

Abstract
Reactive oxygen species (ROS) are known to cause many types of DNA lesions that could be converted into cancer-promoting genetic alterations. Evidence showed that tumor suppressor p53 plays an important role in regulating the generation of cellular ROS, either by reducing oxidative stress under physiological and mildly stressed conditions, or by promoting oxidative stress under highly stressed conditions. In this report we characterized the effect of oxidative stress on the induction of micronuclei, especially the subclass marked by pan-staining of γ-H2AX or MN-γ-H2AX (+). We found that MN-γ-H2AX (+) were more responsive to hydrogen peroxide (H2O2) than the MN-γ-H2AX (−). In human and mouse cells that are deficient in p53, the frequency of MN-γ-H2AX (+) is significantly elevated, but can be attenuated by antioxidant N-acetylcysteine (NAC). Depletion of p53-regulated antioxidant gene SESN1 by RNA interference also resulted in an elevation of MN-γ–H2AX (+). Furthermore, we found that in cells that were depleted of p400 by RNAi, and therefore were experiencing increased ROS, the frequency of MN-γ-H2AX (+), but not that of MN-γ-H2AX (−), was significantly induced. We further demonstrated that the induction of MN-γ–H2AX (+) by replication stress can also be attenuated by NAC, and that H2O2 also leads to increased phosphorylation of Chk1 and Rad17 that mimics replication stress, suggesting that replication stress and oxidative stress are intertwined and may reinforce each other in driving genomic instability. Our findings illustrate the importance of p53-regulated redox level in the maintenance of genomic stability.
Keywords: micronuclei, genomic instability, p53, oxidative stress, replication stress
1. Introduction
Reactive oxygen species (ROS), which include O2•− (superoxide anion radical), •OH (hydroxyl radical) and H2O2 (hydrogen peroxide), can be generated by endogenous sources, such as mitochondria and NADPH oxidase [1]. ROS also mediates the deleterious effects of ultraviolet (UV) radiation [2, 3], ionizing radiation [4], inflammatory cytokines [5, 6] and arsenic exposure [7]. There are multiple antioxidant defense mechanisms, including enzymatic systems (catalase, superoxide dismutase and glutathione peroxidase) and non-enzymatic mechanisms (glutathione, vitamins C and vitamins E) [8, 9]. Maintenance of a homeostatic level of ROS is vital for cell proliferation and various other cellular functions. When ROS overwhelms the antioxidant capacity, damage to cellular macromolecules such as lipids, protein, and DNA ensues [1, 10]. ROS is known to cause at least 100 different types of DNA lesions, including base modifications, singe-strand breaks and double-strand breaks and interstrand crosslinks [10]. Such lesions may also lead to an increased mutant frequency at reporter loci [11–13]. Because oxidative stress is associated with a variety of chronic diseases as well as aging, it remains important to delineate the full spectrum of genomic instability it may cause.
Tumor suppressor p53 plays a critical role in regulating the level of cellular ROS [14, 15]. It upregulates the expression of antioxidant enzymes, such as sestrins and GPX, under physiological conditions. Lack of p53 led to elevated levels of ROS, oxidatively damaged DNA and mutations at the HPRT reporter locus [16]. However, it exhibits prooxidative activities that further increase the levels of stress under high levels of oxidative stress [14, 15]. Mutation within or lack of p53 has long been recognized to lead to increased genomic instability [17–20]. Because p53 may contribute to the maintenance of genomic integrity in multiple pathways, it remains to be determined to what extent each of the p53-regulated pathways contributes.
Micronuclei (MN) in mammalian cells serve as a reliable biomarker of genomic instability and genotoxic exposure [21–23]. Phosphorylation of H2AX, or γ-H2AX, serves as a sensitive marker of DNA damage [24–26]. We observed that a large proportion of MN showed uniform and pan-staining by antibodies against γ-H2AX. We designated them as a MN-γ-H2AX (+), to distinguish them from the MN-γ-H2AX (−) that lack uniform labeling of γ-H2AX in the MN [27]. We had shown that the formation of MN-γ-H2AX (+) probably involves active clustering and disposal of DNA DSBs before the onset of mitosis, and demonstrated that MN-γ-H2AX (+) can be preferentially induced by DNA replication stress.
In this study we studied whether ROS would influence the formation of MN-γ-H2AX (+) and whether p53 function is involved. We found that the frequency of MN-γ-H2AX (+) was significantly increased in cells treated with H2O2, and in mutant cells with reduced p53 function. We also observed that oxidative stress and replication stress are mutually reinforcing in inducing MN-γ-H2AX (+).
2. Materials and Methods
2.1. Cell Culture
Human osteosarcoma cells U2OS were obtained from the American Type Culture Collection (Manassas, VA). HEK-293 cells were obtained from the Institute of Basic Medical Sciences, Chinese Academy of Medical Sciences (Beijing). The immortalized cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 mg/ml penicillin and 100 mg/ml streptomycin. Primary mouse skin (ear) fibroblasts (MSF) were prepared as described [28]. Briefly, the mouse ears were minced in a well of 12-well plate, treated with collagenase D/dispase at 37°C for 45 min, incubated in 1ml DMEM/10% FBS medium overnight. Cells were obtained by passing the cell suspension through a Cell Strainer (70 mm,Falcon), centrifuged and then incubated in growth medium (DMEM supplemented with 10% FBS, 100 mg/ml penicillin, 100 mg/ml streptomycin and 2.5 mg/ml amphotericin B). All cells were cultured in a humidified atmosphere of 5% CO2 at 37°C. N-acetylcysteine (NAC) was purchased from Sigma Chemical Co (St. Louis, MO). Hydrogen peroxide (H2O2) was from Sangon Biotech. Pifithrin-α was purchased from Beyotime.
