases especially where excess glutamate is presumed pathogenic (Rojas et al., 2011; Bařinka et al., 2012). Although potent and selective, 2-PMPA is not orally available perhaps due to its extreme hydrophilic nature and limited permeability through the gastrointestinal tract.
Replacement of the phosphonomethyl group with a thioalkyl group led to discovery of 2-MPPA [2-(3-mercaptopropyl) pentanedioic acid] (Majer et al., 2003), which was found to be orally available and was the first GCP II inhibitor to be administered to man (van der Post et al., 2005), where it was well tolerated and reached exposure levels similar to those required for therapeutic effects in animal models. To develop a dosing regimen for clinical use, the pharmacokinetics and pharmacodynamics were examined in more detail in animals to determine the required concentration and duration of exposure.
Materials and Methods
Animals.
Male Sprague-Dawley rats (Charles River Laboratories, Inc., Wilmington, MA) weighing 200–250 g were used. Animals were housed in suspended polycarbonate cages under a 12-hour light/dark cycle. Food (Teklab; Harlan, Indianapolis, IN) and water (filtered and delivered via an automatic watering system) were provided ad libitum. All procedures were conducted in compliance with the laws, regulations, and guidelines of the National Institutes of Health (NIH) and with approval from the local animal care and use committee.
Drugs.
2-MPPA was dissolved in 50 mM HEPES buffered saline and brought to neutral pH with NaOH. Dosing solution was prepared fresh every 2–3 days and stored under refrigeration (2–8°C).
Pharmacokinetic Studies.
2-MPPA was administered to rats as a solution via the oral gavage or intravenous (caudal vein) route at doses of 1, 3, 10, 30, 50, 100, 500, or 1000 mg/kg. At various time-points after dosing, rats were sacrificed. At each time point, plasma was derived from whole blood collected and centrifuged at 4°C in plasma separator tubes for 10 minutes. In some studies, tissue was also removed. All samples were stored at –80°C until subsequent analysis. Quantification of 2-MPPA using high-performance liquid chromatography with tandem mass spectrometry (LC-MS/MS) was performed as described in the next section.
Bioanalysis of 2-MPPA.
2-MPPA was quantified in plasma and tissue samples following derivatization with N-ethylmaleimide (NEM) and analyzed via LC-MS/MS as described previously (Rais et al., 2012). In brief, a 180-μl plasma sample (or 160 μl of matrix blank and 20 μl of stock for standards) was reacted with 10 μl of 100 mM NEM for 30 minutes along with structurally similar internal standard at 5 μg/ml in acetonitrile. The derivatized samples were extracted with methanol followed by vortexing and centrifugation at 10,000 rpm for 5 minutes. The supernatant (50 μl) was transferred to liquid chromatography vial and a volume of 5 μl used for LC-MS/MS. The tissue samples were processed in a manner similar to plasma. In brief, tissue samples were weighed, followed by addition of phosphate-buffered saline buffer (also containing 10 μl of 100 mM NEM), and volume was adjusted to obtain all samples equal per gram of tissue. The samples were homogenized, vortexed, and extracted following the same procedure as described for plasma. The calibration range was 1–4000 ng/ml for 2-MPPA in plasma and 50–50,000 ng/ml in tissue.
Samples were analyzed on an Agilent 1100 HPLC (Agilent Technologies, Santa Clara, CA) coupled to API 3000 mass spectrometer (Applied Biosystems/MDS Sciex, Toronto, ON, Canada) Derivatized-2-MPPA was separated on a Luna C18 (2 mm) 30 × 4.60 mm, 5 μM column (Phenomenex, Torrance, CA). The mobile phase consisted of acetonitrile (A), and 0.1% formic acid in Milli-Q water (B). Separation was achieved using a gradient run, with 40% (A) going to 90% (A) over a period, maintaining at 90% for 3 minutes, and then re-equilibrating over 3 minutes (40% A) at a flow rate of 350 μl/min and total run time of 10 minutes. The mass spectrometry instrument was operated in a negative ion mode. The multiple reaction monitoring (MRM) transition of derivatized 2-MPPA was 330 > 205 (Q1/Q3) and for the internal standard, 645.3 > 323.3 with a declustering potential of 40 V, entrance potential 10 V, and collision energy of 21 V. The curtain gas, ion-spray voltage, temperature, nebulizer gas (GS1), and auxiliary gas (GS2) were set at 8 psi, 5500 V, 350°C, 8 psi, and 4 psi, respectively, and the interface heater was on.
Pharmacokinetic Analysis.
Initial pharmacokinetic studies with intense sampling were conducted to determine the plasma concentration-time profile of 2-MPPA. Subsequent studies were conducted using time points from the tissue distribution study described above and from single plasma samples obtained from animals evaluated in the chronic constrictive injury model. The entire dataset was analyzed using a two-compartment model with first order absorption. A population pharmacokinetic model was developed utilizing both intense and sparse sampling paradigms (WinNonmix, version 2.0.1; Pharsight Corporation, Cary, NC). AUC and mean peak plasma concentration (Cmax) for each animal were the primary outputs for the population pharmacokinetic analysis.
