

Abstract
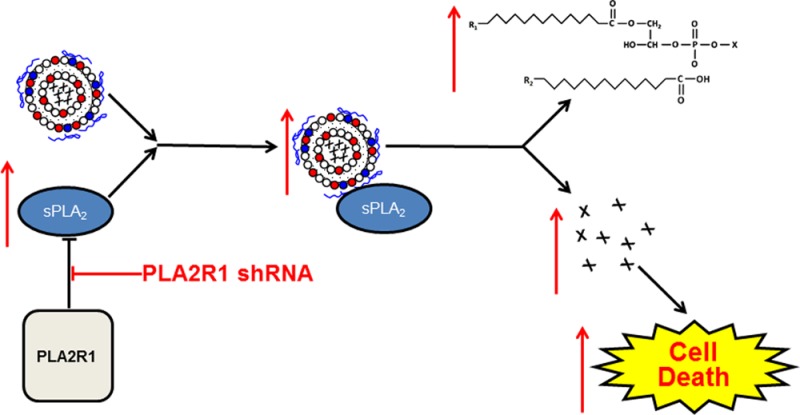
The M-type phospholipase A2 receptor (PLA2R1) is a member of the C-type lectin superfamily and can internalize secreted phospholipase A2 (sPLA2) via endocytosis in non-cancer cells. sPLA2 itself was recently shown to be overexpressed in prostate tumors and to be a possible mediator of metastasis; however, little is known about the expression of PLA2R1 or its function in prostate cancers. Thus, we examined PLA2R1 expression in primary prostate cells (PCS-440-010) and human prostate cancer cells (LNCaP, DU-145, and PC-3), and we determined the effect of PLA2R1 knockdown on cytotoxicity induced by free or liposome-encapsulated chemotherapeutics. Immunoblot analysis demonstrated that the expression of PLA2R1 was higher in prostate cancer cells compared to that in primary prostate cells. Knockdown of PLA2R1 expression in PC-3 cells using shRNA increased cell proliferation and did not affect the toxicity of cisplatin, doxorubicin (Dox), and docetaxel. In contrast, PLA2R1 knockdown increased the in vitro toxicity of Dox encapsulated in sPLA2 responsive liposomes (SPRL) and correlated with increased Dox and SPRL uptake. Knockdown of PLA2R1 also increased the expression of Group IIA and X sPLA2. These data show the novel findings that PLA2R1 is expressed in prostate cancer cells, that PLA2R1 expression alters cell proliferation, and that PLA2R1 modulates the behavior of liposome-based nanoparticles. Furthermore, these studies suggest that PLA2R1 may represent a novel molecular target for controlling tumor growth or modulating delivery of lipid-based nanomedicines.
Keywords: phospholipase A2 receptor, secreted phospholipase A2, prostate cancer, drug delivery, nanoparticles, liposomes, targeting
Introduction
Secreted phospholipase A2 (sPLA2) cleave glycerophospholipids at the sn-2 ester bond.1 They are excreted at the extracellular side of the plasma membrane and are overexpressed in a variety of tumors, e.g., up to 22-fold in prostate cancer.2 These enzymes have been hypothesized to be targets to control drug release from nanoparticles, such as liposomes.3
sPLA2 are regulated by PLA2 receptors including the M-type receptor, otherwise known as PLA2R1.4 While several studies have reported on the expression of sPLA2 in cancer cells, few have examined the expression of PLA2R1 and fewer have determined its role in cell physiology or cancer pathology. Most interestingly, we have not identified any studies that examined the effect of PLA2R1 on the disposition of lipid-based nanomedicines, including those that are targeted or sensitive to expression of sPLA2.
PLA2R1 is a membrane-bound glycoprotein expressed on the extracellular surface and can also exist as a soluble secreted protein.4b This location permits its interaction with free sPLA2 in the extracellular space. The internalization of sPLA2 by PLA2R1 may, but does not always, inactivate sPLA2 by degradation, which is followed by the recycling of PLA2R1 to the membrane. PLA2R1 was initially characterized in rabbit muscle tissues, but it is also found in the lung, spleen, and kidney, and in breast and colon cancers.4−6 sPLA2 binding to the PLA2R1 is species-dependent and calcium-insensitive.4a,5,7 Human PLA2R1 does not preferentially bind Group III sPLA2 (bee venom) or Naja venom (cobra venom) and has weak-to-no binding affinity for Group IB, IIA,and V sPLA2.4c Mouse PLA2R1 has a high affinity for Group X sPLA2.4b,4c,8
sPLA2 binding to PLA2R1 is reported to alter cell invasion, proliferation, and MAPK activation.4a,4c,9 Studies also suggest alterations in lipid metabolism and increases in lipid signaling.4a A recent report in breast cancer cells suggested that PLA2R1 could act as a tumor suppressor.10
We recently demonstrated that engineering liposomes to interact with sPLA2 increased payload release and enhanced intracellular uptake compared to that for pegylated, long-circulating sterically stabilized (SSL) Dox liposomes, which are similar to the clinically approved DOXIL.3b These liposomes, termed sPLA2 responsive liposomes (SPRL), were also more effective at slowing tumor growth in a xenograft model of human prostate cancer. Interestingly, the effectiveness of SPRL formulations was not altered by inhibitors of sPLA2 activity. This suggested that sPLA2-mediated lipid degradation of SPRL and drug (Dox) release may not be the only mechanism for the enhanced antitumor activity. Thus, we hypothesized that PLA2R1 may mediate the disposition of SPRL and other lipid-based nanomedicines.
