

Abstract
Rationale
Aortic stiffening commonly occurs in hypertension and further elevates systolic pressure. Hypertension is also associated with vascular inflammation and increased mechanical stretch. The interplay between inflammation, mechanical stretch and aortic stiffening in hypertension remains undefined.
Objective
To determine the role of inflammation and mechanical stretch in aortic stiffening.
Methods and Results
Chronic angiotensin II infusion caused marked aortic adventitial collagen deposition, as quantified by Masson’s Trichrome Blue staining and biochemically by hydroxyproline content, in wild-type (WT) but not in Recombination Activation Gene-1 deficient (RAG-1−/−) mice. Aortic compliance, defined by ex-vivo measurements of stress-strain curves, was reduced by chronic angiotensin II infusion in WT mice (p<0.01) but not in RAG-1−/− mice (p<0.05). Adoptive transfer of T cells to RAG-1−/− mice restored aortic collagen deposition and stiffness to values observed in WT mice. Mice lacking the T cell derived cytokine IL-17a were also protected against aortic stiffening. In additional studies, we found that blood pressure normalization by treatment with hydralazine and hydrochlorothiazide prevented angiotensin II-induced vascular T cell infiltration, aortic stiffening and collagen deposition. Finally, we found that mechanical stretch induces expression of collagen 1α1, 3α1 and 5a1 in cultured aortic fibroblasts in a p38 MAP kinase-dependent fashion, and that inhibition of p38 prevented angiotensin II-induced aortic stiffening in vivo. IL-17a also induced collagen 3a1 expression via activation of p38 MAP kinase.
Conclusions
Our data define a pathway in which inflammation and mechanical stretch lead to vascular inflammation that promotes collagen deposition. The resultant increase in aortic stiffness likely further worsens systolic hypertension and its attendant end-organ damage.
Keywords: Inflammation, mechanical stretch, collagen deposition, aortic stiffening, vascular remodeling
INTRODUCTION
The capacitance property of the aorta normally blunts blood pressure elevation during systole and maintains diastolic pressure and tissue perfusion during diastole. Loss of this Windkessel function of the proximal aorta causes an increase in systolic pressure, a decline in diastolic pressure and an increase in pulse wave velocity.1 The augmentation of systolic pressure caused by aortic stiffening increases the incidence of stroke, renal failure and myocardial infarction. Aortic stiffening is associated with aging, insulin resistance, diabetes, atherosclerosis and hypertriglyceridemia.2–5 Importantly, hypertension per se causes aortic stiffening, leading to progressive elevation of systolic pressure. Thus, aortic stiffening not only contributes to hypertension but also portends cardiovascular morbidity and mortality.6, 7
The precise mechanisms underlying aortic stiffening remain undefined. Clinical studies suggest that inflammation and arterial stiffness are related.8–11 Patients with inflammatory diseases such as lupus erythematosus, rheumatoid arthritis and psoriasis have increased pulse wave velocity.12–14 Data from our laboratory and others have shown that T cells and T cell-derived cytokines are important in development of hypertension.15, 16 We have previously found that Recombination Activation Gene-1 deficient (RAG-1−/−) mice develop blunted hypertension in response to angiotensin II, DOCA-salt challenge and norepinephrine.17 The RAG-1 gene encodes a gene responsible for recombining the variable regions of the T cell receptor and immunoglobulins and in its absence mice fail to develop either B cells or T cells. Adoptive transfer of T cells restores hypertension in RAG-1−/− mice, indicating a critical role of these cells. Recently, deletion of the RAG-1 gene in Dahl Salt-sensitive rats has been shown to lower blood pressure and to reduce renal injury upon salt feeding.18 Other studies have shown that T cell-derived cytokines also contribute to hypertension, likely by promoting vascular dysfunction and renal injury.16, 19, 20 One such cytokine is interleukin 17a (IL-17a), which is produced by a subset of pro-inflammatory CD4+ T cells referred to as TH17 cells. Mice lacking IL-17a have blunted hypertension and reduced aortic production of reactive oxygen species (ROS) following angiotensin II infusion. Recent studies have also shown that administration of IL-17a to mice causes hypertension and reduces endothelium-dependent vasodilatation, at least in part by activating Rho kinase.21 IL-17a also promotes collagen deposition and contributes to fibrosis in other tissues and conditions.22–24
In the present study we sought to examine mechanisms responsible for aortic stiffening in hypertension. In particular we examined the role of adaptive immunity mediated by T cells and their cytokines and the direct effects of mechanical stimulation by blood pressure lowering and by exposing aortic fibroblasts to hypertensive levels of stretch. We identify a novel pathway that promotes striking aortic adventitial collagen deposition and vascular stiffening.
METHODS
Animals
Male wild type, RAG-1−/−, CD4−/−, CD8−/− and IL-17a−/− mice were studied at 3 months of age. Hypertension was induced by infusion of angiotensin II (490 ng/kg/min) via osmotic minipumps for two weeks unless otherwise indicated. For measurement of blood pressure, mice were implanted with telemetry units and ten days later osmotic minipumps were placed. Blood pressure was recorded for 10 minutes every hour from three days prior to osmotic minipump implantation until the end of angiotensin II infusion at Day 14. For studies of vascular stiffness, the descending thoracic aorta was mounted on cannulas at the in situ length in calcium-free buffer. Intraluminal pressure was increased in a step-wise fashion while video microscopy was used to follow outer and inner diameter. Diameters were recorded with every increment of 25 mmHg from 0 to 200 mmHg. Stress-strain relationships were calculated as described by Baumbach et al.25 In some experiments, RAG-1−/− mice underwent adoptive transfer of T cells or their subsets as described previously.26 The Institutional Animal Care and Use Committee approved all experimental protocols.
Flow cytometry
Single cell suspensions were prepared from aortas as previously described.27 Leukocytes were further isolated using a Percoll gradient. The antibodies used were: FITC anti-CD45; PerCP anti-CD45; PE anti-CD3e; FITC anti-CD3e; APC anti-CD3; APC anti-CD4; PerCP anti-CD8a; PE-Cy7 anti-CD8a.
Measurement of aortic collagen and elastin
Aortic collagen was visualized by Masson’s Trichrome staining and quantified by measurement of tissue hydroxyproline as described previously.28 Elastin was visualized by Verhoeff Van Gieson staining and quantified as previously described, 29, 30 with minor modifications as outlined in the supplemental data section.
Cell culture
Low passage mouse aortic fibroblasts were studied at 90% confluence. The media was changed from 5% to 0.5% FBS for 24 hours prior to experiments. Cells were exposed to either cyclical stretch or to IL-17a (100 ng/mL) for 36 hours.
Real-time PCR and Western Blotting
RNA was extracted from aortas or cultured fibroblasts using the Qiagen RNeasy mini kit. PCR array was performed to screen for matrix and inflammatory genes and real-time PCR was employed to examine selected fibrotic genes. Western blotting was performed using specific rabbit anti-mouse monoclonal primary antibodies and goat anti-rabbit polyclonal secondary antibodies. Western blots were quantified by densitometry.
Statistical analysis
Data in the manuscript are expressed as the mean ± SEM. Comparisons of blood pressure over time were made using one-way ANOVA for repeated measures, followed by a Student Newman Keuls post hoc test when significance was indicated. Compliance curves and stress-strain curves were also compared using ANOVA for repeated measures. To compare the effect of angiotensin II on parameters other than blood pressure, two-way ANOVA was employed, as indicated. The effects of adoptive transfer of pan T cells or antihypertensive treatment on collagen deposition or aortic inflammation were compared using one-way ANOVA. P values are reported in the figures.
