

Abstract
Despite vast improvements in treatment of Philadelphia chromosome–positive chronic myeloid leukemia (CML) in chronic phase (CP), advanced stages of CML, accelerated phase or blast crisis, remain notoriously difficult to treat. Treatments that are highly effective against CML-CP produce disappointing results against advanced disease. Therefore, a primary goal of therapy should be to maintain patients in CP for as long as possible, by (1) striving for deep, early molecular response to treatment; (2) using tyrosine kinase inhibitors that lower risk of disease progression; and (3) more closely observing patients who demonstrate cytogenetic risk factors at diagnosis or during treatment.
Keywords: myeloid leukemias and dysplasias, tyrosine kinase inhibitor, drug resistance
INTRODUCTION
The availability of selective BCR-ABL tyrosine kinase inhibitors (TKIs) for the treatment of chronic myeloid leukemia (CML) marked a major advance in the treatment of this cancer by prolonging survival in patients across all stages of disease severity: chronic phase (CP), accelerated phase (AP), and blast crisis (BC) [1,2,3]. Despite this, however, survival rates continue to be better for patients in CP than for patients in advanced stages of disease, AP or BC [1]. Decades of clinical and molecular research have led to the development of therapies that are highly effective in CML-CP, yet treatment options for CML-AP and -BC are limited and the clinical outlook for patients who progress to AP/BC remains bleak. Advances in the treatment of patients with CML-AP and -BC are hampered in part by factors such as the lack of uniform clinical criteria that define AP/BC and the array of genetic defects implicated in disease progression, which underscores the complexity of the disease in its advanced stages.
This review outlines the various definitions for CML-AP and CML-BC, describes potential mechanisms of disease transformation to AP/BC, discusses clinical factors associated with risk of disease progression, and provides an overview of clinical data on protection from progression and treatment options for patients in AP/BC.
DEFINITIONS OF CML-AP AND CML-BC
Over the years, several groups have outlined staging classification criteria that define CML-AP and CML-BC [4–8] (Table I). The set of criteria most often used in clinical studies of CML are those from the European Leukemia Net (ELN), which sets the threshold blast count for CML-BC at 30% [8]. World Health Organization criteria set the threshold blast count for CML-BC at 20%, in alignment with the clinical definition of acute myeloid leukemia (AML)[7]. Because patients with 20%–29% blasts have significantly better rates of complete cytogenetic response (CCyR) and 3-year overall survival (OS) than do patients with ≥30% blasts [9], they are classified as having CML-AP by criteria of both the ELN and the M. D. Anderson Cancer Center, Houston, TX [8,9]. Regardless of the criteria used to define BC, it should be recognized that blast count thresholds are arbitrary values, because blast count is a continuous variable [9]. Until a biological, evidence-based definition of BC is established, most clinicians will likely continue defining BC according to their own experiences [3].
Table I.
| Disease stage | ELN criteria | MDACC criteria | WHO criteria |
|---|---|---|---|
| Chronic phase |
|
High risk
Low risk
|
|
| Accelerated phase |
|
|
|
| Blast crisis |
|
|
|
Abbreviations: ELN, European Leukemia Net; MDACC, MD Anderson Cancer Center; WHO, World Health Organization
POTENTIAL MECHANISMS OF DISEASE PROGRESSION IN CML
Disease progression to CML-BC is characterized by increased cell proliferation and arrested cell differentiation and apoptosis, pathologic features that must be accounted for by any model of disease progression. Although the role of BCR-ABL tyrosine kinase activity in the pathogenesis of CML is firmly established [10,11], its role in driving disease progression is less clear. Not all mechanisms implicated in disease progression in CML stem directly from BCR-ABL activity, although its involvement in the process is highly plausible. Here, we summarize potential mechanisms of disease progression in CML; a more comprehensive review of the topic can be found in Perrotti et al [12].
BCR-ABL Expression
The level of BCR-ABL gene expression has been shown to be associated with disease progression. An increase in BCR-ABL gene expression can arise from either an up regulation of mRNA levels or an increase in the number of CML cells in the blood. Studies of BCR-ABL transcript kinetics have not demonstrated conclusively that increased BCR-ABL expression is a causal event in the progression of CML to AP/BC [13,14]. Because increased BCR-ABL expression precedes the manifestation of clinical or laboratory signs of AP/BC, however, it is thought to be at least an early event in disease progression.
BCR-ABL–Mediated Genetic Instability
The accumulation of chromosomal abnormalities and genetic defects is a hallmark of disease progression in CML. Among patients with demonstrable hematologic resistance or recurrence, additional cytogenetic abnormalities were more frequent in CML-BC patients than in CML-CP or CML-AP patients (73% versus 52% versus 50%, respectively) [15]. The greater prevalence of genetic instability in leukemic cells of patients with advanced disease arises from enhanced DNA damage and reduced capacity to repair that damage. Both BCR-ABL–dependent [16,17] and –independent [18] mechanisms of generating reactive oxygen species can contribute to overall genomic instability by inducing oxidative DNA damage such as double-strand breaks [19]. Furthermore, in vitro experiments have demonstrated that expression of BCR-ABL protein sensitizes cells to ionizing radiation [20] and causes cells to accumulate drug-induced DNA damage [21]. Indeed, BCR-ABL activity has been shown to (1) disrupt proteins involved in repair of DNA double-strand breaks [20] [22] [23]; (2) up regulate expression of BCL-xL [21], an antiapoptotic protein; and (3) cause cell-cycle arrest in G2/M in cells treated with DNA-damaging agents [21], actions that together can promote greater genomic instability.
This environment of genomic instability may explain the emergence of nonrandom chromosomal abnormalities (i.e., clonal evolution) commonly observed with CML disease progression, although the precise etiology of these abnormalities is not known. The more common “major route” chromosomal abnormalities identified in patients in CML-BC include trisomy 8 (~40%), double Philadelphia (Ph) chromosome (~38%), and isochromosome i (17q) (~21%). Other common chromosomal abnormalities include trisomy 19 (~16%), trisomy 21 (~9%), monosomy 7 (~5%), and monosomy 17 (~4%)[24,25]. Although the pathologic link between these abnormalities and CML disease progression is not fully elucidated, there is some evidence that duplication/amplification of known oncogenes (eg, MYC on chromosome 8[26]) or loss of known tumor suppressor genes (eg, TP53 on chromosome 17[27])that reside on affected chromosomes may contribute to disease progression.
RNA-Binding Proteins
The RNA-binding protein hnRNP A1 is over expressed in myeloid progenitor cells expressing BCR-ABL and in cells from patients with CML-BC [28]. In vitro experiments have shown hnRNP A1 to bind the mRNA of genes whose protein products are also regulated by BCR-ABL activity, suggesting a role for mRNA metabolism in leukemogenesis. The mRNA of SET, a phosphoprotein implicated in acute leukemia [29]is bound by hnRNP A1[30]. Importantly, SET is a potent inhibitor of protein phosphatase 2A (PP2A) [31], a serine/threonine phosphatase that functions as a tumor suppressor in CML (reviewed in Perrotti and Neviani [32]). In vitro studies have demonstrated both BCR-ABL–dependent inhibition of PP2A activity, mediated by upregulated protein expression of SET [30]and PP2A-dependent regulation of BCR-ABL activity and leukemogenic potential. The mutual antagonism of PP2A and BCR-ABL suggests that inactivation of PP2Amight be an early step in the blastic transformation of CML cells. In addition to SET, hnRNP A1 also binds to the transcription factor E2F3 in BCR-ABL–expressing cell lines and cells from patients with CML-BC [33]. Furthermore, E2F3 activity is required in vitro and in vivo for BCR-ABL oncogenic activity. Taken together, these findings highlight the role that mRNA metabolism plays in the post-transcriptional regulation of genes as a mechanism of disease progression in CML.
Centrosomal Aberrations
The centrosome is a eukaryotic cellular organelle that organizes the mitotic spindle and ensures the faithful bipolar separation of sister chromatids during mitosis and meiosis [34]. Defects in centrosome function can lead to chromosome missegregation, representing another potential cause of genomic instability observed in CML [35–37]. The prevalence of centrosomal aberrations correlates with disease stage in CML and with the degree of aneuploidy found in leukemic cells. These observations, viewed in light of evidence that BCR-ABL protein interacts with the centrosomal protein, pericentrin, suggests that BCR-ABL activity may in part cause and/or propagate centrosomal aberrations that result ultimately in loss of chromosomal integrity. Interestingly, treatment of CML patients with BCR-ABL TKIs and exposure of cells to TKIs can induce centrosomal aberrations in disease-unrelated cells, even as TKI treatment suppresses BCR-ABL activity in leukemic cells, suggesting that multiple pathways may be involved in the destabilization of centrosome activity [38]. At present, however, there is no conclusive evidence that TKI therapy in CML increases the risk of secondary cancers [39].
Telomere Length
Telomere length naturally decreases with cycles of cell division. When telomeres become critically short, they may be mistaken for double-strand DNA breaks, so normal somatic cells enter replicative senescence to prevent genomic instability [40]. In cancerous cells, telomere shortening and inappropriate telomerase activation are frequently observed [41]. The average telomere length of patients with CML is significantly shorter than that of age-matched healthy individuals [42,43]. Because average telomere length of patients in CML-AP/BC is significantly shorter than that of patients in CP [42,43] and telomere length significantly correlates with duration of CP [42], telomere shortening has been implicated in disease progression in CML. At present, the prognostic utility of telomere length as a predictive biomarker of disease progression in CML is under study (ClinicalTrials.gov identifier NCT01061177).
Studies of telomere function in mouse models and in primary cancer cells derived from patients show that telomerase dysfunction and telomere shortening promote chromosomal instability [44], [45]. Thus, defects in telomere maintenance could potentially amplify other mechanisms of chromosome instability in CML stemming from BCR-ABL activity. Because telomere length can be partially restored with imatinib treatment [46], a more direct role for BCR-ABL in regulating telomere length cannot be discounted.
Differential Gene Expression
Gene expression profiling studies have sought to identify a set of differentially expressed genes associated with disease stage and therefore likely responsible for progression, manifestation of phenotypes and genotypes characteristic of advanced disease, or both. As expected, expression of genes functionally related to cell-cycle regulation, transcription, myeloid differentiation, intracellular signal transduction, cell adhesion, apoptosis, and immune response have been found to be differentially altered in samples derived from patients in CML-BC compared with CML-CP [47–49]. Despite variations in methodology used in different studies (eg, sources of samples, array platforms), a number of genes were identified in multiple studies as BC-specific, and the functional roles of progression-related genes were similar across studies, giving credence to the overall findings of these studies in aggregate.
