

Abstract
Oxidative stress has been implicated in the pathogenesis of Huntington's disease (HD), however, the origin of the oxidative stress is unknown. System xc- plays a role in the import of cystine to synthesize the antioxidant glutathione. We found in the STHdhQ7/Q7 and STHdhQ111/Q111 striatal cell lines, derived from neuronal precursor cells isolated from knock-in mice containing 7 or 111 CAG repeats in the huntingtin gene, that there is a decrease in system xc- function. System xc- is composed of two proteins, the substrate specific transporter, xCT, and an anchoring protein, CD98. The decrease in function in system xc- that we observed is caused by a decrease in xCT mRNA and protein expression in the STHdhQ111/Q111 cells. In addition we found a decrease in protein and mRNA expression in the transgenic R6/2 HD mouse model at 6 weeks of age. STHdhQ111/Q111 cells have lower basal levels of GSH and higher basal levels of ROS. Acute inhibition of system xc- causes greater increase in oxidative stress in the STHdhQ111/Q111 cells than in the STHdhQ7/Q7 cells. These results suggest that a defect in the regulation of xCT may be involved in the pathogenesis of HD by compromising xCT expression and increasing susceptibility to oxidative stress.
Introduction
Huntington's disease (HD) is an autosomal dominant neurodegenerative disease caused by an expansion of the CAG region in exon 1 of the huntingtin gene (Htt) (The Huntington's Disease Collaborative Research Group, 1993) affecting approximately 10 in 100,000 people (Rawlins, 2010). The pathogenesis of HD is unknown, however, excitotoxicity (DiFiglia, 1990; Fan and Raymond, 2007; Raymond et al., 2011), oxidative stress (Li et al., 2010), and transcriptional dysregulation (Cha, 2007; 2000; Cui et al., 2006) all appear to play a role.
Disruption of glutamate homeostasis has been implicated in HD (Ferrante et al., 2002; Miller et al., 2008; Petr et al., 2013) and other triplet repeat diseases (Custer et al., 2006). Glutamate transport, thought to be mediated primarily by a family of 5 genes (eaat1-5) is important for clearing extracellular glutamate, with important consequences for excitatory signaling and neuronal survival (Rosenberg and Aizenman, 1989; Rosenberg et al., 1992). Recent work suggests that a defect in trafficking to the plasma membrane (Li et al., 2010) and expression (Petr et al., 2013) of the neuronal cysteine and glutamate transporter EAAC1 (EAAT3) is impaired in HD, compromising the ability of neurons to synthesize glutathione. Decreased GSH levels may be an important source of oxidative stress in HD (Browne, 2008; Chen, 2011; Ribeiro et al., 2012). Another glutamate transport system that is important for providing cysteine for synthesis of glutathione is system xc-, comprised of the transporter xCT and the glycoprotein CD98 (Sato et al., 1999). Under physiological conditions system xc- imports one cystine and exports one glutamate in a Na+-independent manner (Bannai, 1986). Interestingly, system xc- also appears to play a critical role in glutamate homeostasis (Bridges et al., 2012; Kalivas, 2009; Lewerenz et al., 2012). xCT is expressed throughout the brain in both neurons and glia cells, and xCT has the highest expression along border areas of the brain where cerebrospinal fluid is present (Burdo et al., 2006).
We sought to characterize abnormalities in glutamate homeostasis in HD taking advantage of the STHdhQ111/Q111 neuronal cell line as a model system for the disease (Trettel et al., 2000). Initially we confined ourselves to high affinity sodium dependent glutamate transport and transporters (Petr et al., 2013). In the course of that study, we observed a sodium independent component of glutamate transport in these cells. Here, we identify the sodium independent component of glutamate transport in the STHdh cells as system xc-, and find that both the function and expression of system xc- are compromised by the expression of mutant huntingtin, both in the cell lines and in an in vivo model. This defect in xCT expression and function has a direct impact on GSH levels and oxidative stress in cells expressing mutant huntingtin.
Materials and Methods
Mice
The colony was maintained by the breeding of an ovarian transplanted R6/2 female (BCBA-Tg(HDexon1)62Gbp/1j) with CBA/C57VL/6 males (Jackson Laboratories, Bar Harbor, ME, USA). All mice were maintained at the Children's Hospital Boston Animal Care Facility under standard conditions (12 hr light cycle from 7:00 AM to 7:00 PM) with ad libitum access to food and water. All experiments were performed in accordance with NIH guidelines and were approved by the Children's Hospital Boston Institutional Animal Care and Use Committee.
STHdh cells
The STHdh cells were generously provided by Dr. Marcy MacDonald (Massachusetts General Hospital, Boston, MA) and have been previously described (Trettel et al., 2000). The cells were grown in 10 cm dishes at 33°C with 5% CO2 in DMEM media supplemented with 10% (v/v) heat-inactivated FBS, 4.5 g/L L-glucose, 110 mg/mL sodium pyruvate, 4 mM L-glutamine, 0.5mg/mL Geneticin, and 100 units/mL penicillin/streptomycin (all Invitrogen, Carlsbad, CA, USA). Cells were used at passage numbers nine to thirteen for all experiments from cells that were frozen at passage seven.
Glutamate Uptake Assay
The cells were plated at 2×105 cells/well on poly-D-lysine coated 24-well plates. The following day, the cells were washed twice with warm uptake buffer (2.5 mM KCl, 1.2 mM CaCl2, 1.2 mM MgCl2, 1.2 mM K2HPO4, 10 mM HEPES, 5 mM Tris, 10 mM D-glucose, 140 mM choline chloride) and then 0.5 mL/well of the uptake solution was applied for 10 minutes [the uptake solution is the uptake buffer with 0.5 μM L-glutamic acid, 0.022 μM L-[3H]-glutamate (PerkinElmer; Waltham, MA, USA), and/or inhibitors]. In some experiments, the concentrations of L-glutamic acid were varied from 0.5 to 300 μM. The following inhibitors were added to the uptake solution in other experiments: 1 mM L-homocysteic acid (HCA), 250 μM sulfasalazine (SSZ), 10 μM (S)-4-carboxyphenylglycine (CPG) (Tocris Bioscience, Ellisville, MO, USA), 100 μM L-cystine, and 200 μM L-cystine (L-CySS). In other experiments, we assayed the effect of upregulation of xCT using 30 μM salubrinal (Enzo Life Sciences, Farmingdale, NY, USA) or 100 μM diethyl maleate (DEM) for 24 hours. The uptake was stopped by washing three times with ice cold 1% BSA in uptake buffer. The cells were then lysed in 0.1 mM NaOH. The protein concentration was determined using DC Protein Assay (Biorad, Hercules, CA, USA) and the radioactivity was measured by liquid scintillation (TRI-CARB 2200CA, Δ PACKARD, Long Island Scientific, Inc.). All chemicals came from Sigma-Aldrich (St. Louis, MO, USA) unless otherwise noted.
Western Blots
The STHdh cells were washed twice with ice-cold PBS then lysed in 20 mM Tris-HCl pH 7.5, 2.5 mM EDTA, 1% Triton-X 100, 1% deoxycholate, 0.1% SDS, 50 mM NaF, 2 mM sodium orthovanadate, Protease Inhibitor Cocktail, Phosphatase Inhibitor Cocktail II, and Phosphatase Inhibitor Cocktail III (Sigma-Aldrich, St. Louis, MO, USA). Brain cell lysates were created from a coronal slice in which the striatum was separated from the overlying cortex. The tissue was then lysed in 1% SDS, 50 mM phosphate buffer pH 7.4 with protease and phosphatase inhibitors. The tissue was homogenized using a dounce hand-held homogenizer. Samples were heated at 95°C for 10 minutes with Laemmli Sample Buffer containing 400 mM 2-mercaptoethanol. 20 μg of lysate was loaded per well in a 4-20% SDS-PAGE gel (Pierce, Thermo Fisher Scientific, Rockford, IL, USA). The blots were probed with antibodies to xCT (Dun et al., 2006) (1:500), β-actin AC-74 (Sigma-Aldrich, St. Louis, MO, USA, A5316) (1:100,000), and β-III-tubulin (Millipore, Billerica, MA, USA, MAB1637)(1:5,000) in 5% milk in tris buffered saline with Triton-X (TBST) (25 mM Trizma base, 140 mM NaCl, 2.5 mM KCl, pH 7.4, 0.1% Triton-X 100). A blocking peptide for xCT antibody (MVRKPVVATISKGGY) was used at 50 μM to determine the xCT specific bands and was synthesized by Genscript (Piscataway, NJ, USA).