2.2. Immunofluorescence staining of phospho-H2AX (Ser139)
Antibodies against phospho-H2AX (Ser139) (DAM1661039) were purchased from Millipore (Billerica, MA). TRITC-conjugated Goat anti-mouse secondary antibody was from Jackson Immuno Research Laboratories (West Grove, PA, USA). Cells were grown on coverslips in 6-well plates. After various treatments, cells were washed in PBS, and fixed in 4% paraformaldehyde for 20 min. They were washed three times in PBS for 5 min each and permeabilized for 20 min in 0.2% Triton X-100. The cells were blocked in 10% normal goat serum overnight at 4 ° C. After being washed in PBS, the coverslips were incubated with anti-phospho-H2AX antibody overnight at 4° C. The cells were then washed in PBS, and incubated with TRITC-conjugated secondary antibody for 1 h at room temperature. Cells were washed in PBS four times and counterstained with 4-6-diamidino-2-phenylindole (DAPI). The coverslips were mounted on slides for examination.
2.3. RNA Interference
The construction containing p53-shRNA (shp53) and the negative control (shNeg) were purchased from GenePharma (Shanghai, China). For stable transfection, U2OS cells were maintained in culture medium free of antibiotics in a 24-well plate. After overnight incubation, transfections were performed using Lipofectamine 2000 reagent (Invitrogen) according to the manufacturer’s instructions. Cells were incubated in the medium (containing selection drug) 24 h after transfection. The siRNA duplexes of SESN1 and p400 and negative control siRNA duplexes were purchased from sigma. U2OS cells were transfected with siRNA duplexes (20 µM) using lipofectamine2000 (Invitrogen) for 24 h. Transfected cells were tested by real-time quantitative RT-PCR assay and Western blotting analysis, and then used for further study.
The oligonucleotide sequences were as follows:
shp53: GACTCCAGTGGTAATCTAC
shNeg: GTTCTCCGAACGTGTCACGT
sip400: CUGAUGAGGAGUUUCAACATT
siSESN1: GUUUGGAUAACAUUACGUUTT
siNeg: UUCUCCGAACGUGUCACGUTT
2.4. RNA Isolation and Real-time Quantitative RT-PCR Array
Total RNA was isolated using Trizol reagent (Invitrogen) according to the manufacturer’s introduction. cDNA was obtained by reverse transcription-PCR of 1 µg of total RNA using the AMV reverse transcriptase (Promega). Real-time quantitative RT-PCR was performed using the SYBR Green PCR Master Mix (Applied Biosystems) in an ABI Prism 7500 sequence detection system (Applied Biosystems). Human GAPDH gene was used as control. The real-time PCR was carried out in a final volume of 10 µl. The samples were loaded in quadruples, and the results of each sample were normalized to GAPDH. The primers for SYBR were from primer bank of Harvard Medical School (http://pga.mgh.harvard.edu/primerbank/).
Primer sequences are described as follow:
- SESN1
- 5’- AAGTGGGGAGTGAAGACGC -3’ (Forward)
- 5’- GGCCCATCCATTTGCAGTAGA -3’ (Reverse)
- p400
- 5’- GAAGCACAGTAGAGACGGACC -3’ (Forward)
- 5’- CTGGAAAACTACCCCTTGGTG -3’ (Reverse)
- GAPDH
- 5’- CAGAACATCATCCCTGCCTCTAC -3’ (Forward)
- 5’- TTGAAGTCAGAGGAGACCACCTG -3’ (Reverse)
2.5. Western Blotting Analysis
Cells were harvested and lysed with cell lysis buffer for Western and IP (Beyotime) according to the manufacturer’s instructions. Protein concentration was determined using the BCA assay. Equal amounts of protein were separated by 12% SDS-PAGE, transferred to PVDF membrane (Millipore, Billerica, MA). After blocking with 5% skimmed milk, the membrane was incubated with specific primary antibodies overnight at 4 °C. Antibodies against p53 (sc-126), p-RAD17 (sc-32843) and RAD17 (sc-5613) were purchased from Santa Cruz Biotechnology. SESN1 (ab103121) and p400 (ab5201) were from Abcam (Cambridge, MA). phospho-Chk1 (Ser345) (2348) and Chk1 (2360) were from Cell Signaling Technology Inc. (Beverly, MA). β-actin was from Sigma Chemical Co. (St. Louis, MO). Proteins of interest were detected with appropriate horseradish peroxidase-conjugated secondary antibody for 1 h and visualized by ECL Western Blotting Substrate (Thermo Scientific).
2.6. Determination of Cellular ROS by Flow Cytometry
To evaluate intracellular ROS levels, a GENMED kit was used. Briefly, cells were washed with PBS, trypsinized and then collected. The cells were then incubated with CM-H2DCFDA in the dark for 20 min at 37 °C. After washing, cells were analyzed by flow cytometry, which was performed using the FACSCalibur flow cytometer (BD Biosciences). The data were analyzed using the FCSExpress V3 program (De Novo Software).
2.7. Scoring of MN
The samples were coded and examined with an Olympus DP71 fluorescence microscope. MN and MN-γ-H2AX (+) were scored as previously reported [27]. Briefly, nuclei were scored first for MN using a 100× objective under DAPI filter. MN were then examined for the presence or absence of γ-H2AX signals under Texas Red filter. At least 1000 cells per sample were scored.
2.8. Statistical analysis
Statistical calculations were performed using the SPSS 13.0. The statistical significance of the differences in micronuclei data was analyzed using the Chi-square Pearson test. P value < 0.05 was considered statistically significant.
