

Abstract
Compounds capable of light-triggered cytotoxicity are appealing potential therapeutics, because they can provide spatial and temporal control over cell killing to reduce side effects in cancer therapy. Two simple homoleptic Ru(II) polypyridyl complexes with almost-identical photophysical properties but radically different physiochemical properties were investigated as agents for photodynamic therapy (PDT). The two complexes were identical, except for the incorporation of six sulfonic acids into the ligands of one complex, resulting in a compound carrying an overall −4 charge. The negatively charged compound exhibited significant light-mediated cytotoxicity, and, importantly, the negative charges resulted in radical alterations of the biological activity, compared to the positively charged analogue, including complete abrogation of toxicity in the dark. The charges also altered the subcellular localization properties, mechanism of action, and even the mechanism of cell death. The incorporation of negative charged ligands provides a simple chemical approach to modify the biological properties of light-activated Ru(II) cytotoxic agents.
Short abstract
Two Ru(II) polypyridyl complexes with essentially identical photophysical properties but different charges and hydrophilicites were explored as potential agents for photodynamic therapy. Remarkably, the complex carrying a −4 overall charge exhibited excellent light-dependent cytotoxicity while remaining inactive in the dark. In contrast, the complex that was +2 charged caused significant cell death in the absence of the light trigger, likely because of its localization in the mitochondria and destruction of the membrane potential.
Introduction
Metal complexes have been studied for decades as potential cytotoxic agents, because of the unprecedented and continued success of cisplatin as a chemotherapeutic.1,2 Investigations into Ru(II) polypyridyl complexes have been particularly extensive, because of their ease of synthesis, appealing chemical, physical, and photophysical characteristics, and their high affinities for nucleic acids. Most early studies focused on characterizing the in vitro interactions of these complexes with DNA,3−6 and quantifying the potencies of the compounds both as traditional cytotoxic agents and as light-activated agents for photodynamic therapy (PDT) or phototherapy. In recent years, attention has shifted to understanding the cellular localization7−10 properties of Ru(II) complexes, along with their mechanisms of cellular uptake10−15 and cytotoxicity, providing a deeper understanding of how these compounds elicit their biological activities.
An attractive feature of Ru(II) polypyridyl complexes that makes them particularly useful for applications as biological probes and effectors is the diversity of the chemical structures that are readily available through modifications of the coordinated ligands.16 However, most biological studies have focused on complexes that carry an overall charge of +2 or greater. This significantly limits the chemical structures and physical properties of the molecules under investigation. A question that occurred to us was this: To what degree could the biological properties of a chemically inert Ru(II) complex be tuned by chemical modification of the ligands surrounding the metal center?
To address this question, we have investigated the biological activities, cellular uptake, localization, and mechanism of cell killing of two simple Ru(II) complexes that are commonly used dyes for solar cell research or biological staining, but have not been previously explored as PDT agents. Ru(bathophenanthroline)3 (1; see Chart 1) is a hydrophobic molecule with a high DNA binding affinity,17,18 while Ru(bathophenanthroline disulfonate)3 (2) is hydrophilic and possesses a high affinity for proteins.19−21 Both complexes are efficient singlet oxygen (1O2) generators with the same quantum yields for 1O2 production (ΦΔ) and similar molar extinction coefficients (ε).22 Both are luminescent, allowing for analysis by fluorescence microscopy. However, although the photophysical properties of the compounds are almost identical, the physical properties of the two Ru(II) compounds are quite dissimilar (see Table 1). Compound 1 carries an overall charge of +2, while 2 has an overall charge of −4. They also have very different hydrophilicities, as indicated by their partition coefficient or log P values. Given the radically different physical properties of the complexes, we anticipated differences in their biological effects that could provide information for future rational design of light-activated cytotoxic agents.
Chart 1. Structures of Compounds 1 and 2.

Table 1. Physical and Photophysical Properties of 1 and 2.
Considering the established dogma of the field, it would be expected that the negatively charged compound 2 would not enter cells23 and would suffer low efficacy, while the positively charged, DNA-binding compound 1 would prove the more effective PDT agent. Instead, our findings run counter to this prediction. Here, we show large differences in potency, cellular uptake, localization, and mechanism of cytotoxicity of these agents, illustrating that radical modulation of biological properties is possible with ligand modifications of simple homoleptic Ru(II) polypyridyl complexes that are substitutionally inert. Most importantly, our results also prove that a significantly greater range of charge states and physical properties of Ru(II) complexes are compatible with potential application as PDT agents.
Results
DNA Damage and Cytotoxicity
As both 1 and 2 are efficient catalysts for the light-activated generation of 1O2 (ΦΔ = 0.42, 0.43),22 it was expected that the two compounds would act as sensitizers for PDT. Accordingly, their DNA damaging properties were assessed with pUC19 plasmid DNA and analyzed by gel electrophoresis (Figure 1). Each compound was incubated with plasmid and irradiated with 40 J/cm2 of visible light (>400 nm) or kept in the dark. Compound 1 is known to bind strongly with DNA, and precipitation of the DNA with the complex was observed at concentrations above 31.3 μM both in the light and in the dark (Figure 1A). In contrast, 2 is a much more hydrophilic molecule, and the negatively charged sulfonate functional groups were anticipated to cause electrostatic repulsions between the complex and the negatively charged backbone of the DNA. Consistent with low DNA affinity,26 no DNA precipitation or smearing was observed with up to 500 μM of 2 (Figure 1B). When exposed to light, both 1 and 2 induced single strand DNA breaks, creating relaxed circular plasmid. This is likely due to the photogeneration of 1O2 that mediates the DNA damage. However, for 2, the amount of relaxed circle plasmid did not exhibit any concentration dependence above 125 μM, suggesting either a reduction in ΦΔ as the concentration of the complex is increased, or alternative quenching mechanisms that impede DNA damage.27
Figure 1.