Quantitative PCR
RNA was isolated using TRIzol reagent following the manufacturer's recommended procedure (Invitrogen, Carlsbad, CA, USA). The isolated RNA was then treated with RQ1 RNase-Free DNase according to the manufacturer's protocol to remove any traces of DNA (Promega, Madison, WI, USA). Single stranded cDNA was created from total mRNA by reverse transcription using iScript Select cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). The sequences of the specific primers were as follows: 5′-GTC TAA TGG GGT TGC CCT TGG-3′ (sense) on exon 5 and 5′-CGC ACT GAT GGT GGT AAA ATA GG-3′ (antisense) on exon 7 for mouse xCT (NM_011990.2). For a control mouse hypoxanthineguanine phosphoribosyltransferase (HPRT) (NM_013552) was used (Gomez et al., 2006). The primers were 5′-GAT CAG TCA ACG GGG GAC ATA-3′ (sense) on exon 4, 5′-GGG GCT GTA CTG CTT AAC CAG G-3′ on exon 6. The qPCR was carried out using iQ SYBR Green Supermix (Bio-Rad, Hercules, CA, USA) following the manufacturer's protocol. The PCR cycling parameters were: denaturation at 95°C for 3 min, amplification and quantification program repeated 40 times (95°C for 5 sec, 56°C for 10 sec, 72°C for 30 sec), melting curve program (65-95°C with a heating rate of 0.5°C per second) with a continuous single fluorescent measurement, carried out in the C100 Thermal Cycler and analyzed using CFX96 Manager Software (Bio-Rad, Hercules, CA, USA).
Glutathione Measurements
Cells were seeded at 2×105 cells/well on 24-well poly-D-lysine coated plates. On the following day, cells were treated with 1 mM L-homocysteic acid, 100 μM sulfasalazine, 100 μM tert-butylhydroquinone (tBHQ) or vehicle for 2, 8, or 24 hours. Total GSH was measured using a kinetic assay (Back et al., 1998; Tietze, 1969). Cell growth media was aspirated and then washed with Hank's Balanced Salt Solution (Invitrogen, Carlsbad, CA). 200 μL of 0.3 M perchloric acid (PCA) was added followed by gentle shaking on ice for 15 minutes to lyse the cells. The PCA solution was then transferred to a 1.5 mL microcentrifuge tube containing 50 μL of 3 M potassium bicarbonate, used to neutralize the PCA, and incubated on ice for 30 minutes. The pellet portion of the neutralized solution was resuspended in RIPA buffer and used to determine total protein concentration using Dc Protein Assay. After incubation the solution was centrifuged at 13,200 rpm at 4°C for 5 minutes. 50 μL of the supernatant was added to a well on a 96-well plate containing: 50 μL of 0.3 M PCA previously neutralized with 3 M potassium bicarbonate, 50 μL of 2.4 mM 5,5′-dithiobis-2-nitrobenzoic acid, and 50 μL of 40 μg/mL glutathione reductase (Roche Applied Science, Indianapolis, IN) in 0.1 M sodium phosphate buffer (pH 7.6) and 5 mM EDTA solution. Immediately after adding 50 μL of 0.8 mM NADPH the rate of reaction was determined by taking the absorbance at 405 nm every 15 seconds for 6 minutes (Thermomax microplate reader; Molecular Devices, Sunnyvale, CA). Total glutathione content was determined from reference to a standard curve and normalized to total protein concentration.
Reactive Oxygen Species Measurement
Cells were seeded at 5×104 cells/well on 24-well coverslipped poly-D-lysine coated plates and treated the same as the GSH assay. After treatment cell growth medium containing 5 μM CellROX Deep Red Reagent (Invitrogen, Carlsbad, CA, USA) was applied for 30 min at 33°C. The cells were then washed 3x with PBS and fixed in 4% PFA (w/v) PBS solution for 10 min at room temperature. The nuclei were stained with 1:1000 DAPI in PBS. Images were taken using a Nikon Eclipse 800 microscope and Spot Advanced software.
Cell Viability Assay
Cells were seeded at 1×105 cells/well on poly-D-lysine coated 24-well plates. After overnight incubation cells were treated with various concentrations of diethyl maleate (DEM) (0, 1, 3, 10, 30, 100, 300, 1000 μM) for 24 hours. The growth media was removed, replaced with 350 μL of Alamar Blue solution (10x Alamar Blue in Earle's buffer) (TREK Diagnostic Systems, Cleveland, OH) per well, and incubated at 33°C for 2 hours. 100 μL of the Alamar Blue solution was then transferred into a 96-well plate and the fluorescence was measured at Ex/Em 530/590 nm (Thermomax microplate reader, Molecular Devices, Sunnyvale, CA).
Statistical Methods
All data analysis was performed in GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA, USA). Saturation analysis: a best fit line based on the Michaelis-Menton equation was fit to the data to obtain the Vmax and Km of xCT in both the STHdhQ&/Q7 and STHdhQ111/Q111 cells. Three separate experiments were performed; significance was determined using an unpaired t-test followed by non-parametric Mann-Whitney test. Inhibition assay: three separate experiments were performed and statistical significance was determined using a two-way ANOVA followed by Tukey's multiple comparison post hoc test. We also performed the non-parametric Kruskal-Wallis test followed by Dunn's multiple comparison post hoc test. Western blots: band intensity was measured in ImageJ and normalized to loading control, all experiments were replicated three times and statistical significance was determined using an unpaired t-test followed by non-parametric Mann-Whitney test. RT-qPCR: four separate cell cultures were used for each STHdhQ7/Q7 and STHdhQ111/Q111 experiment, and eight animals in each WT and HD were used to determine xCT mRNA levels. Statistical significance was determined using an unpaired t-test followed by non-parametric Mann-Whitney test. ROS assay: fluorescent intensity was measured using the full field of view in ImageJ for three separate experiments with 50 field of views used. Significance was determined using a two-way ANOVA and Tukey's multiple comparison post hoc test. In addition, the non-parametric Kruskal-Wallis test followed by Dunn's multiple comparison post hoc test were performed. GSH assay: total GSH was normalized to total protein levels. Significance was determined using a two-way ANOVA followed by Tukey's multiple comparison post hoc test. In addition, the non-parametric Kruskal-Wallis test followed by Dunn's multiple comparison post hoc test were performed. Cell viability assay: the LD50 was determined using a best fit line. Four separate experiments were performed. Significance of effects was determined using Student's unpaired t-test as well as the non-parametric Mann-Whitney test.
Results
L-glutamate Na+-independent uptake is decreased in STHdhQ111/Q111 cells
We began by studying the transport of L-glutamate in STHdh cells, which are striatal derived cell lines. The STHdhQ111/Q111 cell line is from a knock in mouse containing homozygous mouse Huntingtin (HTT) loci with a chimeric (mouse-human) exon 1 with 111 polyglutamine repeats (Wheeler et al., 2000). The STHdhQ7Q/7 cell line contains the 7 CAG repeats of the wild-type huntingtin gene (Trettel et al., 2000). There are two primary known forms of glutamate transport: Na+-dependent, mediated by the EAATs, and Na+-independent, mediated by system xc- (McBean, 2002). We examined the carrying capacity (Vmax) and the affinity (Km) of L-glutamate uptake under Na+-dependent and Na+-independent conditions in STHdh cells using 3[H] L-glutamate at varying concentrations of total L-glutamate to test the hypothesis that glutamate homeostasis is altered in HD. L-glutamate uptake under Na+-independent conditions measures primarily system xc- activity (Bannai, 1986; Bridges et al., 2001; Lewerenz and Maher, 2009). A representative saturation analysis of both Na+-dependent and Na+-independent transport of glutamate can be see in Figure 1A. We observed an increase in Na+-dependent uptake in the STHdhQ111/Q111, which is the subject of another publication (Petr et al., 2013), and a decrease in Na+-independent uptake (Fig. 1A). We chose for this study to focus on the Na+-independent L-glutamate uptake. The STHdhQ111/Q111 cells have approximately one third the carrying capacity of STHdhQ7/Q7 cells (unpaired Student's t test, t(4)=12.2, p<0.001; Mann-Whitney test, p<0.05) (Fig. 1C). The affinity for L-glutamate is unchanged between the two cell lines (unpaired Student's t-test, t(4)=1.5, p>0.05; Mann-Whitney test, p>0.05) (Fig. 1D). A representative saturation analysis of the Na+-independent L-glutamate uptake can be seen in Figure 1B. These results indicate a loss of activity (Vmax) of Na+-independent L-glutamate transport in the STHdhQ111/Q111, but no change in affinity for L-glutamate.
Figure 1.