3. Results
3.1. Induction of MN-γ-H2AX (+) by H2O2
Hydrogen peroxide (H2O2) is frequently used as an inducer of oxidative stress. We treated U2OS cells with increasing concentrations of H2O2 (50 µM, 100 µM, 150 µM) for different durations (2 h, 6 h, 12 h, 24 h), and examined the frequencies of the MN-γ-H2AX (+) at 48 h after H2O2 was washed out. We previously showed that the frequency of MN-γ-H2AX (+) usually peaked at 48 h after drug removal [27]. As shown in Figure 1A, at the lowest concentration tested (50 µM), H2O2 treatment led to a significant increase in the frequency of MN-γ-H2AX (+) in all the treatment durations. Taking H2O2 treatment for 2 h as an example, the frequency of MN-γ-H2AX (+) was increased to 3.32×10−2, from 1.77×10−2 in the control (P<0.05). At this concentration, H2O2 −24 h caused the most pronounced increase, a 3.8 fold increase over the control (P <0.05). In comparison, the frequency of MN-γ-H2AX (−) did not show a significant increase in any of the increasing treatment durations.
Fig. 1.

Induction of MN by H2O2 and its attenuation by NAC. U2OS cells were pretreated with or without the antioxidant NAC (5mM) for 90 min, and then were treated with 50µM H2O2 (A), 100µM H2O2 (B) or 150µM H2O2 (C) for different durations (2h, 6h, 12h, 24h). Cells were processed for examination of the MN-γ-H2AX (+) and MN-γ-H2AX (−) at 48h after H2O2 was washed out. At least 1000 cells were examined for each sample. * P < 0.05, ** P < 0.01.
With increasing H2O2 concentrations, the increase in the frequency of MN-γ-H2AX (+) became more striking (Figure 1B, 1C). However, the higher concentrations of H2O2 also caused significant increase in the frequency of MN-γ-H2AX (−) (Figure 1B, 1C), albeit to a lesser extent in terms of fold increase over the baseline. These results suggest that MN-γ-H2AX (+) are more responsive to H2O2 than MN-γ-H2AX (−).
To confirm that the H2O2 –induced increase in the frequency of MN was specifically caused by increased oxidative stress, we pretreated cells with antioxidant NAC for 90 min, and then exposed them to H2O2. Pre-incubation with NAC significantly attenuated the induction of MN-γ-H2AX (+) by H2O2 in 50 µM-24h, 100 µM-(2 to 24h) and 150 µM-(2 to 24h) groups (Figure 1A, 1B, 1C). In comparison, NAC significantly attenuated the induction of MN-γ-H2AX (−) only in the 100 µM−24h and 150 µM-(4 to 24h) groups (Figure 1B, 1C). These results suggest that induction of MN-γ-H2AX (+) by H2O2 was mediated by increased oxidative stress.
3.2. Elevation of MN-γ-H2AX (+) in p53-deficient cells
We next tested whether p53 deficiency is associated with an elevation in MN-γ-H2AX (+). To this end, we established a p53 knockdown cell line by RNA interference, designated as U2OS-shp53, and a control counterpart as U2OS-shNeg. The downregulation of p53 was confirmed by western blotting (Supplementary Figure S1). As shown in Table 1, 43 of the 1009 U2OS-shp53 cells contained MN-γ-H2AX (+), in comparison to 21 of 1062 U2OS-shNeg cells harboring them (P <0.01), indicating that lack of p53 is associated with increased frequency of MN-γ-H2AX (+).
Table 1.
Elevation of MN-γ-H2AX (+) in cells with dysfunctional p53
| MN frequency (×10−2) |
MN-γ-H2AX(+) /MN(%) |
MN-γ-H2AX (+) | MN-γ-H2AX (−) | |||
|---|---|---|---|---|---|---|
| Frequency (×10−2) |
Fold change over 0µM |
Frequency (×10−2) |
Fold change over 0µM |
|||
| U2OS-shNeg | 5.18 | 38.18 | 1.98 | 1 | 3.2 | 1 |
| U2OS-shp53 | 8.92 | 47.78 | 4.26** | 2.16 | 4.66 | 1.45 |
| U2OS-Neo | 5.99 | 38.33 | 2.3 | 1 | 3.7 | 1 |
| U2OS-mtp53 | 9.91 | 42 | 4.16* | 1.81 | 5.75 | 1.55 |
| MSF-WT | 3.95 | 16.26 | 0.64 | 1 | 3.31 | 1 |
| MSF-p53mt | 8.26 | 26.19 | 2.16** | 3.37 | 6.1 | 1.84 |
P < 0.05,
P < 0.01
We also examined the frequency of MN-γ-H2AX (+) in a previously established a U2OS cell line expressing a dominant negative mutant p53 (U2OS-mtp53). As shown in Table 1, U2OS-mtp53 cells also exhibited a higher frequency of MN-γ-H2AX (+) than in the control U2OS-neo cells (P <0.05).
When U2OS cells were subjected to pifithrin-α, a p53 inhibitor, for 48h and then assayed for MN, we also noted a two-fold increase in the frequency of MN-γ-H2AX (+), from 1.29×10−2 in the control to 2.67×10−2 in the 60 µM treatment group (P <0.05) (Supplemental Table S1). In contrast, the frequency of MN-γ-H2AX (−) remained unchanged.
Similarly, primary mouse skin fibroblasts derived from p53 null mice were observed to have a higher frequency of MN-γ-H2AX (+) than those from p53 wild-type mice (Table 1, P < 0.01). The frequency of MN-γ-H2AX (−) cells showed a less pronounced difference with regard to p53 status (P > 0.05). Together, these results indicate that reduction in or absence of p53 function is preferentially associated with increased production of MN-γ-H2AX (+).
We next tested whether the increased formation of MN-γ-H2AX (+) in U2OS-mtp53 cells is because of ROS. While the frequency of MN-γ-H2AX (+) was significantly higher in U2OS-mtp53 cells than in U2OS-neo cells, the difference became narrow and statistically insignificant when the cells were pre-incubated with NAC (Table 2).
Table 2.