The purpose of the work herein was to determine the expression of PLA2R1 in non-cancerous and cancerous prostate cells and to determine the role of PLA2R1 in prostate cancer cell growth. We also determined the role of PLA2R1 in chemotherapeutic-induced cytotoxicity using free and liposome-encapsulated drug. These findings are important as they provide insights into the roles of PLA2R1, its potential as a chemotherapeutic target for controlling tumorigenesis, and its impact on intracellular delivery of lipid-based nanomedicines for the treatment and identification of aggressive vs indolent disease.
Materials and Methods
Materials
Distearoylphosphatidylcholine (DSPC), distearoylphosphatidylethanolamine (DSPE), and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[poly(ethylene glycol) 2000] (DSPE-PEG) were purchased from Avanti Polar Lipids Inc. (Alabaster, AL). Cell lines derived from primary cultures of human prostate cells (PCS-440-010) and those from prostate cancer tumors (LNCaP, DU-145, and PC-3 cells) were purchased from ATCC (Manassas, VA). Doxorubicin (Dox) was purchased from Toronto Research Chemicals (North York, ON, Canada). 3,3′-Dioctadecyloxacarbocyanine perchlorate (DiO), iScript cDNA synthesis kit, and Universal SYBR Green Master Mix were purchased from BioRad (Hercules, CA). PCR primers specific for PLA2R1, sPLA2 Group IB, IIA, V, X, and GAPDH were purchased from Integrated DNA Technologies (IDT, Coralville, IA). PLA2R1 shRNA lentiviral particles, control shRNA plasmids, polybrene, and puromycin dihydrochloride were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). An alternate set of PLA2R1 and control shRNA plasmids was purchased from GeneCopoeia (Rockville, MD). All other reagents were of analytical quality and purchased from Sigma-Aldrich (St. Louis, MO).
Cell Culture
All cell media and supplements, including antibiotics and serum, were purchased from ATCC. PCS-440-010 (PCS) cells were grown in supplemented prostate epithelial cell basal medium according to the manufacture’s recommendations. LNCaP, DU-145, and PC-3 were cultured in 10% FBS and 1% penicillin/streptomycin supplemented RPMI-1640, EMEM, and F12K, respectively. All cell cultures were incubated in 95% humidity and 5% CO2 at 37 °C.
qRT-PCR
mRNA was isolated from cells using EZNA total RNA kit I (Promega, Madison, WI) according to the manufacturer’s specifications. The quantity and integrity of the RNA was checked using a NanoDrop (Life Science Technology, NY). RNA (1 μg) was converted to cDNA using the iScript cDNA synthesis kit (BioRad, Hercules, CA). cDNA (100 ng) was used for qRT-PCR to analyze the expression of PLA2R1 (F: 5′-TAAATCGGTTCTGACCCTGGA-3′ and R: 5′- GCCACCGTAAGGAAACGAG-3′, 182 bp), Group IB sPLA2 (F: 5′-TGCCAGACACATGACAACTG-3′ and R: 5′-ACGAGTATGAATAGGTGTGGGT-3′, 97 bp), Group IIA sPLA2 (F: 5′-GAAGTTGAGACCACCCAGCA-3′ and R: 5′- GTTGCATCCTTGGGGGATCCTCTG-3′, 201 bp), Group V sPLA2 (F: 5′- GACCCGTTACTGAACCTCTTTG-3′ and R: 5′-AATTCCTGCTGTGTGAAATCCT-3′, 145 bp), Group X sPLA2 (F: 5′-GACCGGCAGAGAACAAATGC-3′ and R: 5′-TTGTACTCAGTTTGGGCTAAGC-3′, 88 bp), and GAPDH (F: 5′-AAGGTCGGAGTCAACGGAT-3′ and R: 5′-TGGAAGATGGTGATGGGATT-3′, 221 bp), as a house-keeping gene. qRT-PCR was performed using a BioRad iCycler. Relative expression values were calculated by ΔΔCt using GAPDH as an internal control and PCS-440-010 cells as a standard of comparison.
Western Blot Analysis
Proteins from different cell lines were collected in RIPA buffer supplemented with 1% (v/v) protease inhibitor cocktail (Sigma, St. Louis, MO). The protein concentrations were determined using the BCA assay, and protein samples (40 μg) were separated on 4%/12% stacked SDS-PAGE gels and transferred to nitrocellulose membranes. The membranes were blocked in 5% (w/v) milk powder TBS-T for 2 h and then exposed to antibodies. Four different antibodies targeting human PLA2R1 were used: a rabbit polyclonal antibody from Abcam (Cambridge, MA), a rabbit polyclonal antibody from Proteintech (Chicago, IL), a rabbit polyclonal antibody from Atlas Antibodies11 (Stockholm, Sweden), and a guinea pig polyclonal antibody raised against rabbit PLA2R112 at 1:500, 1:500, 1:1000, and 1:1000 dilutions in 1% BSA TBS-T, respectively. Samples for immunoblot analysis using the guinea pig polyclonal antibody were prepared under nonreducing conditions. Incubation with these antibodies was performed overnight at 4 °C. Antibodies against GAPDH (Santa Cruz Biotechnology Inc., Santa Cruz, CA) and GFP (Genecopoeia, Rockville, MD) were used at 1:200 and 1:500 dilutions in 1% BSA TBS-T, respectively, for 1 h at room temperature. Membranes were incubated with a relevant peroxidase-conjugated secondary antibody (1:2500 dilution) (Promega, Madison, WI) or a guinea pig HRP-conjugated secondary antibody (1:5000 dilution) (Sigma, St. Louis, MO) for 2 h. Bands were visualized using SuperSignal Chemiluminescent substrate (Thermo Scientific, Waltham, MA), and intensities were visualized and quantified using an Alpha Innotech FluorChem HD2 system (ProteinSimple, Santa Clara, CA).