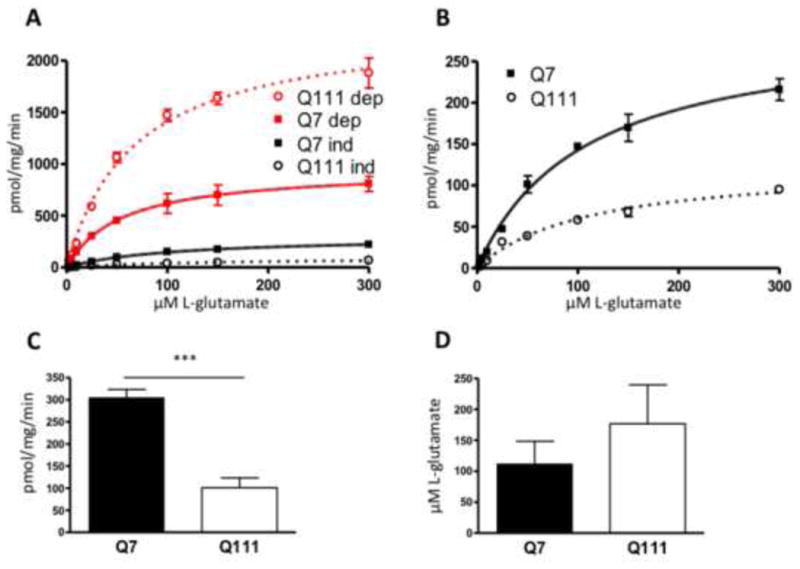
Na+-dependent and independent L-glutamate uptake in STHdh cells, showing the relative contribution of each to total glutamate uptake. A, Glutamate uptake was measured using 3[H] L-glutamate in the presence and absence of sodium, in which case sodium was replaced by choline. Sodium dependent uptake represents total uptake with uptake in the absence of sodium subtracted. Sodium independent uptake is the uptake in the choline medium with background subtracted. A representative saturation analysis is shown. Best fit line was established using the Michaelis-Menten equation. Error bars represent SEM, n=3. B, Na-independent glutamate uptake. Data from A, expanded scale. C. The Vmax values for Na-independent glutamate uptake in the Q7 and Q111 cells were determined from the saturation analysis, error bars represent SEM; unpaired Student's t test: t(4)=12.2 *** p<0.001, n=3; Mann-Whitney test, p<0.05. D, The Km values for Q7 and Q111 cells were also determined from the saturation analysis. Error bars represent SEM, n=3; unpaired Student's t test t(4)=1.5, p>0.05; Mann-Whitney test, p>0.05.
We used known system xc- inhibitors to confirm that the decrease in Na+- independent L-glutamate uptake is due to system xc-: L-homocysteic acid (HCA) (Gochenauer and Robinson, 2001; Murphy et al., 1990), sulfasalazine (SSZ) (Chung et al., 2005), (S)-4-carboxyphenylglycine (CPG) (Gochenauer and Robinson, 2001; Ye and Sontheimer, 1999), and L-cystine (L-CySS) (Bannai and Kitamura, 1980; Murphy et al., 1990). All the inhibitors significantly inhibited the Na+-independent L-glutamate uptake in both cell lines (Fig. 2A, two-way ANOVA main effect of inhibitors: F(5, 132)=212.3, p<0.001; followed by the post hoc Tukey's multiple comparison test when comparing inhibitors to control, p<0.001; Kruskal-Wallis and post hoc Dunn's test p<0.0001). The extent of inhibition is the same between the two cell lines when compared to control levels (Fig. 2B, two-way ANOVA main effect of cell type: F(1, 132)=0.4, p>0.05; followed by the Tukey's multiple comparison test comparing inhibition treatments between cell type, p>0.05, F(1,132)=0.41. These results confirm that the 3[H]-L-glutamate uptake measured in the absence of sodium in the STHdh cells is mediated by system xc-. The decrease in system xc- transport may be due to a decrease in total protein levels, decrease in trafficking to the plasma membrane or post-translational modification.
Figure 2.
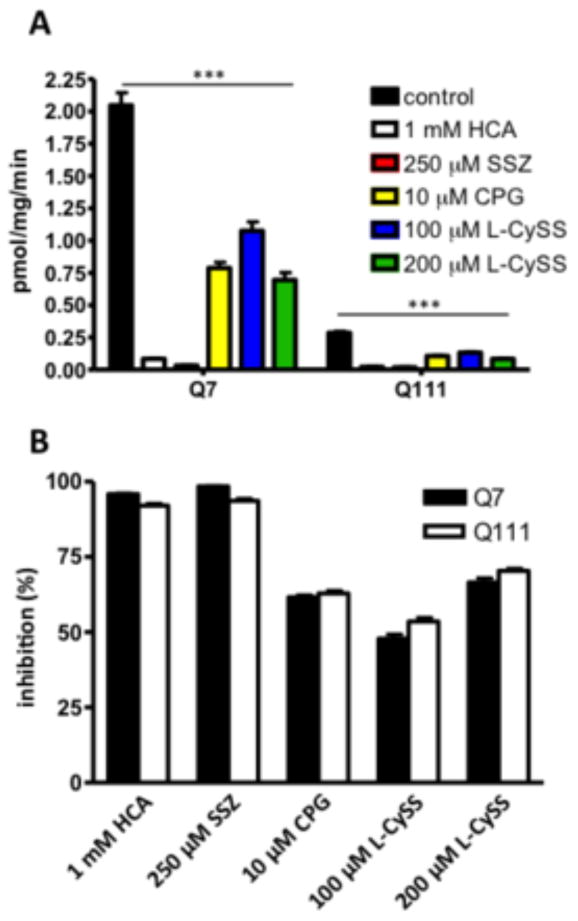
System xc inhibitors inhibited Na+-independent L-glutamate. A, Glutamate uptake was measured using 3[H] L-glutamate in the absence of sodium (choline buffer). Inhibitors or vehicle were added to the uptake buffer: L-homocysteic acid (HCA; 1 mM), sulfasalazine (SSZ; 250 μM), 4-(S)-carboxyphenylglycine (CPG; 10 μM), L-cystine (L-CySS) at 100 and 200 μM (blue and green bars). Error bars represent SEM; two-way ANOVA main effect of treatment: F(5, 132)=212.3, p<0.001; main effect of cell type: F(1, 132)=835.4, p<0.001; interaction: F(5, 132)=125.2, p<0.001; followed by Tukey's multiple comparison test comparing each treatment to control for both cell types: *** p<0.001; Kruskal-Wallis followed by Dunn's post hoc test, p<0.0001. B, Percent of Na+-independent uptake inhibited compared to control, error bars represent SEM, two-way ANOVA main effect of cell type: F(1, 132)=0.4, p>0.05; followed by Tukey's multiple comparison test comparing between cell types of each treatment, p=0.5218, n=3.
xCT protein levels are decreased in STHdhQ111/Q111 cells
Next we examined the cause of the decrease in Vmax in the STHdhQ111/Q111 cells. xCT is the substrate specific transporter of system xc- (Sato et al., 1999), and therefore changes in the expression of this protein could account for the changes in function. To assay protein, we used an antibody against xCT that has been previously characterized (Dun et al., 2006). A blocking peptide corresponding to the immunogenic portion of the xCT antibody was used to determine which of the bands recognized by the antibody represents xCT. xCT bands appeared at ∼50 and ∼100 kDa, demonstrated by the loss of immunoreactivity with the blocking peptide (arrows; Fig. 3A, B). The larger of the two bands most likely is a dimer of monomeric xCT. There is 25.3% the amount of xCT in STHdhQ111/Q111 cells compared to STHdhQ7/Q7 cells when quantified using densitometry and normalization to βactin (unpaired Student's t-test, t(4)=11.8, p<0.001; Mann-Whitney test, p<0.05)(Fig. 3A, C). The loss of function observed in the STHdhQ111/Q111 cells is most likely accounted for by the decrease in protein expression.
Figure 3.
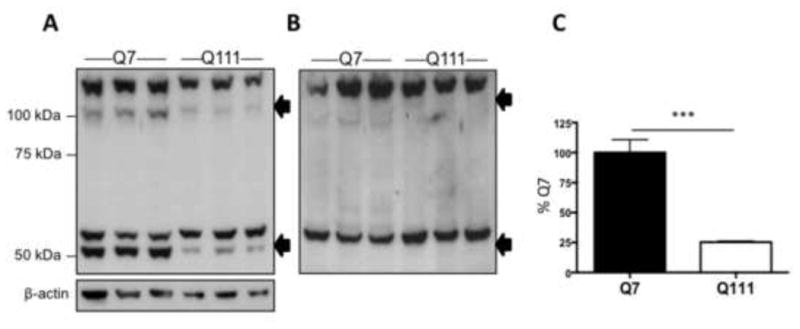
xCT protein expression was decreased in STHdhQ111/Q111 cells. A, Western blot of xCT protein expression in STHdhQ7/Q7 (lanes 1-3) and STHdhQ111/Q111 (lanes 4-6); specific bands (see “B”) are indicated by arrows at ∼50 and ∼100 kDa. B, Western blot with anti- xCT antibody pre-adsorbed using the immunogenic peptide; arrows indicate the position of bands at ∼50 and ∼100 kDa lost with pre-adsorption. C, Densitometry of total xCT protein (bands at ∼50 and ∼100 kDa) normalized to β-actin in the STHdh cells. Error bars represent SEM; unpaired Student's t test: t(4)=11.8, *** p<0.001, n=3; Mann-Whitney test, p<0.05.
xCT protein levels are decreased in the striatum of the R6/2 HD mice
We also looked at the levels of xCT in the striatum and cortex of 6 week old R6/2 mice, a transgenic mouse model containing exon 1 of human mHtt with 141-157 CAG repeats (Mangiarini et al., 1996). Similar to the STHdh cells there are bands corresponding to xCT at ∼50 and ∼100 kDa which disappear with peptide block (Fig. 4B, E). We observed no change in xCT levels normalized to βIII tubulin in the cortex (unpaired Student's t-test, t(4)=1.1, p>0.05, Mann-Whitney test, p>0.05) (Fig. 4A, C), but a 34% decrease in the striatum (unpaired Student's t-test, t(4)=4.8, p<0.05, Mann-Whitney test, p<0.05) (Fig. 4D, F).