Agarose gel electrophoresis of pUC19 with increasing concentrations of (A) 1 and (B) 2 in the dark or irradiated (λ > 400 nm). Lanes 1 and 12, DNA molecular weight standard; lane 2, linear (reaction with EcoR1); lane 3, relaxed circle (reaction with Cu(phen)2); lanes 4–11, 0, 8.25, 16.5, 31.3, 62.5, 125, 250, and 500 μM compound. Cytotoxicity dose response of (C) 1 and (D) 2 in the dark (open squares) or irradiated (closed circles). HL60 cells were incubated for 72 h with compound prior to quantification of viability.
As both compounds are capable of light-induced DNA damage, the cytotoxicity of 1 and 2 were evaluated in the A549 human non-small cell lung cancer, the HL60 human promyelocytic leukemia, and the Jurkat human T lymphoblastoid cell lines in the presence and absence of 7 J/cm2 of >400 nm light. The IC50 values across the cell lines for 1 ranged from 0.62 to 3.75 μM in the dark. Upon irradiation, potency was increased to a range of 0.075 to 0.35 μM, resulting in an average phototoxicity index (PI = IC50 (dark)/IC50 (light) of 10- to 20-fold (see Figure 1, Table 2)).
Table 2. Cytotoxicity IC50 Values (μM) in Various Cell Linesa.
| IC50 Value (μM) |
|||||||
|---|---|---|---|---|---|---|---|
| compound | HL60, light | HL60, dark | A549, light | A549, dark | Jurkat, light | Jurkat, dark | PIb Jurkat |
| 1 | 0.35 ± 0.18 | 3.75 ± 0.18 | 0.11 ± 0.02 | 0.62 ± 0.08 | 0.075 ± 0.004 | 1.63 ± 0.11 | 21.7 |
| 2 | 9.81 ± 1.09 | >300 | 17.25 ± 9.82 | >300 | 3.31 ± 0.36 | >300 | >90 |
| cisplatin | 3.1 ± 0.2 | 3.1 ± 0.2 | 3.4 ± 0.6 | 3.5 ± 0.2 | 0.5 ± 0.07 | 0.5 ± 0.07 | 1 |
IC50 values are averages from three measurements.
The phototoxicity index (PI) is the ratio of the dark and light IC50 values.
In marked contrast to the high toxicity of 1 in the absence of light, no toxicity was observed in the dark with 2 across all cell lines at concentrations up to 300 μM. However, compound 2 was effective in killing cells when irradiated, with IC50 values ranging from 3.3 μM to 17.3 μM, consistent with the concentrations required for in vitro DNA damage. This provides for a large therapeutic window, as no cell death is observed for samples in the absence of irradiation.
Surprisingly, not only was compound 1 toxic to cells upon irradiation, it also induced cell death far more rapidly in HL60 cells than traditional DNA damaging agents such as cisplatin. Complete cell death was observed within 2 h of irradiation with 1 (see Figure S1 in the Supporting Information). The compound also induced cell death in the dark, but more slowly, with ∼30% viable cells remaining at 24 h. In marked contrast to 1, compound 2 induced cell death after irradiation only after a long delay, with 70% viable cells remaining at 24 h and 55% remaining at 48 h.
The significant disparity in the potency, phototoxicity index (PI), and rates of cell killing for the two compounds despite their equivalent abilities to sensitize 1O2 strongly suggested that they were acting through different cellular mechanisms, possibly by interacting with different biological targets. As the compounds are substitutionally inert, they are unlikely to covalently modify biomolecules. This complicates target identification by isolation of protein or nucleic acid components of the cells, so the subcellular localization was investigated instead.
Cellular Uptake and Subcellular Localization
Both 1 and 2 are emissive, allowing for direct visualization in cells. Flow cytometry and fluorescence microscopy were used to provide relative uptake values, the time dependence of compound uptake, and information on the subcellular localization of the compounds. To optimize signal intensity, 1 was assayed at 5 μM while 20 μM was required for 2. Flow cytometry with A549 cells revealed greater uptake of 1 compared with 2 at time points of 2 and 24 h, with an 11.6-fold and 8.2-fold difference in signal, respectively (see Figure S2 in the Supporting Information). Between the time points of 2 and 24 h, the average emission of cells incubated with 1 increased by 2.8-fold while the amount of 2 increased by 4-fold. This data was supported by direct quantification of the number of ruthenium atoms per cell using inductively coupled plasma–optical emission spectroscopy (ICP-OES; see Tables S1 and S2 in the Supporting Information). The relative amount of compound 1 in cells versus the cell media increased ∼4-fold from 2 h to 24 h, from 5.4% to 22%.28 Much less of compound 2 entered the cells, with a maximal uptake of 0.7% at 24 h. While low, this degree of uptake of the −4-charged 2 is comparable to cisplatin, a neutral compound, dosed at the same concentration (20 μM, 0.8% at 24 h).
The emission of 1 and 2 was also measured in A549 cells using an ApoTome microscope (the adherent cell line was chosen to facilitate the required wash steps to allow for the use of fluorescent reporters). The relative rates of uptake of each of the complexes were analyzed as a function of time, and both 1 and 2 were visible inside cells as early as 2 h after compound addition (see Figure S3 in the Supporting Information), consistent with the flow cytometry and ICP-OES results. Images were taken at 2, 8, 18, and 24 h, and intracellular levels of both complexes appeared to plateau at 8 h. The uptake data showed good agreement between the three techniques and two cell lines, suggesting similar behavior in adherent and suspension cell lines.