Formulation of SSL and SPRL
SSL and SPRL were prepared as described previously.3b,13 On the basis of our previous studies,3c we prepared SPRL containing 10% DSPE. These SPRL were chosen because they exhibited sensitivity to sPLA2 with improved tumor growth inhibition in a xenograft model of prostate cancer.3b The individual formulations used in this study are described in Table 1.
Table 1. Liposome Compositions.
| formulation | lipid composition | cholesterol |
|---|---|---|
| SSL | 1,2-distearoyl-sn-glycero-3-phosphocholine | 5 μmol/mL |
| (DSPC) | ||
| 9 μmol/mL | ||
| 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[poly(ethylene glycol) 2000] | ||
| (DSPE-PEG) | ||
| 1 μmol/mL | ||
| SPRL | 1,2-distearoyl-sn-glycero-3-phosphocholine | 5 μmol/mL |
| (DSPC) | ||
| 8 μmol/mL | ||
| 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[poly(ethylene glycol) 2000] | ||
| (DSPE-PEG) | ||
| 1 μmol/mL | ||
| 1,2-distearoyl-sn-glycero-3-phosphoethanolamine | ||
| (DSPE) | ||
| 1 μmol/mL |
Dox- and DiO-Labeled Liposomes
Dox-loaded liposomes were prepared by remote loading using a drug concentration and pH gradient as described previously.14 Briefly, lipids and cholesterol in chloroform were mixed and dried under vacuum using a rotary evaporator. The resulting lipid film was hydrated in 250 mM ammonium sulfate, pH 5.5. This dispersion underwent seven freeze–thaw cycles and was extruded through double-stacked polycarbonate, 80 nm, filters, n > 5 (Lipex, Northern Lipids, ON). Following extrusion, the liposomes were placed immediately on ice for 10 min and then dialyzed overnight with isotonic 10% (w/v) sucrose solution with three changes to remove unencapsulated ammonium sulfate. Drug loading was performed by adding Dox (10% sucrose, pH 8.5) to the dialyzed liposomes at a 0.2:1.0 drug/lipid molar ratio. The formulation was mixed and incubated for 1 h at 65 °C with periodic vortexing and immediately put on ice for 15 min. The loaded liposomes were then dialyzed overnight using a 12–14 kD MWCO membrane (Spectrum Laboratories, Rancho Dominguez, CA) in a 10% (w/v) sucrose solution to remove unencapsulated drug. Dox loading was quantified spectroscopically in acidified (0.2 N HCl) ethanol (1:1 v/v), and lipid concentration was determined using an assay for inorganic phosphate.3c,15
Fluorescent DiO-labeled liposomes were prepared according to the method of Kamps et al., with slight alterations.16 Briefly, lipids and cholesterol in chloroform were mixed, and 1 mol % DiO was added before the solution was evaporated to a lipid film. The resulting film was then rehydrated in PBS or ammonium sulfate depending on whether Dox would subsequently be loaded. The rest of the procedure was the same as above.
shRNA Transfection
PC-3 cells were transfected with various titers of PLA2R1 shRNA lentiviral vectors in media containing 5 μg/mL polybrene, whereas the control cells were transfected with scrambled plasmid. After 24 h, the media was aspirated and replaced with growth media and incubated overnight. The following day, clonal selection was performed using 10–20 μg/mL puromycin. Individual cells that were able to grow under puromycin selection conditions were expanded. Selected cells were grown in media containing puromycin (4 μg/mL). Similar transfection processes were performed with a second set of PLA2R1 and GFP control shRNA vectors (GeneCopoeia, Rockville, MD). GFP was included as a reporter with the alternative shRNA to assist the clonal selection process.
Measurement of MTT Staining
Scrambled and PLA2R1 knockdown PC-3 cells were seeded into 48-well plates, in 50,000 cells/mL and allowed to adhere for 24 h. Cells were then treated with free Dox, liposome-encapsulated Dox, or chemotherapeutics. After 24, 48, and 72 h, 0.25 mg/mL MTT was added. The plates were then incubated for 2 h before media were aspirated and replaced with DMSO. Plates were shaken vigorously for 15 min to dissolve all precipitates, and absorbance was determined at 590 nm with a FLUOstar OPTIMA plate reader (BMG Lab Technologies, Inc., Durham, NC).
Measurement of Crystal Violet
PC-3 cells expressing either scrambled or PLA2R1 shRNA were seeded in 6 cm dishes (4000 cells per dish) or in 48-well plates (50,000 cells/mL). Crystal violet staining was analyzed at 0, 24, and 72 h for 48-well plates or at 0, 7, and 11 days for 6 cm dishes. Cells were fixed with 10% formalin (Sigma, St. Louis, MO) for 10 min and stained with 1% (w/v) crystal violet for 15 min followed by three washes with water. The crystal violet stain was dissolved in methanol (Fisher, Waltham, MA), and the absorbance was determined at 540 nm with a FLUOstar OPTIMA plate reader.
Uptake of Liposomes and Dox in PLA2R1 Knockdown Cells
The uptake of Dox and liposomes into cells was determined using flow cytometry as described in our recent studies.3b Scrambled PC-3 and PLA2R1 knockdown cells were seeded in 12-well plates at 7.0–8.0 × 104 cells/well and allowed to attach for 24 h. Cells were then treated with PBS, free Dox, empty liposomes, empty DiO-labeled liposomes, Dox-loaded liposomes, or DiO-labeled Dox-loaded liposomes. Free drug and formulations containing drug were dosed at equal Dox equivalents. Assays for inorganic phosphate were performed to determine lipid concentrations, and empty and DiO-labeled liposomes were dosed at equal lipid concentrations compared to Dox loaded equivalents (∼10 nmol lipid/mL). At 24, 48, and 72 h post dosing, cells were washed three times with ice-cold PBS, released from the plate using trypsin/EDTA, and pelleted. Pellets were washed again with PBS and suspended in PBS supplemented with 1 mg/mL glucose. Samples were analyzed immediately using a CyAn flow cytometer (Beckman Coulter, Brea, CA). Samples were excited with a 488 nm argon laser, and emission was determined at 575 and 613 nm. Only whole cells were analyzed, as determined by forward and side scatter, and at least 5000 events were counted per run. Data was analyzed using FlowJo software, v. 9.7.4 (Tree Star, Inc., Ashland, OR).