Attenuation of MN-γ-H2AX (+) by antioxidant NAC in U2OS-mtp53 cells
| MN frequency (×10−2) |
MN-γ-H2AX(+) /MN(%) |
MN-γ-H2AX (+) | MN-γ-H2AX (−) | |||
|---|---|---|---|---|---|---|
| Frequency (×10−2) |
Fold change over 0µM |
Frequency (×10−2) |
Fold change over 0µM |
|||
| U2O2-Neo | 6.62 | 37.31 | 2.47 | 1.00 | 4.15 | 1.00 |
| U2OS-mtp53 | 10.28 | 40.45 | 4.16** | 1.68 | 6.12 | 1.48 |
| U2OS-Neo+NAC(5mM) | 7.24 | 30.45 | 2.21 | 1.00 | 5.04 | 1.00 |
| U2OS-mtp53+NAC(5mM) | 8.12 | 34.96 | 2.84 | 1.29 | 5.28 | 1.05 |
P < 0.05,
P < 0.01.
We further tested the effect of NAC on the formation of MN in three pairs of primary mouse skin fibroblasts (MSF) that were p53 wild-type and p53 null, respectively. MSF cells were incubated in the absence or presence of 5mM NAC and then assayed for MN. As shown in Supplementary Figure S2, while the frequency of MN-γ-H2AX (+) was significantly higher in p53-deficient MSF than in the wild-type controls (P < 0.01), elevation of MN-γ-H2AX (+) in p53-deficient MSF was greatly attenuated by NAC. These results strongly support the idea that the elevation of MN-γ-H2AX (+) in p53 knockdown or null cells is mediated by increased oxidative stress.
3.3. Elevation of MN-γ-H2AX (+) in SESN1 knockdown cells
We next wanted to test whether any of the p53 target genes is responsible for reducing ROS and formation of MN-γ-H2AX (+). Among the genes activated by p53, sestrins (encoded by SESN1 and SESN2, respectively) are known to reduce the spontaneous level of ROS [16, 29]. Thus, we tested the role of SESN1 by knocking down SESN1 in HEK-293 cells (Figure 2A). As expected, SESN1 knockdown led to an increase in the level of ROS (Figure 2B). Examination of MN-γ-H2AX (+) showed that SESN1 depletion led to a significant increase in the frequency of MN-γ–H2AX (+) (Figure 2C), from 1.89% in mock siRNA transfected cell to 2.72% in siSESN1 transfected cells (P <0.05). The frequency of MN-γ-H2AX (−), on the other hand, did not show statistically significant increase. These results suggest that the p53 downstream target SESN1 is one of the factors responsible for reducing spontaneous ROS and MN-γ-H2AX (+).
Fig. 2.
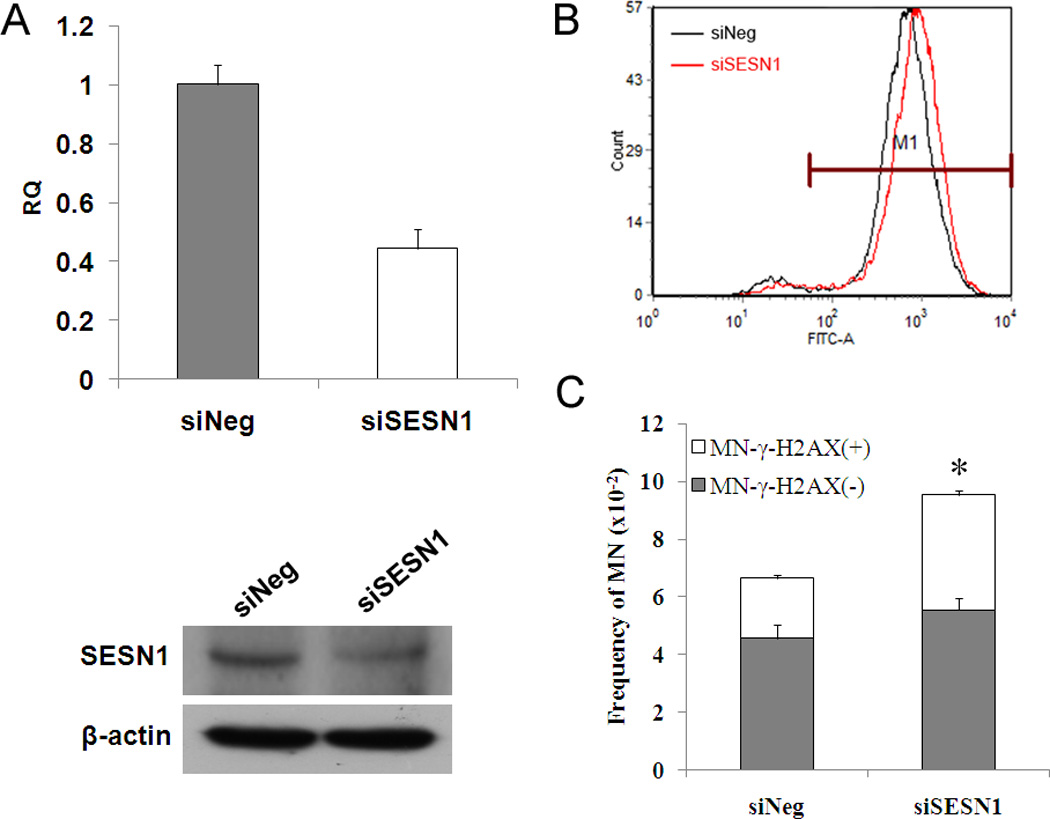
Elevation of MN-γ-H2AX (+) in SESN1 knockdown HEK-293 cells. A. The expression of SESN1 mRNA and protein levels in HEK-293 cells. The level of SESN1 mRNA was quantified by real-time quantitative RT-PCR assay. SESN1 protein level in siSESN1 HEK-293 cells was determined using the antibodies against SESN1. B. Measurement of ROS in SESN1 knockdown cells. HEK-293 cells were transfected using the indicated siRNA. Cells were collected and ROS levels were measured by flow cytometry 24h following transfection. C. Increased MN-γ-H2AX (+) in siSESN1 HEK-293 cells. *P<0.05, when compared with siNeg HEK-293 cells.