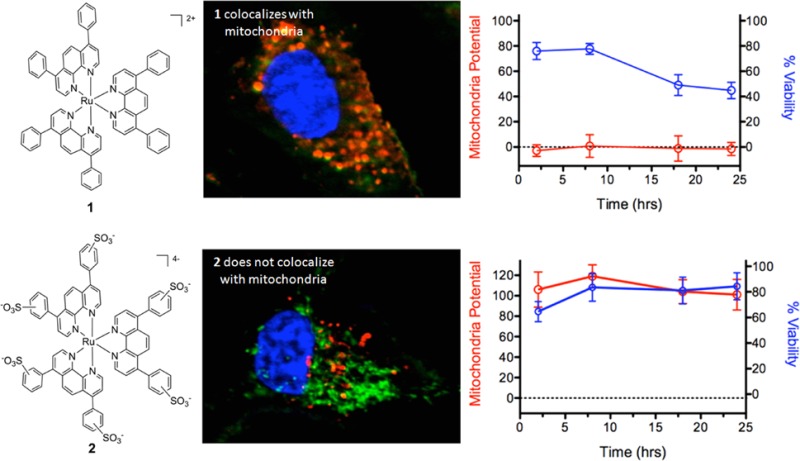
Differences in the intracellular localization of 1 and 2 were evaluated by determining colocalization of luminescence of the compounds and fluorescent markers of organelles in A549 cells. Overlap in signals between the compounds, cellular nucleus, mitochondria, and lysosome was measured over a 24 h period. Compounds 1 and 2 have very different localization profiles, as exemplified by the imaging 8 h after dosing (Figure 2). Compound 1 substantially localized to lysosomes and the mitochondria in the absence of light, while 2 remained primarily in the cytosol. Exposure to light did have an impact on compound localization, where 1 induced nuclear localization of both the mitochondrial and lysosome markers (see Figures 2A and 2B, top), indicating that photoinduced damage mediated by 1 reduced the integrity of the nuclear membrane. In contrast, 2 was primarily observed in lysosomes after irradiation, and did not co-localize with mitochondria (Figures 2A and 2B, bottom). In addition, irradiation in the presence of 2 did not result in the appearance of organelle markers in the nucleus, suggesting the nuclear membrane remained intact. Neither 1 nor 2 was found to associate with the plasma or nuclear membranes.
Figure 2.
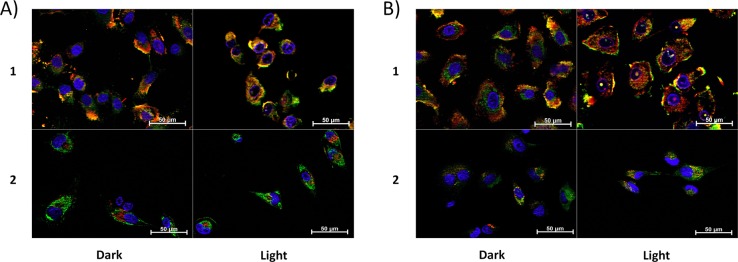
ApoTome microscopy showing subcellular localization of 1 and 2 at 8 h. Co-localization of 1 and 2 in mitochondria or lysosomes is indicated by the apparent yellow emission. (A) Mitotracker Green FM was used to image mitochondria. (B) Lysotracker Green DND-26 was used to image lysosomes. Red color denotes intrinsic emission of 1 and 2, whereas blue color denotes Hoechst staining of the nucleus. The yellow color occurs due to overlap of the red emission from the ruthenium complexes and green emission of the organelle-specific dyes, indicating colocalization. Compound 1 localizes in both the mitochondria and the lysosomes while 2 was not predominantly found in either organelle.
Mitochondrial Function and Time Dependence for Cell Death
As compound 1 appeared to localize within mitochondria, it was hypothesized that this may account for its high toxicity in the absence of irradiation. To determine if either 1 or 2 caused a reduction in mitochondrial function, mitochondrial membrane potential was measured using tetramethylrhodamine ethyl ester (TMRE) (see Figure 3). TMRE is a cationic dye that accumulates in active mitochondria as a result of the negative membrane potential (Δψm). Inactive or depolarized mitochondria exhibit a decreased membrane potential, and TMRE does not localize in these organelles. Compound 1 induced rapid and complete depolarization of mitochondria both in the dark and upon irradiation (Figure 3B, 3C). However, surprisingly, cell viability did not parallel mitochondrial potential. While mitochondrial function was completely impaired at 2 h post treatment with 1 in the dark, viability decreased slowly, with 44% ± 6% viable cells remaining after 24 h. In contrast, both mitochondrial function and cell viability fell to 0% within 2 h of irradiation. Thus, while 1 completely impedes mitochondrial function within 2 h even in the absence of light, irradiation induced additional damage that results in rapid cell death, along with the loss of mitochondrial function.
Figure 3.
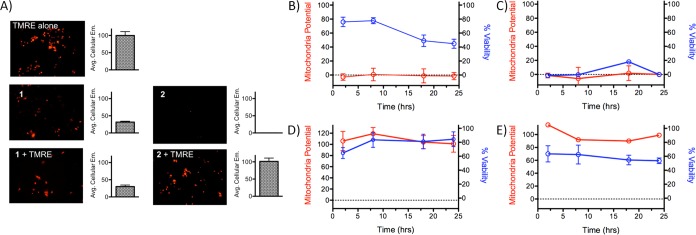
(A) Example images and quantification of the emission of tetramethylrhodamine ethyl ester (TMRE) in the presence and absence of 1 and 2. Compound 1 does not show increased TMRE emission over background emission of 1, while compound 2 does not add to the TMRE emission. Mitochondrial potential and cell viability of A549 cells as a function of time for (B) 1, in the dark; (C) 1, irradiated; (D) 2, in the dark; and (E) 2, irradiated. TMRE was used to quantify membrane potential; values are relative to a no-compound control value of 100.
In contrast to compound 1, 2 did not significantly reduce the mitochondrial potential over a 24 h period either when irradiated or kept in the dark. Irradiation did reduce cell viability to 70% ± 12% at 2 h and 59% ± 4% after 24 h, but this occurred without a significant decrease in the relative mitochondrial potential (Figure 3E), indicating that 2 does not act through inhibition of mitochondrial function. These results strongly suggest that mitochondrial failure plays a role in the dark toxicity of 1, and the lack of mitochondrial localization and inhibition may explain the comparatively low dark toxicity of 2.
