

Abstract
Neurodegenerative diseases are characterized by chronic microglial over-activation and oxidative stress. It is now beginning to be recognized that reactive oxygen species (ROS) produced by either microglia or the surrounding environment not only impact neurons but also modulate microglial activity. In this review, we first analyze the hallmarks of pro-inflammatory and anti-inflammatory phenotypes of microglia and their regulation by ROS. Then, we consider the production of reactive oxygen and nitrogen species by NADPH oxidases and nitric oxide synthases and the new findings that also indicate an essential role of glutathione (γ-glutamyl-l-cysteinylglycine) in redox homeostasis of microglia. The effect of oxidant modification of macromolecules on signaling is analyzed at the level of oxidized lipid by-products and sulfhydryl modification of microglial proteins. Redox signaling has a profound impact on two transcription factors that modulate microglial fate, nuclear factor kappa-light-chain-enhancer of activated B cells, and nuclear factor (erythroid-derived 2)-like 2, master regulators of the pro-inflammatory and antioxidant responses of microglia, respectively. The relevance of these proteins in the modulation of microglial activity and the interplay between them will be evaluated. Finally, the relevance of ROS in altering blood brain barrier permeability is discussed. Recent examples of the importance of these findings in the onset or progression of neurodegenerative diseases are also discussed. This review should provide a profound insight into the role of redox homeostasis in microglial activity and help in the identification of new promising targets to control neuroinflammation through redox control of the brain. Antioxid. Redox Signal. 21, 1766–1801.
I. Introduction
Thanks to the use of antibiotics and improved lifestyles, infectious diseases are no longer a primary cause of death in developed countries, and it is hoped that this situation will extend to the whole world before long. However, improved life expectancy has increased the prevalence of chronic diseases, including neurodegenerative diseases. Old and new maladies share a common immune response, but in chronic degenerative diseases the role of the innate immune system appears to be particularly relevant. In the central nervous system (CNS), the innate immune system is represented by a type of macrophage, known collectively as microglia, initially described by Ramón y Cajal and Pío del Río Hortega as a member of the reticulo-endothelial system at a time, during the 1920s, when it was believed that the brain was composed of three cellular elements: neurons, “neuroglia,” representing astroglia, and “the third element,” which was identified as microglia (67, 68). Although present in all brain regions, microglia are mainly in the gray matter, achieving the highest concentration in hippocampus, olfactory telencephalon, basal ganglia, and substantia nigra, and they comprise 0.5–16% of total nerve cells (143).
During embryogenesis, microglia migrate into the brain parenchyma and retain a low turn-over rate (141), constituting what for many years was believed to be the only phagocytic cell population of the CNS. During development, they participate in the clearance of the huge quantity of neurons that are produced in large excess in the embryo and later die of apoptosis (278). Under normal healthy conditions, microglial cells continuously survey nerve tissue, monitor the integrity of synapses (328), fight infectious agents, and remove apoptotic and necrotic cells with subsequent suppression or promotion of neuroinflammation (84).
While microglia-driven neuroinflammation, undoubtedly, has a beneficial effect on scavenging cell debris, tissue healing, and repair, it is also becoming clear that chronic activation of this cell type leads to noxious effects on neurons and, thus, participates in the pathophysiology of neurodegenerative diseases (233). A key determinant of microglial neurotoxicity is the release of excitotoxins, including glutamate (14, 241), quinolinate (102), d-Serine (343), cyclooxygenase (COX)-derived prostaglandins (47), and, of particular relevance to this review, reactive oxygen species (ROS) and reactive nitrogen species (RNS). These molecules not only influence the surrounding environment but also modulate microglial dynamics between classical pro-inflammatory and alternative wound-healing phenotypes and participate in weakening of the blood–brain barrier (BBB) permeability, thereby facilitating the recruitment of peripheral immune cells at sites of inflammation.
Here, we will review our current knowledge on microglial regulation with a particular focus on the role of ROS. This review will illustrate the relevance of ROS not only as a part of microglial function but also as participants in a feed-forward mechanism of activation that may contribute to chronic unremitting inflammation in neurodegenerative diseases.
II. Microglial Profiles and the Influence of Redox Dynamics
A. Description of microglial phenotypes
Under physiological conditions, microglia display small cell bodies and thin processes that extend and branch several times (Fig. 1). These processes move around, continuously surveying the surrounding environment in search of pathogens, sick cells, and cellular debris (289, 314), or participate in remodeling and maintenance of synapses (226, 328). This “surveillance” phenotype is maintained, in part, through neuron-derived signals, including CX3C-chemokine ligand 1 (CX3CL1), CD47, CD200, and CD22, which act through their cognate receptors expressed at the microglial plasma membrane (211). In addition, microglia have an array of pattern recognition receptors/toll-like receptors (TLRs) that enable them to sense and respond to a large variety of exogenous agents (144, 271).