Pharmacokinetic parameters following single doses of 2-MPPA to rats were calculated using noncompartmental methods (WinNonlin, version 5.1; Pharsight Corporation). Bioavailability was calculated using the following equation:
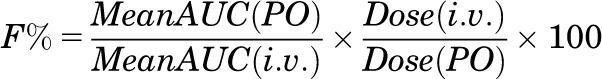 |
Chronic Constrictive Injury Model.
The methods used were as previously described (Bennett and Xie, 1988) using male Sprague-Dawley rats. In brief, the common sciatic nerve was exposed and four ligatures (4.0 chromic gut) were tied loosely around it with 1-mm spacing. Hyperalgesia testing was initiated 10 days postsurgery. Thermal sensitivity was assessed by determining withdrawal latencies to a constant infrared stimulus applied to the plantar surface of the hind paw using a Basile plantar apparatus (Ugo Basile, Varese, Italy) according to the method described by Hargreaves et al. (1988). Withdrawal latency was measured as the time taken for the rat to withdraw its paw from the heat source to the nearest 0.1 second. The “difference score” was calculated by subtracting the average latency of the nonligated versus ligated side. Statistical analyses were conducted using the Student’s t test (when two groups including vehicle were compared) or ANOVA with post-hoc Dunnett’s test (when three or more groups were compared).
Open-Field Locomotion.
Animals were dosed with vehicle or 10, 50, or 100 mg/kg 2-MPPA by orally and placed in open-field locomotor counter monitoring infrared beam interruptions (Columbus Instruments International, Columbus, OH). Locomotion was counted at 5-minute intervals for 1 hour starting from 1 hour after administration of vehicle or 2-MPPA.
Once Daily Dose-Response Studies.
2-MPPA or vehicle at various doses was given daily by oral gavage in a solution volume of 1 ml/kg. Testing was initiated from the day before treatment onset and then on test days from day 1 of treatment, twice a week, and 1 hour after dosing. In follow-up studies investigating the efficacy time course, testing was conducted at 1, 4, or 16 hours postdose.
Dose Fractionation Studies.
2-MPPA or vehicle was administered once, twice, or three times daily by oral gavage. The 5 mg/kg dose was administered twice daily with a 5-hour interval between the two administrations. The 3.3 mg/kg dose was administered three times a day with 4-hour intervals between the administrations. Behavioral testing was always conducted 1 hour after the final daily dose, regardless of the regimen.
Results
Pharmacokinetics of 2-MPPA in Rats.
After a bolus of 10 mg/kg i.v., Cmax (extrapolated to zero) was 27.0 μg/ml, which declined in a biphasic manner, with the initial rapid decline within the first hour. Average clearance, terminal elimination half-life (T1/2elim) and mean residence time were 4.0 l/h per kilogram, 1.0 hour, and 0.2 hours, respectively. Likewise, the oral absorption following a 10 mg/kg dose was rapid with a similar biphasic decline (Fig. 1).
Fig. 1.

Pharmacokinetic profile of 2-MPPA after intravenous and oral administration in rats. Plot of mean plasma concentration versus time following an oral gavage or intravenous (caudal vein) administration of 10 mg/kg.
A single-dose, dose-proportionality study was conducted in nonfasted male and female Dawley rats with 2-MPPA at oral doses of 1, 3, 10, 30, 50, 100, 500, and 1000 mg/kg (Table 1). After oral administration, plasma pharmacokinetic profiles were biphasic with terminal t1/2 ranging from 2.4 to 9.2 hours. Although Cmax increased approximately proportionally with dose, AUC increased supra-proportionally with dose over the range of 1 to 1000 mg/kg. Bioavailability was high (82%) based on the 10 mg/kg i.v. and PO administrations.
TABLE 1.
Pharmacokinetic parameters following single oral dose of 2-MPPA in rats
All values are mean ± S.D. Parameters were derived using noncompartmental analysis. Bioavailability is calculated from the 10 mg/kg i.v. dose and expressed as a percentage.
| Dose | Cmax | T*max | AUC | Cl/Fa | Terminal Half-Life | Bioavailability |
|---|---|---|---|---|---|---|
| ng/ml | h | ng*h per milliliter | l/h per kilogram | h | % | |
| 1 mg/kg | 600 ± 400 | 0.08 | 300 ± 100 | 4.5 ± 1.7 | 6.8 ± 4.4 | 102 |
| 3 mg/kg | 1000 ± 400 | 0.08 | 500 ± 200 | 6.7 ± 2.7 | 4.9 ± 2.1 | 66 |
| 10 mg/kg | 3200 ± 1300 | 0.08 | 2,100 ± 700 | 5.3 ± 1.9 | 9.2 ± 4.2 | 82 |
| 30 mg/kg | 11,100 ± 3900 | 0.08 | 8,200 ± 1,060 | 3.7 ± 0.6 | 4.9 ± 1.3 | 107 |
| 50 mg/kg | 20,300 ± 10,900 | 0.50 | 14,300 ± 4,900 | 3.8 ± 1.0 | 4.1 ± 1.7 | 112 |
| 100 mg/kg | 40,300 ± 9,500 | 0.50 | 40,500 ± 8,200 | 2.6 ± 0.5 | 4.8 ± 1.9 | 158 |
| 500 mg/kg | 110,200 ± 37,900 | 0.08 | 231,100 ± 41,000 | 2.2 ± 0.4 | 6.8 ± 8.4 | 181 |
| 1000 mg/kg | 203,800 ± 76,700 | 0.08 | 537,000 ± 160,500 | 2.0 ± 0.6 | 2.4 ± 0.3 | 210 |
Apparent clearance defined as clearance as a function of oral bioavailabilty (F).