A detailed description of the materials and methods can be found in the on-line supplement.
RESULTS
Effect of angiotensin II-induced hypertension on aortic collagen and elastin content: Role of T cells
Two matrix components that predominantly modulate tissue compliance are collagen and elastin. We performed initial studies to determine how hypertension affects these and to understand the role of T cells in this response. Using Masson’s Trichrome staining, we observed that collagen is predominantly localized to the adventitia of the aorta of sham infused mice. During angiotensin II infusion, a striking accumulation of collagen in the aortic adventitia occurred. The thickness of this adventitial collagen deposition was often equivalent to the media (Figure 1A and 1B). By planimetry, adventitial collagen area was increased from 3.8×104 μm2 to 13.6×104 μm2 by angiotensin II treatment. To directly quantify aortic collagen, we measured hydroxyproline content, which was 19 ± 7 ng/μg of total protein in WT mice and was doubled by chronic angiotensin II infusion (Figure 1C). Thus, by both histochemical staining and biochemical analysis, we found that angiotensin II-induced hypertension is associated with a 2–3 fold increase of aortic collagen content, predominantly localized to the adventitia.
Figure 1. Role of T cells in aortic collagen deposition in hypertension.
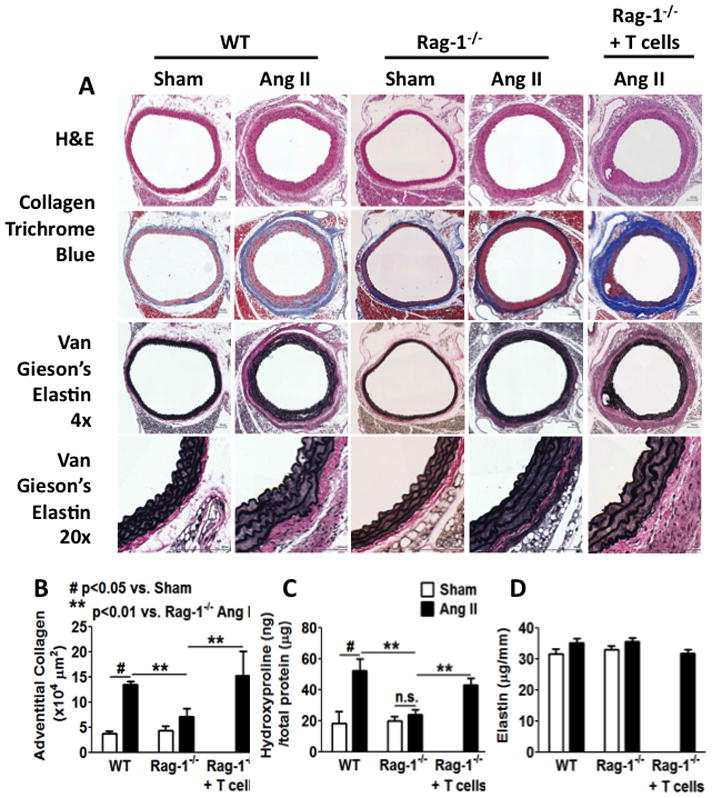
A, Effects of angiotensin II-induced hypertension on vascular collagen and elastin in WT and RAG-1−/− mice. Perfusion-fixed sections of the thoracic aortas were sectioned (6 mm) and stained with Hematoxylin and Eosin, Masson’s trichome and Van Gieson’s Elastica staining to highlight collagen (blue) and elastin (black). Scale bar indicates 100 μm. B, Adventitial collagen area was quantified by planimetry. C, Aortic collagen quantification by hydroxyproline assay. Isolated thoracic aortas were digested in 6 N HCl at 105 °C for 48 hours before hydroxyproline was quantified. D, Elastin was quantified biochemically by initial separation in 0.1 N NaOH at 90 °C for 30 min and subsequently by ninhydrin assay. Ang II, angiotensin II. Data were analyzed using two-way ANOVA, n=6–9.
We and others have established a role of T cells in hypertension.15,16 Moreover, the T cell derived cytokine IL-17 contributes to hypertension and has been shown to promote collagen deposition in experimental scleroderma.17 We therefore examined the role of T cells in aortic stiffening. As in previous studies, we found that the hypertensive response to angiotensin II is blunted in RAG-1−/− mice (Supplemental figure I A and B). In addition, the increase in aortic collagen content caused by angiotensin II, as detected by either Masson’s trichrome staining or by hydroxyproline assay, was blunted in RAG-1−/− mice (Figure 1A–C). Five weeks following adoptive transfer of pan T cells, flow cytometry documented a stable population of these cells in RAG-1−/− mice (Supplemental figure I C). In keeping with our prior studies, T cell adoptive transfer restored the hypertensive response to angiotensin II in RAG-1−/−mice (Supplemental figure I D and I E). Importantly, adoptive transfer of T cells to RAG-1−/− mice also restored the aortic collagen deposition caused by angiotensin II to levels observed in WT mice (Figure 1A–C). Despite these fibrotic changes in the aorta, we did not observe collagen deposition in the mesenteric arteries of WT or RAG-1−/− mice treated with angiotensin II (Supplemental figure II).
Van Gieson’s Elastica staining did not reveal a qualitative difference in elastin between sham or angiotensin II-treat mice or in RAG-1−/− mice (Figure 1A). Angiotensin II-induced hypertension was associated with hypertrophy of medial cells, causing a greater separation of elastin laminae. Biochemical analysis also did not reveal differences in the absolute amount of elastin between sham and angiotensin II-treated WT or RAG-1−/− mice, or an effect of T cell adoptive transfer (Figure 1D). When normalized to total protein, relative elastin levels were decreased by angiotensin II in both mouse strains (Supplemental figure I F), but this could be attributed to the striking increase in total aortic protein associated with angiotensin II-induced aortic hypertrophy (Supplemental figure I G).
Angiotensin II-induced hypertension causes dramatic changes in the expression of the aortic matrix genes
In addition to collagen and elastin, numerous other matrix proteins can influence vascular stiffness. We therefore performed a PCR array to characterize expression of 84 aortic matrix genes related to fibrosis and remodeling. In response to angiotensin II infusion, 17 out 84 genes were significantly upregulated while no genes were down regulated (Supplemental Table I). Among these, collagen 1a1 and MMP-2 were upregulated by more than 8 fold and collagen 3a1 by 7 fold. MMP-11, thrombospondin-1, thrombospondin-2 and secreted acidic cysteine rich glycoprotein were upregulated by 4–5 fold. We also found more than 3 fold upregulation for collagen 5a1 and MMP-14. Surprisingly, the potent pro-fibrotic cytokine, TGFβ1, was not altered in angiotensin II-induced hypertension.
Hypertension reduces aortic compliance – Role of T cells
Additional studies were performed to measure compliance of isolated segments of the descending thoracic aorta. Chronic angiotensin II infusion caused marked stiffening of the thoracic aorta in WT mice as evidenced by a downward shift of the compliance curve (Figure 2A) and a leftward shift of the stress-strain relationship (Figure 2B). In contrast, in RAG-1−/− mice, chronic angiotensin II infusion had essentially no effect on aortic stiffness. Adoptive transfer of T cells to RAG-1−/− mice restored the increase in aortic stiffening caused by angiotensin II (Figure 2C and 2D). Analysis of the changes in vascular inner and outer diameter showed that these increased in parallel in vessels from sham treated mice as intraluminal pressure was increased. In contrast, following angiotensin II infusion, the increase in outer diameter was constrained in aortas from WT mice, but not in aortas from RAG-1−/− mice (Figure 2E and 2F).