FIG. 1.

Microglial phenotypes are tightly controlled by ROS/RNS levels. Under surveillance mode, microglial cells exhibit low levels of ROS/RNS that might be properly managed by the antioxidant machinery. After an inflammatory challenge, microglial cells execute an M1 program that is characterized by a rapid and high increase in ROS/RNS levels mainly derived from the NOX and NOS activities. During this phase, ROS/RNS act as second messengers increasing the phosphorylation levels of the kinases that control NF-κB to further up-regulate the pro-inflammatory M1 gene profile. However, other transcription factors, including NRF2, are increased in response to ROS/RNS but probably in a retarded fashion. NRF2 will restore redox homeostasis and attenuate M1 in favor of M2 phenotypes. NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NOS, nitric oxide synthase; NOX, NADPH oxidase; NRF2, nuclear factor (erythroid-derived 2)-like 2; RNS, reactive nitrogen species; ROS, reactive oxygen species.
Hazardous signals detected by these receptors polarize microglia toward a classical pro-inflammatory phenotype termed M1 (Fig. 1; Table 1). The M1 activation profile is a rapid response of microglia that is characterized by hypertrophic bodies, with fewer, thicker, and shorter processes than those of surveillance microglia. These cells release highly pro-inflammatory cytokines such as tumor necrosis factor (TNF), interleukin (IL)-1, IL-6, IL-12, and IL-23, as well as chemokines and proteases and present antigens through MHCII proteins. Some of these cytokines may be released also from neurons, as is the case with IL-1 (316). The role of nitric oxide (NO) released by inducible isoform of nitric oxide synthase (iNOS) in the acquisition of M1 phenotype is not so clear. iNOS is not expressed at a relevant level in microglia in situ in the brain but can be induced in astrocytes at least in the rodent brain and only cultured microglia express iNOS. With regard to prostaglandins, COX1 is expressed in microglia and COX2 is expressed in neurons (312). All of these events help defend the tissue from acute injuries caused by external or internal agents. Thus, M1 microglia are generally considered potent effector cells that kill and engulf micro-organisms as well as tumor or otherwise damaged cells. This response is rapidly reinforced by cytoactive factors, including ROS and RNS, released by microglia themselves and by surrounding cells.
Table 1.
Characteristic Antigens of the Microglial Phenotypes
| Phenotype | Cytokines/chemokines | Receptors | Enzymes | Other markers |
|---|---|---|---|---|
| Surveilling | Low | CXCR2 | Low iNOS | |
| CD172a | low ARG1 | |||
| CD200R1 | ||||
| Low IBA-1 | ||||
| Low F4/80 | ||||
| M1-profile | IL-6 | MHCII | iNOS | |
| IL-1 | C-type lectins | COX1 | ||
| IL-12p40 | CAT2 | |||
| TNF | CD8 | |||
| IFN | F4/80 | |||
| IBA-1 | ||||
| MARCO | ||||
| RAGE | ||||
| M2-profile | IL-4 | MHCII | ARG1 | YM-1 |
| IL-5 | C-type lectins | Lack of iNOS | FIZZ1 | |
| IL-13 | CAT2 | sphk1/2 | SOCS3 | |
| IL-10 | MR | |||
| TGF | DC-SIGN | |||
| CCL-18 | IL-4Rα | |||
| IL-1Rα | ||||
| CD36 | ||||
| CD136 |
ARG1 arginase 1; CAT2, cationic amino-acid transporter 2; CCL-18, chemokine 18; CD172a, receptor for CD47; CD200R1, receptor for CD200; CXCR2, receptor for CXC3-chemokine ligand-1; DC-SIGN, dendritic cell-specific intracellular adhesion molecule 3-grabbing integrin; FIZZ1, found in inflammatory zone; IBA-1, ionized calcium-binding adapter molecule 1; IFN, interferon; IL-1, interleukin 1; IL-1Ra interleukin 1 receptor antagonist; IL-4Rα, interleukin 4 receptor α; IL-5, interleukin 5; IL-6, interleukin 6; IL-10, interleukin 10; IL-12p40, interleukin 12 p40 subunit; IL-13, interleukin 13; iNOS, inducible nitric oxide synthase; MARCO, macrophage receptor with collagenous structure; MHCII, major histocompatibility antigen II; MR, manose receptor; RAGE, receptor for advanced glycation endproducts; SOCS3, suppressor of cytokine signaling 3; sphk, sphingosine kinase; TGF-b, tumor growth factor b; TNF, tumor necrosis factor; YM-1, mouse homolog chitinase 3 like 1 (CHI3L1).