Statistical Analysis
All experiments were completed at least three times (n = 3/study). Results are shown as the average of all replicates ± SEM. Results were compared using a Student’s t test or a one-way ANOVA followed by a Tukey’s test, considering p < 0.05 to be significant.
Results
Expression of PLA2R1 in Prostate Cells
PLA2R1 expression was determined in cell cultures derived from normal prostate tissue (PCS-440-010) and in PC-3, LNCaP, and DU-145 human prostate cancer cell lines using immunoblot analysis (Figure 1). PLA2R1 protein expression was lower in PCS-440-010 cells compared to that in all prostate cancer cell lines studied. These findings were validated using three additional antibodies, including those demonstrated to recognize PLA2R1 in several different cell types17 (Supporting Information Figure 1A).
Figure 1.
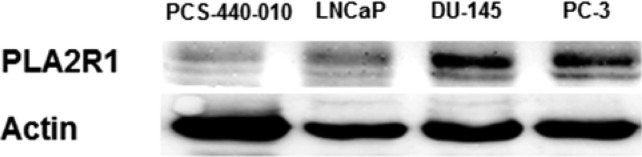
Expression of PLA2R1 in prostate cells. PLA2R1 protein expression was assessed using immunoblot analysis. The data are indicative of at least three separate experiments.
Effect of PLA2R1 Inhibition on Prostate Cancer Cell Growth
Transfection of PC-3 cells with shRNA plasmids significantly decreased PLA2R1 mRNA to levels approximately 25% of that seen in cells transfected with scrambled shRNA (Figure 2A). Decreased mRNA expression correlated to decreased protein expression (Figure 2B). Cells with decreased expression of PLA2R1 appeared to grow more rapidly than those expressing scrambled shRNA. This hypothesis was supported by increased MTT staining (Figure 2C) and was verified by crystal violet staining (Figure 2D and Supporting Information Figure 2). Crystal violet staining also showed that the increase in cell growth was not dependent on the initial cell number, as similar effects were seen if cells were seeded at low density (4000 cells per plate in 6 cm dishes, Figure 2D) or at high density (50,000 cell/mL in 48-well plates, Supporting Information Figure 2). Similar results were also observed when PLA2R1 expression was inhibited using shRNA purchased from GeneCopoeia (Supporting Information Figure 3).
Figure 2.

Effect of PLA2R1 inhibition on PC-3 cell growth. PLA2R1 expression was inhibited using shRNA. Scrambled shRNA was used as a control. Knockdown of PLA2R1 was verified by qRT-PCR (A) and immunoblot analysis (B). GAPDH was used as a house-keeping gene. Assessment of cell growth was performed using MTT assays (C) and crystal violet staining (D). Data in panels A–C are represented as the mean ± SEM of at least three separate experiments (n = 3/study). *Indicates a significant difference (p < 0.05) as compared to control cells. Data in panel D are indicative of at least three separate experiments.
Effect of PLA2R1 Inhibition on Chemotherapeutic-Induced Prostate Cancer Cytotoxicity
Prior to examining the role of PLA2R1 on liposome-induced cell death, we determined the role of PLA2R1 on cytotoxicity induced by a variety of chemotherapeutics, including docetaxel, cisplatin, and Dox. As expected, all three chemotherapeutics induced concentration-dependent decreases in MTT staining after 72 h of exposure (Figure 3A–C). Inhibition of PLA2R1 did not alter the sensitivity of PC-3 cells to any of these chemotherapeutics. Similar results were seen at 24 and 48 h and in cells containing the scrambled shRNA (Supporting Information Figure 4A–C). The absence of effect of PLA2R1 inhibition on Dox-induced cytotoxicity was confirmed using cell morphology (Figure 3D).
Figure 3.

Effect of PLA2R1 knockdown on chemotherapeutic-induced toxicity in PC-3 cells. Cells were treated with docetaxel (A), cisplatin (B), or Dox (C) for 72 h. MTT assays were used to determine the effect of knocking down PLA2R1 on chemotherapeutic-induced cytotoxicity. The effect of PLA2R1 on doxorubicin-induced toxicity was further assessed at 72 h using phase contrast microscopy at 10× magnification (D). Data in panels A–C are represented as the mean ± SD of at least three separate experiments (n = 3/study). Data in panel D are indicative of at least three separate experiments.
Effect of PLA2R1 Inhibition on the Activity of SSL and SPRL
Our recent studies3b,3c demonstrated that sPLA2-targeted liposomes (SPRL) had greater carrier uptake and drug delivery in prostate cancer cells in vitro and increased efficacy against tumor growth in vivo compared to that of SSL. These studies also suggested that the activity of some of these liposome formulations was not dependent on sPLA2 activity.3b We hypothesized that mechanisms other than sPLA2-mediated lipid degradation may be mediating SPRL activity. Therefore, we determined the effect of PLA2R1 knockdown on the cytotoxicity and uptake of SPRL. As shown in Figure 4, PLA2R1 inhibition did not alter decreases in MTT staining caused by Dox-loaded SSL, but did decrease MTT staining in cells treated with Dox-loaded SPRL (Figure 4A). The decrease in MTT staining correlated to a reduction in cell number and alteration in cellular morphology (Figure 4B–E). Interestingly, knockdown of PLA2R1 appeared to have a greater effect on cell morphology than on MTT. Similar results were observed using a different set of shRNA (GeneCopoeia, Supporting Information Figure 4D).