Figure 4.
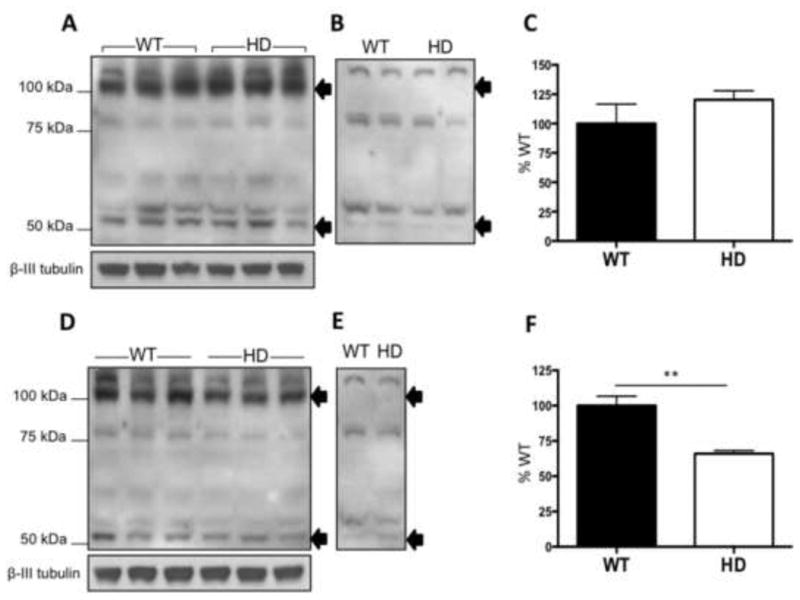
xCT protein expression was decreased in the striatum of 6 week old R6/2 mice. Western blot of xCT protein expression in the cortex (A) and striatum (D), of WT (lanes 1-3) and HD (lanes 4-6) littermates. Bands corresponding to xCT are indicated by arrows at ∼50 and ∼100 kDa. B,E, Western blot with pre-absorbed xCT antibody using immunogenic peptide for the antibody; arrows indicate the loss of xCT specific bands at ∼50 and ∼100 kDa. Densitometry of total xCT protein levels normalized to β-III tubulin in the cortex (C) and the striatum (F). Error bars represent SEM: unpaired Student's t test: striatum t(4)=4.8, * p<0.05; cortex t(4)=1.1, p>0.05, n=3; Mann-Whitney test for striatum, p<0.05.
xCT mRNA levels are decreased in STHdhQ111/Q111 cells and in the striatum of R6/2 mice
A decrease in protein levels may be due to a decrease in translation, increased degradation, or decreased transcription. There is no reported evidence in the literature of decreased translation in HD. Impairment of the ubiquitin-proteasome pathway has been reported in HD (Bennett et al., 2007). Transcriptional dysregulation has also been widely reported in HD (Cha, 2007) and specifically in the STHdhQ111/Q111 cells (Lee et al., 2007) and in the R6/2 mice (Crocker et al., 2006). We found that xCT mRNA expression is decreased in the STHdhQ111/Q111 cells (unpaired Student's t-test, t(7)=7.4, p<0.005; Mann-Whitney test, p<0.001) (Fig. 5A). We also discovered a significant decrease in xCT mRNA expression in the striatum of 6 week old R6/2 mice (unpaired Student's t-test, t(12)=2.6, p<0.05; Mann-Whitney test, p<0.05) and an insignificant decrease in the cortex (unpaired Student's t-test, t(12)=1.5, p>0.05; Mann-Whitney test, p>0.05) when compared to littermate controls (Fig. 5B).
Figure 5.
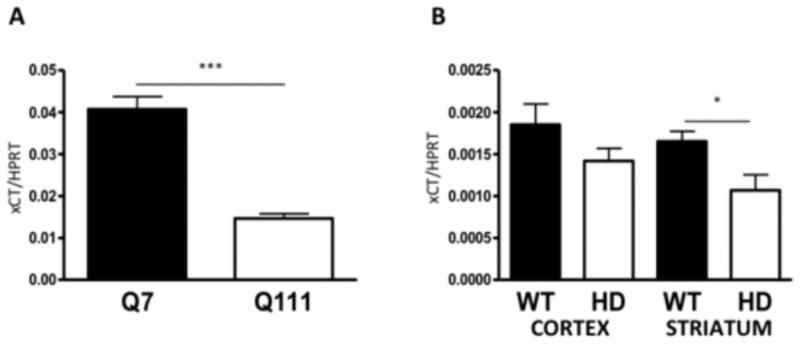
xCT mRNA expression was decreased in STHdhQ111/Q111 cells and in 6 week old R6/2 mice. A, xCT mRNA expression normalized to HPRT in STHdh cells. Error bars represent SEM; unpaired Student's t test: t(7)=7.4, *** p<0.001, n=8; Mann-Whitney test p<0.05. B, xCT mRNA expression in the cortex (lanes 1-2) and striatum (lanes 3-4) in WT and HD littermates. Error bars represent SEM; unpaired Student's t test: striatum t(12)=2.6, * p<0.05; cortex t(12)=1.5, p>0.05, n=6; Mann-Whitney test for striatum, p<0.05.
GSH levels are decreased and ROS are increased in STHdhQ111/Q111 cells
We next observed the pathophysiological consequences of decreased xCT in the STHdhQ111/Q111 cells. Since GSH is necessary for the inactivation of ROS within the cell, we also observed cellular ROS accumulation using the CellROX Deep Red Reagent. Basal GSH levels are decreased in the STHdhQ111/Q111 cells (two-way ANOVA main effect for treatment by cell type: F(7, 24)=281.55, p<0.001; followed by Tukey's multiple comparison test comparing control STHdhQ7/Q7 and STHdhQ111/Q111 at all time points, p<0.01; Kruskal-Wallis test followed by post-hoc Dunn's test, p<0.01) (Fig. 6C). Basal ROS levels are increased in the STHdhQ111/Q111 cells correlating with the decrease in GSH levels (Fig. 6ARow 1). We then wanted to determine if xCT plays a direct role in the oxidative stress in the STHdh cells by observing changes in GSH and ROS levels upon system xc- inhibition. We tested the effect of inhibiting system xc- with HCA and SSZ, and also increasing oxidative stress with the electrophilic agent tert-butylhydroquinone (tBHQ) (Kraft et al., 2004) (Fig. 6A). In the STHdhQ7/Q7 cells, oxidative stress was not detectably increased by inhibition of system xc- (Fig. 6A, 6B; two-way ANOVA main effect of treatment: F(7, 785)=532.3, p<0.001; main effect of time: F(2, 785)=160.4, p<0.001; interaction: F(14, 785)=30.37, p<0.001; followed by Tukey's multiple comparison test for STHdhQ7/Q7 HCA and SSZ treatment effect not significant when compared to control at all time points, p>0.05), even when GSH levels were significantly brought down to the levels observed in STHdhQ111/Q111 cells (Fig. 6C; two-way ANOVA main effect of treatment: F(7, 24)=281.5, p<0.001; main effect of time: F(2, 24)=75.6, p<0.001; interaction: F(14, 24)=59.4, p<0.001; followed by Tukey's multiple comparison test for STHdhQ7/Q7 HCA and SSZ treatments compared to control at 24 hr time point, p<0.01; Kruskal-Wallis test followed by post-hoc Dunn's test, p<0.01), suggesting that decreased GSH is not sufficient in these cells to increase ROS. In contrast, STHdhQ111/Q111 cells had elevated basal levels of oxidative stress, (Fig. 6A, 6B) (two-way ANOVA main effect of treatment by cell type: F(7, 785)=532.2, p<0.001; main effect of time: F(2, 785)=160.4, p<0.001; interaction: F(14, 785)=30.4, p<0.001; followed by Tukey's multiple comparison post hoc test comparing control STHdhQ7/Q7 and STHdhQ111/Q111 at all time points, p<0.05; Kruskal-Wallis test followed by post-hoc Dunn's test, p<0.01) and accumulation of ROS was exacerbated by HCA and SSZ (two-way ANOVA main effect of treatment by cell type: F(7, 785)=532.2, p<0.001; main effect of time: F(2, 785)=160.4, p<0.001; interaction: F(14, 785)=30.4, p<0.001; followed by Tukey's multiple comparison post hoc test comparing HCA and SSZ treatment of STHdhQ111/Q111 cells to control at all time points, p<0.01; Kruskal-Wallis test followed by post-hoc Dunn's test, p<0.01), suggesting that STHdhQ111/Q111 cells are more sensitive to decreased GSH levels than STHdhQ7/Q7 cells, perhaps because of increased ROS generation in these cells. The STHdhQ111/Q111 cells were observed to contain a lower level of GSH than STHdhQ7/Q7 cells in the basal state; HCA and SSZ treatment reduced levels of GSH in both types of cell (Fig. 6C, two-way ANOVA main effect of treatment: F(7, 24)=281.5, p<0.001; main effect of time: F(2, 24)=75.6, p<0.001; interaction: F(14, 24)=59.4, p<0.001; followed by Tukey's multiple comparison post hoc test comparing HCA and SSZ treatment of STHdhQ111/Q111 to control at all time points, p<0.01; Kruskal-Wallis test followed by post-hoc Dunn's test, p<0.01) indicating that system xc- is needed to synthesize glutathione. With tBHQ treatment increased oxidative stress was observed in both STHdh cell lines (Fig. 6A, row 4), and increased GSH levels were also observed in both cell lines exposed to tBHQ (two-way ANOVA main effect of treatment: F(7, 24)=281.5, p<0.001; main effect of time: F(2, 24)=75.6, p<0.001; interaction: F(14, 24)=59.4, p<0.001; followed by Tukey's multiple comparison post hoc test comparing tBHQ treatment of both cell types to control at all time points, p<0.01; Kruskal-Wallis test followed by post-hoc Dunn's test, p<0.01).