3.4. Depletion of p400 increases the frequency of MN-γ-H2AX (+)
It was recently reported that E1A binding protein p400 is involved in the regulation of ROS homeostasis, and knockdown of p400 led to an increase in intracellular ROS [30]. To test whether downregulation of p400 would cause more MN-γ-H2AX (+), we knocked down p400 in U2OS and HEK-293 cells, respectively, and examined the frequency of MN-γ-H2AX (+). As shown in Figure 3A and 3B, p400 could be efficiently downregulated by RNAi in both cell lines. Consistent with previously reported (30), p400 knockdown led to an elevation in ROS levels in both cell lines (Figure 3C). Moreover, p400 knockdown cells (sip400) exhibited a higher frequency of MN-γ-H2AX (+) when compared to control (siNeg) cells. In each case, the frequency of MN-γ-H2AX (+) was about two fold higher in sip400 than in siNeg cells (Figure 3D), with P <0.05 and P <0.01 for U2OS and HEK-293 cells, respectively. The frequency of MN-γ-H2AX (−), on the other hand, remained unchanged. These results further show that MN-γ-H2AX (+) is more responsive to change in ROS than the MN-γ-H2AX (−).
Fig. 3.

Elevation of MN-γ-H2AX (+) in p400 knockdown cells. U2OS cells and HEK-293 cells were transfected with siRNA duplexes specific to p400 or negative oligo in serum-free medium for 5h, and then replaced with complete medium for 24 h. The expression of p400 mRNA and protein levels in U2OS cells (A) and HEK-293 cells (B) were measured by real-time quantitative RT-PCR assay and Western blotting, respectively. C. The levels of ROS in sip400 U2OS cells and sip400 HEK-293 cells were analyzed by flow cytometry. D. Increased MN-γ-H2AX (+) formation in sip400 U2OS cells and sip400 HEK-293 cells cells. *P<0.05, ** P < 0.01, when compared with each siNeg cells.
3.5. Reciprocal reinforcement of ROS and replication stress in inducing MN-γ-H2AX (+)
We reported previously that MN-γ-H2AX (+) were preferentially induced by DNA replication stress [27]. If ROS and replication stress can both preferentially induce MN-γ-H2AX (+), then how are the two factors related to each other in the induction of MN-γ-H2AX (+) ? We first tested whether cells experiencing oxidative stress would exhibit signs of replication stress. To this end, we measured the phosphorylation of Chk1 and RAD17, which each reflects replication stress [31–34], in cells treated with H2O2. We subjected U2OS cells and HEK-293 cells to different doses of H2O2 for 24 h, and then examined the levels of p-Chk1, Chk1, p-RAD17 and RAD17. As shown in Figure 4A, H2O2 treatment resulted in a marked dose-dependent increase in the level of p-Chk1 in both cell lines. The level of p-RAD17 was also increased in a dose-dependent manner, albeit to a lesser extent. This result suggests that ROS may mimic replication stress. Our results is consistent with a recent report showing that H2O2 can lead to ATR-dependent activation of Chk1 in Xenopus egg extracts [35].
Fig. 4.
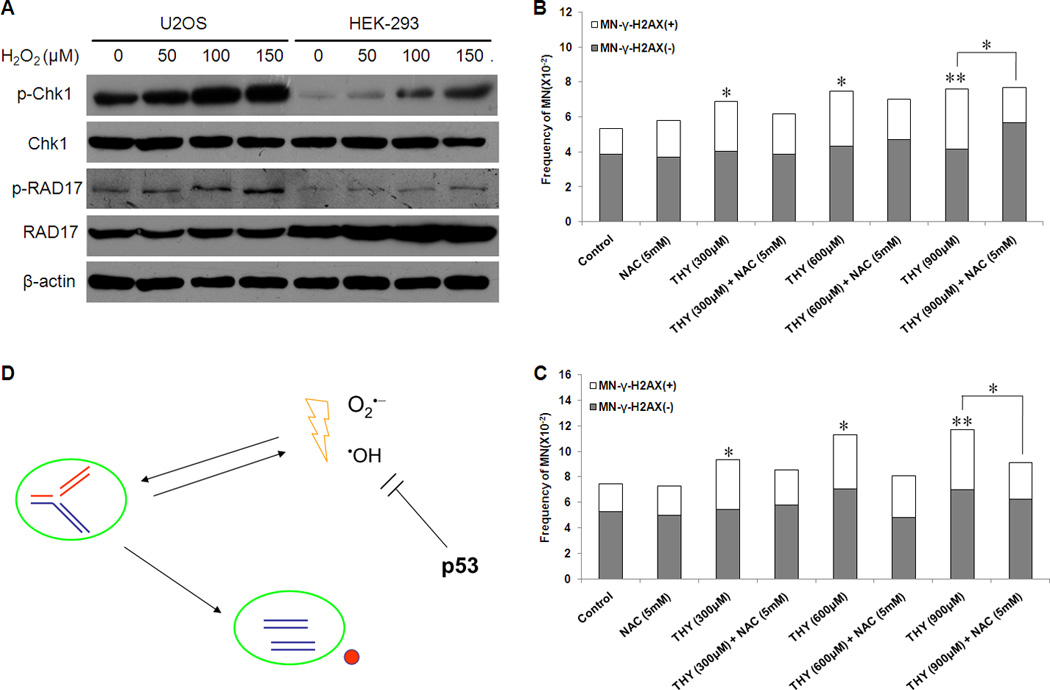
Mutual reinforcement of oxidative stress and replication stress in inducing micronuclei. A. Evaluation of markers reflecting replication stress. U2OS cells and HEK-293 cells were treated with indicated H2O2 concentrations for 24h. Cell extracts were analyzed by Western blot using antibodies to detect p-Chk1, Chk1, p-RAD17 and RAD17. β-actin was used as a loading control. Shown is a representative of three independent experiments. B and C. Attenuation of replication stress-induced MN-γ-H2AX (+) by NAC in U2OS cells (B) and MCF-7 cells (C). Cells were pretreated with or without the antioxidant NAC (5mM) for 90 min, and then were co-incubated with different concentrations of thymidine (THY) for 24h. The frequency of MN were examined at 48h after THY was washed out. * P < 0.05, # P < 0.01. D. A schematic model showing the connection between oxidative stress and replication stress in inducing MN-γ-H2AX (+).