The pro-inflammatory polarization of microglia is often followed by a long-lasting repair stage known as the alternative or M2 phase in which microglia display hypertrophic cell bodies with thick and ramified processes and high phagocytic capacity (Fig. 1; Table 1). The M2 program is activated by anti-inflammatory cytokines such as IL-4, IL-13, and IL-10, immunoglobulin complexes/TLR, transforming growth factor-β (TGF-β), and glucocorticoids. In addition, M2-microglia express low levels of pro-inflammatory signals, but produce IL-4, IL-5, IL-10, and IL-13. Moreover, the expression of wound-healing genes, such as those coding arginase-1 (ARG1), mannose receptors (MMC and Mrc2c), dectin-1, found in inflammatory zone 1 (FIZZ1), chitinase-3-like-1 (Ym1 in rodents), scavenger receptors, CD36, CD163, MARCO, nerve and insulin growth factors, and PPAR-γ, is high in M2-microglia (52). In peripheral tissues, M2-polarized macrophages are further divided into three subsets, including M2a (activated by IL-4 or IL-13), M2b (activated by immune complexes plus IL-1 or lipopolysaccharide [LPS]), and M2c (activated by TGF-β, glucocorticoids or IL-10) (182). However, this classification is less clearly established for microglia. The M2 response is crucial for restoration of normal tissue homeostasis, because it leads to the switch-off of the pro-inflammatory response, scavenges debris, and restructures the damaged extracellular matrix (ECM) (175).
B. The microglial M1/M2 balance is modulated by the redox status
Redox homeostasis influences the acquisition of the final microglial phenotype by a variety of mechanisms. During M1 execution, the normally low levels of ROS/RNS associated with surveillance microglia are dramatically elevated, due to activation of NADPH oxidase (NOX) and nitric oxide synthase (NOS) enzymes (250). Under these conditions, ROS/RNS act as second messengers that are capable of modulating gene expression by inducing signaling kinases or inhibiting signaling phosphatases (327, 373). As we will describe next, transcription factor nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) is particularly sensitive to ROS and is central to acquisition of the pro-inflammatory M1 polarization. In contrast, signaling cascades involved in M2 acquisition are less well understood, yet cytokines such as IL-4 that signal via STAT3 and STAT6 appear to be important (27, 272, 285). Considering that ROS/RNS should be lowered after the initial pro-inflammatory response to prevent further tissue damage, it is reasonable to suggest that induction of anti-oxidant and cytoprotective genes will be a signature of M2 polarization and indeed, as will be discussed later, novel findings indicate that the transcription factor nuclear factor (erythroid-derived 2)-like 2 (NRF2), master regulator of redox homeostasis, favors an M2 compared with an M1 phenotype.
The connection between microglial plasticity and redox state has been analyzed in a range of experimental models. For instance, Rossi-George et al. showed that Cu(I) has the capacity to shift LPS-activated M1-microglia toward an M2 phenotype, but only in the presence of NO (264). Data from in vivo models have established that redox homeostasis of microglia depends mainly on NOX activity. Accordingly, Choi et al. (48) showed that pharmacological inhibition of NOX, or genetic deletion of its functional p47phox subunit, changed microglia from an M1 to M2 phenotype. The authors observed that the microglial marker IBA-1 was significantly reduced in hippocampus from p47phox−/− mice along with a significant reduction of pro-inflammatory factors such as TNF, CCL2, and CCR2. In contrast, IL-4 and IL-4Rα messenger RNA levels were elevated after an LPS injection in p47phox−/− mice compared with wild-type littermates. In addition, expression of the genes encoding Ym1 and FIZZ1 exhibited a marked induction in p47phox−/− mice compared with wild type. Moreover, in the 6-hydroxydopamine (6-OHDA)-induced Parkinson's disease (PD) mouse model, Hernandes et al. (100) recently described that mice lacking the catalytic subunit of NOX2, gp91phox−/− are completely protected against glial M1 over-activation, as determined by reduced release of IL-1, interferon-γ (IFN-γ), and TNF.
Evidence for a role of NRF2 in the modulation of microglial dynamics between M1 and M2 phenotypes was reported by Cuadrado's group in the context of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinsonism (116, 262). Compared with wild-type littermates, NRF2-deficient mice exhibited increased levels of pro-inflammatory M1 markers (COX-2, iNOS, IL-6, and TNF) and reduced levels of alternative M2 markers (FIZZ-1, YM-1, ARG1, and IL-4) in response to this neurotoxin.