FACTORS SHOWN TO PREDICT DISEASE PROGRESSION
According to the German CML Study Group experience, the cumulative incidence of CML-BC before the advent of imatinib (1983–2002) ranged from 12% to 61%, depending on the treatment modality, and has fallen to 4% since 2002 [3]. First-line treatment of CML-CP with TKI therapy significantly lowers the rate of progression to AP/BC, compared with non-TKI therapy (Table II). In the International Randomized Study of Interferon and STI571 (IRIS), the rate of freedom from progression to AP/BC at 18 months was significantly higher in the imatinib group than in the interferon (IFN)-α plus cytarabine comparator group (96.7% versus 91.5%, P<0.001) [50]. Long-term follow-up of patients in the imatinib group shows that most progression events occurred within the first 3 years of treatment [50–53] (Figure 1).
Table II.
Progressions reported in clinical studies of imatinib, nilotinib, and dasatinib
| Study | Drug treatment(s) | Patients, N | Timepoint | Cumulative progressions to AP/BC, n |
|---|---|---|---|---|
| IRIS [50–53] | Imatinib 400 mg vs IFN-α + cytarabine | IM: 553 IFN + cytarabine: 553 |
12 months | 8* 33 |
| IM: 461 | 2 years | 22 | ||
| IM: 431 | 3 years | 29 | ||
| IM: 409 | 4 years | 33 | ||
| IM: 382 | 5 years | 35 | ||
| IM: 364 | 6 years | 35 | ||
| NA | 7 years | 35 | ||
| IM: 304 | 8 years | 36 | ||
| German CML Study IV [54] | Imatinib 400 mg vs imatinib 800 mg vs imatinib + IFN-α | IM 400 mg: 325 IM 800 mg: 338 IM + IFN: 351 |
Median follow-up: IM 400 mg: 43 months IM 800 mg: 28 months IM + IFN: 48 months |
IM 400 mg: 16† IM 800 mg: 21† IM + IFN: 12† |
| ENESTnd [55–57] | Nilotinib 300 mg BID vs nilotinib 400 mg BID vs imatinib 400 mg | NIL 300 mg: 282 NIL 400 mg: 281 IM: 283 |
≥12 months of follow-up | NIL 300 mg: 2‡ NIL 400 mg: 1‡ IM: 11 |
| NIL 300 mg: 261 NIL 400 mg: 266 IM: 261 |
≥24 months of follow-up | NIL 300 mg: 2§ NIL 400 mg: 3§ IM: 12 |
||
| NIL 300 mg: 200 NIL 400 mg: 207 IM: 175 |
≥3 years of follow-up | NIL 300 mg: 2# NIL 400 mg: 3# IM: 12 |
||
| MDACC [58,59] | Nilotinib 400 mg BID | 61 | Median 17.3 months of follow-up | 2 |
| 100 | Median 24 months of follow-up | 2 | ||
| GIMEMA [60,61] | Nilotinib 400 mg BID | 73 | Median 15 months of follow-up | 1 |
| 73 | Median 48 months of follow-up | 1 | ||
| ICORG 0802 [62–64] | Nilotinib 300 mg BID | 15 | Median 70 days of follow-up | 0 |
| 27 enrolled 18 evaluable |
8 months | 0 | ||
| 61 enrolled 31 evaluable |
12 months | 0 | ||
| DASISION [65–67] | Dasatinib 100 mg vs imatinib 400 mg | DAS: 259 IM: 260 |
≥12 months of follow-up | DAS: 5 IM: 9 |
| DAS: 199 IM: 194 |
≥24 months of follow-up | DAS: 6 IM: 13 |
||
| MDACC [68] | Dasatinib 100 mg QD or 50 mg BID | 62 | Median 24 months of follow-up | 0 |
| SWOG 0325 [69] | Dasatinib 100 mg vs imatinib 400 mg | DAS: 123 IM: 123 |
Median 3 years of follow-up | DAS: 1 IM: 2 |
| BELA [70] | Bosutinib 500 mg vs imatinib 400 mg | BOS: 250 IM: 252 |
≥12 months of follow-up | BOS: 4 IM: 10 |
Abbreviations: AP, accelerated phase; BC, blast crisis; BID, twice daily; BOS, bosutinib; DAS, dasatinib; IFN, interferon; IM, imatinib; NIL, nilotinib
P< 0.001 versus control arm.
Progression defined as disease progression to AP/BC or death from any cause.
Time to progression to AP/BC significantly lower versus control arm.
Probability of progressing to AP/BC significantly lower versus control arm.
Rate of progression to AP/BC significantly lower versus control arm.
Figure 1.
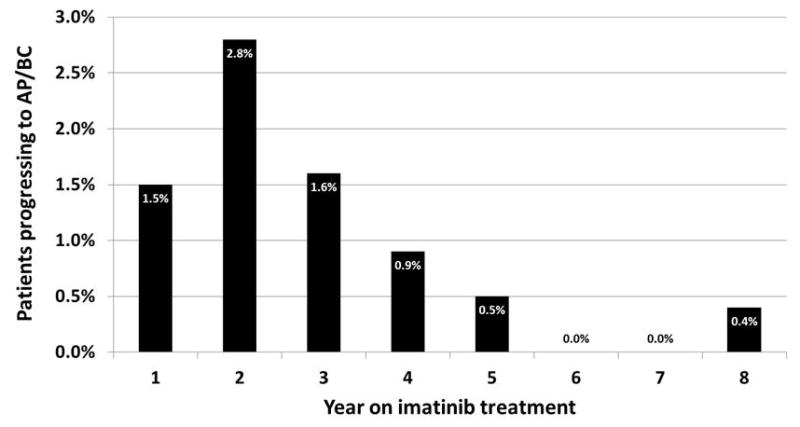
Annual rate of progression to CML in accelerated phase or blast crisis for patients treated with imatinib in the IRIS study [51–53].
It is not unexpected that imatinib, a potent BCR-ABL inhibitor [71], averts disease progression significantly better than did previous standard therapy [3], given that BCR-ABL activity likely plays a role in promoting disease progression. By extrapolation, TKIs that provide greater clinical inhibition of BCR-ABL than imatinib might be expected to provide even greater protection from disease progression. Indeed, 3-year follow-up of the Evaluating Nilotinib Efficacy and Safety in Clinical Trials – Newly Diagnosed Patients (ENESTnd) study shows that the number of progressions (defined by ELN criteria for AP/BC) per year is significantly lower with nilotinib than with imatinib [55–57] (Table II), and recently presented 4-year follow-up data are consistent with this finding [72]. In addition, 2-year follow-up of the Dasatinib versus Imatinib in Patients with Newly Diagnosed CML-CP (DASISION) study shows that the number of progressions (defined by ELN criteria for AP/BC) per year is lower with dasatinib than with imatinib [65–67] (Table II). Preliminary 3-year follow-up data of the DASISION study confirm this [65]. Furthermore, with a minimum of 12 months’ follow-up in the Bosutinib Efficacy and Safety in CML (BELA) study of bosutinibin newly diagnosed patients with CML-CP, four and 10 on-treatment progression events (defined by ELN criteria for AP/BC)were observed with bosutinib and imatinib, respectively [70]. Bosutinib, however, is currently approved only for treatment of CML after failure of prior therapy. Whether a reduction in the incidence of disease progression with approved first-line TKIs, nilotinib and dasatinib will significantly improve OS remains to be determined, and longer-term follow-up of the ENESTnd and DASISION studies are needed.
Treatment Response
Studies correlating the level of response to imatinib with long-term survival outcome have found that patients who respond favorably to imatinib, especially those who respond early in the treatment course, have significantly lower probabilities of an event (eg, death, loss of response, or progression to AP/BC) and of transformation to AP/BC than patients who do not respond as well to imatinib [51–53,73–79]. A correlation between level of response to TKI treatment and long-term outcome holds true for patients in late CP and AP, in terms of OS at 2 years [80] and relapse-free survival over more than 5 years of follow-up [81]. These findings imply that improving response rates and/or the degree of response—for example, with first-line nilotinib or dasatinib—could potentially improve progression outcome [76]. Although longer follow-up of clinical studies with the newer TKIs is needed to confirm this hypothesis, preliminary reports of landmark analyses of the ENESTnd study [82] and the DASISION study [83] indicate that the probability of disease progression to AP/BC is lower and the probability of achieving major molecular response (MMR)is higher for patients achieving BCR-ABL transcript level ≤10% versus >10% at 3 months.
BCR-ABL Mutational Status
The presence of non-silent genetic mutations that alter the BCR-ABL protein sequence has been linked to an increased probability of disease progression, most likely related to acquired resistance to TKI therapy. In a study of patients with imatinib failure who subsequently underwent allogeneic stem cell transplant, patients with baseline BCR-ABL mutation were significantly more likely to progress to AP/BC at the time of imatinib failure and have lower rates of event-free survival (EFS) and OS at 2 years than patients without [84]. The number of BCR-ABL mutations detected at the time of imatinib failure carries prognostic value in patients with CML-CP; rates of 4-year EFS were 56%, 49%, and 0% for patients with no mutations, one mutation, and more than one mutation, respectively [85]. The site of genetic mutation affects progression risk as well. Patients with mutations in the ATP phosphate-binding loop (P-loop) were at higher risk of disease progression to AP/BC [86] and death due to disease progression [87], and had worse estimates of progression-free survival (PFS) [86], than patients with BCR-ABL mutations outside of the P-loop. Given that imatinib, nilotinib, and dasatinib binding to BCR-ABL affects the conformation of the P-loop, genetic mutations in the P-loop that allow bypass of TKI-mediated inhibition could restore BCR-ABL kinase activity [88] and account for the observed increase in progression risk associated with P-loop mutations.
Early concerns that treatment with the newer TKIs nilotinib and dasatinib would lead to selection for TKI-resistant BCR-ABL mutations have been largely assuaged. In the ENESTnd study, post-baseline mutational analysis done on patients with lack of response or loss of response and at the end of treatment detected newly emergent BCR-ABL mutations in 11 patients in the nilotinib 300 mg twice daily (BID)group, 11 in the nilotinib 400 mg BID group, and 21 in the imatinib group. Among these patients, the multi-TKI–resistant T315I mutation was detected in three, two, and three patients, respectively [89]. In the DASISION study, post-baseline mutational analysis was done only in patients who discontinued treatment for any reason. New BCR-ABL mutations were detected in10 patients each in the dasatinib and imatinib arms. Of these, seven of 10 patients in the dasatinib arm carried the T315I mutation, compared with no patients in the imatinib arm [67].