Daily Administration of 2-MPPA Attenuates Thermal Hyperalgesia.
2-MPPA administered at doses of 10 and 30 mg/kg per day PO attenuated thermal hyperalgesia compared with vehicle-treated controls, reaching significance on days 8 through 15 of dosing (Fig. 2, A and B). Repeated daily dosing was required for the effect to develop. Animals receiving 3 mg/kg PO per day 2-MPPA displayed a strong trend toward reduction of hyperalgesia, although the effect did not achieve statistical significance (Fig. 2C). Animals receiving 1 mg/kg PO per day 2-MPPA daily showed no drug effect (Fig. 2D). The sensitivity to thermal stimulation on the nonligated (i.e., normal) paw was unaltered by 2-MPPA treatment at all doses tested. As representative, Fig. 2E depicts the sham-operated side responses after the two highest doses evaluated (10 and 30 mg/kg).
Fig. 2.

Time-course and dose response of 2-MPPA reduction of hyperalgesia in the rat CCI model. Each panel (A–D) shows the difference in withdrawal latency (in seconds) between the ligated and nonligated sides (mean ± S.E.M.) as observed over the course of the experiment. Note that the lowest doses were observed over a longer period of time to allow development of analgesia. Statistical analyses were conducted with comparisons made to vehicle-treated animals. Significance criteria of P < 0.05 were used for all experiments and are noted with an asterisk (*). (E) Daily dosing of 10 or 30 mg/kg 2-MPPA did not affect the response latency to the thermal stimulus applied to the sham-operated side compared with vehicle.
Administration of 2-MPPA Has No Effect on Open-Field Locomotor Activity.
2-MPPA administered to rats at doses of 30, 50, and 100 mg/kg PO had no significant effect on locomotion measured in an open-field compared with vehicle-treated controls (Fig. 3), suggesting no direct suppression of motor function.
Fig. 3.

Lack of effect of 2-MPPA on locomotion in open-field at doses up to 100 mg/kg PO. Total ambulatory scores in rats treated with 30, 50, or 100 mg/kg 2-MPPA were not different from vehicle-treated rats.
2-MPPA Does Not Accumulate in Tissue following Multiple Dosing.
The observation that the speed of onset is dependent on daily exposure can be explained either by an indirect pharmacological mechanism in which effect accumulates over time or a deep compartment where drug accumulates even though there is no accumulation in plasma of this rapidly eliminated drug. To investigate whether this effect was due to tissue accumulation, a single- and multiple-dose oral tissue distribution study was performed.
2-MPPA (10 mg/kg PO per day) was distributed throughout the body within 5 minutes after dosing on day 1. The Cmax in sciatic nerve was 0.018 μg/g, equivalent to 87 nM based on wet weight of tissue, above the Ki for GCP II inhibition by 2-MPPA of 30 nM. The Cmax on both days 5 and 11 was achieved within 30 minutes in sciatic nerve without evidence of accumulation. Data for plasma and sciatic nerve on days 1 and 5 are shown in Fig. 4.
Fig. 4.

Time course of sciatic nerve tissue distribution following single- or 5-day administration of 10 mg/kg 2-MPPA PO. Note that sampling on day 1 continued to 250 minutes while on day 5 it was limited to 120 minutes, but profiles during both periods are similar.
The time course of enzyme inhibition on each day of dosing was determined by measuring enzyme activity in sciatic nerve 30 minutes or 6, or 12 hours following dosing. In sciatic nerve removed from vehicle-treated animals, GCP II activity was 2230 ± 198 fmol/mg protein per hour (mean ± S.E.M.). As predicted by tissue concentrations, enzyme was significantly inhibited after 30 minutes (1363 ± 120 fmol/mg protein per hour, P < 0.05), returning to baseline at 6 hours (2312 ± 283 fmol/mg protein per hour, nonsignificant). This suggests that while enzyme is rapidly inhibited in a concentration-dependent manner, the behavioral effects on hyperalgesia are achieved more gradually.
2-MPPA Efficacy Reduced by Divided Dosing.