Mechanism of Cell Death
Most compounds used for PDT that generate singlet oxygen induce apoptosis. Given the different cellular localization properties and time profiles for cell death induced by 1 and 2, the mechanism of cell death was investigated. Indicators of apoptotic cell death (activation of PARP and caspase 3 through proteolysis) were determined in HL60 cells treated with either 1 or 2 (Figure 4A). The known apoptotic-inducing compounds, cisplatin and doxorubicin, were run in parallel. Compound 1 induced the proteolytic activation of both PARP and caspase 3 within 2 h of irradiation (Figure 4A). In the absence of light, PARP and caspase 3 were observed with 1, but only after 18 and 24 h, and to a lesser degree. In the absence of irradiation, the amount of inactive procaspase 3 did not change.
Figure 4.

(A) Western blot analysis of cleaved PARP, caspase 3, and pro-caspase 3. GAPDH is used as a loading control. (B) Agarose gel electrophoresis of genomic DNA harvested from HL60 cells after treatment with various agents. Lanes 1 and 9, ladder; lane 2, no compound control; lane 3, 10% EtOH, 24 h (necrosis control); lane 4, cisplatin, 24 h (apoptosis control); lane 5, 1, irradiated, 8 h; lane 6, 1, in the dark, 8 h; lane 7, 2, irradiated, 24 h; and lane 8, 2, in the dark, 24 h.
Exposure of 2 to light also induced PARP cleavage as early as 2 h, but unlike 1, increasing amounts of cleaved PARP were observed over the course of 24 h (Figure 4A). The increase in the level of activated caspase 3 also occurred on a slower time scale than PARP cleavage, with the protein initially observed at 18 and 24 h. This suggests that the irradiated samples undergo apoptosis that is not primarily signaled through caspase 3. Conversely, cells treated with 2 and protected from light did not display PARP or caspase 3 cleavage, which is consistent with viability measurements indicating no cytotoxicity in the absence of light. The level of procaspase 3 also did not change over 24 h, further confirming a lack of cytotoxicity under these conditions.
Since 2 showed strong PARP induction without significant caspase 3 activation, as compared to 1 and cisplatin or doxorubicin, a mechanism of cell death through necrosis was explored. The level of an alternate 55 kDa PARP fragment was determined by immunoblot as a marker for necrosis (see Figure S6 in the Supporting Information), with 10% (v/v) ethanol used as a positive control.29 Exposure of HL60 cells to 1 produced this fragment at significant levels both when protected from light and when irradiated, which is consistent with necrosis. Cisplatin and doxorubicin also produced this cleavage product, indicating that some cells had progressed into necrosis (Figure S6 in the Supporting Information). In contrast, cells exposed to 2, both in the presence and absence of irradiation, produced a lower level of the 55 kDa PARP fragment, similar to the untreated cells, suggesting necrosis is not a significant cell death pathway for this compound.
To support the assessment of the disparate mechanisms of cell death induced by 1 and 2, the degradation pattern of genomic DNA was investigated. DNA laddering is observed as a result of DNA fragmentation stemming from the executionary phase of apoptosis. In contrast, necrotic cell death lacks this characteristic laddering effect, allowing differentiation between these two mechanisms. HL60 cells were exposed to the Ru(II) complexes, cisplatin, and 10% ethanol, followed by genomic DNA isolation and resolution by gel electrophoresis. As expected, the apoptosis inducer, cisplatin, initiated DNA fragmentation, resulting in a laddering pattern on the gel (Figure 4B). This laddering was absent in the cells treated with ethanol and compounds 1 and 2 in the dark. However, both compounds 1 and 2 displayed similar laddering patterns as cisplatin when irradiated, suggesting apoptosis is a contributing cell death pathway for both compounds when irradiated. In contrast, given the cytotoxicity of 1 in the dark and the presence of the 55 kDa PARP fragment, it appears that necrotic cell death is a significant pathway for 1. For compound 2, the absence of DNA laddering is a result of the lack of cytotoxicity of the negatively charged compound.
Flow cytometry was also employed to further corroborate the assessment of the mechanism of cell death using Annexin V/propidium iodide (PI) or Annexin V/Hoechst staining to differentiate apoptotic versus necrotic cell death. Fluorescent Annexin V conjugates recognize the translocation of phosphatidylserine to the outer leaflet of the cell membrane during apoptosis. PI and Hoechst were used as nuclear stains to distinguish between live and dead cells; while PI is most commonly used, Hoechst was also applied, because of spectral interference of 1 and 2 with PI. The presence of significant populations of PI positive cells in the absence of Annexin V staining for 1 both in light and in the dark demonstrate that necrosis is a significant cell death pathway for this compound (see Figures S7 and S8 in the Supporting Information; 5% and 14%, respectively, at 2 h). In contrast, compound 2 produced large populations of Annexin V positive cells (55% at 24 h), showing an apoptotic pathway (see Figures S9 and S10 in the Supporting Information). Similarly, Hoechst staining was consistent with necrosis as a contributing pathway for 1 in the dark and in light, while 2 induced apoptosis.
Discussion
The goal of phototherapy is to achieve cell death in cancerous or abnormal tissues that are irradiated, while protecting healthy tissues that are not exposed to light. As a result, the compounds developed for this application should possess large “therapeutic windows” where the toxicity in the dark is minimized. This has proven to be challenging for many inorganic agents developed for PDT applications. The rational design of new and more efficacious compounds would be facilitated by (1) a better understanding of the mechanisms of action that induce dark toxicity for promising PDT agents, and (2) the identification of chemical features that eliminate dark toxicity. The investigations of the biological activities of these two simple Ru(II) complexes provide guidance for both approaches toward improved PDT agents.