Figure 4.

Effect of PLA2R1 knockdown on the toxicity of doxorubicin encapsulated in SSL and SPRL in PC-3 cells. PLA2R1 knockdown cells and those expressing scrambled shRNA were treated with 2.5 μM concentrations of Dox encapsulated in SSL or SPRL. MTT assays (A) and phase contrast microscopy at 40× magnification (B) were used to determine the effect of PLA2R1 knockdown on cytotoxicity. Data in panel A are represented as the mean ± SEM of at least three separate experiments (n = 3/study). *Indicates a significant difference (p < 0.05) as compared to cells transfected with scrambled shRNA. Data in panel E are indicative of at least three separate experiments.
Effect of PLA2R1 Inhibition on the Uptake of Dox and SSL and SPRL
To examine the importance of PLA2R1 in the uptake of SSL and SPRL formulations, PLA2R1 knockdown cells were exposed to Dox-loaded SSL or SPRL formulations labeled with DiO, and the intracellular uptake of both drug and nanoparticle was determined using flow cytometry3b (Figure 5). As previously reported, SSL and SPRL were taken up by PC-3 cells (Figure 5A,C), and Dox uptake was greatest in cells incubated with SPRL vs SSL formulations (Figure 5B,D). Knockdown of PLA2R1 did not affect the uptake of the SSL formulation (i.e., DiO uptake), but it did slightly increase Dox uptake at 48 and 72 h (Figure 5A,B). In comparison, knockdown of PLA2R1 significantly increased the intracellular uptake of SPRL (i.e., DiO) at 48 and 72 h (Figure 5C) and increased the uptake of Dox at all time points studied (Figure 5D). Similar results were obtained with alternate shRNA (data not shown).
Figure 5.

Effect of PLA2R1 knockdown on DiO and doxorubicin uptake from SSL and SPRL in PC-3 cells. PC-3 cells were treated with liposomes containing Dox and DiO for 24 to 72 h. The efficiency of DiO and drug uptake via SSL (A, B) and SPRL (C, D) was quantified using flow cytometry. Data are represented as the mean ± SEM of at least three separate experiments (n = 3/study). *Indicates a significant difference (p < 0.05) as compared to cells expressing scrambled shRNA.
Effect of PLA2R1 Inhibition on the Expression of sPLA2
It has been shown previously that PLA2R1 functions as a negative regulator of sPLA218 by binding to and removing sPLA2 from the extracellular side of the cell membrane.4 We showed that addition of different sPLA2 isoforms, including Group IIA, increased payload release and liposome degradation in vitro.3c We hypothesized that increased SPRL activity in cells in which PLA2R1 was inhibited may result from the upregulation of sPLA2. We tested this hypothesis by assessing changes in the expression of various sPLA2 isoforms in PC-3 cells after PLA2R1 inhibition. Knockdown of PLA2R1 increased the expression of Group IIA sPLA2, as compared to that in cells expressing the scrambled control shRNA (Figure 6A). A slight increase in Group X sPLA2 protein expression was also detected. To determine if these increases correlated to increased mRNA levels, we performed qRT-PCR analysis, which also allowed us to further assess changes in expression of other sPLA2 isoforms including Groups IB and V (Figure 6B). In agreement with the immunoblot analysis, inhibition of PLA2R1 increased sPLA2 Group IIA mRNA levels. In contrast, no increases in sPLA2 Group IB, V, or X mRNA were detected. Collectively, these data suggest that inhibition of PLA2R1 increases the expression of select sPLA2 isoforms, which correlates to the increase in cell growth as well increased drug (Dox) and liposome (DiO) uptake.
Figure 6.

Effect of PLA2R1 knockdown on sPLA2 expression in PC-3 cells. PLA2R1 expression was inhibited in PC-3 cells using shRNA, and changes in the expression of various sPLA2 isoforms was determined by immunoblot analysis (A) and qRT-PCR (B). Data in panel A are indicative of at least three separate experiments. Data in panel B are represented as the mean ± SEM of at least three separate experiments (n = 3/study). *Indicates a significant difference (p < 0.05) as compared to cells expressing scrambled shRNA.
Discussion
PLA2R1 is expressed in several tissues,19 including skeletal muscle,20 kidney,19,21 spleen,8 breast,10 and pancreas.19 The expression of PLA2R1 is species-dependent, with significantly different tissue profiles being reported for its mRNA expression in mouse, rat, and human.19 To our knowledge, these studies are the first to show protein expression of PLA2R1 in prostate cancer cells.
PLA2R1 protein expression was higher in PC-3 and DU145 cells compared to that in LNCaP cells. One of the only other studies that examined PLA2R1 expression in cancer cells did so in breast cancer cells11 and showed that mRNA expression was decreased in cancer cells compared to that in non-cancerous cell lines. However, this study did not compare the expression of PLA2R1 protein between the non-cancerous and cancerous cells.
PCS-440-010 cells are derived from prostate tissue and represent multiple prostate cell types, whereas the prostate cancer cell lines used here are actually derived from prostate tumors that metastasized to distal sites. This may account for differential expression between PCS-440-010 and the prostate cancer cells used in this study. Regardless of the mechanisms involved, the data clearly suggest that the protein expression of PLA2R1 is higher in the prostate cancer cell lines than in non-cancerous prostate cells.