To confirm that the slower behavioral effect was due to daily brief inhibition of GCP II and not a loading of a deep tissue compartment, the effect of divided doses was examined. If the effect was dependent on maximal exposure (e.g., Cmax) or a time-above-a-threshold concentration, divided doses would be less effective. If, on the other hand, total exposure (e.g., AUC) was occurring, overall exposure would be more important and divided doses would be just as effective.
A dose of 10 mg/kg was chosen because it was the minimally effective daily dose of 2-MPPA in the chronic constriction injury model. As shown in Fig. 5, drug effect was completely lost when this single daily dose was divided into two doses of 5 mg/kg or three doses of 3.3 mg/kg each. This suggests that AUC alone does not determine the pharmacodynamic effect, i.e., there is a minimal plasma concentration threshold of 2-MPPA that must be achieved each day for attenuation of hyperalgesia.
Fig. 5.

Loss of effect of 2-MPPA with divided dosing. When the minimal-effect dose of oral daily 10 mg/kg 2-MPPA was distributed throughout the day by divided dosing as either 5 mg/kg BID or 3.3 mg/kg TID, the analgesic effect was lost. In each case, testing was conducted at 1 hour following the last 2-MPPA dose of the day. Statistical analyses were conducted with comparisons made to vehicle-treated animals. Significance criteria of P < 0.05 were used and are noted by an asterisk (*).
2-MPPA Efficacy Persists after Daily Dosing Is Stopped.
To confirm that daily exposure acted through a long lasting mechanism, the time course of effect related to plasma concentration was further examined. The duration of neuropathic pain alleviation following administration of 2-MPPA was tested 1, 4, and 16 hours after 10 mg/kg 2-MPPA. As shown in Fig. 6, on day 11 animals tested 1 hour after the daily dose showed a significant reduction in thermal pain perception. A strong trend was also observed at 4 or 16 hours but did not attain statistical significance. After 15 days of dosing, significant improvement was also observed 4 or 16 hours after the daily dose of 10 mg/kg 2-MPPA. These data suggest that once effective neuropathic pain alleviation is observed in animals, the effect can be maintained for up to 16 hours following drug administration.
Fig. 6.

Analgesic effect of 2-MPPA in the CCI model up to 16 hours following daily dosing. All animals were dosed with 2-MPPA at 10 mg/kg PO once daily. Different groups were subsequently evaluated for thermal withdrawal latency at 1, 4, or 16 hours after administration on the days noted. Analgesia was established at day 11, and by day 15, it was maintained for up to 16 hours after dose. Statistical analyses were conducted with comparisons made to vehicle-treated animals. Significance criteria of P < 0.05 were used and are noted with an asterisk (*).
Maintenance of effect was further examined by cessation of dosing after analgesic effect was well established after 15 days of daily dosing. As shown in Fig. 7, significant attenuation of hyperalgesia continued to be observed for an additional 17 days without further administration of 2-MPPA, confirming that enzyme inhibition is not necessary once the reduction in hyperalgesia is established.
Fig. 7.

Sustained effect of 2-MPPA on hyperalgesia after cessation of dosing. Once analgesia was achieved after 15 days of dosing, drug administration was stopped. However, withdrawal latency measurements were continued. The analgesic efficacy of 2-MPPA persisted up to 17 days after drug administration was halted. Statistical analyses were conducted with comparisons made to vehicle-treated animals. Significance criteria of P < 0.05 were used and are noted with an asterisk (*).
Discussion
Neuropathic pain is thought to be attributed to aberrant neuronal responses along pain pathways from the dorsal root ganglion (DRG) to spinal cord, thalamus, and cortex with both central and peripheral mechanisms likely involved (Zhuo et al., 2011).
2-MPPA is a potent and specific inhibitor of GCP II (Majer et al., 2003) that has safely been administered to man (van der Post et al., 2005). We show that 2-MPPA efficacy in the CCI model of neuropathic pain is dependent upon, but not simply correlated with, dose and tissue exposure. Lack of general sedative effects of 2-MPPA at doses up to 100 mg/kg acutely on locomotor activity, as well as unaltered sensitivity to thermal stimulation on nonligated (i.e., normal) paws in CCI rats after multiple days of treatment suggest a specific effect of GCP II inhibition on decreasing nociceptive input and not decreasing motor outflow. Similar nonsedative effects have been reported for other GCP II inhibitors (Lukawski et al., 2008; Olszewski et al., 2008). The analgesic effects took at least 8 days of daily dosing to become significant even though inhibitory concentrations of the drug were achieved in the nerve rapidly on the first day of dosing and there was no accumulation of drug in plasma or tissue after multiple daily dosing. Divided doses lost effectiveness, suggesting the effect is more dependent on Cmax than AUC. Thus the effect is not simply due to total daily exposure, consistent with an indirect mechanism of action caused by daily enzyme inhibition, rather than tissue loading with drug. In addition, the half-life of 2-MPPA in plasma was short, yet the analgesic effect once established was sustained. The analgesia outlasted the short plasma exposure on testing days and was maintained for days even after dosing was halted. This lack of direct correlation of pharmacodynamic with pharmacokinetic time profile is also typical of an indirect pharmacological effect.