Suppressors of cytokine signaling (SOCS) are a group of inducible proteins that prevent neuroinflammation by inducing feedback inhibition of cytokine signaling at the level of the JAK/STAT axis (12). The SOCS family includes eight members (cytokine-inducible SH2 protein, CIS, and SOCS1-7) that orchestrate distinct reactions by antagonizing STAT activation. They bind to phosphorylated tyrosine residues on cytokine receptors and to the SH2 domain of JAKs. This physical interaction switches off JAKs by acting as pseudosubstrates and recruiting the ubiquitination machinery via the SOCS-box domain (350). At least SOCS1 and SOCS3 are expressed in microglia. SOCS1 is induced by various cytokines, including IFN-γ, IL-4, and TNF, and down-regulates the signals induced by these cytokines, mainly IFN-γ, in a feedback loop. SOCS3 inhibits signaling by the IL-6 family of cytokines and meditates the anti-inflammatory effects of IL-10. It has been reported that an additional mechanism of up-regulation of SOCS expression is the response to ROS/RNS and the fact that they repress the M1 program (249). Thus, the protein levels of SOCS1 and SOCS3 are drastically elevated in response not only to TNF but also to hydrogen peroxide (H2O2) in Jurkat T cells and mouse splenocytes (218). With regard to microglia, electrophilic compounds such as a caffeamide derivative (163) and gemfibrozil (89) have been reported to exert anti-neuroinflammatory functions through induction of SOCS3. The role of SOCS in neuroinflammation has been studied mainly in the context of multiple sclerosis (MS) where they are proposed to be putative targets to modulate chronic inflammation (78).
It has been suggested that M1 and M2 phenotypes are mutually independent. Thus, anti-inflammatory cytokines IL-4 or IL-13 prevent the induction of pro-inflammatory mediators such as iNOS, COX-2, IL-6, and TNF (52, 165, 244). In the mouse model of amyotrophic lateral sclerosis (ALS), based on the expression of mutant Cu/Zn-superoxide dismutase (SOD1), microglia isolated from animals in their early stage of disease exhibit an M2 phenotype that is protective to motoneurons; whereas microglia isolated from end-stage mice have adopted an M1 phenotype with exacerbated NOX2 activity, supporting the dual phenotypes of microglia and their transformation during progression of neurodegenerative diseases (154). Aging is associated with a progressive increase in ROS, and, therefore, it is possible that those changes might be due, in part, to ROS-mediated reinforcement of the M1 program.
The protein lipocalin 2 (LCN2) is secreted by astroglia and microglia under pro-inflammatory conditions (147, 148) and elevated ROS levels (294) and strengthens the expression of M1-markers (IL-12, IL-23, iNOS, TNF, and CXCL10) without affecting M2-markers. LCN2 plays a central role in iron transport and prevents oxidant Fenton reactions, as LCN2-deficient mice exhibit elevated intracellular labile iron and exacerbated sensibility to endotoxin (292). In contrast to wild-type microglia, cells from LCN2-deficient mice failed to induce M1-markers in response to combined LPS plus IFN-γ treatment. However, their capacity to induce M2 markers in response to IL-4 was increased. LCN2 inhibited phosphorylation of STAT6 in IL-4-stimulated microglia, suggesting that LCN2 acts as a brake for the M2 program.
The differential expression of iNOS and ARG1 in M1 compared with M2 profiles provides an attractive model for the molecular basis of redox control of microglial phenotypes (Fig. 2). Both enzymes use l-Arginine as a substrate, but iNOS produces NO, favouring nitrosative and oxidative stress and the M1 phenotype, whereas ARG1 generates ornithine. Ornithine aminotransferase generates glutamate, proline, and hydroxyproline. Hydroxyproline is an essential component of collagen synthesis and, therefore, contributes to maintenance of the ECM, reinforcing the M2 program. In this program, l-Arginine is transformed to ornithine by ARG1, because (i) ARG1 is highly expressed; (ii) iNOS expression is silenced, in part, due to the actions of IL-4 and IL-13; and (iii) although iNOS is catalytically more effective than ARG1 (KmiNOS=3–10 μM; KmARG1=3–10 mM), the Vmax of ARG1 is 1500-fold greater (52). In the M1-program, the expression of each enzyme is reversed, enabling high NO generation through iNOS activity.
FIG. 2.

Dual use of l-Arginine by iNOS and ARG1. l-Arginine enters microglia through CAT and becomes a substrate of both pro-M1 iNOS and pro-M2 ARG1. A fine tuning between these two activities participates in microglial polarization. ARG1, arginase-1; CAT, cationic amino acid transporter; iNOS, inducible isoform of nitric oxide synthase.