While compound 1 demonstrated notable potency when irradiated (0.075–0.35 μM, depending on cell line), the toxicity of the compound in the dark (0.62 to 3.75 μM) reduces its potential as a PDT agent. Imaging studies showed both mitochondrial and lysosomal localization, and assessment of mitochondrial potential indicated that 1 immediately inhibits mitochondrial function. However, the disconnect between the time dependence of the inhibition of mitochondrial function and the reduction in cell viability in the dark shows that disruption of mitochondrial membrane potential does not lead to rapid cell death. It is possible that the cells treated with 1 and kept in the dark survive for several hours, despite the complete abrogation of mitochondrial function, due to the Warburg effect, where cancer cells exhibit a reduced reliance on oxidative phosphorylation and increased dependence on glycolysis for energy production.
After 72 h, cell death is complete for 1 in the dark. The obliteration of mitochondrial function would explain the observation of necrotic cell death in this case.30 In contrast, when 1 is irradiated, it appears that a combination of necrotic and apoptotic pathways are activated. Cell death is so rapid (with most cells undergoing death at 2 h, as indicated by Trypan Blue staining) that necrosis is likely to be a primary pathway for a majority of cells. The breakdown of membrane integrity is apparent not only for the plasma membrane, but also for the membranes of organelles, as cells treated with 1 and irradiated showed nonspecific nuclear localization of both Mitotracker and Lysotracker. The high dark toxicity, mitochondrial localization, and induction of necrotic death pathways may reduce the potential of compounds structurally similar to 1 for PDT.
In marked contrast, compound 2 was found to possess several features that encourage further exploration of derivatives or similar compounds. While uptake was low, it was comparable to that of cisplatin, despite the overall charge of −4. Most importantly, 2 exhibited IC50 values on the order of 3.3–17.3 μM when exposed to light, with no observed toxicity in the dark. A slight increase in intracellular accumulation was observed upon irradiation, possibly due to induction of plasma membrane damage that facilitated compound uptake. Once inside the cell, the compound remained in the cytosol, with no observable localization to the mitochondria or inhibition of mitochondrial function. Furthermore, the light-induced cell death mediated by 2 occurred by apoptosis, in contrast to the mitochondrial targeting of 1, which resulted in necrosis. One possibility to explore is that PDT compounds that avoid mitochondrial localization will exhibit lower toxicity in the absence of light than those that associate with the mitochondria. It is anticipated that structural modifications that result in a modest increase in cellular uptake could sufficiently drive down potency to make improved derivatives of 2 that maintain large therapeutic windows.
Conclusions
Given the high binding affinities of most Ru(II) complexes for DNA (Kb > 106), it was previously believed that the compounds would preferentially localize in the nucleus, and indeed several do.7,8,31−33 However, recent fluorescence and electron microscopy studies have shown localization of several Ru(II) compounds in the mitochondria, suggesting it is a common target.34,35 Other reports indicate membrane accumulation and disruption,10,36 along with apoptosis pathways that are mediated by the mitochondria.37,38 Gasser and co-workers have shown in a recent report that this mitochondrial localization was, in fact, required for cytotoxicity for a lead compound in a structure–activity relationship study of a family of Ru(II) polypyridyl complexes.39
While mitochondrial accumulation results in cytotoxicity, this mechanism is not likely to be compatible with a PDT-type approach where such redox-active compounds are required to be essentially nontoxic in the absence of photons. Alternatively, targeting moieties such as nuclear localization signals can be conjugated to the coordinated ligands to affect the affinity of the complex for biological molecules and regulate cellular uptake and subcellulation localization.14,39,40 This approach requires significant chemical modifications, and the targeting often fails to increase cytotoxicity.
While microscopy is a powerful tool to assess compound localization, imaging experiments can cause relocalization of compounds that induce production of 1O2 or perform other photochemical reactions when exposed to light.41 Previous studies on porphyrins used for PDT applications also have demonstrated this phenomenon,42 including uptake and relocalization of an anionic tetrasulfonated porphyrin.43 For this reason, it is important to perform imaging using a minimum of light exposure, and to probe for compound relocalization by comparing to conditions where the treated cells have been exposed to significant light doses.
This current study shows that simple ligand modifications produce complexes with divergent physical properties and, correspondingly, different biological activities. Compound 1, despite its high DNA affinity, localizes to the mitochondria and induces rapid membrane depolarization and necrotic cell death. It is possible that this will be a common problem for compounds containing the bathophenanthroline ligand. Compound 2, despite its overall charge of −4, is taken up into cancer cells to a sufficient degree to mediate light-induced cell death through an apoptotic pathway. The absence of mitochondrial localization may be the factor that eliminates the dark toxicity of this Ru(II) complex. The incorporation of the sulfonic acids into the ligands is likely responsible for the alteration in subcellular localization, suggesting a possible general approach to reducing dark toxicity for other Ru(II) complexes developed for applications in phototherapy.
Experimental Section
Materials
Ru(bathophenanthroline)3 (1) and Ru(bathophenanthroline disulfonate)3 (2) were synthesized using previously established procedures.20,44 All cell lines were purchased from ATCC. Cell culture media, heat-inactivated fetal bovine serum (FBS), 4–20% tris-glycine precast gels, Dulbecco’s phosphate buffered saline (DPBS), penicillin-streptomycin solution (pen-strep), and 0.4% Trypan Blue solution were from Invitrogen. 35 mm wide, 4-compartment CELLview cell culture dishes were obtained from USA Scientific. Serum supreme was from Lonza. Hoechst 33342, Lysotracker Green DND-26 and Mitotracker Green FM were purchased from Invitrogen. Propidium iodide (PI) and FITC-Annexin V were obtained from BD Science. Trimethylrhodamine ethyl ester (TMRE) was purchased from Sigma–Aldrich. Antibodies for PARP-1, procaspase 3, and GAPDH were from Santa Cruz Biotechnology, Inc., while cleaved PARP and cleaved caspase 3 was from Cell Signaling Technology. RIPA buffer was purchased from Santa Cruz Biotechnology, Inc. Clarity Western ECL Substrate was from Bio-Rad. An apoptotic DNA-ladder kit was purchased from Roche Applied Science.