It is noteworthy that while we observed a delayed analgesia onset in our model of neuropathic pain, more rapid effects have been observed in other peripheral pain models such as those induced by carrageenan and formalin (Yamamoto et al., 2007). The time course of action may depend on site of injury, participation of central mechanisms, or degree of GCP II inhibition. There may also be two analgesic mechanisms, one acute and one chronic. Analgesic effects might involve indirect drug effect in either or both depending on the model.
Drug accumulation in a deep tissue compartment can sometimes explain a lack of direct temporal correlation between plasma concentration and analgesic effect. In cases of deep compartment loading, slow onset of effect occurs as drug slowly enters an effect compartment, and the loss of effect occurs over time due to slow efflux of drug. Such a site would have to be hydrophilic due to the nature of 2-MPPA and of small volume to explain lack of accumulation in tissue samples.
Our measurements of enzyme inhibition in tissue exclude tissue accumulation as an explanation for the delayed onset and persistent effects of daily administration. The concentration of 2-MPPA in sciatic nerve, a possible site of action, reached levels consistent with GCP II inhibition (greater than 0.018 μg/g for at least 8 hours). This was confirmed by measurement of enzyme activity in tissue, further excluding the possibility of persistent enzyme inhibition. We also excluded accumulation of an active metabolite by measuring the time course of enzyme inhibition in tissue and showing direct correlation to plasma drug levels. Since enzyme inhibition was well predicted by tissue drug levels, we conclude that after an oral dose, enzyme is rapidly inhibited but the analgesic effects outlast the period of enzyme inhibition. Thus, the magnitude of the pharmacodynamic effect is dependent on daily exposure but not directly correlated with its time course.
GCP II has been localized to both peripheral and central nervous systems. In the periphery, GCP II is found in satellite cells of the dorsal root ganglia, the cytoplasm of Schwann cells, and the surface of nerve fibers (Berger and Lassner, 1994; Carozzi et al., 2008a,b). Enzymatic activity can be found at nerve endings in human skin (Rojas et al., 2011). In the brain and spinal cord, GCP II has been localized to astrocytes by both in situ hybridization (Ghose et al., 2004) and immunochemistry (Berger et al., 1999; Sacha et al., 2007). Overall, this localization is typical of an enzyme involved in neuronal and glial signaling and is not consistent with the presence of a deep anatomic compartment to explain delayed onset and prolonged effect. In addition, NAAG is expressed in millimolar levels in the spinal cord (Fuhrman et al., 1994) and intrathecal administration of GCP II inhibitors induces an analgesic response to inflammatory pain in the hindlimb (Yamamoto et al., 2001). Likewise, introduction of GCP II inhibitors directly into the ipsilateral lateral ventricle reduced responses to footpad inflammation (Yamamoto et al., 2008). GCP II inhibition has also been shown to reduce induction of contralateral hindlimb allodynia 24 hours after an inflammatory insult (Adedoyin et al., 2010).
Overall, the underlying pharmacological mechanism involved in GCP II inhibition attenuating pain is not established. Analgesia induced by systemically administered GCP II inhibitors appears to be mediated both via spinal cord and brain. GCP II inhibitors have effects both in the peripheral nervous system, decreasing ectopic discharges from injured nerves (Chen et al., 2002), and in the central nervous system, reversing central sensitization in the spinal cord (Yamamoto et al., 2001, 2004; Carpenter et al., 2003).
It is likely that the slow onset and prolonged effect of 2-MPPA in the CCI model is due to an indirect effect at the site of injury or on the function of pain pathways. Neuropathic pain itself is thought to be dependent on neuroplasticity and persistent changes in pain pathway structure and function (Besson, 1999; Woolf, 2011). Therefore, it is possible that GCP II inhibition reverses neuropathic pain-inducing mechanisms in the periphery and/or the brain and spinal cord and that the effects outlast inhibition of the enzyme. Since GCP II inhibition attenuates excessive glutamatergic activity in a number of pathologic models, the effect might be produced at multiple points at which excessive glutamatergic activity triggers long-term changes in pain sensitivity. This might occur in a manner analogous to the delayed onset of effect of antidepressants that depend on neuroplasticity in brain circuits (Danzer, 2012; Quide et al., 2012; Schloesser et al., 2012). In addition, GCP II inhibition might act at the site of nerve injury. This is suggested by finding that GCP II inhibition prevents or reverses damage in models of diabetic- (Zhang et al., 2006) and chemotherapy-induced peripheral neuropathy (Carozzi et al., 2008a).