Figure 2. T cells mediate angiotensin II-induced aortic stiffening.

Freshly-isolated aortas were mounted on a myograph system in Ca2+-free buffer to determine pressure-diameter relationships. A–D, compliance curves and stress-strain relationships were constructed from inner diameter and outer diameter. E and F, inner and outer diameter of WT and RAG-1−/− mice measured at 25 mmHg step changes in pressure from 0–200 mmHg. Ang II, angiotensin II. Data were analyzed using one-way ANOVA with repeated measures, n=6–9.
To determine the relative contribution of T cell subtypes, we examined the effect of angiotensin II infusion in CD4−/− and CD8−/− mice. The increase in collagen caused by angiotensin II was blunted in mice lacking either T cell subtype, but was most striking in mice lacking CD8+ T cells as evidenced by both Masson’s trichrome staining and by hydroxyproline assay (Supplemental Figure III A–C). In addition, CD4−/− and CD8−/− mice were both partially protected against the development of aortic stiffening caused by angiotensin II (Supplemental Figure III D–G). In additional studies, we performed adoptive transfer of CD4+ T cells or CD8+ T cells to RAG-1−/− mice and then infused angiotensin II three weeks later. Neither cell type alone increased aortic stiffness or collagen content significantly (Supplemental Figure IV). These data indicate that both cell types are required and likely interact to mediate hypertension-related aortic stiffening.
We have previously shown that that mice lacking IL-17a are protected against the development of hypertension, aortic inflammation and angiotensin II-induced vascular oxidative stress.16 In the present study, we found that IL-17a−/− mice are also protected against aortic collagen deposition (Figure 3A–C) and aortic stiffening (Figure 3D and E) in response to chronic angiotensin II infusion.
Figure 3. Role of IL-17a in aortic stiffening.
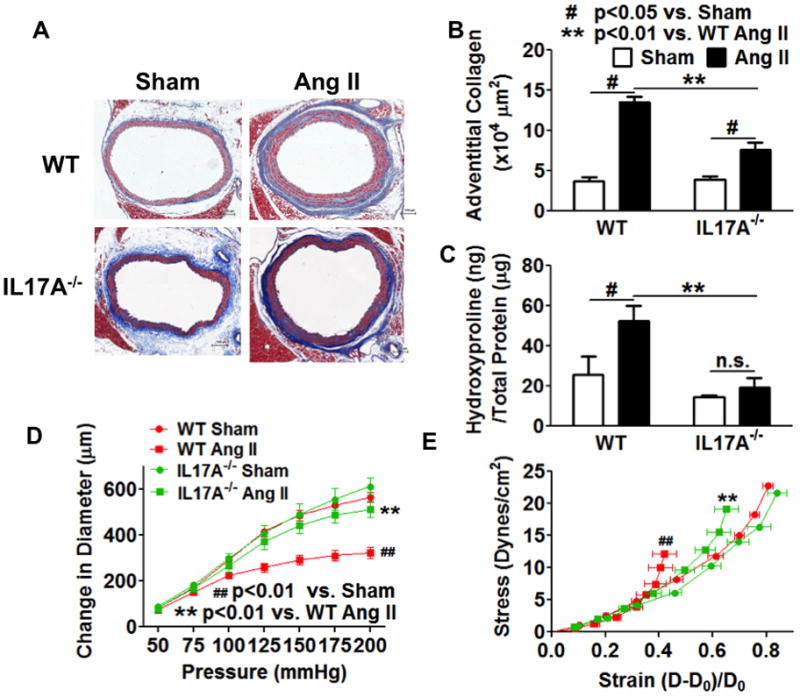
IL-17a deficient mice were infused with chronic angiotensin II (490ng/kg/min) for 14 days. A and B, the thoracic aortas were fix-perfused for Masson’s trichrome staining and quantified for adventitia collagen staining. Scale bar indicates 100 μm. C, segments of the thoracic aorta were digested in 6N HCl at 105 °C for 48 hours and measured for hydroxyproline concentration. Data analyzed using two-way ANOVA. D and E, compliance curves and stress-strain curves for IL-17a−/− mice. Ang II, angiotensin II. Data analyzed using one-way ANOVA with repeated measures, n=6–8.
We have previously shown that T cells play a role in models of hypertension other than angiotensin II infusion, including DOCA-salt hypertension and norepinephrine-induced hypertension.15, 17 To determine if the reduced adventitial collagen deposition and aortic stiffening in RAG-1−/− mice are specific to angiotensin II, we also studied deoxycorticosterone acetate (DOCA)-salt-induced hypertension, which is characterized by low levels of circulating angiotensin II. As shown in supplemental figure V, while WT mice developed marked collagen deposition and aortic stiffening in response to DOCA-salt challenge, RAG-1−/− mice were protected from these changes. These data suggest that T cells contribute to aortic stiffening in forms of hypertension other than that caused by angiotensin II.
Aortic stiffening is dependent on blood pressure elevation
In clinical studies, aortic stiffening is commonly associated with hypertension; however, the causal relationship remains unclear. To address the possible role of pressure elevation in the genesis of aortic stiffening, we normalized blood pressure in angiotensin II-infused mice by adding hydralazine (320 mg/L) and hydrochlorothiazide (60 mg/L) to the drinking water. As shown in Figure 4A and 4B, concurrent hydralazine and hydrochlorothiazide treatment completely prevented the elevation of blood pressure. This treatment also protected against adventitial collagen deposition (Figure 4C–E). More importantly, the shifts in compliance and stress-strain curves were prevented by this treatment regimen (Figure 4F and 4G). Interestingly, we found that hydralazine and hydrochlorothiazide also abrogated T cell infiltration and inflammation in the aortas of angiotensin II-infused mice (Figure 4H–L). Collectively, these data suggest that aortic stiffening and vascular inflammation are at least in part due to mechanical effects of hypertension.
Figure 4. Antihypertensive treatment (antiHBP) prevents collagen deposition, aortic stiffening and T cell infiltration.

Mice were treated with hydralazine and hydrochlorothiazide (320 mg/L and 60 mg/L in the drinking water) concurrently with angiotensin II infusion. Hyd, Hydralazine, HCTZ, hydrochlorothiazide. A and B, telemetry recording of blood pressures. D, day; N, night. The effects of blood pressure normalization on collagen depostion (C–E), aortic stiffening (F and G) and aorfic inflammatory cell infiltration (H–L) are shown. Scale bar indicates 100 μm. Ang II, angiotensin II. Data for blood pressure and aortic stiffness were analyzed using one-way ANOVA with repeated measurements (n=8). Collagen deposition and flow cytometry were analyzed using one-way ANOVA (n=8).
To determine whether normalization of blood pressure in established hypertension could reverse collagen deposition and aortic stiffening, hydralazine and hydrochlorothiazide treatment was administered from day 15 to day 28 during a 4-week angiotensin II infusion. Although this successfully normalized blood pressure during the last two weeks of angiotensin II infusion, it failed to reverse adventitial collagen deposition and aortic stiffening (Supplemental Figure VI). These data suggest that hypertension might cause irreversible large vessel fibrosis and remodeling.