The importance of oxidative stress in M1/M2 polarization goes beyond the acquisition of either phenotype and may participate in the development of endotoxin tolerance (ET). In ET, the exposure of an organism to an inflammatory stimulus (for example, the endotoxin LPS as a “first hit”) results in a low immune response to a subsequent inflammatory insult (“second hit”). The pathological consequence is an impaired capacity to respond to new infections. Although ET is best characterized in the acute inflammatory reactions of sepsis, we speculate that it may be a general adaptive mechanism that could affect chronic diseases in which prolonged low-grade inflammation might act as the first hit. Taking Alzheimer's disease (AD) as a model, clinical observations indicate that these patients show a greater susceptibility to infections compared with age-matched healthy individuals (24). Considering that the BBB is weakened in AD, enabling bidirectional transit of molecules, and that both neurons and glia release several modulators of inflammation, it is plausible that low-grade chronic neuroinflammation in AD might result in a state of reduced immunological response or “tolerance” to peripheral infections. Systems analyses have demonstrated that activation of transcription factor ATF3 is an early response to endotoxin in macrophages, which represses the production of IL-6, TNF, and IFN-γ (105). Since ROS deplete the levels of the tripeptide glutathione (γ-glutamyl-l-cysteinylglycine) (GSH), and GSH depletion enhances endotoxin-induced ATF3 expression, it follows that oxidative stress will also modulate ET. In fact, ROS-mediated induction of ATF3 caused increased susceptibility to bacterial and fungal infections through the suppression of IL-6 in wild-type but not in Atf3-knock-out mice (105). Pena et al., (232), who also used a systems biology approach and bioinformatics analysis, determined that gene and protein expression during ET is similar to that found during M2 polarization. While these observations point to redox control as a crucial element in the development of ET, it is not yet clear how this impacts microglia and brain tolerance to either acute infections or progressive neurodegeneration. In fact, experimental evidence suggests that innate immune cells in the brain do not become tolerant to repeated systemic administration of LPS, but, on the contrary, lead to prolonged and damaging cytokine production which may have a profound effect on the onset or progression of pre-existing neurodegenerative disease (248).
C. Brain aging and neurodegenerative diseases alter microglial dynamics and the cross-talk between neurons and microglia
There is a strong correlation between brain aging and neurodegeneration and redox imbalance, as well as low-grade chronic inflammation. One would predict that, over time, microglia would become increasingly susceptible to accumulative oxidative damage, due to their limited capacity to divide (4, 90, 192) and to the need to remove copious amounts of ROS/RNS after M1 activation. Consistently, microglia from old or degenerated brains undergo morphological changes that resemble the M1 phenotype, with decreased branching and beading of their processes (295, 296). In addition, these microglial cells present a hyper-inflammatory response (294) with increased M1-markers (MHCII, CD86, CD68, TLRs, and pro-inflammatory cytokines such as IL-1, TNF, IL-6, and CD11) and a parallel decrease in anti-inflammatory mediators (IL-10, BDNF, inhibitor of NF-κB [IκB] inhibitor, and MAD3A) [reviewed in refs. (83, 215)].
As will be discussed later, GSH is a major low-molecular-weight antioxidant that is used for the maintenance of microglial redox balance, and its levels decrease with aging. For example, the brain of aged rodents exhibits a decrease in GSH accompanied with higher glutathione disulfide (GSSG) levels compared with young animals (258). Microglia from aged mice constitutively secrete more TNF and IL-6 relative to microglia from young mice and are less responsive to stimulation (213). Furthermore, microglia from aged mice have lower GSH levels and internalise less amyloid beta (Aβ) peptide than young mice. It has long been recognized that there is an age-related increase in activities of GSH-consuming enzymes, including glutathione peroxidase (GPx), glutathione S-transferase (GST), and γ-glutamyl transpeptidase (γGT) in rat brain (110). Similarly, in AD patients, up-regulation of a specific isoform of GST (hGST3) has been reported (306). On the other hand, γGluCys ligase expression and activity has been widely reported to decline with age (157, 332, 374), thus contributing to a failure of cells to maintain adequate GSH levels for anti-oxidant protection.