In human endothelial cells, S-phase arrest caused by aphidicolin, an inhibitor of DNA polymerase α and δ, is accompanied by an increase in the level of ROS [36]. Persistent DNA breaks were also shown to be sufficient to elevate the level of ROS [30]. We next tested whether the replication stress-induced formation of MN-γ-H2AX (+) can be attenuated by antioxidant NAC. We previously showed that thymidine could serve as an effective inducer of replication stress and MN-γ-H2AX (+) [27]. Here we pretreated U2OS and MCF-7 cells with NAC (5mM) for 1.5 h before they were treated with thymidine at different concentrations for 24 h. Consistent with our previous report, thymidine caused a significant increase in the frequency of MN-γ-H2AX (+) in both cell lines. Pretreatment with NAC tended to offset the effect of thymidine. It effectively attenuated the induction of MN-γ-H2AX (+) by thymidine at 900 µM (Figure 4B and 4C). Taken together, our results suggest that replication stress and oxidative stress are intertwined and can reinfornce each other in the induction of MN-γ-H2AX (+).
4. Discussion
MN-γ–H2AX (+) are distinguished from MN-γ–H2AX (−) by the way they emerge. We previously showed that MN-γ–H2AX (+) are formed in S-phase, are preferentially induced by replication stress and are probably derived from aggregated DNA breaks [27]. A recent study showed that this subtype of MN may occur when their nuclear envelopes collapse and MN without intact envelope are incapable of DNA replication, DNA repair and transcription[37]. In this study, we demonstrated that the MN-γ–H2AX (+) can also be preferentially induced by H2O2. We showed that both H2O2 treatment and genetic manipulations that raise the endogenous level of ROS are much more effective for the induction of MN-γ–H2AX (+) than for that of MN-γ–H2AX (−). Application of the antioxidant NAC could significantly attenuate the induction of MN-γ–H2AX (+). Importantly, we showed that p53 function is more important for preventing the formation of MN-γ–H2AX (+) than for that of MN-γ–H2AX (−). Our results provide an important insight into the mechanism by which genomic instability arises when p53 function is lacking or reduced. Besides protecting cells against oxidative stress under physiological and mildly stressed conditions, p53 also functions in other cellular processes, including cell cycle checkpoints, DNA repair and cell death. While failure in any of those processes when p53 is dysfunctional can potentially lead to genomic instability, our results showed that it is the surge in oxidative stress that accounts for the bulk of the elevated MN-γ–H2AX (+) in p53 mutant cells. Considering that human TP53 germline mutation carriers exhibit a higher level of oxidative metabolism [38], our results may bear an implication in understanding cancer development as well as other clinical aspects associated with p53 dysfunction.
Our results also suggest that oxidative stress mimics replication stress in the induction of MN-γ-H2AX (+). We showed previously that replication stress can preferentially induce MN-γ-H2AX (+) [27]. We here showed that oxidative stress is also involved in the formation of MN-γ-H2AX (+) in cells experiencing replication stress, since antioxidant NAC can effectively neutralize the effect of replication stressor thymidine (Figure 4B and 4C). It was shown that thymine glycol, the most common oxidative thymine lesion, can stall replicative DNA polymerase [39]. The S-phase arrest caused by resveratrol in human endothelial cells was also shown to be mediated by an increase in the level of ROS [36]. Consistently, we observed that cells treated with H2O2 exhibited increased phosphorylation of Chk1 and RAD17 (Figure 4A), which mimics replication stress. A recent study showed that H2O2 can lead to ATR-dependent activation of Chk1 in Xenopus egg extracts [35]. It is proposed that APE2 (apurinic/apyrimidinic endonuclease 2), a key component in base excision repair pathway, may generate a stretch of single-stranded DNA that can bind RPA and lead to the activation of ATR-Chk1 pathway. Beacause ATR can phosphorylate H2AX, it is possible that in addition to inducing MN-γ-H2AX (+) indirectly by reinforcing replication stress, oxidative stress may also directly induce MN-γ-H2AX (+). Thus, oxidative stress and replication stress may reinforce each other in exerting their effect as an inducer of MN-γ-H2AX (+) (Figure 4D). Interestingly, DNA replication is restricted to the reductive phase (glycolytic phase) to protect genome integrity in the yeast, and an impediment in metabolic cycle–directed restriction of cell division leads to increased mutation rate [40]. Mammalian cells in S-phase are considerably more sensitive to H2O2 than in G1 phase [41]. Because replication stress is a major driver of genomic instability [27, 42–44], reducing the level of ROS during replication would understandably reduce replication stress and protect genome integrity. p53 appears to play a critical role in keeping the spontaneous level of ROS in check. In doing so, it alleviates replication stress and maintains genomic integrity.
Supplementary Material
Highlights.
A subtype of micronuclei (MN) is preferentially induced by oxidative stress.
Induction of MN subtype by p53 dysfunction is mediated by oxidative stress.
Oxidative and replication stresses reinforce each other in generating MN.
Acknowledgements
This study was supported by National Basic Research Program of China (973 Program) grant (2011CB966200), National Natural Science Foundation Research Grants (81372241 and 81171968), State Program of National Natural Science Foundation of China for Innovative Research Group (81321061) and National Institutes of Health grant R01ES011633.