Figure 6.
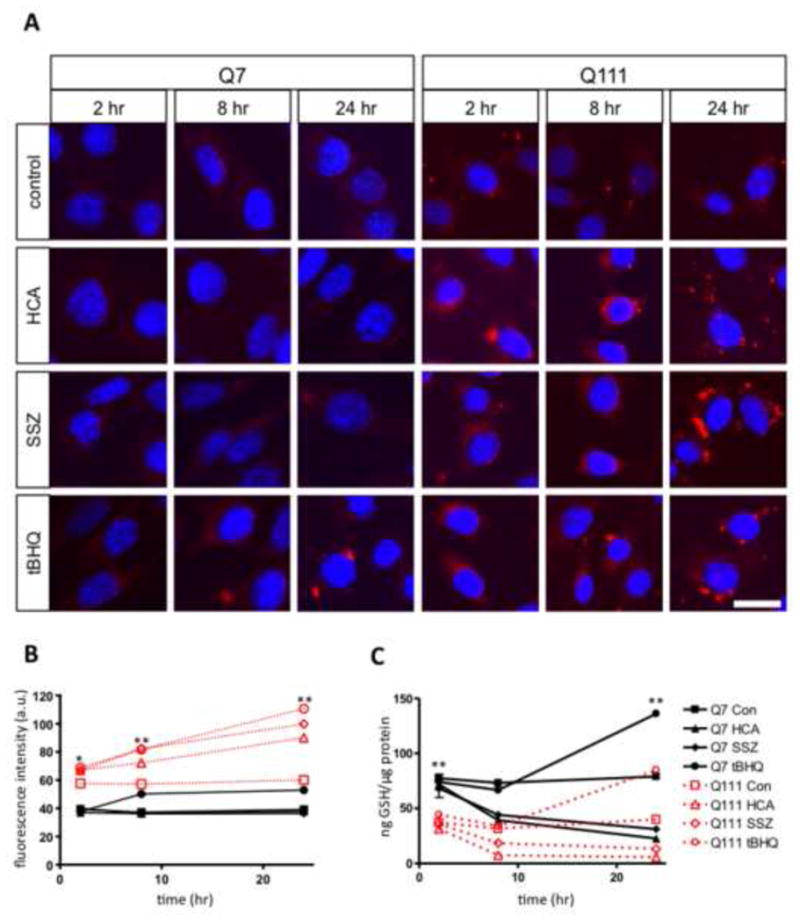
GSH content was decreased and ROS expression was increased in STHdhQ111/Q111 cells. A, STHdh cells treated with vehicle (control), 1 mM HCA, 100 μM SSZ, and 100 μM tBHQ for 2, 8, and 24 hours. Nuclei were stained with DAPI and ROS with CellROX Deep Red Reagent; scale bar 25 μm. B, Quantification of fluorescence intensity from cells treated with CellROX Deep Red Reagent. Errors bars represent SEM; two-way ANOVA main effect of treatment by cell type: F(7, 785)=532.3, p<0.001; main effect of time: F(2, 785)=160.4, p<0.001; interaction: F(14, 785)=30.4, p<0.001) followed by Tukey's multiple comparison test: * p<0.05 (comparing STHdhQ7/Q7 control cells to STHdhQ111/Q111 control cells), ** p<0.01 (comparing STHdhQ7/Q7 control to tBHQ treated cells; and STHdhQ111/Q111 control to HCA, SSZ, and tBHQ treated cells), n=50; Kruskal-Wallis followed by Dunn's post hoc test, p<0.001. C, Total GSH levels in STHdh cells treated with vehicle (control), 1 mM HCA, 100 μM SSZ, and 100 μM tBHQ for 2, 8, and 24 hours. Error bars represent SEM; n=3 two-way ANOVA main effect of treatment by cell type: F(7, 24)=281.5, p<0.001; main effect of time: F(2, 24)=75.6, p<0.001; interaction: F(14, 24)=59.4, p<0.001) followed by Tukey's multiple comparison test, **p<0.01 (comparing basal GSH levels in control STHdh cells; and treated and control STHdh cells at 24 hrs); Kruskal-Wallis followed by Dunn's post hoc test, p<0.01.
STHdhQ111/Q111 cells are more susceptible to oxidative cell death
We then observed cell survival of the STHdh cells in response to oxidative stress. Diethyl maleate (DEM) is an agent that induces oxidative stress by conjugating with GSH and rapidly reducing GSH availability (Weber et al., 1990). The STHdhQ111/Q111 cells are more sensitive to cell death caused by DEM compared to the STHdhQ7/Q7 cells upon increasing concentrations of DEM (Fig. 7A). The LD50 of DEM in causing cell death was for STHdhQ7/Q7 cells 297.9±5.7 μM and for the STHdhQ111/Q111 cells 9.9±0.1 μM DEM (unpaired Student's t-test, t(6)=6.1, p<0.001, n=4; Mann-Whitney test, p<0.05)(Fig. 7B). Thus, STHdhQ111/Q111 cells were 30 times more sensitive to the toxic effects of DEM.
Figure 7.
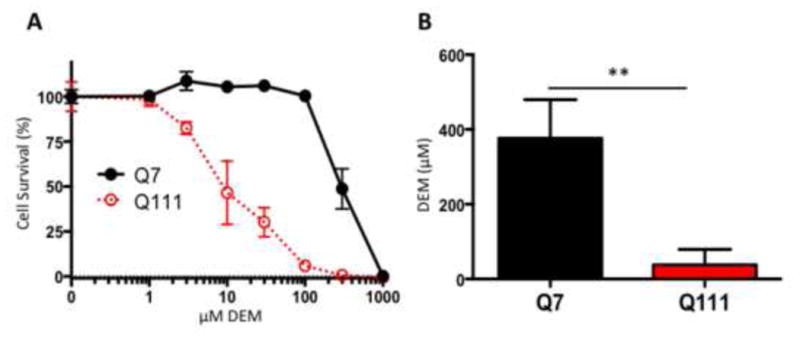
STHdhQ111/Q111 are more susceptible to oxidative cell death. A, STHdh cells treated with 0, 1, 3, 10, 30, 100, 300, 1000 μM DEM for 24 hours. Error bars represent SEM; n=4. B, LD50 of STHdh cells treated with DEM. Error bars represent SEM; unpaired Student's t-test: t(6)=6.1, p<0.001, n=4; Mann-Whitney test, p<0.05.
Discussion
In this study we show decreased system xc- mediated L-glutamate transport capacity (Vmax) in STHdhQ111/Q111 cells that is caused by a decrease in xCT mRNA and protein expression. A defect in expression of xCT protein and mRNA was also found in vivo in the striatum of the R6/2 HD mouse model at 6 weeks of age. The defect we observed in basal levels of xCT would be expected to compromise the import of cystine and production of cysteine for glutathione synthesis. We observed both a decrease in cellular GSH content and increased oxidative stress in the STHdhQ111/Q111 cells that are likely to be consequences of impaired xCT expression.
System xc- acts to import cystine for the synthesis of GSH (Bannai, 1986). Cystine uptake into cells in vitro is crucial for the synthesis of GSH. Oxidative glutamate toxicity is produced by the inhibition of cystine transport by glutamate and other competitive inhibitors of system xc- mediated cystine transport resulting in decreased GSH levels (Coyle and Puttfarcken, 1993). Overexpression of xCT in neurons increases cell survival under conditions of oxidative stress (Lewerenz et al., 2006). Depletion of GSH in cultured astrocytes upregulates system xc- activity (Seib et al., 2011). These studies demonstrate that system xc- may play a role in alleviating oxidative stress by directly impacting GSH levels. The decrease in system xc- function we have observed (Fig. 1) is associated with decreased basal GSH levels (Fig. 6).