Aortic fibroblasts express collagen in response to IL-17a and hypertensive mechanical stretch
The above experiments suggest that both inflammation and mechanical forces contribute to aortic stiffening. To differentiate between the direct effect of mechanical stretch and the inflammatory cytokine IL-17a, we performed additional studies using cell culture. Because we found aortic collagen deposition in hypertension occurs largely in the adventitia, we studied aortic fibroblasts, which represent the predominant cell type of the adventitia. We focused these studies on the collagen subtypes identified in the real-time PCR array. Fibroblasts were exposed to either 5% or 10% stretch, mimicking levels of mechanical stretch observed in the setting of normal and elevated blood pressures, or were exposed to IL-17a (100 ng/mL) for 36 hours. We found that while IL-17a had no effect on the mRNA expression of collagen 1a1, 10% mechanical stretch doubled expression of this subtype beyond that observed with 5% stretch (Table 1). Collagen 3a1 was increased by more than 4-fold by IL-17a and by more than 3-fold in response to 10% stretch. Collagen 5a1 mRNA expression was increased approximately 2-fold by IL-17a and by 10% stretch, but was not further increased by the combination of this cytokine and stretch. The addition of angiotensin II had minimal effect on these responses except for collagen 3a1, where it doubled the effect of 10% stretch. These experiments show that the hypertensive milieu, which includes increased vascular stretch, inflammatory cytokines such as IL-17a and angiotensin II interact to modulate aortic fibroblast collagen production.
Table 1.
Effects of IL-17a, angiotensin II and mechanical stretch on collagen expression by mouse aortic fibroblasts
| Treatment | Col 1a1 | Col 3a1 | Col 5a1 | Angiotensin II (100 nM)
|
||
|---|---|---|---|---|---|---|
| Col 1a1 | Col 3a1 | Col 5a1 | ||||
| Control | 1.00±0.05 | 1.00±0.34 | 1.00±0.12 | 1.03±0.02 | 1.59±0.08 | 1.24±0.25 |
| IL-17a (100 ng/mL) | 1.20±0.07 | 4.63±0.66 ** | 1.89±0.10 * | 1.12±0.12 | 4.28±0.33 ** | 1.74±0.20 |
| 5% Stretch | 1.33±0.25 | 1.98±0.20 | 1.39±0.21 | NA | NA | NA |
| 10% Stretch | 2.24±0.24** | 3.40±0.53 ** # | 2.45±0.37 ** | 2.80±0.34 ** | 7.50±1.43 ** | 2.68±0.62** |
| IL-17a (100 ng/mL) + 10% Stretch | 2.16±0.13* * | 3.67±0.41 ** | 2.32±0.43 ** | NA | NA | NA |
Primary mouse aortic fibroblasts were exposed to 5% or 10% cyclic stretch or exposed to IL-17a (100 ng/mL) in the presence or absence of angiotensin II (100 nM) for 36 hours. Relative quantity of mRNA was calculated based on the ΔΔCt value normalized to control. Data are expressed as mean ± SE. The effects of IL17a and mechanical stretch are analyzed with one-way ANOVA, while the effect of angiotensin II by two-way ANOVA (n=3–6),
p<0.05,
p<0.01 vs. control;
p<0.05 vs. angiotensin II. NA, not assessed.
Activation of p38 MAPK is required for collagen expression and aortic collagen deposition induced by stretch and inflammation in vitro and in vivo
p38 MAPK has been implicated in the induction of collagen expression and fibrosis in different hypertensive models.31–34 Interestingly, both 10% stretch and IL-17a at 100 ng/mL induced p38MAP kinase phosphorylation in aortic fibroblasts (Figure 5A–C). Moreover, inhibition of p38 MAP kinase with SB203580 prevented the increase in mRNA for collagens 1a1, 3a1 and 5a1 caused by stretch (Figure 5D–5F). SB203580 also inhibited IL-17a-induced expression of collagen 3a1 but not collagen 5a1 (Figure 5G–I). We also found that angiotensin II-induced hypertension increased p38 MAPK phosphorylation in thoracic aortas, and that this was prevented by the normalization of blood pressure (Figure 6A–C). To gain insight into the role of p38 MAPK in vivo, we treated mice with SB203580 (10 mg/kg/day, i.p.) concurrently with angiotensin II. SB203580 reduced the hypertension caused by angiotensin II (Figure 6D and E). More importantly, SB203580 prevented collagen deposition (Figure 6F–H) and prevented the shift in aortic compliance and stress strain relationships caused by angiotensin II (Figure 6I and J). These data show that p38 MAP kinase is responsive to stretch and inflammation, and thus plays a central role in mediating collagen expression in cultured fibroblasts and in inducing aortic stiffness in vivo.
Figure 5. Effect of cyclical stretch and IL-17a on aortic fibroblasts and role of p38 MAP kinase.
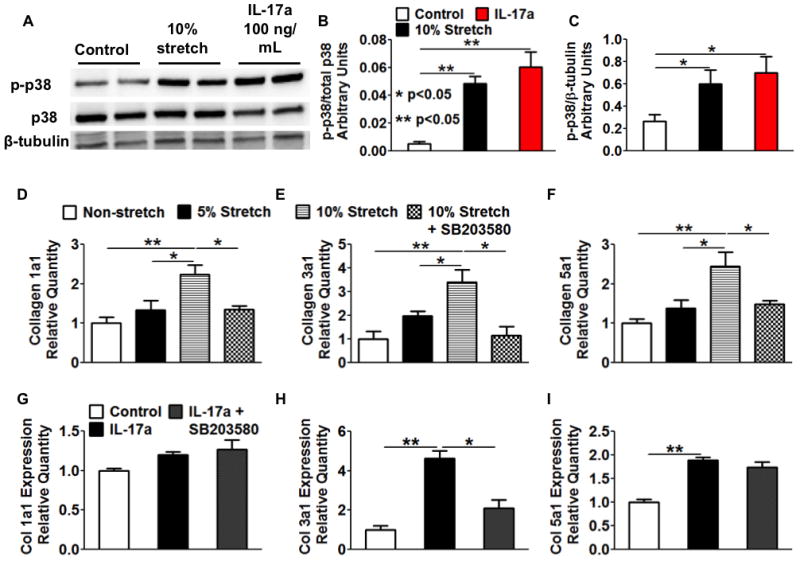
Mouse aortic fibrobasts were exposed to various degrees of cyclical stretch or the T cell cytokine IL-17a (100 ng/mL) in culture for 36 hours. A–C, both hypertensive mechanical stretch and IL-17a activated p38 MAPK in cultured mouse aortic fibroblasts. D–F, The effects of p38 inhibitor SB203580 (10 ng/mL) on mechanical stretch-induced expression of collagen subtypes in fibroblasts. G–I, The effects of p38 inhibition on IL-17a-induced collagen expression. Data analyzed using one-way ANOVA, n=3–6.
Figure 6. P38 MAP kinase mediates collagen deposition and aortic stiffening in angiotensin II-induced hypertension.