Consistent with the expression of NAAG immunoreactivity in large and some mid-size spinal sensory neurons (Cangro et al., 1987), expression of mGluR3 by these neurons (Carlton and Hargett, 2007), and the analgesic efficacy of Group II mGluR agonists on peripheral neurites (Yang and Gereau, 2003), NAAG and GCP II inhibitors were shown to be analgesic when injected directly into the hindpaw before induction of an inflammatory insult. In each of these studies, analgesia induced by peptidase inhibition was blocked by coadministration of the group II mGluR3 antagonist LY341495. In addition, analgesic effects of locally administered GCP II inhibitors were also attenuated by mGluR3 antagonism (Yamamoto et al., 2007), supporting the conclusion that the process is mediated by NAAG activation of mGluR3. The slow onset and prolonged effect of inhibition might be due to effects on pain-generating mechanisms at the site of injury. This would be consistent with the observation that hyperalgesia did not return for days even after dosing was halted once analgesia had been achieved.
Although we have interpreted the delayed analgesic effect of GCP II inhibition as indirect pharmacology, alternative possibilities exist. The CCI ligation model has temporal alterations in its mechanisms including an initial neuroinflammatory component followed by more central plastic changes (Bennett and Xie, 1988). For example, it is possible that new mechanisms of neuropathic pain exquisitely sensitive to GC II inhibition might be emerging after the initiation of the treatment at 10 days. These developing neuropathic mechanisms may be responsible, in part, for the delayed onset of effect. Further experiments exploring the exact timing of effect would be necessary to evaluate this alternative hypothesis.
The prolonged effects of 2-MPPA suggest that GCP II inhibition would be a valuable addition to currently available pain therapies. Such a therapy would be long lasting and with a mechanism distinct from available analgesics, it would be useful for combination approaches. In the clinic, 2-MPPA was well tolerated in man at doses achieving plasma exposures equivalent to those shown to be effective in the current animal models (van der Post et al., 2005). These experiments suggest that GCP II inhibition may have therapeutic utility in neuropathic pain and that efficacy might not be expected in acute administration but rather might take multiple administrations for effect and have to be sustained for some time even without further drug administration. Thus, clinical trials could explore once per day dosing even for a short half-life compound.
Abbreviations
- 2-MPPA
2-(3-mercaptopropyl) pentanedioic acid
- CCI
chronic constriction injury
- GCP II
glutamate carboxypeptidase II
- LC-MS/MS
liquid chromatography-mass spectrometry/ mass spectrometry
- NAAG
N-acetyl-aspartyl glutamate
- NEM
N-ethylmaleimide
Authorship Contributions
Participated in research design: Vornov, Wozniak, Rojas, Slusher.
Conducted experiments: Wozniak, Wu.
Performed data analysis: Wozniak, Wu, Rais.
Wrote or contributed to the writing of the manuscript: Vornov, Wozniak, Rais, Slusher.
Footnotes
This work was supported by and performed at Eisai Inc. and Johns Hopkins Brain Science Institute, Baltimore, Maryland.
References
- Adedoyin MO, Vicini S, Neale JH. (2010) Endogenous N-acetylaspartylglutamate (NAAG) inhibits synaptic plasticity/transmission in the amygdala in a mouse inflammatory pain model. Mol Pain 6:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bařinka C, Rojas C, Slusher B, Pomper M. (2012) Glutamate carboxypeptidase II in diagnosis and treatment of neurologic disorders and prostate cancer. Curr Med Chem 19:856–870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. (1988) A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33:87–107 [DOI] [PubMed] [Google Scholar]
- Berger A, Lassner F. (1994) Peripheral nerve allografts: survey of present state in an experimental model of the rat. Microsurgery 15:773–777 [DOI] [PubMed] [Google Scholar]
- Berger UV, Luthi-Carter R, Passani LA, Elkabes S, Black I, Konradi C, Coyle JT. (1999) Glutamate carboxypeptidase II is expressed by astrocytes in the adult rat nervous system. J Comp Neurol 415:52–64 [DOI] [PubMed] [Google Scholar]
- Besson JM. (1999) The neurobiology of pain. Lancet 353:1610–1615 [DOI] [PubMed] [Google Scholar]
- Cangro CB, Namboodiri MA, Sklar LA, Corigliano-Murphy A, Neale JH. (1987) Immunohistochemistry and biosynthesis of N-acetylaspartylglutamate in spinal sensory ganglia. J Neurochem 49:1579–1588 [DOI] [PubMed] [Google Scholar]