An essential aspect of normal brain function is the bidirectional communication between neurons and neighboring glia, including microglia. With regard to the role of ROS in this review, it will be implicit that activated microglia may release several factors, including superoxide anion (O2−) and H2O2, that impact the surrounding brain parenchyma damaging neurons and leading to a vicious cycle of microglial activation and neuron damage. With regard to signaling from neurons to microglia, recent studies are uncovering a mechanism used by the neurons to shut down microglial over-activation. Several groups have shown that the chemokine fractalkine (CX3CL1) is released by injured neurons and interacts with its cognate receptor, which in the brain is exclusively expressed in microglia. Cuadrado's group (142) has reported that CX3CL1 released by TAU-injured neurons up-regulates in microglia the transcription factor NRF2, which as will be discussed later, is a master regulator of redox homeostasis. By this mechanism, neurons limit the extent of ROS release and, thus, prevent over-activation of microglia.
III. Sources of ROS in Microglia: NOX and NOS Enzymes
As with any other aerobic eukaryotic cells, incomplete reduction of molecular oxygen in mitochondrial respiration in microglia leads to generation of the O2−. Despite the marvelous design of complex IV of the mitochondrial respiratory chain, which is admirably suited to quench O2 until it is fully reduced to water, it is estimated that 1–2% of molecular oxygen generates O2− (30). While O2− is not a very reactive molecule, it undergoes spontaneous or enzymatic dismutation by Mn-SOD (SOD2) in mitochondria and generates H2O2, which is arguably the most important ROS signaling molecule. One potential effect of O2− in the mitochondria is the release of Fe3+ from 4Fe-4S clusters of complex I and complex II, leading, on the one hand, to loss of energy production and, on the other, to the generation of the very reactive hydroxyl radical (•HO) by the Fenton reaction (246). ROS production by mitochondria is a part of their physiological function. While this source of ROS generation is not specific to microglia, it may be relevant to microglial function, particularly during the aging process as discussed earlier. In addition to mitochondrial respiration, ROS production by NOX in microglia occurs mainly at the plasma membrane, as discussed in the next section.
A. NOX system
The main source of ROS in microglia is the activation of NOX (28). Earlier investigations demonstrated a relevant role of NOX in the acute inflammatory response by neutrophils, participating in a respiratory burst that is aimed at killing pathogens (66, 238). Later studies demonstrated that monocytes/macrophages, including microglia, also express NOX, and it is now accepted that NOX activity is found in both phagocytic and non-phagocytic cells, such as astroglia and neurons (49, 251). Therefore, in addition to a role in pathogen defence, NOX participate in multiple cellular processes such as host defence, migration, changes in morphology of microglia, pro-inflammatory gene expression (251), up-regulation of different markers in response to inflammatory stimuli, and microglia-induced neurotoxicity (265). An excellent review on the role of NOX enzymes in the CNS has been recently published (207) and here, we will only provide a brief description of this system.
NOX is a multi-subunit enzyme complex that transfers electrons from NADPH and, to a lesser extent, NADH (23) to molecular oxygen, resulting in the formation of O2− anion as the primary product, but in some NOX isoforms such as NOX4, O2− dismutation is so fast that H2O2 is the end product detected (212, 281). Under resting conditions, the different subunits of this complex are localized in the cytosol (p40phox, p47phox, and p67phox) and in the cell membrane (cytochrome b558, comprising p22phox and gp91phox) and on stimulation, the catalytically active complex is assembled in the plasma membrane (5, 207).
Seven different NOX isoforms exist in humans: NOX1, NOX2 (also known as gp91phox), NOX3, NOX4, NOX5, dual oxygenase (DUOX)1, and DUOX2, which differ in subcellular localization and regulatory mechanisms (23, 207). Microglia express at least NOX1, NOX2, and NOX4 isoforms, but most studies have focused on NOX2 (291). NOX2 levels are very low at the surveillance state and increase in the M1 state in both humans (86) and mice (341).
Although ROS generated by NOX in microglia will act on surrounding cells, in the context of this review, it is particularly relevant that their main target is the microglial cell itself, due to the proximity of their site of formation and their high degree of reactivity. This autocrine effect has received little attention, despite the fact that it will have a profound effect on microglial activity. For instance, increased expression of NOX4 leads to constitutive release of IL-6 and ROS generation in these cells (151). Therefore, NOX activation and subsequent ROS generation play a critical role in the regulation of microglial fate, including cell proliferation, induction of apoptosis during the embryonic phase, release of neurotransmitters, and pro-inflammatory cytokine production (99, 117, 179). This pro-inflammatory M1 response may play a key role in the pathological over-activation of microglia that has been reported in several neurodegenerative diseases.
Thus, NOX2-deficient mice that over-express the Swedish mutation of APP(Tg2576) exhibited a significant reduction in oxidative stress and behavioral deficits (228, 229), while the accumulation of Aβ fragments was not affected. In line with these observations, the inhibition of NOX2 with the peptide gp91ds-tat attenuates oxidative stress and behavioral deficits in mice over-expressing APP (229). Based on these preclinical studies, the inhibition of NOX2 could be considered a potential target to develop new drugs for the treatment of AD.