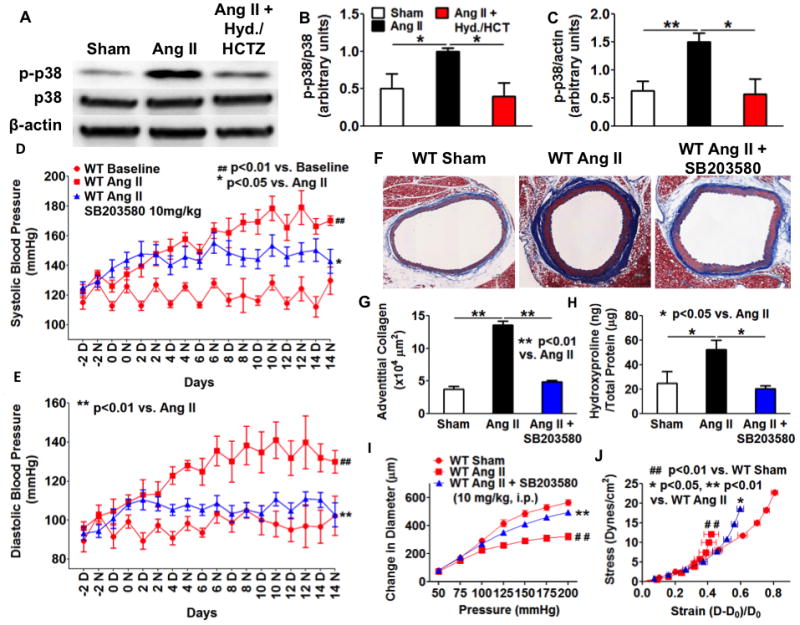
A–C, Normalization of blood pressure prevented the activation of p38 MAP kinase in angiotensin II-infused mouse aortas. Data analyzed with one-way ANOVA (n=4–6). D and E, Effect of SB203580 on blood pressure. D, day; N, night. F–H, Effect of SB203580 on aortic collagen deposition. Data analyzed using one-way ANOVA (n=6–8). Mice received intraperitoneal injections of SB203580 (10 mg/kg/day) during angiotensin II infusion. Scale bar indicates 100 μm. I and J, Effect of SB203580 on aortic stiffening. Ang II, angiotensin II. Blood pressure and aortic stiffness were analyzed using one-way ANOVA with repeated measures (n=6–8).
DISCUSSION
In the present study, we identified novel mechanisms of aortic stiffening associated with hypertension as delineated in the scheme shown in Figure 7. Our data indicate that elevation of blood pressure and the attendent increase in vascular stretch leads to an inflammatory process characterized by infiltration of T cells and activation of p38 MAP kinase. These events lead to a striking deposition of collagen in the aortic adventitia, with scattered amounts of collagen in the media. This is associated with a marked alteration in aortic complaince, characterized by a leftward shift in the stress strain relationship and a downward relationship between aortic distending pressure and diameter. In vivo, this increase in aortic stiffness leads to a loss of the Windkessel or capacitance function of the aorta, increasing systolic pressure and promoting hypertension-related end-organ damage.
Figure 7. Pathway showing interactions of mechanical stretch and inflammation in aortic stiffening.
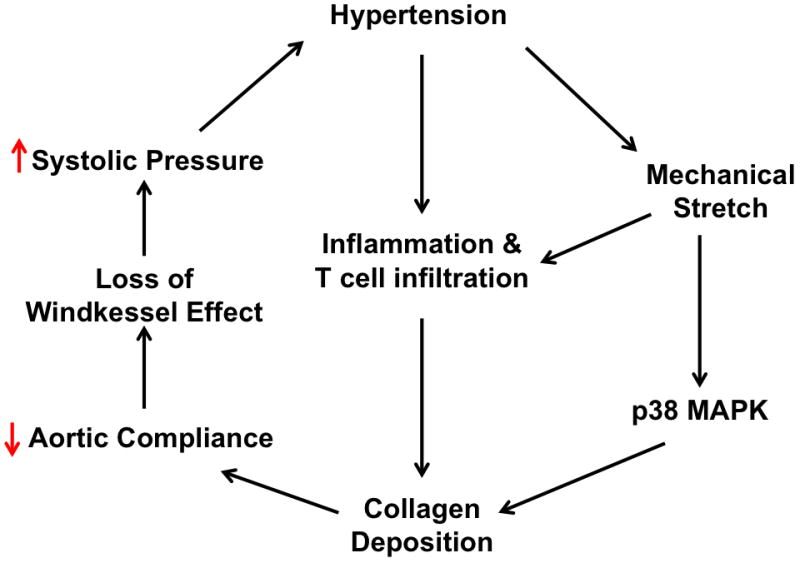
Increased mechanical stretch promotes perivascular T cell infiltration and activates p38 MAP kinase, leading to adventitial collagen deposition. This reduces aortic compliance and ultimately leads to loss of windkessel effect in large capacitance arteries such as the aorta.
Interestingly, we found that T cells and in particular the T cell-derived cytokine IL-17a contribute to aortic stiffening. We have previously shown that mice lacking IL-17a are protected against hypertension.16 More recently, IL-17 has been shown to increase Rho kinase-mediated phosphorylation of the endothelial nitric oxide synthase, to promote endothelial dysfunction and to elevate blood pressure when administered parenteraly.21 In keeping with a role of inflammation in collagen deposition, we found that RAG-1−/− mice and IL-17a−/− mice are protected against aortic stiffening and collagen depostion during angiotensin II infusion, and that adoptive transfer of T cells to RAG-1−/− mice restores these abnormalities. We cannot exclude a role of cytokines other than IL-17a. As an example, TNFα often acts synergistically with IL-17 to promote inflammatory responses. Recent studies showed that the TNFα antagoinist etanercept reduces pulse wave velocity and aortic stiffness in patients with rheumatoid arthritis.35, 36 We have also found that mice treated with the TNFα antagonist etanercept and mice lacking IL-17a are protected against these vascular alterations.15 Thus, it is possible that this and other cytokines act in concert with IL-17 to promote aortic stiffening.
It is likely that both CD4+ and CD8+ T cells contribute to aortic stiffening, as mice lacking either one alone had an intermediate phenotype. In particular, mice lacking CD8+ T cells had a reduction in aortic collagen accumulation caused by angiotensin II. Likewise, adoptive transfer of only CD4+ cells or only CD8+ T cells to RAG-1−/− mice failed to restore aortic stiffening in response to angiotensin II. While CD8+ T cells are generally considered cytotoxic, it is now clear that CD8+ T cells produce substantial amounts of cytokines and contribute to the inflammatory millieu.37, 38 In preliminary studies, we have found that angiotensin II infusion induces IL-17a production in several T cell subtypes in vivo.
Our data also indicate that the mechanical effect of increased blood pressure also contributes to aortic stiffening. Prevention of hypertension with hydralazine and hydrochlorothiazide prevented collagen deposition and the increase in aortic stiffness caused by chronic angiotensin II infusion. These data do not separate mechanical effects from inflammation, because blood pressure lowering also prevented aortic leukocyte iniltration. In keeping with this finding, there are numerous mechanisms by which mechanical stretch could affect vascular inflammation. As examples, increased cyclical strain, similar to that observed in hypertension, activates the NADPH oxidase and NF-κB, increases expression intracellular adhesion molecule-1 (ICAM1), and enhances adhesion of monocytes to the endothelium.39,40
Substantial research has focused on the role of TGFβ in tissue fibrosis and collagen deposition. In several preliminary studies, we found that mRNA for TGFβ1 is not increased in aortas from angiotensin II-treated mice nor in cultured fibroblasts exposed to stretch. This does not exclude the possibility that TGFβ activity is increased at a post-translational level in response to hypertension. Latent TGFβ is processed to its active form by components of the inflammatory milieu, including matrix metalloproteinases, reactive oxygen species, thrombospondin-1 and integrins.41,42 Related to this, our array analysis of the genes affected by hypertension provides insight. This analysis showed that hypertension led to a 9-fold increase in MMP-2 and a 5-fold increase in thrombospondin-1. These could contribute to post-translational activation of latent TGFβ in the absence of changes in its expression.