- Carlton SM, Hargett GL. (2007) Colocalization of metabotropic glutamate receptors in rat dorsal root ganglion cells. J Comp Neurol 501:780–789 [DOI] [PubMed] [Google Scholar]
- Carozzi V, Marmiroli P, Cavaletti G. (2008a) Focus on the role of Glutamate in the pathology of the peripheral nervous system. CNS Neurol Disord Drug Targets 7:348–360 [DOI] [PubMed] [Google Scholar]
- Carozzi VA, Canta A, Oggioni N, Ceresa C, Marmiroli P, Konvalinka J, Zoia C, Bossi M, Ferrarese C, Tredici G, et al. (2008b) Expression and distribution of ‘high affinity’ glutamate transporters GLT1, GLAST, EAAC1 and of GCPII in the rat peripheral nervous system. J Anat 213:539–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter KJ, Sen S, Matthews EA, Flatters SL, Wozniak KM, Slusher BS, Dickenson AH. (2003) Effects of GCP-II inhibition on responses of dorsal horn neurones after inflammation and neuropathy: an electrophysiological study in the rat. Neuropeptides 37:298–306 [DOI] [PubMed] [Google Scholar]
- Chen SR, Wozniak KM, Slusher BS, Pan HL. (2002) Effect of 2-(phosphono-methyl)-pentanedioic acid on allodynia and afferent ectopic discharges in a rat model of neuropathic pain. J Pharmacol Exp Ther 300:662–667 [DOI] [PubMed] [Google Scholar]
- Coyle JT. (1997) The nagging question of the function of N-acetylaspartylglutamate. Neurobiol Dis 4:231–238 [DOI] [PubMed] [Google Scholar]
- Danzer SC. (2012) Depression, stress, epilepsy and adult neurogenesis. Exp Neurol 233:22–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrman S, Palkovits M, Cassidy M, Neale JH. (1994) The regional distribution of N-acetylaspartylglutamate (NAAG) and peptidase activity against NAAG in the rat nervous system. J Neurochem 62:275–281 [DOI] [PubMed] [Google Scholar]
- Ghose S, Weickert CS, Colvin SM, Coyle JT, Herman MM, Hyde TM, Kleinman JE. (2004) Glutamate carboxypeptidase II gene expression in the human frontal and temporal lobe in schizophrenia. Neuropsychopharmacology 29:117–125 [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. (1988) A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain 32:77–88 [DOI] [PubMed] [Google Scholar]
- Jackson PF, Tays KL, Maclin KM, Ko YS, Li W, Vitharana D, Tsukamoto T, Stoermer D, Lu XC, Wozniak K, et al. (2001) Design and pharmacological activity of phosphinic acid based NAALADase inhibitors. J Med Chem 44:4170–4175 [DOI] [PubMed] [Google Scholar]
- Long JB, Yourick DL, Slusher BS, Robinson MB, Meyerhoff JL. (2005) Inhibition of glutamate carboxypeptidase II (NAALADase) protects against dynorphin A-induced ischemic spinal cord injury in rats. Eur J Pharmacol 508:115–122 [DOI] [PubMed] [Google Scholar]
- Lukawski K, Kamiński RM, Czuczwar SJ. (2008) Effects of selective inhibition of N-acetylated-alpha-linked-acidic dipeptidase (NAALADase) on mice in learning and memory tasks. Eur J Pharmacol 579:202–207 [DOI] [PubMed] [Google Scholar]
- Majer P, Jackson PF, Delahanty G, Grella BS, Ko YS, Li W, Liu Q, Maclin KM, Poláková J, Shaffer KA, et al. (2003) Synthesis and biological evaluation of thiol-based inhibitors of glutamate carboxypeptidase II: discovery of an orally active GCP II inhibitor. J Med Chem 46:1989–1996 [DOI] [PubMed] [Google Scholar]
- Neale JH, Olszewski RT, Zuo D, Janczura KJ, Profaci CP, Lavin KM, Madore JC, Bzdega T. (2011) Advances in understanding the peptide neurotransmitter NAAG and appearance of a new member of the NAAG neuropeptide family. J Neurochem 118:490–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszewski RT, Wegorzewska MM, Monteiro AC, Krolikowski KA, Zhou J, Kozikowski AP, Long K, Mastropaolo J, Deutsch SI, Neale JH. (2008) Phencyclidine and dizocilpine induced behaviors reduced by N-acetylaspartylglutamate peptidase inhibition via metabotropic glutamate receptors. Biol Psychiatry 63:86–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quidé Y, Witteveen AB, El-Hage W, Veltman DJ, Olff M. (2012) Differences between effects of psychological versus pharmacological treatments on functional and morphological brain alterations in anxiety disorders and major depressive disorder: a systematic review. Neurosci Biobehav Rev 36:626–644 [DOI] [PubMed] [Google Scholar]
- Rais R, Hoover R, Wozniak K, Rudek MA, Tsukamoto T, Alt J, Rojas C, Slusher BS. (2012) Reversible disulfide formation of the glutamate carboxypeptidase II inhibitor E2072 results in prolonged systemic exposures in vivo. Drug Metab Dispos 40:2315–2323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rojas C, Stathis M, Polydefkis M, Rudek MA, Zhao M, Ebenezer GJ, Slusher BS. (2011) Glutamate carboxypeptidase activity in human skin biopsies as a pharmacodynamic marker for clinical studies. J Transl Med 9:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sácha P, Zámecník J, Barinka C, Hlouchová K, Vícha A, Mlcochová P, Hilgert I, Eckschlager T, Konvalinka J. (2007) Expression of glutamate carboxypeptidase II in human brain. Neuroscience 144:1361–1372 [DOI] [PubMed] [Google Scholar]