Oxidative stress has been implicated in HD, but the origins are still unknown (Browne and Beal, 2006; Chen, 2011; Valencia et al., 2013; 2012). The oxidative stress marker 8-hydroxydeoxyguanosine is increased in post-mortem tissue samples from the caudate and parietal cortex from HD patients (Browne et al., 1997; Polidori et al., 1999). In the R6/2 mouse model there is increased lipid peroxidation in the striatum compared to littermate controls (Browne and Beal, 2006). The basal increase in ROS levels in the STHdhQ111/Q111 cells is consistent with the literature suggesting increased oxidative stress in HD (Fig. 7) (Ribeiro et al., 2012). GSH is a major antioxidant in the brain and depletion of GSH has been linked with oxidative stress induced cell death in vitro (Back et al., 1998; Dringen, 2000; Dringen and Hirrlinger, 2003; Li et al., 1997; Wüllner et al., 1999). Increasing the amount of L-cysteine via L-cystamine in the R6/2 model has been shown to be neuroprotective by protecting against oxidative stress (Fox et al., 2004). Decreased GSH due to decreased surface expression of the glutamate and cysteine transporter EAAC1 has been correlated with oxidative stress in neurons derived from the Q140 mouse model (Li et al., 2010). Decreased GSH levels in the STHdhQ111/Q111 cells most likely contributes to the increased oxidative stress we have observed (Fig. 6). These results are in agreement with another study that found STHdhQ111/Q111 cells have increased oxidative stress (Ribeiro et al., 2012). In addition, that study showed decreased expression of GSH synthetic enzymes. Perplexingly, that study also found increased glutathione levels, apparently related to decreased export of GSH by the multidrug resistance protein 1 (Mrp1), a transport protein that mediates cellular export of glutathione disulfide and glutathione conjugates. It is not clear why our results differ from this previous study regarding absolute levels of GSH, but the two studies together suggest that there may be multiple reasons to explain low GSH levels in the STHdhQ111/Q111 cells. Multiple published studies have examined GSH levels in R6/1 and R6/2 mice, and interestingly in these studies either no change or increases were found, rather than decreases when compared to control animals (Choo et al., 2005; Fox et al., 2004; Petersén et al., 2001; Tkác et al., 2007). In contrast, decreased GSH levels were found in neurons in culture derived from the Q140 mouse model that have a defect in trafficking of the cysteine transporter EAAC1 to the plasma membrane (Li et al., 2010). It is not clear how to reconcile these data with our data. One possibility, as has been suggested before, is that the levels measured in R6 mice are in part determined by compensatory processes, and that the defects observed in xCT expression (as well as the decreases in EAAC1 trafficking noted by Li et al) impairs the ability of HD cells to respond adequately to oxidative stress by upregulating GSH synthesis.
EAAC1 is a high affinity sodium dependent glutamate transporter that also transports cysteine into neurons (Zerangue and Kavanaugh, 1996). Of great interest, knockout of EAAC1 is associated with a chronic neurodegenerative disorder that is associated with decreased GSH levels and is treatable by providing N-acetylcysteine in the diet (Aoyama et al., 2006). In addition, a spontaneously occurring mutation that inactivates xCT also produces a neurodegenerative disorder with prominent striatal atrophy (Shih et al., 2006).
We observed a decrease in basal levels of GSH and increased basal levels of ROS in the STHdhQ111/Q111 cells (Fig. 6). We wanted to further test the association between xCT levels and ROS and GSH levels in the normal and mutant cell lines. Using system xc- inhibitors we observed in the STHdhQ111/Q111 cells an increase in ROS levels with HCA and SSZ treatment, but no such increase in the STHdhQ7/Q7 cells (Fig. 6), even though GSH was reduced by both inhibitors in STHdhQ7/Q7 cells to the levels as seen in the STHdhQ111/Q111 cells. HCA interacts with glutamate receptors in addition to glutamate transporters; therefore oxidative stress produced by HCA might in part be due to excitotoxicity (Flott-Rahmel et al., 1998) However, the fact that no increase in oxidative stress was seen in the Q7 cells makes this unlikely. Furthermore, no such cross-reactivity has been documented for sulfasalazine, which also causes increased oxidative stress in the Q111 cells.
The difference in basal ROS accumulation might be because in the basal state, the rate of ROS production in the STHdhQ111/Q111 cells is greater than in the STHdhQ7/Q7 cells and outstrips the ability of STHdhQ111/Q111 cells to generate GSH. Even with inhibition of xCT, STHdhQ7/Q7 cells are still resistant to accumulating ROS, but STHdhQ111/Q111 cells show a marked increase in ROS accumulation. These results show that system xc- is crucial for maintaining GSH within the STHdh cell lines. The difference in the basal ROS levels and response to xCT inhibition is most likely due to increased production of ROS in the STHdhQ111/Q111, evident in Fig. 6A, and possibly due to increased production by NAPDH oxidase (Valencia et al., 2013), overall making the STHdhQ111/Q111 more susceptible to limitations in the production of GSH. From these data it is possible to speculate that neuronal injury in HD might be due to two hits—1) a limitation of GSH production caused by impaired function and expression of transporters that supply the rate-limiting substrate for GSH production, cystine/cysteine and perhaps deficiency in other mechanisms of antioxidant defense, such as PGC1α(Cui et al., 2006; St-Pierre et al., 2006; Tsunemi et al., 2012; Weydt et al., 2006), and 2) an increase in ROS production (Valencia et al., 2013; 2012). In both STHdh cell lines the application of electrophilic agent tBHQ increased GSH levels indicating that the mechanism for producing GSH in response to oxidative stress is still intact in the STHdhQ111/Q111 cells.
We also studied the effect of a GSH depleting agent, DEM, on STHdh cell survival. GSH concentrations represent steady state levels, and thus reflect a balance between synthesis and utilization. We found that STHdhQ111/Q111 cells were more sensitive to the toxic effects of DEM (Fig. 7). This increased sensitivity of the STHdhQ111/Q111 cells is likely due to the decrease in basal levels of GSH compared to STHdhQ7/Q7 cells, possibly caused by an impaired capacity to synthesize GSH by decreased basal expression of xCT which supplies the rate limiting precursor, cystine/cysteine. Previous studies have shown a decreased expression of GSH synthetic enzymes that may contribute to the decreased GSH levels (Ribeiro et al., 2012). In addition, the lower GSH levels might reflect increased utilization, for example, if STHdhQ111/Q111 cells had increased levels of oxidative stress and demands on antioxidant systems that consume GSH. These experiments demonstrate the importance of system xc- expression in regulating GSH levels and ultimately STHdh cell survival.
Global transcriptional dysregulation has been widely documented in HD (Cha, 2000), and specifically in the STHdhQ111/Q111 cells (Lee et al., 2007) and in the R6/2 mouse model (Crocker et al., 2006). We observed a decrease in xCT mRNA levels in both the STHdhQ111/Q111 cells and the R6/2 mice (Fig. 5), which is consistent with a transcriptional defect. xCT has two known transcription factors ATF4 and Nrf2 (Sasaki et al., 2002; Sato et al., 2004). ATF4 has been shown to regulate the basal expression levels of xCT (Lewerenz and Maher, 2009). ATF4 is the master regulator of the ISR, and expression of ATF4 is translationally controlled by phosphorylation of eIF2αat serine 51 (Kilberg et al., 2009). Interestingly, in some cells, knockdown of ATF4 has shown to be protective against oxidative stress (Lange et al., 2008), indicating a complex relationship between ATF4 expression and cellular redox status. Recently, another study using an overexpression model for Huntington's disease in vitro found phospho-eIF2α levels are upregulated in HD (Leitman et al., 2013). The ISR involves genes mediating amino acid transport, glutathione biosynthesis, and resistance to oxidative stress, all of which are thought to be impaired in HD (Faideau et al., 2010; Klepac et al., 2007; Valencia et al., 2012). Further studies need to be performed to determine the role of Nrf2 and ATF4 regulation of xCT in HD. Our results suggest that there is a transcriptional defect in xCT leading to an increase in oxidative stress in STHdhQ111/Q111 cells and one possible cause is a global defect in expression of ISR genes.
Highlights.
Na+-independent glutamate uptake is decreased in STHdhQ111/Q111 cells.
xCT mRNA and protein levels are decreased in STHdhQ111/Q111 cells.
xCT mRNA and protein are decreased in the R6/2 striatum at 6 weeks.
ROS levels are increased and GSH levels are decreased in STHdhQ111/Q111 cells.
Acknowledgments
This work was supported by the Hereditary Disease Foundation, the Harvard NeuroDiscovery Center, National Institutes of Health grants P30 HD018655, R01 NS066019 (P.R.) and R01 EY014560 (S.S.), and Boehringer Ingelheim Fonds (E.B.). We thank Dr. Marcy MacDonald at Massachusetts General Hospital for the gift of the STHdh cells, and Dr. Kush Kapur for advice regarding statistical treatment of the data.