In an effort to separate the direct effects of stretch and inflammation, we exposed cultured murine aortic fibrobasts to either cyclical stetch or IL-17a. We selected fibroblasts because these represent the predominant cell in the adventital region where collagen deposition occurs. Our data indicate that both of these stimuli induce mRNA of various collagen subtypes, all of which could contribute to aortic stiffening. Our data also indicate that p38 MAPK likely plays an important role in collagen expression both in cultured fibroblasts and in vivo. Several studies has shown that inhibition of p38 blocks the pro-fibrotic effects of mechanical stretch both in vitro and in vivo. Park et al have shown that inhibition of p38 MAPK reduces cardiac and renal fibrosis in double transgenic rats harboring the human renin and angiotensinogen genes.32 Similarly p38 inhibition decreases cyclical stretch-induced collagen expression in isolated smooth muscle cells from WKY and SHR rats.34 Of note, p38 MAPK has been shown to bypass TGFβ signaling by transactivation of Smad-2/3 in mouse models of Marfan’s Syndrome and systemic inhibition of p38 blocks these effects. In addition, increased activation of p38 MAPK due to loss of MAPK phosphatase-1 activity induces expression of microRNA-21 (miR-21),43 a critical regulator of TGF-β signaling involved in renal and myocaridal fibrosis.44,45,46 MiR-21 inhibits Smad-7 and augments TGF-β signalling, enhancing its pro-fibrotic effects.47 Interestingly, miR-21 also contributes to renal fibrosis by regulating MMP9/TIMP-1. Thus, p38 MAPK plays a pivital role in the interaction between vascular inflammation and mechanical stretch.
It would be desirable to differentiate the causal relationship bewteen aortic stiffening and hypertension in vivo. Recent data from the Framingham study indicate that increases in pulse wave velocity precede the development of overt hypertension in the general population.48 Our study does not refute this, but suggests that once hypertension is initiated, aortic stiffening is markedly exagerated. In keeping with this, in mice subjected to transverse aortic constriction, adventitial collagen deposition is observed proximal to the constriction, where pressures are elevated, but not distal to the constriction where pressures are normal.49 Likewise, a recent finding suggest that mild hypertension in children predisposes to increasesd pulse wave velocity in adulthood.50 No matter the cause, stiffening of the proximal aorta leads to the loss of Windkessel effect, further elevating systolic blood pressure. Therefore, in the long run, aortic stiffening and hypertension likely promote one another, ultimately leading to progressive elevations in systolic blood pressure.
In the present study, the absolute amount of elastin present in the aorta was not affected by angiotensin II infusion, while the relative amount of elastin, expressed as a percent of total protein was reduced by about 50%. This relative loss of elastin might also contribute to the reduced aortic compliance observed in angiotensin II-induced hypertension. Loss of aortic elastin leads to aortic stiffening and hypertension in mice that are haploinsufficient for the elastin gene.51 It is also possible that while total elastin is not changed, elastin damage might occur in the setting of angiotensin II-induced hypertension. Of note, MMP3, which was increased by 2.5 fold by angiotensin II infusion in our gene array analysis, has elastase activity, and could contribute to elastin fragmentation in hypertension.
The striking deposition of collagen in the adventitial layer might not only promote vascular stiffness, but could also increase compression of vascular smooth muscle cells during systole. Our in vitro studies showed that when intraluminal pressure is increased in a normal aorta, there is relatively equal expansion of both the lumen and the outer vascular media, thus preserving medial thickness. In contrast, increasing intraluminal pressure in aortas from angiotensin II-treated mice led to an increase in the lumen that was substantially greater than that of the outer media, leading to compression of the media. This could lead to a condition in which vascular smooth muscle cells undergo cyclic compression during each cardiac cycle. The impact of this on vascular smooth muscle function remains to be defined.
In summary, this study provides new insight into mechansims of aortic stiffening in hypertension as depicted in Figure 7. An important component of this process is a striking deposition of collagen in the adventitia. Our data indicate that aortic stiffening is mediated by both mechanical factors assoicated with elevated pressures and by T cells and their release of inflammatory cytokines. The p38 MAP kinase likely plays an important signalling role in response to these stimuli. Adventitial collagen deposition and aortic stiffening can not only be considered a form of end-organ damage in hypertension but can also further elevate systolic pressure, leading to progression of this disease.
Supplementary Material
Novelty and Significance.
What Is Known?
Aortic stiffening occurs in hypertension in humans and increases systolic pressure.
Mild hypertension in childhood predisposes to aortic stiffening in adults. Conversely, adults with stiff aortas subsequently develop worse hypertension.
Hypertension increases mechanical stretch of the aorta and induces vascular inflammation.
What New Information Does This Article Contribute?
Angiotensin II and Deoxycorticosterone Acetate (DOCA)-salt hypertension causes formation of a “rind” of collagen that surrounds the aorta, which causes marked aortic stiffening.
Inflammation and mechanical stretch seem to promote the production of collagen in the adventitial cells of the aorta, and this seems dependent on the p38 MAP kinase.
The normal aorta is compliant and able to expand with each heartbeat, accommodating a portion of the stroke volume in a capacitance fashion. In a variety of common conditions like aging, diabetes, obesity, atherosclerosis and smoking, the aorta becomes stiff such that the stroke volume from each heartbeat is rapidly ejected to the distal tissues. This raises systolic blood pressure and predisposes to stroke, renal failure and heart failure. The exact causes of aortic stiffening are unknown. In this study we show that inflammation, in the form of T cells and cytokines like interleukin 17 act in concert with the increased mechanical stretch caused by elevated blood pressure to induce aortic collagen production and aortic stiffening. We found that the MAP kinase p38 plays a central role in signaling these processes. Efforts to decrease aortic mechanical stretch and to reduce vascular inflammation might be effective in preventing aortic stiffening.
Acknowledgments
We are grateful to the Transitional Pathology Shared Resource (TPSR) core and the Epithelial Biology Center at Vanderbilt University for the preparation, imaging and quantification of immunohistochemical staining.
SOURCES OF FUNDING
This work was supported by NIH R01 Grants HL105294 and HL039006 and Program Project Grants P01 HL58000 and GM015431, an American Heart Association pre-doctoral fellowship to Jing Wu and a postdoctoral fellowship award to Dr. Kirabo.
Nonstandard Abbreviations and Acronyms
- RAG-1
Recombination activating gene-1
- DOCA
Deoxycorticosterone acetate
- IL-17a
Interleukin 17a
- ROS
Reactive oxygen species
- TH17 cells
T helper cells producing interleukin 17
- CD
Cluster of differentiation
- FBS
Fetal bovine serum
- ANOVA
Analysis of variance
- WT
Wild-type
- MMP
Matrix metalloproteinase
- TGFβ1
Transforming growth factor beta1
- HCTZ
Hydrochlorothiazide
- Hyd
Hydralazine
Footnotes
DISCLOSURES
None.