Abbreviations
- ROS
reactive oxygen species
- H2O2
hydrogen peroxide
- NAC
N-acetylcysteine
- MN
micronuclei
- DSBs
double-strand breaks
- MSF
primary mouse skin fibroblasts
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest:
The authors declare no conflict of interest.
Reference
- 1.Klaunig JE, Kamendulis LM. The role of oxidative stress in carcinogenesis. Annual review of pharmacology and toxicology. 2004;44:239–267. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 2.Kvam E, Tyrrell RM. Induction of oxidative DNA base damage in human skin cells by UV and near visible radiation. Carcinogenesis. 1997;18:2379–2384. doi: 10.1093/carcin/18.12.2379. [DOI] [PubMed] [Google Scholar]
- 3.Sander CS, Chang H, Hamm F, Elsner P, Thiele JJ. Role of oxidative stress and the antioxidant network in cutaneous carcinogenesis. International journal of dermatology. 2004;43:326–335. doi: 10.1111/j.1365-4632.2004.02222.x. [DOI] [PubMed] [Google Scholar]
- 4.Clutton SM, Townsend KM, Walker C, Ansell JD, Wright EG. Radiation-induced genomic instability and persisting oxidative stress in primary bone marrow cultures. Carcinogenesis. 1996;17:1633–1639. doi: 10.1093/carcin/17.8.1633. [DOI] [PubMed] [Google Scholar]
- 5.Babbar N, Casero RA., Jr Tumor necrosis factor-alpha increases reactive oxygen species by inducing spermine oxidase in human lung epithelial cells: a potential mechanism for inflammation-induced carcinogenesis. Cancer research. 2006;66:11125–11130. doi: 10.1158/0008-5472.CAN-06-3174. [DOI] [PubMed] [Google Scholar]
- 6.Federico A, Morgillo F, Tuccillo C, Ciardiello F, Loguercio C. Chronic inflammation and oxidative stress in human carcinogenesis, International journal of cancer. Journal international du cancer. 2007;121:2381–2386. doi: 10.1002/ijc.23192. [DOI] [PubMed] [Google Scholar]
- 7.Hei TK, Filipic M. Role of oxidative damage in the genotoxicity of arsenic. Free radical biology & medicine. 2004;37:574–581. doi: 10.1016/j.freeradbiomed.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Nordberg J, Arner ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free radical biology & medicine. 2001;31:1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 9.Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proceedings of the National Academy of Sciences of the United States of America. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cadet J, Berger M, Douki T, Ravanat JL. Oxidative damage to DNA: formation, measurement, and biological significance. Reviews of physiology, biochemistry and pharmacology. 1997;131:1–87. doi: 10.1007/3-540-61992-5_5. [DOI] [PubMed] [Google Scholar]
- 11.Hsie AW, Recio L, Katz DS, Lee CQ, Wagner M, Schenley RL. Evidence for reactive oxygen species inducing mutations in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America. 1986;83:9616–9620. doi: 10.1073/pnas.83.24.9616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chiera F, Meccia E, Degan P, Aquilina G, Pietraforte D, Minetti M, Lambeth D, Bignami M. Overexpression of human NOX1 complex induces genome instability in mammalian cells. Free radical biology & medicine. 2008;44:332–342. doi: 10.1016/j.freeradbiomed.2007.09.018. [DOI] [PubMed] [Google Scholar]
- 13.Rani V, Neumann CA, Shao C, Tischfield JA. Prdx1 deficiency in mice promotes tissue specific loss of heterozygosity mediated by deficiency in DNA repair and increased oxidative stress. Mutation research. 2012;735:39–45. doi: 10.1016/j.mrfmmm.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu B, Chen Y, St Clair DK. ROS and p53: a versatile partnership. Free radical biology & medicine. 2008;44:1529–1535. doi: 10.1016/j.freeradbiomed.2008.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu D, Xu Y. p53, oxidative stress, and aging. Antioxidants & redox signaling. 2011;15:1669–1678. doi: 10.1089/ars.2010.3644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sablina AA, Budanov AV, Ilyinskaya GV, Agapova LS, Kravchenko JE, Chumakov PM. The antioxidant function of the p53 tumor suppressor. Nature medicine. 2005;11:1306–1313. doi: 10.1038/nm1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yin Y, Tainsky MA, Bischoff FZ, Strong LC, Wahl GM. Wild-type p53 restores cell cycle control and inhibits gene amplification in cells with mutant p53 alleles. Cell. 1992;70:937–948. doi: 10.1016/0092-8674(92)90244-7. [DOI] [PubMed] [Google Scholar]
- 18.Bouffler SD, Kemp CJ, Balmain A, Cox R. Spontaneous and ionizing radiation-induced chromosomal abnormalities in p53-deficient mice. Cancer research. 1995;55:3883–3889. [PubMed] [Google Scholar]
- 19.Fukasawa K, Wiener F, Vande Woude GF, Mai S. Genomic instability and apoptosis are frequent in p53 deficient young mice. Oncogene. 1997;15:1295–1302. doi: 10.1038/sj.onc.1201482. [DOI] [PubMed] [Google Scholar]
- 20.Shao C, Deng L, Henegariu O, Liang L, Stambrook PJ, Tischfield JA. Chromosome instability contributes to loss of heterozygosity in mice lacking p53. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:7405–7410. doi: 10.1073/pnas.97.13.7405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heddle JA, Cimino MC, Hayashi M, Romagna F, Shelby MD, Tucker JD, Vanparys P, MacGregor JT. Micronuclei as an index of cytogenetic damage: past, present, and future. Environmental and molecular mutagenesis. 1991;18:277–291. doi: 10.1002/em.2850180414. [DOI] [PubMed] [Google Scholar]
- 22.Bonassi S, Ugolini D, Kirsch-Volders M, Stromberg U, Vermeulen R, Tucker JD. Human population studies with cytogenetic biomarkers: review of the literature and future prospectives. Environmental and molecular mutagenesis. 2005;45:258–270. doi: 10.1002/em.20115. [DOI] [PubMed] [Google Scholar]