DNA Gel Electrophoresis
Compounds were mixed with 40 μg/mL pUC19 plasmid DNA in 10 mM potassium phosphate buffer, pH 7.4. To determine the effect of light, samples were irradiated with light (>400 nm) from a 200 W light source for total light doses of 40 J/cm2. Samples were then incubated for 12 h at room temperature in the dark. Single- and double-strand DNA break controls were prepared, and the DNA samples were resolved on agarose gels, as described previously.45
In brief, samples were resolved on a 1% agarose gels prepared in tris-acetate buffer with 0.3 μg of plasmid/lane. The gels were stained with 0.5 μg/mL ethidium bromide in tris-acetate buffer at room temperature for 40 min, destained with tris-acetate buffer, and imaged on a ChemiDoc MP System (Bio-Rad).
Cell Cytotoxicity Determination
Human alveolar adenocarcinoma cell line A549, Human promyelocytic leukemia cell line HL60, and Human T lymphocyte cell line Jurkat cells were maintained in media supplemented with 10% FBS and 50 U/mL pen-strep at 37 °C with 5% CO2, with DMEM used for A549 cells and IMDM and RPMI 1640 used for HL60 and Jurkat cells, respectively. Cells were assayed in Opti-MEM supplemented with 1% serum supreme and 50 U/mL pen-strep and seeded into 96 well plates at a density of 1.5 × 103 cells/well for A549, 2 × 104 cells/well for HL60, and 1 × 104 cells/well for Jurkat followed by a 6 h incubation at 37 °C, 5% CO2. Cells were then dosed with serial dilutions of compound and incubated for 18 h. They were then irradiated with 7 J/cm2 light (>400 nm) in 30 s pulses or kept in the dark. Cell viability was determined 72 h later by measuring the conversion of resazurin to resorufin,45 using a SpectraFluor Plus Plate Reader (Tecan).
Intracellular Measurement of Ru Complexes by Flow Cytometry
A549 cells were seeded in Opti-MEM with 1% serum supreme at a concentration of 2 × 105 cells/ml in 25 cm2 cell culture flasks and incubated overnight. A concentration of 5 μM of 1 and 20 μM of 2 were added to the cells and incubated for 2 or 24 h protected from light; the concentration of 20 μM was selected for 2 to correspond with the IC50 of the complex when irradiated with light. A concentration of 5 μM was used for 1 for compatibility with fluorescent imaging. After 2 and 24 h the media was removed, cells were washed twice with DPBS, trypsinized, and collected by centrifugation at 125 × g for 4 min. The cells were resuspended in Opti-MEM, filtered through 40-μm cell strainers, and analyzed on a FACSCalibur (Becton–Dickenson) with an excitation wavelength of 488 nm and emission measured at 650 nm. A minimum of 30,000 events were measured for each sample.
ApoTome Structured Illumination Imaging of Ru Complex Uptake
The ApoTome microscope was used to resolve fine features of cellular structure. This instrument averages the fluorescence of three separate images to greatly reduce out-of-plane fluorescence. A549 cells were seeded in 35 mm, four-compartment CELLview culture dishes at a density of 5 × 104 cells per compartment in a 500 μL volume and incubated for 24 h in Opti-MEM containing 1% FBS, followed by the addition of 5 μM 1 or 20 μM 2 and time points measured at 2, 8, 18, or 24 h. Media was removed at each time point, cells rinsed with DPBS, and incubated in Opti-MEM with 16 μM Hoechst 33342, 16 μM Hoechst and 0.15 μM Lysotracker Green DND-26, or 16 μM Hoechst and 0.2 μM Mitotracker Green FM. Cells were incubated for 30 min then washed three times with DPBS and imaged at 50× magnification using an ApoTome structured illumination fluorescent microscope (Carl Zeiss AG).
Mitochondrial Membrane Potential Measurement
A549 cells were seeded at 2 × 104 cells/well in 24-well plates and incubated for 18 h, followed by the addition of 5 μM 1 or 20 μM 2. They were incubated for an additional 8 h, and then irradiated with light as described for cell cytotoxicity measurements or kept in the dark. The cells were incubated for an additional 2, 8, 18, or 24 h, washed with DPBS, followed by the addition of 0.5 μM TMRE in Opti-MEM, and incubated for 30 min. The cells were then washed twice with DPBS and imaged. The change in TMRE signal was determined by the difference in fluorescence between compound-treated cells in the presence and absence of TMRE. Fluorescence was measured at 10× magnification using an ApoTome microscope in normal fluorescence mode. Images were processed and the average cell fluorescence was calculated using ImageJ software. Samples dosed with 1 and 2 were compared to untreated A549 cells (n = 3) to give the relative mitochondrial potential.
Cell Viability As a Function of Time
HL60 cells were seeded in Opti-MEM at a density of 1 × 106 cells/mL and incubated for 2 h. The cells were then dosed 1 or 2, incubated for 8 h, irritated as above or protected from light, and incubated for 2, 8, 18, 24, or 48 h. As HL60 cells grow in suspension, Trypan Blue staining was employed in place of resazurin to simplify and accelerate cell viability analysis. At each time point, a 10 μL cell suspension was mixed with an equal amount of Trypan Blue solution and cell viability determined by manual counting with a hemocytometer.
Apoptosis Marker Immunoblotting
HL60 cells were cultured, dosed with 1 and 2, then irradiated as above or protected from light. Cells were harvested 0, 2, 8, 18, or 24 h after treatment, pelleted by centrifugation at 124 × g for 5 min, washed twice with DPBS, and lysed in RIPA buffer supplemented with 5 mM sodium pyrophosphate for 15 min on ice. The insoluble fraction was removed by centrifugation at 20,800 × g for 10 min at 4 °C. The supernatant was collected and the protein concentration determined by BCA assay. Ten micrograms (10 μg) of protein was loaded onto 4–20% tris-glycine gels, followed by transfer to nitrocellulose membranes. After blocking with 5% nonfat milk in DPBS with 0.1% Tween20 (PBST) for 1 h at room temperature, the membrane was immunoblotted with PARP-1 at a 1:500 dilution, procaspase 3, cleaved caspase 3, and cleaved PARP at 1:1,000 dilutions, or GAPDH at a 1:2,000 dilution in 5% nonfat milk overnight at 4 °C. Immunoblots were washed with PBST for 10 min four times and incubated for 1 h with secondary antibodies at a 1:10,000 dilution for GAPDH and at a 1:5,000 dilution for all other antibodies. Detection was performed using Clarity Western ECL substrate and imaged with the ChemiDoc MP System.