- Schloesser RJ, Martinowich K, Manji HK. (2012) Mood-stabilizing drugs: mechanisms of action. Trends Neurosci 35:36–46 [DOI] [PubMed] [Google Scholar]
- Slusher BS, Robinson MB, Tsai G, Simmons ML, Richards SS, Coyle JT. (1990) Rat brain N-acetylated alpha-linked acidic dipeptidase activity. Purification and immunologic characterization. J Biol Chem 265:21297–21301 [PubMed] [Google Scholar]
- Slusher BS, Vornov JJ, Thomas AG, Hurn PD, Harukuni I, Bhardwaj A, Traystman RJ, Robinson MB, Britton P, Lu XC, et al. (1999) Selective inhibition of NAALADase, which converts NAAG to glutamate, reduces ischemic brain injury. Nat Med 5:1396–1402 [DOI] [PubMed] [Google Scholar]
- van der Post JP, de Visser SJ, de Kam ML, Woelfler M, Hilt DC, Vornov J, Burak ES, Bortey E, Slusher BS, Limsakun T, et al. (2005) The central nervous system effects, pharmacokinetics and safety of the NAALADase-inhibitor GPI 5693. Br J Clin Pharmacol 60:128–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ. (2011) Central sensitization: implications for the diagnosis and treatment of pain. Pain 152(3, Suppl)S2–S15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak KM, Rojas C, Wu Y, Slusher BS. (2012) The role of glutamate signaling in pain processes and its regulation by GCP II inhibition. Curr Med Chem 19:1323–1334 [DOI] [PubMed] [Google Scholar]
- Wroblewska B, Wroblewski JT, Saab OH, Neale JH. (1993) N-acetylaspartylglutamate inhibits forskolin-stimulated cyclic AMP levels via a metabotropic glutamate receptor in cultured cerebellar granule cells. J Neurochem 61:943–948 [DOI] [PubMed] [Google Scholar]
- Xi ZX, Kiyatkin M, Li X, Peng XQ, Wiggins A, Spiller K, Li J, Gardner EL. (2010a) N-acetylaspartylglutamate (NAAG) inhibits intravenous cocaine self-administration and cocaine-enhanced brain-stimulation reward in rats. Neuropharmacology 58:304–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi ZX, Li X, Peng XQ, Li J, Chun L, Gardner EL, Thomas AG, Slusher BS, Ashby CR., Jr (2010b) Inhibition of NAALADase by 2-PMPA attenuates cocaine-induced relapse in rats: a NAAG-mGluR2/3-mediated mechanism. J Neurochem 112:564–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Nozaki-Taguchi N, Sakashita Y. (2001) Spinal N-acetyl-alpha-linked acidic dipeptidase (NAALADase) inhibition attenuates mechanical allodynia induced by paw carrageenan injection in the rat. Brain Res 909:138–144 [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Hirasawa S, Wroblewska B, Grajkowska E, Zhou J, Kozikowski A, Wroblewski J, Neale JH. (2004) Antinociceptive effects of N-acetylaspartylglutamate (NAAG) peptidase inhibitors ZJ-11, ZJ-17 and ZJ-43 in the rat formalin test and in the rat neuropathic pain model. Eur J Neurosci 20:483–494 [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Saito O, Aoe T, Bartolozzi A, Sarva J, Zhou J, Kozikowski A, Wroblewska B, Bzdega T, Neale JH. (2007) Local administration of N-acetylaspartylglutamate (NAAG) peptidase inhibitors is analgesic in peripheral pain in rats. Eur J Neurosci 25:147–158 [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Kozikowski A, Zhou J, Neale JH. (2008) Intracerebroventricular administration of N-acetylaspartylglutamate (NAAG) peptidase inhibitors is analgesic in inflammatory pain. Mol Pain 4:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Gereau RW., 4th (2003) Peripheral group II metabotropic glutamate receptors mediate endogenous anti-allodynia in inflammation. Pain 106:411–417 [DOI] [PubMed] [Google Scholar]
- Yogeeswari P, Semwal A, Mishra R, Sriram D. (2009) Current approaches with the glutamatergic system as targets in the treatment of neuropathic pain. Expert Opin Ther Targets 13:925–943 [DOI] [PubMed] [Google Scholar]
- Zhang W, Murakawa Y, Wozniak KM, Slusher B, Sima AA. (2006) The preventive and therapeutic effects of GCPII (NAALADase) inhibition on painful and sensory diabetic neuropathy. J Neurol Sci 247:217–223 [DOI] [PubMed] [Google Scholar]
- Zhuo M, Wu G, Wu LJ. (2011) Neuronal and microglial mechanisms of neuropathic pain. Mol Brain 4:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo D, Bzdega T, Olszewski RT, Moffett JR, Neale JH. (2012) Effects of N-acetylaspartylglutamate (NAAG) peptidase inhibition on release of glutamate and dopamine in prefrontal cortex and nucleus accumbens in phencyclidine model of schizophrenia. J Biol Chem 287:21773–21782 [DOI] [PMC free article] [PubMed] [Google Scholar]