- 23.Norppa H, Falck GC. What do human micronuclei contain? Mutagenesis. 2003;18:221–233. doi: 10.1093/mutage/18.3.221. [DOI] [PubMed] [Google Scholar]
- 24.Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. The Journal of cell biology. 1999;146:905–916. doi: 10.1083/jcb.146.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Current biology : CB. 2000;10:886–895. doi: 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 26.Cleaver JE. gammaH2Ax: biomarker of damage or functional participant in DNA repair "all that glitters is not gold!". Photochemistry and photobiology. 2011;87:1230–1239. doi: 10.1111/j.1751-1097.2011.00995.x. [DOI] [PubMed] [Google Scholar]
- 27.Xu B, Sun Z, Liu Z, Guo H, Liu Q, Jiang H, Zou Y, Gong Y, Tischfield JA, Shao C. Replication stress induces micronuclei comprising of aggregated DNA double-strand breaks. PloS one. 2011;6:e18618. doi: 10.1371/journal.pone.0018618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shao C, Deng L, Henegariu O, Liang L, Raikwar N, Sahota A, Stambrook PJ, Tischfield JA. Mitotic recombination produces the majority of recessive fibroblast variants in heterozygous mice. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:9230–9235. doi: 10.1073/pnas.96.16.9230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Budanov AV. Stress-responsive sestrins link p53 with redox regulation and mammalian target of rapamycin signaling. Antioxidants & redox signaling. 2011;15:1679–1690. doi: 10.1089/ars.2010.3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mattera L, Courilleau C, Legube G, Ueda T, Fukunaga R, Chevillard-Briet M, Canitrot Y, Escaffit F, Trouche D. The E1A-associated p400 protein modulates cell fate decisions by the regulation of ROS homeostasis. PLoS genetics. 2010;6:e1000983. doi: 10.1371/journal.pgen.1000983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Molecular and cellular biology. 2001;21:4129–4139. doi: 10.1128/MCB.21.13.4129-4139.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chini CC, Wood J, Chen J. Chk1 is required to maintain claspin stability. Oncogene. 2006;25:4165–4171. doi: 10.1038/sj.onc.1209447. [DOI] [PubMed] [Google Scholar]
- 33.Chini CC, Chen J. Claspin, a regulator of Chk1 in DNA replication stress pathway. DNA repair (Amst) 2004;3:1033–1037. doi: 10.1016/j.dnarep.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 34.Bartkova J, Hamerlik P, Stockhausen MT, Ehrmann J, Hlobilkova A, Laursen H, Kalita O, Kolar Z, Poulsen HS, Broholm H, Lukas J, Bartek J. Replication stress and oxidative damage contribute to aberrant constitutive activation of DNA damage signalling in human gliomas. Oncogene. 2010;29:5095–5102. doi: 10.1038/onc.2010.249. [DOI] [PubMed] [Google Scholar]
- 35.Willis J, Patel Y, Lentz BL, Yan S. APE2 is required for ATR-Chk1 checkpoint activation in response to oxidative stress. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:10592–10597. doi: 10.1073/pnas.1301445110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schilder YD, Heiss EH, Schachner D, Ziegler J, Reznicek G, Sorescu D, Dirsch VM. NADPH oxidases 1 and 4 mediate cellular senescence induced by resveratrol in human endothelial cells. Free radical biology & medicine. 2009;46:1598–1606. doi: 10.1016/j.freeradbiomed.2009.03.013. [DOI] [PubMed] [Google Scholar]
- 37.Hatch EM, Fischer AH, Deerinck TJ, Hetzer MW. Catastrophic nuclear envelope collapse in cancer cell micronuclei. Cell. 2013;154:47–60. doi: 10.1016/j.cell.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang PY, Ma W, Park JY, Celi FS, Arena R, Choi JW, Ali QA, Tripodi DJ, Zhuang J, Lago CU, Strong LC, Talagala SL, Balaban RS, Kang JG, Hwang PM. Increased oxidative metabolism in the Li-Fraumeni syndrome. The New England journal of medicine. 2013;368:1027–1032. doi: 10.1056/NEJMoa1214091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Aller P, Rould MA, Hogg M, Wallace SS, Doublie S. A structural rationale for stalling of a replicative DNA polymerase at the most common oxidative thymine lesion, thymine glycol. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:814–818. doi: 10.1073/pnas.0606648104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen Z, Odstrcil EA, Tu BP, McKnight SL. Restriction of DNA replication to the reductive phase of the metabolic cycle protects genome integrity. Science. 2007;316:1916–1919. doi: 10.1126/science.1140958. [DOI] [PubMed] [Google Scholar]
- 41.Frankenberg-Schwager M, Becker M, Garg I, Pralle E, Wolf H, Frankenberg D. The role of nonhomologous DNA end joining, conservative homologous recombination, and single-strand annealing in the cell cycle-dependent repair of DNA double-strand breaks induced by H(2)O(2) in mammalian cells. Radiation research. 2008;170:784–793. doi: 10.1667/RR1375.1. [DOI] [PubMed] [Google Scholar]
- 42.Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell. 2011;145:435–446. doi: 10.1016/j.cell.2011.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E, Chew SK, Rowan AJ, Schenk A, Sheffer M, Howell M, Kschischo M, Behrens A, Helleday T, Bartek J, Tomlinson IP, Swanton C. Replication stress links structural and numerical cancer chromosomal instability. Nature. 2013;494:492–496. doi: 10.1038/nature11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Toledo LI, Altmeyer M, Rask MB, Lukas C, Larsen DH, Povlsen LK, Bekker-Jensen S, Mailand N, Bartek J, Lukas J. ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell. 2013;155:1088–1103. doi: 10.1016/j.cell.2013.10.043. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.