Cytogenetic Aberrations at Diagnosis
The effect of additional cytogenetic aberrations (ACAs) at diagnosis of CML on treatment response and survival outcomes was studied in 1151 patients enrolled in the German CML Study IV. Stratification of patients by type of cytogenetic aberration—standard t(9;22), variant t(v;22), lack of the Y chromosome (−Y), minor route, and major route—revealed a significant adverse effect of major-route aberrations at diagnosis on 5-year PFS (90% versus 81% versus 88% versus 96% versus 50%, P<0.001)and OS (92% versus 87% versus 91% versus 96% versus 53%, P<0.001) [90]. These findings corroborate others showing similarly that the presence of ACAs, particularly major-route aberrations, at diagnosis is associated with lower rates of PFS and OS at 2 years [91] and at 5 years [78]. Thus patients with major-route aberrations at diagnosis may benefit from closer evaluation or more intensive therapy.
Risk Score at Baseline
Two commonly used formulas to predict prognosis are the Sokal and the Hasford risk scores [92,93]. In the IRIS study, patients in the imatinib group with low Sokal score at baseline had significantly better 6-year survival without progression to AP/BC (93.9% versus 76.3%, P< 0.001) than patients with high Sokal score [53]. In another study of 134 consecutive patients with CML-CP treated at a single center, patients with low/intermediate risk score by either Sokal or Hasford risk stratification at baseline had significantly better PFS during the study than patients with corresponding high risk score [94].
Recently, the European Treatment and Outcome Study for CML (EUTOS) developed a new formula to predict prognosis, the EUTOS score [95]. In the validation study of the EUTOS score, patients with low EUTOS score had significantly better 5-year PFS than patients with high EUTOS score [95]. Others have confirmed the value of the EUTOS score for predicting PFS [96] and EFS [97] in patients with CML treated with first-line imatinib, while others have not [98].
TREATMENT OPTIONS IN CML-AP/BC
Because advanced-phase CML is a complex disease, consensus on an optimal treatment approach has not been achieved. The following information on treatment options is based on that available from clinical practice guidelines, published literature, and our own clinical practices. Treatment options for patients with CML-AP/BC may include TKI therapy, allogeneic hematopoietic stem cell transplant (HSCT), chemotherapy, and other treatment modalities, such as IFN-α. When and in what sequence or combinations these treatment options are administered largely depends on the stage of disease, clinical presentation, and clinician experience.
TKI Therapy
All five currently available TKIs are approved in the US and Europe for treatment of CML-AP, and all but nilotinib are also approved for treatment of CML-BC [99–103]. Although TKI therapy is moderately effective in patients with CML-AP/BC, the overall effectiveness of TKI therapy in advanced CML is less robust than that observed in early-stage disease (Table III). We and others agree that TKI therapy should be considered a “bridge” to get patients with advanced CML to HSCT [3,104]. If HSCT is not offered, we recommend close monitoring by molecular testing every 3 months. Patients demonstrating a confirmed five-fold increase in BCR-ABL transcript levels and/or loss of MMR should be referred for HSCT.
Table III.
Summary of clinical studies of TKIs in CML-AP and/or CML-BC
| Study | TKI treatment | Patients* | CHR, % | CCyR, % | OS and/or PFS |
|---|---|---|---|---|---|
| Kantarjian 2002 [105] | Imatinib 400 or 600 mg | AP; N = 237 (n = 200 evaluable) | 80.0 | 23.5 | 18-month OS: 73% |
| Talpaz 2002 [106] | Imatinib 400 or 600 mg | AP; N = 181 | 33.7** | 16.6 | 12-month OS: 74% |
| Palandri 2009 [107] | Imatinib 600 mg | AP; N = 111 | 71.2** | 20.7 | Median OS: 37 months 7-year OS: 43% |
| Druker 2001 [108] | Imatinib 300–1000 mg | MyBC; n = 38 LyBC or ALL; n = 20 |
MyBC: 10.5 LyBC or ALL: 20.0 |
NR | NR |
| Kantarjian 2002 [109] | Imatinib 300–1000 mg | MyBC; n = 65 LyBC; n = 10 |
MyBC: 23.1 LyBC: 10.0 |
MyBC: 4.6 LyBC: 10.0 |
Median OS: MyBC: 6.5 months LyBC: 7 months |
| Sawyers 2002 [110] | Imatinib 400 or 600 mg | BC; N = 229 | 7.9** | 7.4 | Median OS: 6.9 months |
| Sureda 2003 [111] | Imatinib 600 mg | BC; N = 30 | 33.3** | 0 | 1-year OS: 36% |
| Palandri 2008 [112] | Imatinib 600 mg | MyBC; n = 72 LyBC; n = 20 |
MyBC: 23.6** LyBC: 35.0 |
MyBC: 6.9 LyBC: 20.0 |
Median OS: 7 months |
| le Coutre 2008 [113] | Nilotinib 400 mg BID | AP; N = 119 | 26.1 | 16.0 | 12-month OS: 79% |
| Giles 2010 [114] | Nilotinib 400 mg BID | AP; N = 21† (n = 17 evaluable) | 0 | 0 | 6-month PFS: 57% 6-month OS: 80% 12-month OS: 80% |
| Kantarjian 2006 [115] | Nilotinib up to 1200 mg | AP; n = 56 MyBC; n = 24 LyBC; n = 9† |
AP: 46.4 MyBC: 8.3 LyBC: 0 |
AP: 14.3 MyBC: 4.2 LyBC: 11.1 |
NR |
| Nicolini 2012 [116] | Nilotinib 400 mg BID | AP; n = 181 MyBC; n = 133 LyBC; n = 50 BC‡; n = 7 |
AP: 22.1 MyBC: 6.8 LyBC: 14.0 |
AP: 11.0 MyBC: 8.3 LyBC: 26.0 |
18-month OS: AP: 81% MyBC: 62% LyBC: 66% |
| Giles 2012 [117] | Nilotinib 400 mg BID | MyBC; n = 105 LyBC; n = 31 |
MyBC: 60§ LyBC: 59§ |
MyBC: 30 LyBC: 32 |
Median OS: MyBC: 10.1 months LyBC: 7.9 months |
| Guilhot 2007 [118] | Dasatinib 70 mg BID | AP; N = 107 | 39.3 | 24.3 | 8-month PFS: 76% |
| Apperley 2009 [119] | Dasatinib 70 mg BID | AP; N = 174 | 44.8 | 31.6 | 12-month PFS: 66% 12-month OS: 82% |
| Kantarjian 2009 [120] | Dasatinib 140 mg QD vs 70 mg BID | AP, QD/BID; n = 158/159 | QD/BID: 47.5/51.5 | QD/BID: 32.3/32.7 | Median PFS, QD/BID: 25.1/26.0 months Median OS, QD/BID: not reached/30.7 months |
| Talpaz 2006 [121] | Dasatinib 50–100 mg BID | AP; n = 11 MyBC; n =23 Ly BC or ALL; n = 10† |
AP: 45.5 MyBC: 34.8 LyBC or ALL: 70.0 |
AP: 18.2 MyBC: 26.1 LyBC or ALL: 30.0 |
NR |
| Cortes 2007 [122] | Dasatinib 70 mg BID | MyBC; n = 74 LyBC; n = 42 |
MyBC: 25.7 LyBC: 26.2 |
MyBC: 27.0 LyBC: 42.9 |
Median PFS: MyBC: 5.0 months LyBC: 2.8 months |
| Cortes 2008 [123] | Dasatinib 70 mg BID | MyBC; n = 109 LyBC; n = 48 |
MyBC: 26.6** LyBC: 29.2** |
MyBC: 25.7 LyBC: 45.8 |
Median PFS/OS: MyBC: 6.7/11.8 months LyBC: 3.0/5.3 months |
| Saglio 2010 [124] | Dasatinib 140 mg QD vs 70 mg BID | QD/BID: MyBC; n = 75/74 LyBC; n = 33/28 |
QD/BID: MyBC: 17.3/17.6 LyBC: 21.2/14.3 |
QD/BID: MyBC: 13.9/21.1 LyBC: 37.5/36.0 |
Median OS, QD/BID: MyBC: 7.9/7.7 months LyBC: 11.4/9.0 months |
| Bosutinib 2012 [102] | Bosutinib 500 mg QD | AP; n = 76 BC; n = 64 |
AP: 30.4 BC: 15.0 |
NR | NR |
| Cortes 2012 [103] | Ponatinib 2 mg, 8 mg, 15 mg, 30 mg, 45 mg, or 60 mg QD | AP; n = 9 BC; n = 8 ALL; n = 5 |
AP/BC/ALL (N = 22): CHR: NA Major HR: 36 |
AP/BC/ALL (N = 22): CCyR: 14 |
NR |
| Cortes 2012 [#87 ASCO 2012 #6503] | Ponatinib 45 mg QD | AP; n = 79 BC or ALL; n = 94 |
Major HR: AP: 67 BC or ALL: 37 |
AP: 17 BC or ALL: 28 |
NR |
Abbreviations: ALL, acute lymphoblastic leukemia; AP, accelerated phase; BC, blast crisis; BID, twice daily; CCyR, complete cytogenetic response; CHR, complete hematologic response; LyBC, lymphoid blast crisis; MyBC, myeloid blast crisis; NA, not applicable; NR, not reported; OS, overall survival; PFS, progression-free survival; QD, once daily; TKI, tyrosine kinase inhibitor
All studies of nilotinib, dasatinib, bosutinib, and ponatinib in this table involved patients who had prior imatinib therapy.
Response lasting ≥4 weeks.
This study also included patients with CML-CP, results for whom are not described in this table.
Subtype of BC (myeloid or lymphoid) was unknown for seven patients.
Rate of major hematologic response, which includes CHR and partial hematologic response.
Patients harboring the T315I mutation may benefit from treatment with ponatinib [126]. Preliminary reporting of the phase 2 Ponatinib Ph+ Acute Lymphoblastic Leukemia (ALL) and CML Evaluation (PACE) study showed that CCyR was achieved in 4 of 17 (24%) evaluable patients with CML-AP and in 12 of 41 (29%) evaluable patients with CML-BC or Ph+ ALL with the T315I mutation [125].
Patients who fail treatment with or who develop resistance to currently available TKIs may soon have another BCR-ABL TKI as a treatment option. Rebastinib (formerly DCC-2036) is a BCR-ABL TKI that, like ponatinib, has activity against the T315I mutation [127]. In a phase 1 study of 30 patients, including eight in CML-AP and three in CML-BC, rebastinib elicited hematologic responses in two patients with CML-AP [128].