With regard to PD, a pathological examination of substantia nigra samples from patients and animal models has also revealed an up-regulation of microglial NOX2 (342). Thus, aggregated α-synuclein stimulates microglial ROS production via NOX2 activation and may cause the death of dopaminergic neurons (364). Indeed, in the toxic models of PD such as exposure to MPTP, the deletion of NOX2 caused a marked reduction of dopaminergic neuronal death (365), further evidencing the participation of microglial NOX2.
In ALS, an augmented expression of microglial NOX2 has been reported in patients with the SOD1 mutation (SOD1G93A) (177, 341) and in patients with sporadic form of the disease (341). The relevance of NOX2 in ALS pathogenesis was suggested by Marden et al. (177), who showed that homozygous knockouts of NOX1 and NOX2 significantly increased lifespan of the hybrid B6SJL transgenic SODG93A mice. The greatest increase in survival was achieved in the NOX2 knockout (almost 100 days) compared with NOX1 knockout (around 33 days), indicating that NOX2 plays a predominant role. The extension of survival in the ALS SODG93A mouse on NOX disruption was also reported by Harraz et al. (98). However, results from Wu et al. (341) showed that NOX2 deletion in the ALS mice only increases survival to 15 days. It is obvious that further characterization of the role of NOX2 in ALS is still required.
B. NOS enzymes
Another hallmark of microglial over-activation is the high production of NO, but it should be emphasized that NO production is much more relevant in rodents than in humans. In fact, NOS activation is one of the distinctive characteristics in the inflammatory response between these two species, and this difference has been frequently underestimated at the time of extrapolating results from rodent models to human diseases.
At low concentrations, NO plays a significant role in several aspects of CNS physiology, including neurotransmission, vasodilation, cognition, sleep, appetite, body temperature, and neurosecretion (38, 170). Microglial NO is mainly synthesized from l-Arginine by the iNOS. iNOS functions independently of a rise in intracellular Ca2+ unlike the other two constitutive NOS isoenzymes (endothelial NOS [eNOS] and neuronal NOS [nNOS]) and is transcriptionally induced by inflammatory mediators such as LPS and pro-inflammatory cytokines. Once induced, iNOS continuously produces high levels of NO that may cause neuronal death by mechanisms such as inhibition of mitochondrial cytochrome oxidase in neurons (31), inhibition of mitochondrial respiratory function, and release of exocytotoxic concentrations of glutamate via the N-methyl-D-aspartate receptor (293). Another important factor for NOS-mediated ROS production is the availability of its cofactor tetrahydrobiopterin (BH4). nNOS and eNOS can form dimers in the absence of BH4 (324), whereas iNOS dimerization requires the presence of BH4 (11). In NOS catalysis, BH4 controls coupling of the heme-oxygen intermediate with l-Arginine oxidation, thus controlling the generation of either NO or superoxide (187). When BH4 levels become deficient, the main product of NOS enzymes becomes O2−, produced from the oxygenase domain of the enzyme. Therefore, the lack of BH4 converts iNOS into an O2−-producing enzyme, altering the redox balance and leading to neuronal injury and more neuroinflammation.
Microglial iNOS can be regulated at transcriptional and post-transcriptional levels but as an inducible gene, it is mainly regulated at the transcriptional level. The most important transcription factor involved in iNOS expression is the NF-κB heterodimer p50:p65. All known inducers of iNOS have been shown to recruit NF-κB via one or more kinase pathways, such as tyrosine kinases (JAK, SRC family), MAPKK, protein kinase A (PKA), phosphatidyl inositol-3 kinase (PI3K), and protein kinase C (PKC) (269). In addition to these pathways, iNOS expression is also regulated by intracellular ROS production (269).
C. Cross-talk between RNS and ROS
ROS derived from NOX act as second messengers to induce NO production. In this regard, microglial cells treated with antioxidant compounds such as N-acetylcysteine, pyrrolidine dithiocarbamate, and lycopene reduce their NO content (221, 225). In addition, iNOS activation in response to LPS/IFN-γ or Aβ is prevented by over-expression of either a non-active p47phox or a mutant RAC1 subunit of NOX, respectively (53, 231). Min et al. (193) showed that ganglioside induces the activation of microglia and the production of pro-inflammatory cytokines (IL-1β, TNF), and iNOS was attenuated by the inhibition of NOX system with diphenylene iodonium. Furthermore, Mander and Brown (174) showed that neurodegeneration induced by different pro-inflammatory stimuli, such as TNF, IL-1β, LPS, ATP, or phorbol 12-myristate 13-acetate, is mediated by dual activation of iNOS and NOX. In fact, synergic activation of NOX and NOS in microglia resulted in neuronal apoptosis that was prevented using scavengers of O2− anion or peroxynitrite (ONOO−).