Effect of PLA2R1 Inhibition on Cell Growth
Our data clearly demonstrated that inhibition of PLA2R1 increased MTT and crystal violet staining, supporting the hypothesis that PLA2R1 plays a role in the growth of the prostate cancer cells. This hypothesis is further supported by recent studies in human breast cancer and fibroblasts cells that suggest PLA2R1 mediates replicative senescence, increases colony formation, and possibly acts as a tumor suppressor.10,22
The mechanisms by which PLA2R1 inhibits cell growth are under investigation. The aforementioned studies in breast cancer cells and fibroblasts suggested that PLA2R1 regulates senescence through the p53 pathway.22 Another study, from the same group, demonstrated that PLA2R1 activated JAK2 signaling, which resulted in decreased cellular transformation.10 PC-3 cells do not express p53, suggesting that PLA2R1 may inhibit cell growth by p53-independent mechanisms, possibly involving activation of kinase-mediated pathways.
Another possible mechanism by which PLA2R1 inhibition may increase cell growth is by altering cell death. In support of this hypothesis, increasing PLA2R1 expression in breast cancer cells decreased cell growth and colony formation.11 Cell death was associated with an increase in mitochondrial-mediated reactive oxygen formation. In addition, some cancer cells may overexpress PLA2R1 in order to inactivate sPLA2 activity, helping them to survive under inflammatory conditions. Although the mechanism is not clear, the expression and effect of PLA2R1 in breast and prostate cancer may differ and suggest further studies are needed.
Effect of PLA2R1 Inhibition on Chemotherapeutic-Induced Cell Death
These data suggest that PLA2R1 does not mediate cell death induced by several different types of chemotherapeutics when administered as free drug. The slight increase in cytotoxicity induced by Dox in PLA2R1 knockdown cells is somewhat suggestive, but this was lost when a plasmid control was included (Supporting Information Figure 4A–C). It should be noted that PLA2R1 expression was inhibited in PC-3 cells by approximately 50% compared to control cells. Thus, increased inhibition may be necessary to induce significant effects. Additionally, the role of PLA2R1 in cell death may be more specific to those circumstances where sPLA2 is induced, such as inflammation. Finally, it is also possible that the increase in cell growth induced by PLA2R1 inhibition counteracts any enhanced toxicity that might be seen under the conditions tested in this study.
Effect of PLA2R1 Inhibition on Liposome-Based Drug Delivery and Cytotoxicity
We recently showed that SPRL are superior to SSL at inhibiting tumor growth in vivo and in releasing drug payload in vitro.3b We also showed that SPRL degradation was increased in prostate cancer cells exposed to sPLA2.3c However, subsequent studies investigating the mechanisms involved suggested that the uptake of liposomes and drug was independent of enzymatic activity.3b This suggests that other proteins may mediate SPRL disposition. Data from the current study suggest that PLA2R1 may be one such protein. The binding of sPLA2 to PLA2R1 is independent of sPLA2 activity,4c and this may explain why LY311727 (a commonly used sPLA2 inhibitor) did not alter SPRL activity in our previous study.3b It remains to be seen if sPLA2, liposomes, and PLA2R1 can indeed form a complex at the cell membrane that undergoes endocytosis.
The fact that PLA2R1 inhibition had a minimal effect on the toxicity of Dox-loaded SSL, as compared to that with SPRL, correlates to the fact that PLA2R1 inhibition did not alter SSL delivery. This specificity may be due to the increased preference of SPRL for sPLA2, which may increase its preference for PLA2R1. Regardless, these data suggest that some specificity is inherent in the ability of PLA2R1 to mediate liposome uptake, and they further support our previous findings that SPRL behave differently than SSL. The increase in cytotoxicity with Dox-loaded liposomes required that Dox be present in the liposome, as PLA2R1 inhibition did not increase cytotoxicity when cells were treated with free, unencapsulated Dox.
Our data show that inhibition of PLA2R1 increases the expression of Groups IIA and X sPLA2 protein. This finding, though novel, is not totally surprising. The increase in sPLA2 expression induced by PLA2R1 inhibition may facilitate increased interaction with liposomes and increased delivery. It should be noted that Group IIA sPLA2 is not believed to bind to human PLA2R1 with high affinity.5,7 Thus, the increase in Group IIA sPLA2 induced by PLA2R1 inhibition is probably not a result of decreased uptake of this protein. Rather, it is possible that PLA2R1 is inducing a signaling pathway that leads to increased Group IIA mRNA transcription. Another possibility is that increased degradation of liposomes by sPLA2 may enhance the release of individual DiO labeled lipids, which may be more rapidly incorporated into cellular membranes than whole liposomes.
Accumulation of sPLA2 may increase liposomal degradation, resulting in faster rates of drug release, greater effective concentrations of drug outside of the cell, and greater uptake. This may account for the increased uptake of Dox in PLA2R1 knockdown cells seen with both SSL and SPRL formulations. Our previous studies showed that exogenous sPLA2 degraded both SSL and SPRL, but that SPRL were more sensitive to the effect of sPLA2 and released Dox at a higher level.3b,3c We have shown that the addition of the 10% of DSPE into liposomes enhances their interactions with sPLA2.3b This finding is supported by data in this study, which demonstrate greater increases in Dox release from SPRL, as compared to that from SSL, and at earlier time points. It may also explain the enhanced effect of PLA2R1 inhibition on SPRL.