Footnotes
Conflict of interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- The Huntington's Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-e. [DOI] [PubMed] [Google Scholar]
- Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, Swanson RA. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci. 2006;9:119–126. doi: 10.1038/nn1609. [DOI] [PubMed] [Google Scholar]
- Back SA, Gan X, Li Y, Rosenberg PA, Volpe JJ. Maturation-dependent vulnerability of oligodendrocytes to oxidative stress-induced death caused by glutathione depletion. J Neurosci. 1998;18:6241–6253. doi: 10.1523/JNEUROSCI.18-16-06241.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannai S. Exchange of cystine and glutamate across plasma membrane of human fibroblasts. J Biol Chem. 1986;261:2256–2263. [PubMed] [Google Scholar]
- Bannai S, Kitamura E. Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J Biol Chem. 1980;255:2372–2376. [PubMed] [Google Scholar]
- Bennett EJ, Shaler TA, Woodman B, Ryu KY, Zaitseva TS, Becker CH, Bates GP, Schulman H, Kopito RR. Global changes to the ubiquitin system in Huntington's disease. Nature. 2007;448:704–708. doi: 10.1038/nature06022. [DOI] [PubMed] [Google Scholar]
- Bridges CC, Kekuda R, Wang H, Prasad PD, Mehta P, Huang W, Smith SB, Ganapathy V. Structure, function, and regulation of human cystine/glutamate transporter in retinal pigment epithelial cells. Invest Ophthalmol Vis Sci. 2001;42:47–54. [PubMed] [Google Scholar]
- Bridges RJ, Natale NR, Patel SA. System xc cystine/glutamate antiporter: an update on molecular pharmacology and roles within the CNS. British Journal of Pharmacology. 2012;165:20–34. doi: 10.1111/j.1476-5381.2011.01480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne SE. Mitochondria and Huntington's disease pathogenesis: insight from genetic and chemical models. Annals of the New York Academy of Sciences. 2008;1147:358–382. doi: 10.1196/annals.1427.018. [DOI] [PubMed] [Google Scholar]
- Browne SE, Beal MF. Oxidative damage in Huntington's disease pathogenesis. Antioxid Redox Signal. 2006;8:2061–2073. doi: 10.1089/ars.2006.8.2061. [DOI] [PubMed] [Google Scholar]
- Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, Bird ED, Beal MF. Oxidative damage and metabolic dysfunction in Huntington's disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–653. doi: 10.1002/ana.410410514. [DOI] [PubMed] [Google Scholar]
- Burdo J, Dargusch R, Schubert D. Distribution of the cystine/glutamate antiporter system xc- in the brain, kidney, and duodenum. J Histochem Cytochem. 2006;54:549–557. doi: 10.1369/jhc.5A6840.2006. [DOI] [PubMed] [Google Scholar]
- Cha JHJ. Transcriptional signatures in Huntington's disease. Progress in Neurobiology. 2007;83:228–248. doi: 10.1016/j.pneurobio.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JH. Transcriptional dysregulation in Huntington's disease. Trends Neurosci. 2000;23:387–392. doi: 10.1016/s0166-2236(00)01609-x. [DOI] [PubMed] [Google Scholar]
- Chen CM. Mitochondrial dysfunction, metabolic deficits, and increased oxidative stress in Huntington's disease. Chang Gung Med J. 2011;34:135–152. [PubMed] [Google Scholar]
- Choo YS, Mao Z, Johnson GVW, Lesort M. Increased glutathione levels in cortical and striatal mitochondria of the R6/2 Huntington's disease mouse model. Neuroscience Letters. 2005;386:63–68. doi: 10.1016/j.neulet.2005.05.065. [DOI] [PubMed] [Google Scholar]
- Chung WJ, Lyons SA, Nelson GM, Hamza H, Gladson CL, Gillespie GY, Sontheimer H. Inhibition of cystine uptake disrupts the growth of primary brain tumors. Journal of Neuroscience. 2005;25:7101–7110. doi: 10.1523/JNEUROSCI.5258-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- Crocker SF, Costain WJ, Robertson HA. DNA microarray analysis of striatal gene expression in symptomatic transgenic Huntington's mice (R6/2) reveals neuroinflammation and insulin associations. Brain Research. 2006;1088:176–186. doi: 10.1016/j.brainres.2006.02.102. [DOI] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional Repression of PGC-1α by Mutant Huntingtin Leads to Mitochondrial Dysfunction and Neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Custer SK, Garden GA, Gill N, Rueb U, Libby RT, Schultz C, Guyenet SJ, Deller T, Westrum LE, Sopher BL, La Spada AR. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci. 2006;9:1302–1311. doi: 10.1038/nn1750. [DOI] [PubMed] [Google Scholar]
- DiFiglia M. Excitotoxic injury of the neostriatum: a model for Huntington's disease. Trends Neurosci. 1990;13:286–289. doi: 10.1016/0166-2236(90)90111-m. [DOI] [PubMed] [Google Scholar]
- Dringen R. Glutathione metabolism and oxidative stress in neurodegeneration. Eur J Biochem. 2000;267:4903. doi: 10.1046/j.1432-1327.2000.01651.x. [DOI] [PubMed] [Google Scholar]
- Dringen R, Hirrlinger J. Glutathione pathways in the brain. Biol Chem. 2003;384:505–516. doi: 10.1515/BC.2003.059. [DOI] [PubMed] [Google Scholar]
- Dun Y, Mysona B, Van Ells T, Amarnath L, Ola MS, Ganapathy V, Smith SB. Expression of the cystine-glutamate exchanger (xc-) in retinal ganglion cells and regulation by nitric oxide and oxidative stress. Cell Tissue Res. 2006;324:189–202. doi: 10.1007/s00441-005-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faideau M, Kim J, Cormier K, Gilmore R, Welch M, Auregan G, Dufour N, Guillermier M, Brouillet E, Hantraye P, Déglon N, Ferrante RJ, Bonvento G. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington's disease subjects. Human Molecular Genetics. 2010;19:3053–3067. doi: 10.1093/hmg/ddq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan MMY, Raymond LA. N-methyl-D-aspartate (NMDA) receptor function and excitotoxicity in Huntington's disease. Progress in Neurobiology. 2007;81:272–293. doi: 10.1016/j.pneurobio.2006.11.003. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Andreassen OA, Dedeoglu A, Ferrante KL, Jenkins BG, Hersch SM, Beal MF. Therapeutic effects of coenzyme Q10 and remacemide in transgenic mouse models of Huntington's disease. Journal of Neuroscience. 2002;22:1592–1599. doi: 10.1523/JNEUROSCI.22-05-01592.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flott-Rahmel B, Schürmann M, Schluff P, Fingerhut R, Musshoff U, Fowler B, Ullrich K. Homocysteic and homocysteine sulphinic acid exhibit excitotoxicity in organotypic cultures from rat brain. Eur J Pediatr. 1998;157(Suppl 2):S112–7. doi: 10.1007/pl00014291. [DOI] [PubMed] [Google Scholar]
- Fox JH, Barber DS, Singh B, Zucker B, Swindell MK, Norflus F, Buzescu R, Chopra R, Ferrante RJ, Kazantsev A, Hersch SM. Cystamine increases L-cysteine levels in Huntington's disease transgenic mouse brain and in a PC12 model of polyglutamine aggregation. Journal of Neurochemistry. 2004;91:413–422. doi: 10.1111/j.1471-4159.2004.02726.x. [DOI] [PubMed] [Google Scholar]
- Gochenauer GE, Robinson MB. Dibutyryl-cAMP (dbcAMP) up-regulates astrocytic chloride-dependent L-[3H]glutamate transport and expression of both system xc- subunits. Journal of Neurochemistry. 2001;78:276–286. doi: 10.1046/j.1471-4159.2001.00385.x. [DOI] [PubMed] [Google Scholar]
- Gomez GT, Hu H, McCaw EA, Denovan-Wright EM. Brain-specific factors in combination with mutant huntingtin induce gene-specific transcriptional dysregulation. Mol Cell Neurosci. 2006;31:661–675. doi: 10.1016/j.mcn.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Kalivas PW. The glutamate homeostasis hypothesis of addiction. Nature Publishing Group. 2009;10:561–572. doi: 10.1038/nrn2515. [DOI] [PubMed] [Google Scholar]
- Kilberg MS, Shan J, Su N. ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol Metab. 2009;20:436–443. doi: 10.1016/j.tem.2009.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klepac N, Relja M, Klepac R, Hećimović S, Babić T, Trkulja V. Oxidative stress parameters in plasma of Huntington“s disease patients, asymptomatic Huntington”s disease gene carriers and healthy subjects : a cross-sectional study. J Neurol. 2007;254:1676–1683. doi: 10.1007/s00415-007-0611-y. [DOI] [PubMed] [Google Scholar]
- Kraft AD, Johnson DA, Johnson JA. Nuclear factor E2-related factor 2-dependent antioxidant response element activation by tert-butylhydroquinone and sulforaphane occurring preferentially in astrocytes conditions neurons against oxidative insult. Journal of Neuroscience. 2004;24:1101–1112. doi: 10.1523/JNEUROSCI.3817-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange PS, Chavez JC, Pinto JT, Coppola G, Sun CW, Townes TM, Geschwind DH, Ratan RR. ATF4 is an oxidative stress-inducible, prodeath transcription factor in neurons in vitro and in vivo. J Exp Med. 2008;205:1227–1242. doi: 10.1084/jem.20071460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Ivanova EV, Seong IS, Cashorali T, Kohane I, Gusella JF, MacDonald ME. Unbiased gene expression analysis implicates the huntingtin polyglutamine tract in extra-mitochondrial energy metabolism. PLoS Genet. 2007;3:e135. doi: 10.1371/journal.pgen.0030135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitman J, Ulrich Hartl F, Lederkremer GZ. Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat Commun. 2013;4:2753. doi: 10.1038/ncomms3753. [DOI] [PubMed] [Google Scholar]