Hematopoietic Stem Cell Transplant
Long-term remission is uncommon in advanced CML, but HSCT represents the best option for patients in advanced stages of CML to achieve remission or cure. Unfortunately, HSCT is associated with both considerable morbidity and the need for subsequent immunosuppressive therapy to modulate graft-versus-host disease, so this procedure is generally reserved for patients younger than 65 years of age with relatively good performance status who have an available donor. Considering that the median age of diagnosis of CML is 64 years [129], the role of HSCT is limited. The identification of potential matching donors can be a time-consuming process; thus, clinicians who are considering HSCT for their patients should refer them for evaluation for HSCT before their health status precipitously declines [3,104]. Consistent with observed trends in efficacy with other treatment modalities, long-term outcomes following HSCT are better for patients who undergo the procedure in CML-CP than in CML-AP/BC. In a subanalysis of the German CML Study IV, 3-year survival rate after HSCT was 91% for 56 patients who underwent the procedure while in CML-CP and 59% for 28 patients in CML-AP/BC (25 of 28 patients were in CML-BC)[130]. Notably, another study of outcomes after HSCT found that patients in second CP before transplant had survival outcomes at 3 years that were comparable to that of patients in AP and more favorable than that of patients in BC [131]. Following HSCT, we recommend maintenance therapy with a TKI (selected based on previous response and mutation profile) and molecular monitoring every 3 months.
Newly Diagnosed Advanced CML
For patients who are newly diagnosed with CML-AP/BC or have signs that may indicate high risk of progression to AP/BC (eg, high Sokal or Hasford risk score, unfavorable cytogenetics), were commend that they be treated intensively. Treatment of patients with CML-AP with nilotinib (400 mg BID) or dasatinib (140 mg once daily [QD]) has been recommended [132]. For patients with CML-BC who have been pretreated with conventional therapy, such as IFN-α or hydroxyurea, but not with TKIs, treatment with high-dose imatinib (600–800 mg QD), dasatinib (140 mg QD), or nilotinib (400 mg BID) are all valid options [3]. Patients with CML-AP or CML-BC should be referred for evaluation of eligibility for HSCT [133].
Progression to Advanced CML
For patients who progress to CML-AP/BC while on first-line TKI therapy, median OS is short, approximately 10.5 months for both imatinib and nilotinib [56]. We consider that a switch in treatment to an alternative TKI is appropriate for patients who progress to CML-AP while on imatinib (Table III). The choice of second-line TKI should depend in part on BCR-ABL mutational status. In vitro evidence shows that TKIs have differential binding affinity for common BCR-ABL kinase domain mutations [134], although it should be acknowledged that in vitro binding affinity by itself is insufficient to predict clinical activity [135]. Depending on response to TKI treatment, patients may be considered for HSCT [132].
For patients progressing to CML-BC following prior imatinib, treatment with nilotinib or dasatinib (depending on mutational status) in combination with acute leukemia-type chemotherapy is a viable option [3,132]. Patients with lymphoid BC may be treated with ALL-type induction chemotherapy, and those with myeloid BC with AML-type induction therapy [132]. If TKI-based therapy fails, then treatment with an ALL-type or AML-type induction chemotherapy regimen, depending on the leukemia subtype, or enrollment in a clinical study is recommended [3].
Patients with CML-BC with extramedullary infiltration have particularly poor prognosis [136,137]. There is no standard treatment for CML-BC with extramedullary involvement. Treatment with either chemotherapy regimens or imatinib has been shown to yield comparable outcomes, in terms of response and duration of response [138]. For cases in which extramedullary infiltration involves the central nervous system, however, evidence suggests that imatinib treatment may not be appropriate. Both in humans and other primates, imatinib concentrations in cerebrospinal fluid is much lower than in plasma [139–142], suggesting that imatinib may not cross the blood-brain barrier. At present, studies of the pharmacokinetics of other TKIs in cerebrospinal fluid are lacking.
TKI-Based Combination Regimens
Although allogeneic HSCT remains the gold standard for patients in CML-BC after complete remission with induction chemotherapy and TKIs, a recent report has shown improved OS among patients with Ph+ ALL who achieved MMR or better after induction chemotherapy plus imatinibor dasatinib and did not undergo HSCT [143]. These findings suggest that long-term survival without HSCT may be possible in patients who achieve deep molecular response to induction chemotherapy plus TKIs. In addition, patients who achieved MMR in this study at 3, 6, 9, and 12 months had significantly better OS than patients who did not [143], findings that support the prognostic value of achieving deep response at those timepoints and underscore the importance of molecular monitoring every 3 months in patients with advanced disease.
Various combination regimens of imatinib plus chemotherapy have been evaluated in clinical studies [144–148]. Generally, the rationales for these combinations include: (1) agents in combination may overcome resistance of leukemia cells to single agents; (2) combining agents with distinct mechanisms of action may delay clonal selection of resistant leukemia cells; (3) combination regimens may elicit responses more rapidly than single agents, allowing patients to be referred to HSCT sooner; (4) combination regimens may enhance elimination of quiescent leukemic stem cells.
Several studies have found that TKI-based combination induction regimens followed by HSCT can be effective in prolonging OS. In one study of imatinib plus mitoxantrone/etoposide/cytarabine in 16 patients with CML-BC, seven patients who received the more intensive induction regimen achieved hematologic response, and four of seven subsequently underwent HSCT. Of these four patients, three remained in CCyR and achieved sustained negativity as shown by polymerase chain reaction for at least 9–28 months after HSCT [146]. In another study of imatinib plus omacetaxine/granulocyte colony–stimulating factor in 11 patients with CML-BC after imatinib failure, major cytogenetic response was achieved in 100% of patients. Eight patients proceeded to HSCT, and six patients were alive, including five with complete hematologic response (CHR), a median of 12 months later [147]. Studies of anthracyclines plus imatinib/cytarabine combinations have also shown clinical activity in CML-BC. In one such study, an induction regimen of daunorubicin plus imatinib/cytarabine was evaluated in 36 patients with myeloid CML-BC. Twenty patients achieved CHR, half of whom underwent HSCT thereafter. Median OS for patients who achieved hematologic response was 35.4 months [148]. In another study, treatment of 19 patients with myeloid CML-BC with idarubicin plus imatinib/cytarabine resulted in CHR for nine patients, including CCyR in three patients. Six patients underwent HSCT. Median survival was 5 months [149].
CONCLUSIONS
The advent of BCR-ABL TKI therapy has vastly improved the clinical outlook for patients with CML. Patients with advanced CML fare better today than at any other time in history; nonetheless, there remains considerable room for improvement. The number of effective treatment options is limited in advanced CML, and treatment modalities effective in CML-CP are not nearly so in CML-AP/BC. Clinical studies of currently approved therapies, investigational agents, and combination regimens continue unabated with the goal of expanding the number of treatment options available to patients in CML-AP/BC.
The approval in the US and Europe of nilotinib, dasatinib, bosutinib, and ponatinib for advanced CML provides clinicians with multiple treatment options for patients with CML-AP/BC, although treatment response to TKIs in late-stage disease is not as favorable as in early CP. As such, our view is that a prudent approach to clinical practice is to stably maintain patients in CML-CP for as long as possible [3,104]. The treating clinician can accomplish this goal of therapy by optimizing response to first-line therapy and by monitoring for the presence of clonal evolution, which portends poor prognosis.
Regarding optimal first-line therapy, the higher rates of molecular response and lower rates of progression with nilotinib and dasatinib compared with imatinib indicate that the use of the newer TKIs for first-line treatment may improve overall survival of patients in CML-CP, in part by effectively preventing disease progression. In this regard, longer-term survival data from clinical studies of first-line TKI treatment are eagerly awaited.
Regarding clonal evolution, its presence at diagnosis predicts shortened PFS and OS, and its occurrence during treatment is considered a marker of treatment failure. Thus, clinicians can consider for closer observation and/or more intensive therapy patients who demonstrate high-risk cytogenetic features at diagnosis or inadequate response to first-line treatment. Adopting such a proactive approach in treating patients showing early signs of potential disease progression will reduce the likelihood of disease progression.
Acknowledgments
Financial support for medical editorial assistance was provided by Novartis Pharmaceuticals Corporation. The authors thank Anna Lau, PhD, and Patricia Segarini, PhD, of Percolation Communications LLC for their medical editorial assistance.
Footnotes
Potential Conflicts of Interest
Dr. Jabbour reports honoraria from Bristol-Myers Squibb, Novartis, and Pfizer, outside the submitted work. Dr. Hughes reports honoraria, consultancy, and research support from Bristol-Myers Squibb, Novartis, and Ariad, outside the submitted work. Dr. Cortes reports grants, consultancy, and research support from Ariad, grants and research support from Novartis, consultancy and research support from Pfizer, consultancy and research support from ChemGenex, grants and research support from Bristol-Myers Squibb, and research support from Deciphera, outside the submitted work. Dr. Kantarjian reports research support from Bristol-Myers Squibb, consultancy and research support from Novartis, and research support from Pfizer, outside the submitted work. Dr. Hochhaus reports consultancy, honoraria, and research support from Ariad, Novartis, Pfizer, Bristol-Myers Squibb, and Merck Sharpe & Dohme, outside the submitted work.