The mechanism by which PLA2R1 inhibition leads to increased sPLA2 is under investigation. The most obvious explanation is that the loss of PLA2R1 decreases sPLA2 uptake and degradation. The most obvious explanation is that the loss of PLA2R1 decreases sPLA2 uptake and degradation, but this is unlikely as neither Group IIA or X are believed to be a substrate for human PLA2R1. However, the fact that mRNA levels were increased for sPLA2 Group IIA suggests that a transcriptional component may also be involved, at least for some isoforms. It is also possible that PLA2R1 acts as transcriptional suppressor for sPLA2 Group IIA. It is not known why Group IIA sPLA2 mRNA levels would be increased but not Group X mRNA levels. The increase in Groups IIA and X sPLA2 does correlate to the increase in cell growth in PLA2R1 knockdown cells. This result is similar to the clinical observation that Group IIA sPLA2 expression is greater in more metastatic and aggressive prostate tumors.2,23
In conclusion, this study determined the expression of PLA2R1 in non-cancerous and cancerous prostate cell lines and showed that inhibition of PLA2R1 increased cell growth and the expression of select sPLA2 isoforms. Increases in the expression of sPLA2 correlated to increased uptake of liposomes and drugs. These findings suggest novel roles for PLA2R1 in the regulation of sPLA2 and in the targeting of lipid-based nanoparticles for the treatment of prostate cancer.
Acknowledgments
We thank the UGA Flow Cytometry Core Facility in the Center for Tropical and Emerging Diseases, particularly Julie Nelson for all of her assistance and insight. The authors would also like to acknowledge and give special thanks to both the American Foundation for Pharmaceutical Education (AFPE) and the Achievement Rewards for College Scientists (ARCS) foundation for providing predoctoral fellowship assistance to J.N.M.. This research was funded in part by Georgia Cancer Coalition Distinguished Scholar Grants and NIH NIBIB (R21EB08153 and R01EB0116100) to B.S.C./R.D.A., an Interdisciplinary Toxicology Program Graduate Stipend to N.E.S., the Association pour la Recherche sur le cancer to G.L., and an Auburn University Research Initiative in Cancer Graduate Fellowship to M.W.E.
Glossary
Abbreviations
- DiO
3,3′-dioctadecyloxacarbocyanine perchlorate
- Dox
doxorubicin
- DSPC
distearoylphosphatidylcholine
- DSPE
distearoylphosphatidylethanolamine
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PEG
poly(ethylene glycol)
- PLA2R1
M-type phospholipase A2 receptor
- sPLA2
secreted phospholipase A2
- SPRL
secreted phospholipase A2 responsive liposome
- SSL
sterically stabilized liposomes
Supporting Information Available
Verification of PLA2R1 expression with different antibodies, effect of PLA2R1 inhibition on PC-3 cell growth, effect of PLA2R1 inhibition on PC-3 cell growth with a different PLA2R1 shRNA plasmid, and effect of PLA2R1 knockdown on chemotherapeutic-induced and liposome-induced toxicity in PC-3 cells. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
∥ N.D.Q. and J.N.M. contributed equally to this work
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Andresen T. L.; Jensen S. S.; Kaasgaard T.; Jorgensen K. Triggered activation and release of liposomal prodrugs and drugs in cancer tissue by secretory phospholipase A2. Curr. Drug Deliv 2005, 2, 353–62. [DOI] [PubMed] [Google Scholar]
- Graff J. R.; Konicek B. W.; Deddens J. A.; Chedid M.; Hurst B. M.; Colligan B.; Neubauer B. L.; Carter H. W.; Carter J. H. Expression of group IIa secretory phospholipase A2 increases with prostate tumor grade. Clin. Cancer Res. 2001, 7, 3857–61. [PubMed] [Google Scholar]
- a Andresen T. L.; Jensen S. S.; Madsen R.; Jorgensen K. Synthesis and biological activity of anticancer ether lipids that are specifically released by phospholipase A2 in tumor tissue. J. Med. Chem. 2005, 48, 7305–14. [DOI] [PubMed] [Google Scholar]; b Mock J. N.; Costyn L. J.; Wilding S. L.; Arnold R. D.; Cummings B. S. Evidence for distinct mechanisms of uptake and antitumor activity of secretory phospholipase A2 responsive liposome in prostate cancer. Integr. Biol. 2013, 5, 172–82. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhu G.; Mock J. N.; Aljuffali I.; Cummings B. S.; Arnold R. D. Secretory phospholipase A responsive liposomes. J. Pharm. Sci. 2011, 100, 3146–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lambeau G.; Lazdunski M. Receptors for a growing family of secreted phospholipases A2. Trends Pharmacol. Sci. 1999, 20, 162–70. [DOI] [PubMed] [Google Scholar]; b Hanasaki K. Mammalian phospholipase A2: phospholipase A2 receptor. Biol. Pharm. Bull. 2004, 27, 1165–7. [DOI] [PubMed] [Google Scholar]; c Hanasaki K.; Arita H. Phospholipase A2 receptor: a regulator of biological functions of secretory phospholipase A2. Prostaglandins Other Lipid Mediators 2002, 68–69, 71–82. [DOI] [PubMed] [Google Scholar]
- Rouault M.; Le Calvez C.; Boilard E.; Surrel F.; Singer A.; Ghomashchi F.; Bezzine S.; Scarzello S.; Bollinger J.; Gelb M. H.; Lambeau G. Recombinant production and properties of binding of the full set of mouse secreted phospholipases A2 to the mouse M-type receptor. Biochemistry 2007, 46, 1647–62. [DOI] [PubMed] [Google Scholar]