- Lewerenz J, Hewett S, Huang Y, Lambros M, Gout PW, Kalivas P, Massie A, Smolders I, Methner A, Pergande M, Smith SB, Ganapathy V, Maher P. The Cystine/Glutamate Antiporter System xc- in Health and Disease: From Molecular Mechanisms to Novel Therapeutic Opportunities. Antioxid Redox Signal. 2012 doi: 10.1089/ars.2011.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewerenz J, Klein M, Methner A. Cooperative action of glutamate transporters and cystine/glutamate antiporter system Xc- protects from oxidative glutamate toxicity. Journal of Neurochemistry. 2006;98:916–925. doi: 10.1111/j.1471-4159.2006.03921.x. [DOI] [PubMed] [Google Scholar]
- Lewerenz J, Maher P. Basal levels of eIF2α phosphorylation determine cellular antioxidant status by regulating ATF4 and xCT expression. J Biol Chem. 2009;284:1106–1115. doi: 10.1074/jbc.M807325200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Valencia A, Sapp E, Masso N, Alexander J, Reeves P, Kegel KB, Aronin N, DiFiglia M. Aberrant Rab11-dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington's disease. Journal of Neuroscience. 2010;30:4552–4561. doi: 10.1523/JNEUROSCI.5865-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–463. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- McBean GJ. Cerebral cystine uptake: a tale of two transporters. Trends Pharmacol Sci. 2002;23:299–302. doi: 10.1016/s0165-6147(02)02060-6. [DOI] [PubMed] [Google Scholar]
- Miller BR, Dorner JL, Shou M, Sari Y, Barton SJ, Sengelaub DR, Kennedy RT, Rebec GV. Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington's disease phenotype in the R6/2 mouse. Neuroscience. 2008;153:329–337. doi: 10.1016/j.neuroscience.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Schnaar RL, Coyle JT. Immature cortical neurons are uniquely sensitive to glutamate toxicity by inhibition of cystine uptake. FASEB J. 1990;4:1624–1633. [PubMed] [Google Scholar]
- Petersén A, Hansson O, Puschban Z, Sapp E, Romero N, Castilho RF, Sulzer D, Rice M, DiFiglia M, Przedborski S, Brundin P. Mice transgenic for exon 1 of the Huntington's disease gene display reduced striatal sensitivity to neurotoxicity induced by dopamine and 6-hydroxydopamine. Eur J Neurosci. 2001;14:1425–1435. doi: 10.1046/j.0953-816x.2001.01765.x. [DOI] [PubMed] [Google Scholar]
- Petr GT, Bakradze E, Frederick NM, Wang J, Armsen W, Aizenman E, Rosenberg PA. Glutamate transporter expression and function in a striatal neuronal model of Huntington's disease. Neurochemistry International. 2013;62:973–981. doi: 10.1016/j.neuint.2013.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polidori MC, Mecocci P, Browne SE, Senin U, Beal MF. Oxidative damage to mitochondrial DNA in Huntington's disease parietal cortex. Neuroscience Letters. 1999;272:53–56. doi: 10.1016/s0304-3940(99)00578-9. [DOI] [PubMed] [Google Scholar]
- Rawlins M. Huntington's disease out of the closet? Lancet. 2010;376:1372–1373. doi: 10.1016/S0140-6736(10)60974-9. [DOI] [PubMed] [Google Scholar]
- Raymond LA, André VM, Cepeda C, Gladding CM, Milnerwood AJ, Levine MS. Pathophysiology of Huntington's disease: time-dependent alterations in synaptic and receptor function. Neuroscience. 2011;198:252–273. doi: 10.1016/j.neuroscience.2011.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro M, Rosenstock TR, Cunha-Oliveira T, Ferreira IL, Oliveira CR, Rego AC. Glutathione redox cycle dysregulation in Huntington's disease knock-in striatal cells. Free Radical Biology and Medicine. 2012;53:1857–1867. doi: 10.1016/j.freeradbiomed.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Aizenman E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex. Neuroscience Letters. 1989;103:162–168. doi: 10.1016/0304-3940(89)90569-7. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Amin S, Leitner M. Glutamate uptake disguises neurotoxic potency of glutamate agonists in cerebral cortex in dissociated cell culture. J Neurosci. 1992;12:56–61. doi: 10.1523/JNEUROSCI.12-01-00056.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki H, Sato H, Kuriyama-Matsumura K, Sato K, Maebara K, Wang H, Tamba M, Itoh K, Yamamoto M, Bannai S. Electrophile response element-mediated induction of the cystine/glutamate exchange transporter gene expression. J Biol Chem. 2002;277:44765–44771. doi: 10.1074/jbc.M208704200. [DOI] [PubMed] [Google Scholar]
- Sato H, Nomura S, Maebara K, Sato K, Tamba M, Bannai S. Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochemical and Biophysical Research Communications. 2004;325:109–116. doi: 10.1016/j.bbrc.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455–11458. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- Seib TM, Patel SA, Bridges RJ. Regulation of the system x(C)-cystine/glutamate exchanger by intracellular glutathione levels in rat astrocyte primary cultures. Glia. 2011;59:1387–1401. doi: 10.1002/glia.21176. [DOI] [PubMed] [Google Scholar]
- Shih AY, Erb H, Sun X, Toda S, Kalivas PW, Murphy TH. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. Journal of Neuroscience. 2006;26:10514–10523. doi: 10.1523/JNEUROSCI.3178-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione: applications to mammalian blood and other tissues. Anal Biochem. 1969;27:502–522. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- Tkác I, Dubinsky JM, Keene CD, Gruetter R, Low WC. Neurochemical changes in Huntington R6/2 mouse striatum detected by in vivo 1H NMR spectroscopy. Journal of Neurochemistry. 2007;100:1397–1406. doi: 10.1111/j.1471-4159.2006.04323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trettel F, Rigamonti D, Hilditch-Maguire P, Wheeler VC, Sharp AH, Persichetti F, Cattaneo E, MacDonald ME. Dominant phenotypes produced by the HD mutation in STHdh(Q111) striatal cells. Human Molecular Genetics. 2000;9:2799–2809. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RAV, Lazarowski ER, Damian VA, Masliah E, La Spada AR. PGC-1 α rescues Huntington's disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012;4:142r97. doi: 10.1126/scitranslmed.3003799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia A, Sapp E, Kimm JS, McClory H, Reeves PB, Alexander J, Ansong KA, Masso N, Frosch MP, Kegel KB, Li X, DiFiglia M. Elevated NADPH oxidase activity contributes to oxidative stress and cell death in Huntington's disease. Human Molecular Genetics. 2013;22:1112–1131. doi: 10.1093/hmg/dds516. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Valencia A, Sapp E, Reeves PB, Alexander J, Masso N, Li X, Kegel KB, DiFiglia M. Reagents that block neuronal death from Huntington's disease also curb oxidative stress. NeuroReport. 2012;23:10–15. doi: 10.1097/WNR.0b013e32834d92e6. [DOI] [PubMed] [Google Scholar]
- Weber CA, Duncan CA, Lyons MJ, Jenkinson SG. Depletion of tissue glutathione with diethyl maleate enhances hyperbaric oxygen toxicity. Am J Physiol. 1990;258:L308–12. doi: 10.1152/ajplung.1990.258.6.L308. [DOI] [PubMed] [Google Scholar]
- Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, Gilbert ML, Morton GJ, Bammler TK, Strand AD, Cui L, Beyer RP, Easley CN, Smith AC, Krainc D, Luquet S, Sweet IR, Schwartz MW, La Spada AR. Thermoregulatory and metabolic defects in Huntington“s disease transgenic mice implicate PGC-1α in Huntington”s disease neurodegeneration. Cell Metabolism. 2006;4:349–362. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Wheeler VC, White JK, Gutekunst CA, Vrbanac V, Weaver M, Li XJ, Li SH, Yi H, Vonsattel JP, Gusella JF, Hersch S, Auerbach W, Joyner AL, MacDonald ME. Long glutamine tracts cause nuclear localization of a novel form of huntingtin in medium spiny striatal neurons in HdhQ92 and HdhQ111 knock-in mice. Human Molecular Genetics. 2000;9:503–513. doi: 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]
- Wüllner U, Seyfried J, Groscurth P, Beinroth S, Winter S, Gleichmann M, Heneka M, Löschmann P, Schulz JB, Weller M, Klockgether T. Glutathione depletion and neuronal cell death: the role of reactive oxygen intermediates and mitochondrial function. Brain Research. 1999;826:53–62. doi: 10.1016/s0006-8993(99)01228-7. [DOI] [PubMed] [Google Scholar]
- Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Research. 1999;59:4383–4391. [PubMed] [Google Scholar]
- Zerangue N, Kavanaugh MP. Interaction of L-cysteine with a human excitatory amino acid transporter. J Physiol (Lond) 1996;493(Pt 2):419–423. doi: 10.1113/jphysiol.1996.sp021393. [DOI] [PMC free article] [PubMed] [Google Scholar]