References
- 1.O’Rourke MF. Arterial aging: pathophysiological principles. Vasc Med. 2007;12:329–341. doi: 10.1177/1358863X07083392. [DOI] [PubMed] [Google Scholar]
- 2.Mitchell GF, Guo CY, Benjamin EJ, Larson MG, Keyes MJ, Vita JA, Vasan RS, Levy D. Cross-sectional correlates of increased aortic stiffness in the community: the Framingham Heart Study. Circulation. 2007;115:2628–2636. doi: 10.1161/CIRCULATIONAHA.106.667733. [DOI] [PubMed] [Google Scholar]
- 3.Dietrich T, Schaefer-Graf U, Fleck E, Graf K. Aortic stiffness, impaired fasting glucose, and aging. Hypertension. 2010;55:18–20. doi: 10.1161/HYPERTENSIONAHA.109.135897. [DOI] [PubMed] [Google Scholar]
- 4.Payne RA, Wilkinson IB, Webb DJ. Arterial stiffness and hypertension: emerging concepts. Hypertension. 2010;55:9–14. doi: 10.1161/HYPERTENSIONAHA.107.090464. [DOI] [PubMed] [Google Scholar]
- 5.Mitchell GF, DeStefano AL, Larson MG, Benjamin EJ, Chen MH, Vasan RS, Vita JA, Levy D. Heritability and a genome-wide linkage scan for arterial stiffness, wave reflection, and mean arterial pressure: the Framingham Heart Study. Circulation. 2005;112:194–199. doi: 10.1161/CIRCULATIONAHA.104.530675. [DOI] [PubMed] [Google Scholar]
- 6.Mattace-Raso FU, van der Cammen TJ, Hofman A, van Popele NM, Bos ML, Schalekamp MA, Asmar R, Reneman RS, Hoeks AP, Breteler MM, Witteman JC. Arterial stiffness and risk of coronary heart disease and stroke: the Rotterdam Study. Circulation. 2006;113:657–663. doi: 10.1161/CIRCULATIONAHA.105.555235. [DOI] [PubMed] [Google Scholar]
- 7.Laurent S, Katsahian S, Fassot C, Tropeano AI, Gautier I, Laloux B, Boutouyrie P. Aortic stiffness is an independent predictor of fatal stroke in essential hypertension. Stroke. 2003;34:1203–1206. doi: 10.1161/01.STR.0000065428.03209.64. [DOI] [PubMed] [Google Scholar]
- 8.Abramson JL, Weintraub WS, Vaccarino V. Association between pulse pressure and C-reactive protein among apparently healthy US adults. Hypertension. 2002;39:197–202. doi: 10.1161/hy0202.104270. [DOI] [PubMed] [Google Scholar]
- 9.Yasmin, McEniery CM, Wallace S, Mackenzie IS, Cockcroft JR, Wilkinson IB. C-reactive protein is associated with arterial stiffness in apparently healthy individuals. Arterioscler Thromb Vasc Biol. 2004;24:969–974. doi: 10.1161/01.ATV.zhq0504.0173. [DOI] [PubMed] [Google Scholar]
- 10.Nakhai-Pour HR, Grobbee DE, Bots ML, Muller M, van der Schouw YT. C-reactive protein and aortic stiffness and wave reflection in middle-aged and elderly men from the community. J Hum Hypertens. 2007;21:949–955. doi: 10.1038/sj.jhh.1002255. [DOI] [PubMed] [Google Scholar]
- 11.Tsioufis C, Dimitriadis K, Selima M, Thomopoulos C, Mihas C, Skiadas I, Tousoulis D, Stefanadis C, Kallikazaros I. Low-grade inflammation and hypoadiponectinaemia have an additive detrimental effect on aortic stiffness in essential hypertensive patients. Eur Heart J. 2007;28:1162–1169. doi: 10.1093/eurheartj/ehm089. [DOI] [PubMed] [Google Scholar]
- 12.Selzer F, Sutton-Tyrrell K, Fitzgerald S, Tracy R, Kuller L, Manzi S. Vascular stiffness in women with systemic lupus erythematosus. Hypertension. 2001;37:1075–1082. doi: 10.1161/01.hyp.37.4.1075. [DOI] [PubMed] [Google Scholar]
- 13.Wallberg-Jonsson S, Caidahl K, Klintland N, Nyberg G, Rantapaa-Dahlqvist S. Increased arterial stiffness and indication of endothelial dysfunction in long-standing rheumatoid arthritis. Scand J Rheumatol. 2008;37:1–5. doi: 10.1080/03009740701633238. [DOI] [PubMed] [Google Scholar]
- 14.Gisondi P, Fantin F, Del Giglio M, Valbusa F, Marino F, Zamboni M, Girolomoni G. Chronic plaque psoriasis is associated with increased arterial stiffness. Dermatology. 2009;218:110–113. doi: 10.1159/000182256. [DOI] [PubMed] [Google Scholar]
- 15.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Madhur MS, Lob HE, McCann LA, Iwakura Y, Blinder Y, Guzik TJ, Harrison DG. Interleukin 17 promotes angiotensin II-induced hypertension and vascular dysfunction. Hypertension. 2010;55:500–507. doi: 10.1161/HYPERTENSIONAHA.109.145094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ, Harrison DG. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ Res. 2010;107:263–270. doi: 10.1161/CIRCRESAHA.110.217299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mattson DL, Lund H, Guo C, Rudemiller N, Geurts AM, Jacob H. Genetic mutation of recombination activating gene 1 in Dahl salt-sensitive rats attenuates hypertension and renal damage. Am J Physiol Regul Integr Comp Physiol. 2013;304:R407–414. doi: 10.1152/ajpregu.00304.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marko L, Kvakan H, Park JK, Qadri F, Spallek B, Binger KJ, Bowman EP, Kleinewietfeld M, Fokuhl V, Dechend R, Muller DN. Interferon-gamma signaling inhibition ameliorates angiotensin II-induced cardiac damage. Hypertension. 2012;60:1430–1436. doi: 10.1161/HYPERTENSIONAHA.112.199265. [DOI] [PubMed] [Google Scholar]
- 20.Schrader LI, Kinzenbaw DA, Johnson AW, Faraci FM, Didion SP. IL-6 deficiency protects against angiotensin II induced endothelial dysfunction and hypertrophy. Arterioscler Thromb Vasc Biol. 2007;27:2576–2581. doi: 10.1161/ATVBAHA.107.153080. [DOI] [PubMed] [Google Scholar]
- 21.Nguyen H, Chiasson VL, Chatterjee P, Kopriva SE, Young KJ, Mitchell BM. Interleukin-17 causes Rho-kinase-mediated endothelial dysfunction and hypertension. Cardiovasc Res. 2013;97:696–704. doi: 10.1093/cvr/cvs422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kurasawa K, Hirose K, Sano H, Endo H, Shinkai H, Nawata Y, Takabayashi K, Iwamoto I. Increased interleukin-17 production in patients with systemic sclerosis. Arthritis Rheum. 2000;43:2455–2463. doi: 10.1002/1529-0131(200011)43:11<2455::AID-ANR12>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 23.Molet SM, Hamid QA, Hamilos DL. IL-11 and IL-17 expression in nasal polyps: relationship to collagen deposition and suppression by intranasal fluticasone propionate. Laryngoscope. 2003;113:1803–1812. doi: 10.1097/00005537-200310000-00027. [DOI] [PubMed] [Google Scholar]
- 24.Feng W, Li W, Liu W, Wang F, Li Y, Yan W. IL-17 induces myocardial fibrosis and enhances RANKL/OPG and MMP/TIMP signaling in isoproterenol-induced heart failure. Exp Mol Pathol. 2009;87:212–218. doi: 10.1016/j.yexmp.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 25.Baumbach GL, Siems JE, Heistad DD. Effects of local reduction in pressure on distensibility and composition of cerebral arterioles. Circ Res. 1991;68:338–351. doi: 10.1161/01.res.68.2.338. [DOI] [PubMed] [Google Scholar]