- Gorovetz M.; Schwob O.; Krimsky M.; Yedgar S.; Reich R. MMP production in human fibrosarcoma cells and their invasiveness are regulated by group IB secretory phospholipase A2 receptor-mediated activation of cytosolic phospholipase A2. Front. Biosci. 2008, 13, 1917–25. [DOI] [PubMed] [Google Scholar]
- Cupillard L.; Mulherkar R.; Gomez N.; Kadam S.; Valentin E.; Lazdunski M.; Lambeau G. Both group IB and group IIA secreted phospholipases A2 are natural ligands of the mouse 180-kDa M-type receptor. J. Biol. Chem. 1999, 274, 7043–51. [DOI] [PubMed] [Google Scholar]
- Morioka Y.; Saiga A.; Yokota Y.; Suzuki N.; Ikeda M.; Ono T.; Nakano K.; Fujii N.; Ishizaki J.; Arita H.; Hanasaki K. Mouse group X secretory phospholipase A2 induces a potent release of arachidonic acid from spleen cells and acts as a ligand for the phospholipase A2 receptor. Arch. Biochem. Biophys. 2000, 381, 31–42. [DOI] [PubMed] [Google Scholar]
- Surrel F.; Jemel I.; Boilard E.; Bollinger J. G.; Payre C.; Mounier C. M.; Talvinen K. A.; Laine V. J.; Nevalainen T. J.; Gelb M. H.; Lambeau G. Group X phospholipase A2 stimulates the proliferation of colon cancer cells by producing various lipid mediators. Mol. Pharmacol. 2009, 76, 778–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vindrieux D.; Augert A.; Girard C. A.; Gitenay D.; Lallet-Daher H.; Wiel C.; Le Calve B.; Gras B.; Ferrand M.; Verbeke S.; de Launoit Y.; Leroy X.; Puisieux A.; Aubert S.; Perrais M.; Gelb M.; Simonnet H.; Lambeau G.; Bernard D. PLA2R1 mediates tumor suppression by activating JAK2. Cancer Res. 2013, 73, 6334–45. [DOI] [PubMed] [Google Scholar]
- Augert A.; Vindrieux D.; Girard C. A.; Le Calve B.; Gras B.; Ferrand M.; Bouchet B. P.; Puisieux A.; de Launoit Y.; Simonnet H.; Lambeau G.; Bernard D. PLA2R1 kills cancer cells by inducing mitochondrial stress. Free Radical Biol. Med. 2013, 65, 969–77. [DOI] [PubMed] [Google Scholar]
- Zvaritch E.; Lambeau G.; Lazdunski M. Endocytic properties of the M-type 180-kDa receptor for secretory phospholipases A2. J. Biol. Chem. 1996, 271, 250–7. [DOI] [PubMed] [Google Scholar]
- Zhu G.; Alhamhoom Y.; Cummings B. S.; Arnold R. D. Synthesis of lipids for development of multifunctional lipid-based drug-carriers. Bioorg. Med. Chem. Lett. 2011, 21, 6370–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Haran G.; Cohen R.; Bar L. K.; Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim. Biophys. Acta 1993, 1151, 201–15. [DOI] [PubMed] [Google Scholar]; b Arnold R. D.; Mager D. E.; Slack J. E.; Straubinger R. M. Effect of repetitive administration of doxorubicin-containing liposomes on plasma pharmacokinetics and drug biodistribution in a rat brain tumor model. Clin. Cancer Res. 2005, 11, 8856–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett G. R. Phosphorus assay in column chromatography. J. Biol. Chem. 1959, 234, 466–8. [PubMed] [Google Scholar]
- Kamps J. A.; Morselt H. W.; Swart P. J.; Meijer D. K.; Scherphof G. L. Massive targeting of liposomes, surface-modified with anionized albumins, to hepatic endothelial cells. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 11681–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas J. P.; Lambeau G.; Lazdunski M. Identification of the binding domain for secretory phospholipases A2 on their M-type 180-kDa membrane receptor. J. Biol. Chem. 1995, 270, 28869–73. [DOI] [PubMed] [Google Scholar]
- Yokota Y. [Functional analysis of phospholipase A2 receptor by gene knockout studies]. Yakugaku Zasshi 2001, 121, 23–33. [DOI] [PubMed] [Google Scholar]
- Higashino K.; Ishizaki J.; Kishino J.; Ohara O.; Arita H. Structural comparison of phospholipase-A2-binding regions in phospholipase-A2 receptors from various mammals. Eur. J. Biochem. 1994, 225, 375–82. [DOI] [PubMed] [Google Scholar]
- Lambeau G.; Ancian P.; Barhanin J.; Lazdunski M. Cloning and expression of a membrane receptor for secretory phospholipases A2. J. Biol. Chem. 1994, 269, 1575–8. [PubMed] [Google Scholar]
- Gunnarsson I.; Schlumberger W.; Ronnelid J. Antibodies to M-type phospholipase A2 receptor (PLA2R) and membranous lupus nephritis. Am. J. Kidney Dis. 2012, 59, 585–6. [DOI] [PubMed] [Google Scholar]
- Augert A.; Payre C.; de Launoit Y.; Gil J.; Lambeau G.; Bernard D. The M-type receptor PLA2R regulates senescence through the p53 pathway. EMBO Rep. 2009, 10, 271–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Jiang J.; Neubauer B. L.; Graff J. R.; Chedid M.; Thomas J. E.; Roehm N. W.; Zhang S.; Eckert G. J.; Koch M. O.; Eble J. N.; Cheng L. Expression of group IIA secretory phospholipase A2 is elevated in prostatic intraepithelial neoplasia and adenocarcinoma. Am. J. Pathol. 2002, 160, 667–671. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dong Z.; Liu Y.; Scott K. F.; Levin L.; Gaitonde K.; Bracken R. B.; Burke B.; Zhai Q. J.; Wang J.; Oleksowicz L.; Lu S. Secretory phospholipase A2-IIa is involved in prostate cancer progression and may potentially serve as a biomarker for prostate cancer. Carcinogenesis 2010, 31, 1948–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.