DNA Laddering Gel Electrophoresis
HL60 cells were cultured, dosed with 1 and 2, then photoactivated or protected from light as detailed above. Cells were harvested 2 h after photoactivation for 1 or after 24 h for 2, pelleted by centrifugation at 124 × g for 5 min, washed twice with DPBS, and prepared with an apoptotic DNA-ladder kit as per manufacturer instructions. Gel electrophoresis was carried out using a 1% agarose gel containing 0.5 μg/mL ethidium bromide with 1 μg DNA loaded per lane and run for 90 min at 75 V. Gel imaging was performed with the ChemiDoc MP System.
Quantification of Metal Complex Uptake by ICP-OES
HL60 cells were seeded in Opti-MEM at a density of 1 × 106 cells/mL in 25 cm2 cell culture flasks, cultured overnight, then dosed with 5 μM 1, 20 μM 2, or 20 μM cisplatin. Cells treated with Ru complexes were incubated for 8 h, protected from light before irradiating or kept in the dark. Cells were collected after 2 and 24 h by centrifugation at 124 × g for 5 min. The cell media was transferred to separate 15-mL centrifuge tubes, and cells were washed twice with DPBS. Two milliliters (2 mL) of concentrated HNO3 was added to media samples while cell pellets were resuspended in 5 mL of 20% (v/v) HNO3. All samples were heated at 110 °C for 3 h. After digestion, the volume of all cell samples was adjusted to 5 mL and media samples were adjusted to 10 mL with deionized (DI) water. The metal content was analyzed using a VISTA-PRO CCD simultaneous inductively coupled plasma optical emission spectrometer (Varian, Inc.) with detection at 240.272, 245.657, and 267.876 nm for ruthenium and 214.424, 217.468, and 265.945 nm for platinum, with a replicate reading time of 60 s. Yttrium (1 ppm) in 1% nitric acid was employed as an internal standard. The percentage intracellular metal ratio in 107 cells was calculated by normalizing the metal amount to 107 cells, and then divided by the total metal amount in both media and cells.
Cell Death by Flow Cytometry
HL60 cells were cultured, dosed with 1 and 2, then irradiated or protected from light. Cells were harvested 2 and 24 h after treatment, pelleted by centrifugation at 124 × g for 5 min, washed twice with DPBS, stained for 15 min with FITC-Annexin V and PI or FITC-Annexin V and Hoechst 33342, because of the interference between the emission of 1 and PI. Cells were analyzed with a FACSCalibur (Becton–Dickenson). A minimum of 20,000 events were measured for each sample.
Acknowledgments
This work was supported by the American Cancer Society (RSG-13-079-01-CDD, awarded to E.C.G.). M.D. acknowledges the University of Kentucky Cancer Nanotechnology Training Center (UK-CNTC) postdoctoral traineeship, supported by the NCI/NIH and part of the National Cancer Institute Alliance for Nanotechnology in Cancer (5R25 CA153954). Y.S. acknowledges the University of Kentucky for the Predoctoral Kentucky Opportunity Fellowship. ICP-OES was performed at the University of Kentucky Environmental Research Training Laboratory (ERTL), and flow cytometry was performed at the University of Kentucky Flow Cytometry and Cell Sorting Core Facility. The UK Flow Cytometry & Cell Sorting core facility is supported in part by the Office of the Vice President for Research, the Markey Cancer Center, and an NCI Center Core Support Grant (P30 CA177558) to the University of Kentucky Markey Cancer Center.
Supporting Information Available
Additional figures, tables, and experimental descriptions. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Author Contributions
‡ These authors contributed equally to this work.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Lippert B.Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug; Wiley–VCH: Weinheim, Germany, 1999. [Google Scholar]
- Cohen S. M.; Lippard S. J.. Prog. Nucleic Acid Res. Mol. Biol. 2001, 67, 93–130. [DOI] [PubMed] [Google Scholar]
- Hadjiliadis N.; Sletten E.. Metal Complex–DNA Interactions, 1st Edition; Wiley: Chichester, U.K., 2009. [Google Scholar]
- Moucheron C. New J. Chem. 2009, 332235–245. [Google Scholar]
- Boerner L. J. K.; Zaleski J. M. Curr. Opin. Chem. Biol. 2005, 92135–144. [DOI] [PubMed] [Google Scholar]
- Zeglis B. M.; Pierre V. C.; Barton J. K. Chem. Commun. 2007, 44, 4565–4579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill M. R.; Derrat H.; Smythe C. G.; Battaglia G.; Thomas J. A. ChemBioChem 2011, 126877–880. [DOI] [PubMed] [Google Scholar]
- Gill M. R.; Thomas J. A. Chem. Soc. Rev. 2012, 4183179–3192. [DOI] [PubMed] [Google Scholar]
- Svensson F. R.; Andersson J.; Amand H. L.; Lincoln P. J. Biol. Inorg. Chem. 2012, 174565–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zava O.; Zakeeruddin S. M.; Danelon C.; Vogel H.; Gratzel M.; Dyson P. J. ChemBioChem. 2009, 10111796–1800. [DOI] [PubMed] [Google Scholar]