References
- 1.Kantarjian H, O’Brien S, Jabbour E, et al. Improved survival in chronic myeloid leukemia since the introduction of imatinib therapy: a single-institution historical experience. Blood. 2012;119:1981–1987. doi: 10.1182/blood-2011-08-358135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bjorkholm M, Ohm L, Eloranta S, et al. Success story of targeted therapy in chronic myeloid leukemia: a population-based study of patients diagnosed in Sweden from 1973 to 2008. J Clin Oncol. 2011;29:2514–2520. doi: 10.1200/JCO.2011.34.7146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hehlmann R. How I treat CML blast crisis. Blood. 2012 May 31; doi: 10.1182/blood-2012-03-380147. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 4.Kantarjian HM, Keating MJ, Smith TL, Talpaz M, McCredie KB. Proposal for a simple synthesis prognostic staging system in chronic myelogenous leukemia. Am J Med. 1990;88:1–8. doi: 10.1016/0002-9343(90)90119-x. [DOI] [PubMed] [Google Scholar]
- 5.Savage DG, Szydlo RM, Chase A, Apperley JF, Goldman JM. Bone marrow transplantation for chronic myeloid leukaemia: the effects of differing criteria for defining chronic phase on probabilities of survival and relapse. Br J Haematol. 1997;99:30–35. doi: 10.1046/j.1365-2141.1997.3453159.x. [DOI] [PubMed] [Google Scholar]
- 6.Sokal JE, Baccarani M, Russo D, Tura S. Staging and prognosis in chronic myelogenous leukemia. Semin Hematol. 1988;25:49–61. [PubMed] [Google Scholar]
- 7.Swerdlow SH, Campo E, Harris NL, et al., editors. World Health Organization classification of tumours of haematopoietic and lymphoid tissues. 4. Geneva, Switzerland: WHO Press; 2008. [Google Scholar]
- 8.Baccarani M, Saglio G, Goldman J, et al. Evolving concepts in the management of chronic myeloid leukemia: recommendations from an expert panel on behalf of the European LeukemiaNet. Blood. 2006;108:1809–1820. doi: 10.1182/blood-2006-02-005686. [DOI] [PubMed] [Google Scholar]
- 9.Cortes JE, Talpaz M, O’Brien S, et al. Staging of chronic myeloid leukemia in the imatinib era. Cancer. 2006;106:1306–1315. doi: 10.1002/cncr.21756. [DOI] [PubMed] [Google Scholar]
- 10.Nowell PC, Hungerford DA. Chromosome studies on normal and leukemic human leukocytes. J Natl Cancer Inst. 1960;25:85–109. [PubMed] [Google Scholar]
- 11.Shtivelman E, Lifshitz B, Gale RP, Canaani E. Fused transcript of abl and bcr genes in chronic myelogenous leukaemia. Nature. 1985;315:550–554. doi: 10.1038/315550a0. [DOI] [PubMed] [Google Scholar]
- 12.Perrotti D, Jamieson C, Goldman J, Skorski T. Chronic myeloid leukemia: mechanisms of blastic transformation. J Clin Invest. 2010;120:2254–2264. doi: 10.1172/JCI41246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gaiger A, Henn T, Horth E, et al. Increase of bcr-abl chimeric mRNA expression in tumor cells of patients with chronic myeloid leukemia precedes disease progression. Blood. 1995;86:2371–2378. [PubMed] [Google Scholar]
- 14.Lin F, van Rhee F, Goldman JM, Cross NC. Kinetics of increasing BCR-ABL transcript numbers in chronic myeloid leukemia patients who relapse after bone marrow transplantation. Blood. 1996;87:4473–4478. [PubMed] [Google Scholar]
- 15.Lahaye T, Riehm B, Berger U, et al. Response and resistance in 300 patients with BCR-ABL-positive leukemias treated with imatinib in a single center: a 4. 5-year follow-up. Cancer. 2005;103:1659–1669. doi: 10.1002/cncr.20922. [DOI] [PubMed] [Google Scholar]
- 16.Koptyra M, Falinski R, Nowicki MO, et al. BCR/ABL kinase induces self-mutagenesis via reactive oxygen species to encode imatinib resistance. Blood. 2006;108:319–327. doi: 10.1182/blood-2005-07-2815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sattler M, Verma S, Shrikhande G, et al. The BCR/ABL tyrosine kinase induces production of reactive oxygen species in hematopoietic cells. J Biol Chem. 2000;275:24273–24278. doi: 10.1074/jbc.M002094200. [DOI] [PubMed] [Google Scholar]
- 18.Flis K, Irvine D, Copland M, Bhatia R, Skorski T. Chronic myeloid leukemia stem cells display alterations in expression of genes involved in oxidative phosphorylation. Leuk Lymphoma. 2012 Jun 18; doi: 10.3109/10428194.2012.696313. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 19.Nowicki MO, Falinski R, Koptyra M, et al. BCR/ABL oncogenic kinase promotes unfaithful repair of the reactive oxygen species-dependent DNA double-strand breaks. Blood. 2004;104:3746–3753. doi: 10.1182/blood-2004-05-1941. [DOI] [PubMed] [Google Scholar]
- 20.Deutsch E, Dugray A, AbdulKarim B, et al. BCR-ABL down-regulates the DNA repair protein DNA-PKcs. Blood. 2001;97:2084–2090. doi: 10.1182/blood.v97.7.2084. [DOI] [PubMed] [Google Scholar]
- 21.Slupianek A, Hoser G, Majsterek I, et al. Fusion tyrosine kinases induce drug resistance by stimulation of homology-dependent recombination repair, prolongation of G(2)/M phase, and protection from apoptosis. Mol Cell Biol. 2002;22:4189–4201. doi: 10.1128/MCB.22.12.4189-4201.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Slupianek A, Poplawski T, Jozwiakowski SK, et al. BCR/ABL stimulates WRN to promote survival and genomic instability. Cancer Res. 2011;71:842–851. doi: 10.1158/0008-5472.CAN-10-1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slupianek A, Dasgupta Y, Ren SY, et al. Targeting RAD51 phosphotyrosine-315 to prevent unfaithful recombination repair in BCR-ABL1 leukemia. Blood. 2011;118:1062–1068. doi: 10.1182/blood-2010-09-307256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Johansson B, Fioretos T, Mitelman F. Cytogenetic and molecular genetic evolution of chronic myeloid leukemia. Acta Haematol. 2002;107:76–94. doi: 10.1159/000046636. [DOI] [PubMed] [Google Scholar]
- 25.Mitelman F, Levan G, Nilsson PG, Brandt L. Non-random karyotypic evolution in chronic myeloid leukemia. Int J Cancer. 1976;18:24–30. doi: 10.1002/ijc.2910180105. [DOI] [PubMed] [Google Scholar]
- 26.Jennings BA, Mills KI. c-myc locus amplification and the acquisition of trisomy 8 in the evolution of chronic myeloid leukaemia. Leuk Res. 1998;22:899–903. doi: 10.1016/s0145-2126(98)00097-6. [DOI] [PubMed] [Google Scholar]
- 27.Honda H, Ushijima T, Wakazono K, et al. Acquired loss of p53 induces blastic transformation in p210(bcr/abl)-expressing hematopoietic cells: a transgenic study for blast crisis of human CML. Blood. 2000;95:1144–1150. [PubMed] [Google Scholar]
- 28.Iervolino A, Santilli G, Trotta R, et al. hnRNP A1 nucleocytoplasmic shuttling activity is required for normal myelopoiesis and BCR/ABL leukemogenesis. Mol Cell Biol. 2002;22:2255–2266. doi: 10.1128/MCB.22.7.2255-2266.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Lindern M, van Baal S, Wiegant J, Raap A, Hagemeijer A, Grosveld G. Can, a putative oncogene associated with myeloid leukemogenesis, may be activated by fusion of its 3′ half to different genes: characterization of the set gene. Mol Cell Biol. 1992;12:3346–3355. doi: 10.1128/mcb.12.8.3346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Neviani P, Santhanam R, Trotta R, et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer Cell. 2005;8:355–368. doi: 10.1016/j.ccr.2005.10.015. [DOI] [PubMed] [Google Scholar]
- 31.Li M, Makkinje A, Damuni Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. J Biol Chem. 1996;271:11059–11062. doi: 10.1074/jbc.271.19.11059. [DOI] [PubMed] [Google Scholar]
- 32.Perrotti D, Neviani P. ReSETting PP2A tumour suppressor activity in blast crisis and imatinib-resistant chronic myelogenous leukaemia. Br J Cancer. 2006;95:775–781. doi: 10.1038/sj.bjc.6603317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eiring AM, Neviani P, Santhanam R, et al. Identification of novel posttranscriptional targets of the BCR/ABL oncoprotein by ribonomics: requirement of E2F3 for BCR/ABL leukemogenesis. Blood. 2008;111:816–828. doi: 10.1182/blood-2007-05-090472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bornens M. The centrosome in cells and organisms. Science. 2012;335:422–426. doi: 10.1126/science.1209037. [DOI] [PubMed] [Google Scholar]
- 35.Chan JY. A clinical overview of centrosome amplification in human cancers. Int J Biol Sci. 2011;7:1122–1144. doi: 10.7150/ijbs.7.1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Giehl M, Fabarius A, Frank O, et al. Centrosome aberrations in chronic myeloid leukemia correlate with stage of disease and chromosomal instability. Leukemia. 2005;19:1192–1197. doi: 10.1038/sj.leu.2403779. [DOI] [PubMed] [Google Scholar]
- 37.Patel H, Gordon MY. Abnormal centrosome-centriole cycle in chronic myeloid leukaemia? Br J Haematol. 2009;146:408–417. doi: 10.1111/j.1365-2141.2009.07772.x. [DOI] [PubMed] [Google Scholar]
- 38.Giehl M, Leitner A, Haferlach C, et al. Detection of centrosome aberrations in disease-unrelated cells from patients with tumor treated with tyrosine kinase inhibitors. Eur J Haematol. 2010;85:139–148. doi: 10.1111/j.1600-0609.2010.01459.x. [DOI] [PubMed] [Google Scholar]
- 39.Stein BL. Chronic myeloid leukemia and risk of second malignancy in two eras of treatment. Leuk Lymphoma. 2012;53:1651–1653. doi: 10.3109/10428194.2012.668684. [DOI] [PubMed] [Google Scholar]
- 40.Blackburn EH. Structure and function of telomeres. Nature. 1991;350:569–573. doi: 10.1038/350569a0. [DOI] [PubMed] [Google Scholar]
- 41.Raynaud CM, Sabatier L, Philipot O, Olaussen KA, Soria JC. Telomere length, telomeric proteins and genomic instability during the multistep carcinogenic process. Crit Rev Oncol Hematol. 2008;66:99–117. doi: 10.1016/j.critrevonc.2007.11.006. [DOI] [PubMed] [Google Scholar]
- 42.Brümmendorf TH, Holyoake TL, Rufer N, et al. Prognostic implications of differences in telomere length between normal and malignant cells from patients with chronic myeloid leukemia measured by flow cytometry. Blood. 2000;95:1883–1890. [PubMed] [Google Scholar]