- 26.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, Weyand CM, Harrison DG, Guzik TJ. Inhibition and genetic ablation of the B7/CD28 T-cell costimulation axis prevents experimental hypertension. Circulation. 2010;122:2529–2537. doi: 10.1161/CIRCULATIONAHA.109.930446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hofman K, Hall B, Cleaver H, Marshall S. High-throughput quantification of hydroxyproline for determination of collagen. Anal Biochem. 2011;417:289–291. doi: 10.1016/j.ab.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 29.Mecham RP. Methods in elastic tissue biology: elastin isolation and purification. Methods. 2008;45:32–41. doi: 10.1016/j.ymeth.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Starcher B. A ninhydrin-based assay to quantitate the total protein content of tissue samples. Anal Biochem. 2001;292:125–129. doi: 10.1006/abio.2001.5050. [DOI] [PubMed] [Google Scholar]
- 31.Liang Q, Elson AC, Gerdes AM. p38 MAP kinase activity is correlated with angiotensin II type 1 receptor blocker-induced left ventricular reverse remodeling in spontaneously hypertensive heart failure rats. J Card Fail. 2006;12:479–486. doi: 10.1016/j.cardfail.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 32.Park JK, Fischer R, Dechend R, Shagdarsuren E, Gapeljuk A, Wellner M, Meiners S, Gratze P, Al-Saadi N, Feldt S, Fiebeler A, Madwed JB, Schirdewan A, Haller H, Luft FC, Muller DN. p38 mitogen-activated protein kinase inhibition ameliorates angiotensin II-induced target organ damage. Hypertension. 2007;49:481–489. doi: 10.1161/01.HYP.0000256831.33459.ea. [DOI] [PubMed] [Google Scholar]
- 33.Wang D, Warner GM, Yin P, Knudsen BE, Cheng J, Butters KA, Lien KR, Gray CE, Garovic VD, Lerman LO, Textor SC, Nath KA, Simari RD, Grande JP. Inhibition of p38 MAPK attenuates renal atrophy and fibrosis in a murine renal artery stenosis model. Am J Physiol Renal Physiol. 2013;304:F938–947. doi: 10.1152/ajprenal.00706.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paravicini TM, Montezano AC, Yusuf H, Touyz RM. Activation of vascular p38MAPK by mechanical stretch is independent of c-Src and NADPH oxidase: influence of hypertension and angiotensin II. J Am Soc Hypertens. 2012;6:169–178. doi: 10.1016/j.jash.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 35.Maki-Petaja KM, Elkhawad M, Cheriyan J, Joshi FR, Ostor AJ, Hall FC, Rudd JH, Wilkinson IB. Anti-tumor necrosis factor-alpha therapy reduces aortic inflammation and stiffness in patients with rheumatoid arthritis. Circulation. 2012;126:2473–2480. doi: 10.1161/CIRCULATIONAHA.112.120410. [DOI] [PubMed] [Google Scholar]
- 36.Galarraga B, Khan F, Kumar P, Pullar T, Belch JJ. Etanercept improves inflammation-associated arterial stiffness in rheumatoid arthritis. Rheumatology (Oxford) 2009;48:1418–1423. doi: 10.1093/rheumatology/kep251. [DOI] [PubMed] [Google Scholar]
- 37.Ortega C, Fernandez AS, Carrillo JM, Romero P, Molina IJ, Moreno JC, Santamaria M. IL-17-producing CD8+ T lymphocytes from psoriasis skin plaques are cytotoxic effector cells that secrete Th17-related cytokines. J Leukoc Biol. 2009;86:435–443. doi: 10.1189/JLB.0109046. [DOI] [PubMed] [Google Scholar]
- 38.He D, Wu L, Kim HK, Li H, Elmets CA, Xu H. CD8+ IL-17-producing T cells are important in effector functions for the elicitation of contact hypersensitivity responses. J Immunol. 2006;177:6852–6858. doi: 10.4049/jimmunol.177.10.6852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matsushita H, Lee KH, Tsao PS. Cyclic strain induces reactive oxygen species production via an endothelial NAD(P)H oxidase. J Cell Biochem Suppl. 2001;(Suppl 36):99–106. doi: 10.1002/jcb.1094. [DOI] [PubMed] [Google Scholar]
- 40.Cheng JJ, Wung BS, Chao YJ, Wang DL. Cyclic strain enhances adhesion of monocytes to endothelial cells by increasing intercellular adhesion molecule-1 expression. Hypertension. 1996;28:386–391. doi: 10.1161/01.hyp.28.3.386. [DOI] [PubMed] [Google Scholar]
- 41.Hyytiainen M, Penttinen C, Keski-Oja J. Latent TGF-beta binding proteins: extracellular matrix association and roles in TGF-beta activation. Crit Rev Clin Lab Sci. 2004;41:233–264. doi: 10.1080/10408360490460933. [DOI] [PubMed] [Google Scholar]
- 42.Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, Boivin GP, Bouck N. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93:1159–1170. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- 43.Perdiguero E, Sousa-Victor P, Ruiz-Bonilla V, Jardi M, Caelles C, Serrano AL, Munoz-Canoves P. p38/MKP-1-regulated AKT coordinates macrophage transitions and resolution of inflammation during tissue repair. J Cell Biol. 2011;195:307–322. doi: 10.1083/jcb.201104053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang G, Kwan BC, Lai FM, Chow KM, Li PK, Szeto CC. Urinary miR-21, miR-29, and miR-93: novel biomarkers of fibrosis. Am J Nephrol. 2012;36:412–418. doi: 10.1159/000343452. [DOI] [PubMed] [Google Scholar]
- 45.Wang J, Gao Y, Ma M, Li M, Zou D, Yang J, Zhu Z, Zhao X. Effect of miR-21 on Renal Fibrosis by Regulating MMP-9 and TIMP1 in kk-ay Diabetic Nephropathy Mice. Cell Biochem Biophys. 2013 doi: 10.1007/s12013-013-9539-2. [DOI] [PubMed] [Google Scholar]
- 46.Villar AV, Garcia R, Merino D, Llano M, Cobo M, Montalvo C, Martin-Duran R, Hurle MA, Nistal JF. Myocardial and circulating levels of microRNA-21 reflect left ventricular fibrosis in aortic stenosis patients. Int J Cardiol. 2012 doi: 10.1016/j.ijcard.2012.07.021. [DOI] [PubMed] [Google Scholar]
- 47.Zhu H, Luo H, Li Y, Zhou Y, Jiang Y, Chai J, Xiao X, You Y, Zuo X. MicroRNA-21 in Scleroderma Fibrosis and its Function in TGF-beta- Regulated Fibrosis-Related Genes Expression. J Clin Immunol. 2013 doi: 10.1007/s10875-013-9896-z. [DOI] [PubMed] [Google Scholar]
- 48.Kaess BM, Rong J, Larson MG, Hamburg NM, Vita JA, Levy D, Benjamin EJ, Vasan RS, Mitchell GF. Aortic stiffness, blood pressure progression, and incident hypertension. Jama. 2012;308:875–881. doi: 10.1001/2012.jama.10503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen J, Wu J, Li L, Zou YZ, Zhu DL, Gao PJ. Effect of an acute mechanical stimulus on aortic structure in the transverse aortic constriction mouse model. Clin Exp Pharmacol Physiol. 2011;38:570–576. doi: 10.1111/j.1440-1681.2011.05544.x. [DOI] [PubMed] [Google Scholar]
- 50.Aatola H, Magnussen CG, Koivistoinen T, Hutri-Kahonen N, Juonala M, Viikari JS, Lehtimaki T, Raitakari OT, Kahonen M. Simplified definitions of elevated pediatric blood pressure and high adult arterial stiffness. Pediatrics. 2013;132:e70–76. doi: 10.1542/peds.2012-3426. [DOI] [PubMed] [Google Scholar]
- 51.Faury G, Pezet M, Knutsen RH, Boyle WA, Heximer SP, McLean SE, Minkes RK, Blumer KJ, Kovacs A, Kelly DP, Li DY, Starcher B, Mecham RP. Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Invest. 2003;112:1419–1428. doi: 10.1172/JCI19028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.