- Puckett C. A.; Barton J. K. J. Am. Chem. Soc. 2007, 129146–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puckett C. A.; Barton J. K. Biochemistry 2008, 474511711–11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puckett C. A.; Barton J. K. J. Am. Chem. Soc. 2009, 131258738–8739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puckett C. A.; Barton J. K. Bioorg. Med. Chem. 2010, 18103564–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puckett C. A.; Ernst R. J.; Barton J. K. Dalton Trans. 2010, 3951159–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juris A.; Balzani V.; Barigelletti F.; Campagna S.; Belser P.; Vonzelewsky A. Coord. Chem. Rev. 1988, 84, 85–277. [Google Scholar]
- Barton J. K.; Basile L. A.; Danishefsky A.; Alexandrescu A. Proc. Nat. Acad. Sci. U.S.A. 1984, 8171961–1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldstein B. M.; Barton J. K.; Berman H. M. Inorg. Chem. 1986, 256842–847. [Google Scholar]
- Rabilloud T.; Strub J. M.; Luche S.; van Dorsselaer A.; Lunardi J. Proteomics 2001, 15699–704. [DOI] [PubMed] [Google Scholar]
- Castellano F. N.; Lakowicz J. R. Photochem. Photobiol. 1998, 672179–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamanda A.; Zahn A.; Roder D.; Langen H. Proteomics 2004, 43599–608. [DOI] [PubMed] [Google Scholar]
- Garcia-Fresnadillo D.; Georgiadou Y.; Orellana G.; Braun A. M.; Oliveros E. Helv. Chim. Acta 1996, 7941222–1238. [Google Scholar]
- Alberts B.; Johnson A.; Lewis J.. Molecular Biology of the Cell, 4th Edition; Garland Science: New York, 2002; p 653. [Google Scholar]
- Friedman A. E.; Kumar C. V.; Turro N. J.; Barton J. K. Nucleic Acids Res. 1991, 19102595–2602. [PMC free article] [PubMed] [Google Scholar]
- Zanarini S.; Della Ciana L.; Marcaccio M.; Marzocchi E.; Paolucci F.; Prodi L. J. Phys. Chem. B 2008, 1123310188–10193. [DOI] [PubMed] [Google Scholar]
- Hergueta-Bravo A.; Jimenez-Hernandez M. E.; Montero F.; Oliveros E.; Orellana G. J. Phys. Chem. B 2002, 106154010–4017. [Google Scholar]
- Previous reports have shown that the association with DNA increases the yield of singlet oxygen-mediated single strand breaks, and as a result, compound 2 has a low yield of strand breakage. See ref (26).
- These data are consistent with earlier reports that the bathophenanthroline ligand increases the uptake of Ru(II) complexes; see ref (11).
- Gobeil S.; Boucher C. C.; Nadeau D.; Poirier G. G. Cell Death Differ. 2001, 86588–594. [DOI] [PubMed] [Google Scholar]
- Leist M.; Nicotera P. Biochem. Biophys. Res. Commun. 1997, 23611–9. [DOI] [PubMed] [Google Scholar]
- Gill M. R.; Garcia-Lara J.; Foster S. J.; Smythe C.; Battaglia G.; Thomas J. A. Nat. Chem. 2009, 18662–667. [DOI] [PubMed] [Google Scholar]
- Xu W. C.; Zuo J. R.; Wang L. L.; Ji L. N. A.; Chao H. Chem. Commun. 2014, 50172123–2125. [DOI] [PubMed] [Google Scholar]
- Baggaley E.; Gill M. R.; Green N. H.; Turton D.; Sazanovich I. V.; Botchway S. W.; Smythe C.; Haycock J. W.; Weinstein J. A.; Thomas J. A. Angew. Chem., Int. Ed. Engl. 2014, 53, 3367–3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierroz V.; Joshi T.; Leonidova A.; Mari C.; Schur J.; Ott I.; Spiccia L.; Ferrari S.; Gasser G. J. Am. Chem. Soc. 2012, 1345020376–20387. [DOI] [PubMed] [Google Scholar]
- Wang J. Q.; Zhang P. Y.; Qian C.; Hou X. J.; Ji L. N.; Chao H. J. Biol. Inorg. Chem. 2014, 193335–348. [DOI] [PubMed] [Google Scholar]
- Du Y.; Fu X.; Li H.; Chen B.; Guo Y.; Su G.; Zhang H.; Ning F.; Lin Y.; Mei W.; Chen T. ChemMedChem 2014, 714–718. [DOI] [PubMed] [Google Scholar]
- Chen T. F.; Mei W. J.; Wong Y. S.; Liu J.; Liu Y. N.; Xie H. S.; Zheng W. J. MedChemComm 2010, 1173–75. [Google Scholar]
- Chen T.; Liu Y.; Zheng W. J.; Liu J.; Wong Y. S. Inorg. Chem. 2010, 49146366–6368. [DOI] [PubMed] [Google Scholar]
- Joshi T.; Pierroz V.; Ferrari S.; Gasser G. ChemMedChem. 2014, 971419–1427. [DOI] [PubMed] [Google Scholar]
- Blackmore L.; Moriarty R.; Dolan C.; Adamson K.; Forster R. J.; Devocelle M.; Keyes T. E. Chem. Commun. 2013, 49262658–2660. [DOI] [PubMed] [Google Scholar]
- Alvarez M.; Villanueva A.; Acedo P.; Canete M.; Stockert J. C. Acta Histochem. 2011, 1133363–368. [DOI] [PubMed] [Google Scholar]
- Snyder J. W.; Lambert J. D.; Ogilby P. R. Photochem. Photobiol. 2006, 821177–184. [DOI] [PubMed] [Google Scholar]
- Strauss W. S.; Gschwend M. H.; Sailer R.; Schneckenburger H.; Steiner R.; Ruck A. J. Photochem. Photobiol., B 1995, 282155–161. [DOI] [PubMed] [Google Scholar]
- Garcia-Fresnadillo D.; Orellana G. Helv. Chim. Acta 2001, 84, 2708–2730. [Google Scholar]
- Howerton B. S.; Heidary D. K.; Glazer E. C. J. Am. Chem. Soc. 2012, 134208324–8327. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.