- 43.Drummond M, Lennard A, Brummendorf T, Holyoake T. Telomere shortening correlates with prognostic score at diagnosis and proceeds rapidly during progression of chronic myeloid leukemia. Leuk Lymphoma. 2004;45:1775–1781. doi: 10.1080/10428190410001693542. [DOI] [PubMed] [Google Scholar]
- 44.Wong KK, Chang S, Weiler SR, et al. Telomere dysfunction impairs DNA repair and enhances sensitivity to ionizing radiation. Nat Genet. 2000;26:85–88. doi: 10.1038/79232. [DOI] [PubMed] [Google Scholar]
- 45.Plentz RR, Schlegelberger B, Flemming P, et al. Telomere shortening correlates with increasing aneuploidy of chromosome 8 in human hepatocellular carcinoma. Hepatology. 2005;42:522–526. doi: 10.1002/hep.20847. [DOI] [PubMed] [Google Scholar]
- 46.Brümmendorf TH, Ersoz I, Hartmann U, et al. Normalization of previously shortened telomere length under treatment with imatinib argues against a preexisting telomere length deficit in normal hematopoietic stem cells from patients with chronic myeloid leukemia. Ann N Y Acad Sci. 2003;996:26–38. doi: 10.1111/j.1749-6632.2003.tb03229.x. [DOI] [PubMed] [Google Scholar]
- 47.Radich JP, Dai H, Mao M, et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2006;103:2794–2799. doi: 10.1073/pnas.0510423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nowicki MO, Pawlowski P, Fischer T, Hess G, Pawlowski T, Skorski T. Chronic myelogenous leukemia molecular signature. Oncogene. 2003;22:3952–3963. doi: 10.1038/sj.onc.1206620. [DOI] [PubMed] [Google Scholar]
- 49.Zheng C, Li L, Haak M, et al. Gene expression profiling of CD34+ cells identifies a molecular signature of chronic myeloid leukemia blast crisis. Leukemia. 2006;20:1028–1034. doi: 10.1038/sj.leu.2404227. [DOI] [PubMed] [Google Scholar]
- 50.O’Brien SG, Guilhot F, Larson RA, et al. Imatinib compared with interferon and low-dose cytarabine for newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2003;348:994–1004. doi: 10.1056/NEJMoa022457. [DOI] [PubMed] [Google Scholar]
- 51.Deininger M, O’Brien SG, Guilhot F, et al. International randomized study of interferon vs STI571 (IRIS) 8-year follow up: sustained survival and low risk for progression or events in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) treated with imatinib. Blood (ASH Annual Meeting Abstracts) 2009;114:1126. [Google Scholar]
- 52.Druker BJ, Guilhot F, O’Brien SG, et al. Five-year follow-up of patients receiving imatinib for chronic myeloid leukemia. N Engl J Med. 2006;355:2408–2417. doi: 10.1056/NEJMoa062867. [DOI] [PubMed] [Google Scholar]
- 53.Hochhaus A, O’Brien SG, Guilhot F, et al. Six-year follow-up of patients receiving imatinib for the first-line treatment of chronic myeloid leukemia. Leukemia. 2009;23:1054–1061. doi: 10.1038/leu.2009.38. [DOI] [PubMed] [Google Scholar]
- 54.Hehlmann R, Lauseker M, Jung-Munkwitz S, et al. Tolerability-adapted imatinib 800 mg/d versus 400 mg/d versus 400 mg/d plus interferon-alpha in newly diagnosed chronic myeloid leukemia. J Clin Oncol. 2011;29:1634–1642. doi: 10.1200/JCO.2010.32.0598. [DOI] [PubMed] [Google Scholar]
- 55.Kantarjian HM, Hochhaus A, Saglio G, et al. Nilotinib versus imatinib for the treatment of patients with newly diagnosed chronic phase, Philadelphia chromosome-positive, chronic myeloid leukaemia: 24-month minimum follow-up of the phase 3 randomised ENESTnd trial. Lancet Oncol. 2011;12:841–851. doi: 10.1016/S1470-2045(11)70201-7. [DOI] [PubMed] [Google Scholar]
- 56.Larson RA, Hochhaus A, Hughes TP, et al. Nilotinib vs imatinib in patients with newly diagnosed Philadelphia chromosome-positive chronic myeloid leukemia in chronic phase: ENESTnd 3-year follow-up. Leukemia. 2012 May 18; doi: 10.1038/leu.2012.134. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 57.Saglio G, Kim DW, Issaragrisil S, et al. Nilotinib versus imatinib for newly diagnosed chronic myeloid leukemia. N Engl J Med. 2010;362:2251–2259. doi: 10.1056/NEJMoa0912614. [DOI] [PubMed] [Google Scholar]
- 58.Cortes JE, Jones D, O’Brien S, et al. Nilotinib as front-line treatment for patients with chronic myeloid leukemia in early chronic phase. J Clin Oncol. 2010;28:392–397. doi: 10.1200/JCO.2009.25.4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quintas-Cardama A, Kantarjian HM, Luthra R, et al. Efficacy of frontline nilotinib therapy in patients (Pts) with newly diagnosed Philadelphia chromosome (Ph)-positive chronic myeloid leukemia in early chronic phase (CML-CP) Blood (ASH Annual Meeting Abstracts) 2011;118:454. [Google Scholar]
- 60.Gugliotta G, Castagnetti F, Breccia M, et al. Early CP CML, nilotinib 400 mg twice daily frontline: beyond 3 years, results remain excellent and stable (A GIMEMA CML Working Party Trial) Blood (ASH Annual Meeting Abstracts) 2011;118:2756. [Google Scholar]
- 61.Rosti G, Palandri F, Castagnetti F, et al. Nilotinib for the frontline treatment of Ph(+) chronic myeloid leukemia. Blood. 2009;114:4933–4938. doi: 10.1182/blood-2009-07-232595. [DOI] [PubMed] [Google Scholar]
- 62.O’Dwyer EO, Swords R, Giles F, et al. Nilotinb 300 mg twice daily is effective and well tolerated as first line treatment of Ph-positive chronic myeloid leukemia in chronic phase: updated results of the ICORG 0802 phase study [abstract 0812] Haematologica. 2010;95:340. [Google Scholar]
- 63.O’Dwyer MC, Kent E, Parker M, et al. Nilotinib 300 mg twice daily is effective and well tolerated as first line treatment of Ph-positive chronic myeloid leukemia in chronic phase: preliminary results of the ICORG 0802 phase 2 study. Blood (ASH Annual Meeting Abstracts) 2009;114:3294. [Google Scholar]
- 64.O’Dwyer ME, Swords R, Giles F, et al. Analysis of the GeneXpert System on the international multicentre ICORG 08-02 phase II study of nilotinib 300 mg BID as frontline treatment in patients with early chronic phase chronic myeloid leukemia (ECPCML) Blood (ASH Annual Meeting Abstracts) 2011;118:3774. [Google Scholar]
- 65.Hochhaus A, Shah NP, Cortes JE, et al. Dasatinib versus imatinib (IM) in newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP): DASISION 3-year follow-up. J Clin Oncol. 2012;30:6504. [Google Scholar]
- 66.Kantarjian H, Shah NP, Hochhaus A, et al. Dasatinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia. N Engl J Med. 2010;362:2260–2270. doi: 10.1056/NEJMoa1002315. [DOI] [PubMed] [Google Scholar]
- 67.Kantarjian HM, Shah NP, Cortes JE, et al. Dasatinib or imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: 2-year follow-up from a randomized phase 3 trial (DASISION) Blood. 2012;119:1123–1129. doi: 10.1182/blood-2011-08-376087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cortes JE, Jones D, O’Brien S, et al. Results of dasatinib therapy in patients with early chronic-phase chronic myeloid leukemia. J Clin Oncol. 2010;28:398–404. doi: 10.1200/JCO.2009.25.4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Radich JP, Kopecky KJ, Appelbaum FR, et al. A randomized trial of dasatinib 100 mg vs imatinib 400 mg in newly diagnosed chronic phase chronic myeloid leukemia. Blood. 2012 Aug 28; doi: 10.1182/blood-2012-02-410688. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cortes JE, Kim DW, Kantarjian HM, et al. Bosutinib versus imatinib in newly diagnosed chronic-phase chronic myeloid leukemia: results from the BELA trial. J Clin Oncol. 2012 Sep 4; doi: 10.1200/JCO.2011.38.7522. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buchdunger E, Zimmermann J, Mett H, et al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996;56:100–104. [PubMed] [Google Scholar]
- 72.Kantarjian HM, Kim D-W, Issaragrisil S, et al. Enestnd 4-year (y) update: continued superiority of nilotinib vs imatinib in patients (pts) with newly diagnosed Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia in chronic phase (CML-CP) Blood (ASH Annual Meeting Abstracts) 2012;120:1676. [Google Scholar]
- 73.Alvarado Y, Kantarjian H, O’Brien S, et al. Significance of suboptimal response to imatinib, as defined by the European LeukemiaNet, in the long-term outcome of patients with early chronic myeloid leukemia in chronic phase. Cancer. 2009;115:3709–3718. doi: 10.1002/cncr.24418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hanfstein B, Muller MC, Hehlmann R, et al. Early molecular and cytogenetic response is predictive for long-term progression-free and overall survival in chronic myeloid leukemia (CML) Leukemia. 2012 Mar 26; doi: 10.1038/leu.2012.85. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 75.Hughes TP, Hochhaus A, Branford S, et al. Long-term prognostic significance of early molecular response to imatinib in newly diagnosed chronic myeloid leukemia: an analysis from the International Randomized Study of Interferon and STI571 (IRIS) Blood. 2010;116:3758–3765. doi: 10.1182/blood-2010-03-273979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jabbour E, Kantarjian H, O’Brien S, et al. The achievement of an early complete cytogenetic response is a major determinant for outcome in patients with early chronic phase chronic myeloid leukemia treated with tyrosine kinase inhibitors. Blood. 2011;118:4541–4546. doi: 10.1182/blood-2011-04-348110. quiz 4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marin D, Ibrahim AR, Lucas C, et al. Assessment of BCR-ABL1 transcript levels at 3 months is the only requirement for predicting outcome for patients with chronic myeloid leukemia treated with tyrosine kinase inhibitors. J Clin Oncol. 2012;30:232–238. doi: 10.1200/JCO.2011.38.6565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Marin D, Milojkovic D, Olavarria E, et al. European Leukemia Net criteria for failure or suboptimal response reliably identify patients with CML in early chronic phase treated with imatinib whose eventual outcome is poor. Blood. 2008;112:4437–4444. doi: 10.1182/blood-2008-06-162388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Quintas-Cardama A, Kantarjian H, Jones D, et al. Delayed achievement of cytogenetic and molecular response is associated with increased risk of progression among patients with chronic myeloid leukemia in early chronic phase receiving high-dose or standard-dose imatinib therapy. Blood. 2009;113:6315–6321. doi: 10.1182/blood-2008-07-166694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fava C, Kantarjian HM, Jabbour E, et al. Failure to achieve a complete hematologic response at the time of a major cytogenetic response with second-generation tyrosine kinase inhibitors is associated with a poor prognosis among patients with chronic myeloid leukemia in accelerated or blast phase. Blood. 2009;113:5058–5063. doi: 10.1182/blood-2008-10-184960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Piazza RG, Magistroni V, Franceschino A, et al. The achievement of durable complete cytogenetic remission in late chronic and accelerated phase patients with CML treated with Imatinib mesylate predicts for prolonged response at 6 years. Blood Cells Mol Dis. 2006;37:111–115. doi: 10.1016/j.bcmd.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 82.Hochhaus A, Guilhot F, Al-Ali KH, et al. Early BCR-ABL transcript levels predict future molecular response and long-term outcomes in newly diagnosed patients with chronic myeloid leukemia in chronic phase: analysis of ENESTnd 3-year data [abstract 0584]. 17th Congress of the European Hematology Association; June 14–17, 2012; Amsterdam, the Netherlands. [Google Scholar]
- 83.Hochhaus A, Boqué C, Bradley Garelik M, Manos G, Steegmann JL. Molecular response kinetics and BCR-ABL reductions in patients with newly diagnosed chronic myeloid leukemia in chronic phase (CML-CP) receiving dasatinib vs imatinib: DASISION 3-year follow-up [abstract 0192]. 17th Congress of the European Hematology Association; June 14–17, 2012; Amsterdam, the Netherlands. [Google Scholar]
- 84.Jabbour E, Cortes J, Santos FP, et al. Results of allogeneic hematopoietic stem cell transplantation for chronic myelogenous leukemia patients who failed tyrosine kinase inhibitors after developing BCR-ABL1 kinase domain mutations. Blood. 2011;117:3641–3647. doi: 10.1182/blood-2010-08-302679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Quintas-Cardama A, Kantarjian H, O’Brien S, et al. Outcome of patients with chronic myeloid leukemia with multiple ABL1 kinase domain mutations receiving tyrosine kinase inhibitor therapy. Haematologica. 2011;96:918–921. doi: 10.3324/haematol.2010.039321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Soverini S, Martinelli G, Rosti G, et al. ABL mutations in late chronic phase chronic myeloid leukemia patients with up-front cytogenetic resistance to imatinib are associated with a greater likelihood of progression to blast crisis and shorter survival: a study by the GIMEMA Working Party on Chronic Myeloid Leukemia. J Clin Oncol. 2005;23:4100–4109. doi: 10.1200/JCO.2005.05.531. [DOI] [PubMed] [Google Scholar]
- 87.Branford S, Rudzki Z, Walsh S, et al. Detection of BCR-ABL mutations in patients with CML treated with imatinib is virtually always accompanied by clinical resistance, and mutations in the ATP phosphate-binding loop (P-loop) are associated with a poor prognosis. Blood. 2003;102:276–283. doi: 10.1182/blood-2002-09-2896. [DOI] [PubMed] [Google Scholar]
- 88.Weisberg E, Manley PW, Cowan-Jacob SW, Hochhaus A, Griffin JD. Second generation inhibitors of BCR-ABL for the treatment of imatinib-resistant chronic myeloid leukaemia. Nat Rev Cancer. 2007;7:345–356. doi: 10.1038/nrc2126. [DOI] [PubMed] [Google Scholar]
- 89.Hochhaus A, Saglio G, Larson RA, et al. Nilotinib is associated with a reduced incidence of BCR-ABL mutations vs imatinib in patients with newly diagnosed chronic myeloid leukemia in chronic phase. Blood. 2013;121:3703–3708. doi: 10.1182/blood-2012-04-423418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fabarius A, Leitner A, Hochhaus A, et al. Impact of additional cytogenetic aberrations at diagnosis on prognosis of CML: long-term observation of 1151 patients from the randomized CML Study IV. Blood. 2011;118:6760–6768. doi: 10.1182/blood-2011-08-373902. [DOI] [PubMed] [Google Scholar]
- 91.Verma D, Kantarjian H, Shan J, et al. Survival outcomes for clonal evolution in chronic myeloid leukemia patients on second generation tyrosine kinase inhibitor therapy. Cancer. 2010;116:2673–2681. doi: 10.1002/cncr.25015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hasford J, Pfirrmann M, Hehlmann R, et al. A new prognostic score for survival of patients with chronic myeloid leukemia treated with interferon alfa. Writing Committee for the Collaborative CML Prognostic Factors Project Group. J Natl Cancer Inst. 1998;90:850–858. doi: 10.1093/jnci/90.11.850. [DOI] [PubMed] [Google Scholar]
- 93.Sokal JE, Cox EB, Baccarani M, et al. Prognostic discrimination in “good-risk” chronic granulocytic leukemia. Blood. 1984;63:789–799. [PubMed] [Google Scholar]
- 94.Oyekunle AA, Osho PO, Aneke JC, Salawu L, Durosinmi MA. The predictive value of the Sokal and Hasford scoring systems in chronic myeloid leukaemia in the imatinib era. Journal of Hematological Malignancies. 2012;2:25–32. [Google Scholar]
- 95.Hasford J, Baccarani M, Hoffmann V, et al. Predicting complete cytogenetic response and subsequent progression-free survival in 2060 patients with CML on imatinib treatment: the EUTOS score. Blood. 2011;118:686–692. doi: 10.1182/blood-2010-12-319038. [DOI] [PubMed] [Google Scholar]
- 96.Hoffmann VS, Baccarani M, Lindoerfer D, et al. The EUTOS prognostic score: review and validation in 1288 patients with CML treated frontline with imatinib. Leukemia. 2013 Jun 11; doi: 10.1038/leu.2013.171. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 97.Uz B, Buyukasik Y, Atay H, et al. EUTOS CML prognostic scoring system predicts ELN-based ‘event-free survival’ better than Euro/Hasford and Sokal systems in CML patients receiving front-line imatinib mesylate. Hematology. 2013 doi: 10.1179/1607845412Y.0000000071. [DOI] [PubMed] [Google Scholar]
- 98.Marin D, Ibrahim AR, Goldman JM. European Treatment and Outcome Study (EUTOS) score for chronic myeloid leukemia still requires more confirmation. J Clin Oncol. 2011;29:3944–3945. doi: 10.1200/JCO.2011.37.6962. [DOI] [PubMed] [Google Scholar]
- 99.Sprycel (dasatinib) [prescribing information] Princeton, NJ: Bristol-Myers Squibb Co; 2011. [Google Scholar]
- 100.Gleevec (imatinib) [prescribing information] East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2012. [Google Scholar]
- 101.Tasigna (nilotinib) [prescribing information] East Hanover, NJ: Novartis Pharmaceuticals Corporation; 2012. [Google Scholar]
- 102.Bosulif (bosutinib) [prescribing information] New York, NY: Pfizer Labs; 2012. [Google Scholar]
- 103.Cortes JE, Kantarjian H, Shah NP, et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N Engl J Med. 2012;367:2075–2088. doi: 10.1056/NEJMoa1205127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Radich JP. The biology of CML blast crisis. Hematology Am Soc Hematol Educ Program. 2007:384–391. doi: 10.1182/asheducation-2007.1.384. [DOI] [PubMed] [Google Scholar]
- 105.Kantarjian HM, O’Brien S, Cortes JE, et al. Treatment of Philadelphia chromosome-positive, accelerated-phase chronic myelogenous leukemia with imatinib mesylate. Clin Cancer Res. 2002;8:2167–2176. [PubMed] [Google Scholar]
- 106.Talpaz M, Silver RT, Druker BJ, et al. Imatinib induces durable hematologic and cytogenetic responses in patients with accelerated phase chronic myeloid leukemia: results of a phase 2 study. Blood. 2002;99:1928–1937. doi: 10.1182/blood.v99.6.1928. [DOI] [PubMed] [Google Scholar]
- 107.Palandri F, Castagnetti F, Alimena G, et al. The long-term durability of cytogenetic responses in patients with accelerated phase chronic myeloid leukemia treated with imatinib 600 mg: the GIMEMA CML Working Party experience after a 7-year follow-up. Haematologica. 2009;94:205–212. doi: 10.3324/haematol.13529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–1042. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 109.Kantarjian HM, Cortes J, O’Brien S, et al. Imatinib mesylate (STI571) therapy for Philadelphia chromosome-positive chronic myelogenous leukemia in blast phase. Blood. 2002;99:3547–3553. doi: 10.1182/blood.v99.10.3547. [DOI] [PubMed] [Google Scholar]
- 110.Sawyers CL, Hochhaus A, Feldman E, et al. Imatinib induces hematologic and cytogenetic responses in patients with chronic myelogenous leukemia in myeloid blast crisis: results of a phase II study. Blood. 2002;99:3530–3539. doi: 10.1182/blood.v99.10.3530. [DOI] [PubMed] [Google Scholar]
- 111.Sureda A, Carrasco M, de Miguel M, et al. Imatinib mesylate as treatment for blastic transformation of Philadelphia chromosome positive chronic myelogenous leukemia. Haematologica. 2003;88:1213–1220. [PubMed] [Google Scholar]
- 112.Palandri F, Castagnetti F, Testoni N, et al. Chronic myeloid leukemia in blast crisis treated with imatinib 600 mg: outcome of the patients alive after a 6-year follow-up. Haematologica. 2008;93:1792–1796. doi: 10.3324/haematol.13068. [DOI] [PubMed] [Google Scholar]
- 113.le Coutre P, Ottmann OG, Giles F, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is active in patients with imatinib-resistant or -intolerant accelerated-phase chronic myelogenous leukemia. Blood. 2008;111:1834–1839. doi: 10.1182/blood-2007-04-083196. [DOI] [PubMed] [Google Scholar]
- 114.Giles FJ, Abruzzese E, Rosti G, et al. Nilotinib is active in chronic and accelerated phase chronic myeloid leukemia following failure of imatinib and dasatinib therapy. Leukemia. 2010;24:1299–1301. doi: 10.1038/leu.2010.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Kantarjian H, Giles F, Wunderle L, et al. Nilotinib in imatinib-resistant CML and Philadelphia chromosome-positive ALL. N Engl J Med. 2006;354:2542–2551. doi: 10.1056/NEJMoa055104. [DOI] [PubMed] [Google Scholar]
- 116.Nicolini FE, Masszi T, Shen Z, et al. Expanding Nilotinib Access in Clinical Trials (ENACT), an open-label multicenter study of oral nilotinib in adult patients with imatinib-resistant or -intolerant chronic myeloid leukemia in accelerated phase or blast crisis. Leuk Lymphoma. 2012;53:907–914. doi: 10.3109/10428194.2011.627480. [DOI] [PubMed] [Google Scholar]
- 117.Giles FJ, Kantarjian HM, le Coutre PD, et al. Nilotinib is effective in imatinib-resistant or -intolerant patients with chronic myeloid leukemia in blastic phase. Leukemia. 2012;26:959–962. doi: 10.1038/leu.2011.355. [DOI] [PubMed] [Google Scholar]
- 118.Guilhot F, Apperley J, Kim DW, et al. Dasatinib induces significant hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in accelerated phase. Blood. 2007;109:4143–4150. doi: 10.1182/blood-2006-09-046839. [DOI] [PubMed] [Google Scholar]
- 119.Apperley JF, Cortes JE, Kim DW, et al. Dasatinib in the treatment of chronic myeloid leukemia in accelerated phase after imatinib failure: the START A trial. J Clin Oncol. 2009;27:3472–3479. doi: 10.1200/JCO.2007.14.3339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kantarjian H, Cortes J, Kim DW, et al. Phase 3 study of dasatinib 140 mg once daily versus 70 mg twice daily in patients with chronic myeloid leukemia in accelerated phase resistant or intolerant to imatinib: 15-month median follow-up. Blood. 2009;113:6322–6329. doi: 10.1182/blood-2008-11-186817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Talpaz M, Shah NP, Kantarjian H, et al. Dasatinib in imatinib-resistant Philadelphia chromosome-positive leukemias. N Engl J Med. 2006;354:2531–2541. doi: 10.1056/NEJMoa055229. [DOI] [PubMed] [Google Scholar]
- 122.Cortes J, Rousselot P, Kim DW, et al. Dasatinib induces complete hematologic and cytogenetic responses in patients with imatinib-resistant or -intolerant chronic myeloid leukemia in blast crisis. Blood. 2007;109:3207–3213. doi: 10.1182/blood-2006-09-046888. [DOI] [PubMed] [Google Scholar]
- 123.Cortes J, Kim DW, Raffoux E, et al. Efficacy and safety of dasatinib in imatinib-resistant or -intolerant patients with chronic myeloid leukemia in blast phase. Leukemia. 2008;22:2176–2183. doi: 10.1038/leu.2008.221. [DOI] [PubMed] [Google Scholar]
- 124.Saglio G, Hochhaus A, Goh YT, et al. Dasatinib in imatinib-resistant or imatinib-intolerant chronic myeloid leukemia in blast phase after 2 years of follow-up in a phase 3 study: efficacy and tolerability of 140 milligrams once daily and 70 milligrams twice daily. Cancer. 2010;116:3852–3861. doi: 10.1002/cncr.25123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cortes JE, Kim DW, Pinilla-Ibarz J, et al. PACE: A pivotal phase II trial of ponatinib in patients with CML and Ph+ALL resistant or intolerant to dasatinib or nilotinib, or with the T315I mutation. J Clin Oncol. 2012;15:6503. [Google Scholar]
- 126.O’Hare T, Shakespeare WC, Zhu X, et al. AP24534, a pan-BCR-ABL inhibitor for chronic myeloid leukemia, potently inhibits the T315I mutant and overcomes mutation-based resistance. Cancer Cell. 2009;16:401–412. doi: 10.1016/j.ccr.2009.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Eide CA, Adrian LT, Tyner JW, et al. The ABL switch control inhibitor DCC-2036 is active against the chronic myeloid leukemia mutant BCR-ABLT315I and exhibits a narrow resistance profile. Cancer Res. 2011;71:3189–3195. doi: 10.1158/0008-5472.CAN-10-3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cortes JE, Talpaz M, Kantarjian HM, et al. A Phase 1 study of DCC-2036, a novel oral inhibitor of BCR-ABL kinase, in patients with Philadelphia chromosome positive (Ph+) leukemias including patients with T315I mutation. Blood (ASH Annual Meeting Abstracts) 2011;118:601. [Google Scholar]
- 129.National Cancer Institute. [Accessed July 24, 2012];SEER Stat Fact Sheets: Chronic Myeloid Leukemia. http://seer.cancer.gov/statfacts/html/cmyl.html.
- 130.Saussele S, Lauseker M, Gratwohl A, et al. Allogeneic hematopoietic stem cell transplantation (allo SCT) for chronic myeloid leukemia in the imatinib era: evaluation of its impact within a subgroup of the randomized German CML Study IV. Blood. 2010;115:1880–1885. doi: 10.1182/blood-2009-08-237115. [DOI] [PubMed] [Google Scholar]
- 131.Khoury HJ, Kukreja M, Goldman JM, et al. Prognostic factors for outcomes in allogeneic transplantation for CML in the imatinib era: a CIBMTR analysis. Bone Marrow Transplant. 2012;47:810–816. doi: 10.1038/bmt.2011.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.National Comprehensive Cancer Network. NCCN Clinical Practice Guidelines in Oncology. [Accessed July 24, 2012];Chronic myelogenous leukemia. Version 1.2013. doi: 10.6004/jnccn.2009.0065. http://www.nccn.org/professionals/physician_gls/pdf/cml.pdf. [DOI] [PubMed]
- 133.Baccarani M, Cortes J, Pane F, et al. Chronic myeloid leukemia: an update of concepts and management recommendations of European LeukemiaNet. J Clin Oncol. 2009;27:6041–6051. doi: 10.1200/JCO.2009.25.0779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Soverini S, Hochhaus A, Nicolini FE, et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood. 2011;118:1208–1215. doi: 10.1182/blood-2010-12-326405. [DOI] [PubMed] [Google Scholar]
- 135.Laneuville P, Dilea C, Yin OQ, Woodman RC, Mestan J, Manley PW. Comparative In vitro cellular data alone are insufficient to predict clinical responses and guide the choice of BCR-ABL inhibitor for treating imatinib-resistant chronic myeloid leukemia. J Clin Oncol. 2010;28:e169–171. doi: 10.1200/JCO.2009.26.4945. author reply e172. [DOI] [PubMed] [Google Scholar]
- 136.Cervantes F, Rozman M, Rosell J, Urbano-Ispizua A, Montserrat E, Rozman C. A study of prognostic factors in blast crisis of Philadelphia chromosome-positive chronic myelogenous leukaemia. Br J Haematol. 1990;76:27–32. doi: 10.1111/j.1365-2141.1990.tb07832.x. [DOI] [PubMed] [Google Scholar]
- 137.Inverardi D, Lazzarino M, Morra E, et al. Extramedullary disease in Ph’-positive chronic myelogenous leukemia: frequency, clinical features and prognostic significance. Haematologica. 1990;75:146–148. [PubMed] [Google Scholar]
- 138.Wadhwa J, Szydlo RM, Apperley JF, et al. Factors affecting duration of survival after onset of blastic transformation of chronic myeloid leukemia. Blood. 2002;99:2304–2309. doi: 10.1182/blood.v99.7.2304. [DOI] [PubMed] [Google Scholar]
- 139.Bornhauser M, Jenke A, Freiberg-Richter J, et al. CNS blast crisis of chronic myelogenous leukemia in a patient with a major cytogenetic response in bone marrow associated with low levels of imatinib mesylate and its N-desmethylated metabolite in cerebral spinal fluid. Ann Hematol. 2004;83:401–402. doi: 10.1007/s00277-003-0829-4. [DOI] [PubMed] [Google Scholar]
- 140.Leis JF, Stepan DE, Curtin PT, et al. Central nervous system failure in patients with chronic myelogenous leukemia lymphoid blast crisis and Philadelphia chromosome positive acute lymphoblastic leukemia treated with imatinib (STI-571) Leuk Lymphoma. 2004;45:695–698. doi: 10.1080/10428190310001625728. [DOI] [PubMed] [Google Scholar]
- 141.Neville K, Parise RA, Thompson P, et al. Plasma and cerebrospinal fluid pharmacokinetics of imatinib after administration to nonhuman primates. Clin Cancer Res. 2004;10:2525–2529. doi: 10.1158/1078-0432.ccr-03-0155. [DOI] [PubMed] [Google Scholar]
- 142.Petzer AL, Gunsilius E, Hayes M, et al. Low concentrations of STI571 in the cerebrospinal fluid: a case report. Br J Haematol. 2002;117:623–625. doi: 10.1046/j.1365-2141.2002.03523.x. [DOI] [PubMed] [Google Scholar]
- 143.Ravandi F, Jorgensen JL, Thomas DA, et al. Detection of MRD may predict the outcome of patients with Philadelphia chromosome-positive ALL treated with tyrosine kinase inhibitors plus chemotherapy. Blood. 2013;122:1214–1221. doi: 10.1182/blood-2012-11-466482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Oki Y, Kantarjian HM, Gharibyan V, et al. Phase II study of low-dose decitabine in combination with imatinib mesylate in patients with accelerated or myeloid blastic phase of chronic myelogenous leukemia. Cancer. 2007;109:899–906. doi: 10.1002/cncr.22470. [DOI] [PubMed] [Google Scholar]
- 145.Verma D, Kantarjian HM, Jones D, et al. Chronic myeloid leukemia (CML) with P190 BCR-ABL: analysis of characteristics, outcomes, and prognostic significance. Blood. 2009;114:2232–2235. doi: 10.1182/blood-2009-02-204693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Fruehauf S, Topaly J, Buss EC, et al. Imatinib combined with mitoxantrone/etoposide and cytarabine is an effective induction therapy for patients with chronic myeloid leukemia in myeloid blast crisis. Cancer. 2007;109:1543–1549. doi: 10.1002/cncr.22535. [DOI] [PubMed] [Google Scholar]
- 147.Fang B, Li N, Song Y, Han Q, Zhao RC. Standard-dose imatinib plus low-dose homoharringtonine and granulocyte colony-stimulating factor is an effective induction therapy for patients with chronic myeloid leukemia in myeloid blast crisis who have failed prior single-agent therapy with imatinib. Ann Hematol. 2010;89:1099–1105. doi: 10.1007/s00277-010-0991-4. [DOI] [PubMed] [Google Scholar]
- 148.Deau B, Nicolini FE, Guilhot J, et al. The addition of daunorubicin to imatinib mesylate in combination with cytarabine improves the response rate and the survival of patients with myeloid blast crisis chronic myelogenous leukemia (AFR01 study) Leuk Res. 2011;35:777–782. doi: 10.1016/j.leukres.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 149.Quintas-Cardama A, Kantarjian H, Garcia-Manero G, et al. A pilot study of imatinib, low-dose cytarabine and idarubicin for patients with chronic myeloid leukemia in myeloid blast phase. Leuk Lymphoma. 2007;48:283–289. doi: 10.1080/10428190601075973. [DOI] [PubMed] [Google Scholar]