At a high concentration, NO actively unbalances redox homeostasis due to its reactivity with ROS. NO can react with molecular oxygen and form nitrogen trioxide (N2O3) that not only efficiently nitrosylates thiols and amines, but also combines with O2− to generate ONOO− that rapidly reacts with CO2, producing nitrogen dioxide (NO2) and carbonate (CO3−) which are also responsible for NO-related cytotoxicity. Figure 3 depicts the main ROS and RNS and how they modify several macromolecules. A more detailed description of their impact on lipid modification will be described later (Fig. 7).
FIG. 3.

Cross-talk NOX–NOS: H2O2 directly produced by NOX (NOX4 releases predominantly H2O2) of generated after O2− dismutation can induce cystein oxidation, S-glutathionylation, lipid peroxidation, and a reaction with other peroxides or non-specific oxidation of other molecules. NO, as a result of the transformation of l-arginine by NOS, can react with O2− and produce ONOO−, a molecule that can induce tyrosine nitration, S-glutathionylation of diverse molecules, or reduction of ferric sulphide. H2O2, hydrogen peroxide; NO, nitric oxide.
FIG. 7.

Oxidized lipid by-products formed and detected during chronic neuroinflammatory diseases. During acute neuroinflammation, O2− is formed through the microglial NOX system. As inflammation progresses to the chronic state, iNOS produces nitric oxide. Since these two inflammatory phases can coexist, ONOO− is formed in a reaction of O2− and NO radical. ONOO− can react with carbon dioxide, thus forming highly reactive radicals (nitric dioxide radical, hidroxyl radical, and carbonate radical). Each of them has the ability to initiate lipid peroxidation. As a result, aldehydes–HNE, ACR, and MDA are formed. During the oxidation of AA and DHA, isoprostanes and neuroprostanes are formed. However, IsoPs and NeuroPs are detected only in mild cognitive impairment and AD, ACR, MDA, and HNE can also be found in PD, MS, and Hungtington disease. AA, arachidonic acid; ACR, acrolein; AD, Alzheimer's disease; DHA, doecosahexaenoic acid; HNE, 4-hydroxy-2-nonenal; IsoPs, isoprostanes; MDA, malondialdehyde; MS, multiple sclerosis; NeuroPs, neuroprostanes; PD, Parkinson's disease.
Although the required levels of NO may be rather high to achieve neuronal cell death, an alternative mechanism would be for NO to react with O2− generated via NOX and produce ONOO−, which is potentially more neurotoxic than NO or O2− alone (13, 21). As an example, the study by Wang and co-workers showed that in iNOS-deficient mice, Aβ did not inhibit long-term potentiation (LTP). Moreover, a NOX inhibitor prevented Aβ-mediated inhibition of LTP (365). Therefore, the interplay between iNOS and NOX to produce O2−, NO, and ONOO− seems to be essential in neurodegeneration.
IV. Glutathione and Microglial Redox Regulation
Despite its high dependence on oxidative metabolism, the brain has low levels of antioxidants and is vulnerable to oxidative stress. The activities of catalase, SOD, and GPx are modest in comparison to the liver and kidney (40, 300). GSH is the main low-molecular-weight antioxidant, and it has a crucial role in maintaining redox balance, particularly in astrocytes and microglia. The cytosolic concentration of GSH is typically in the low micromolar range with the majority (>90% in microglia) in the reduced (GSH) form. In culture, microglia maintain a higher concentration of GSH than either astrocytes or neurones (43, 104, 106), which underlies their considerable resistance to oxidative stress. As a strong reducing agent, GSH contributes in several ways to antioxidant defence: (i) rapid non-enzymatic clearance of NO and O2−, which is coupled to oxidation of GSH to GSSG (Fig. 4A); (ii) as an electron donor in GPx-coupled reduction of H2O2 and ONOO− (Fig. 4B). The expression and activity of GPx is higher in human and rat microglia than in neurones or astrocytes (104, 155, 247) and increases in response to oxidative stress. For example, the generation of oxygen-derived radicals after quinolinic acid-induced damage promotes expression of GPx (155). (iii) GSH participates in regeneration of the reduced form of α-tocopherol (vitamin E) (42) and (iv) acts as a co-factor in a number of cellular isomerization reactions (267). (v) GSH functions as both a transport and storage form of the amino acid cysteine.
FIG. 4.
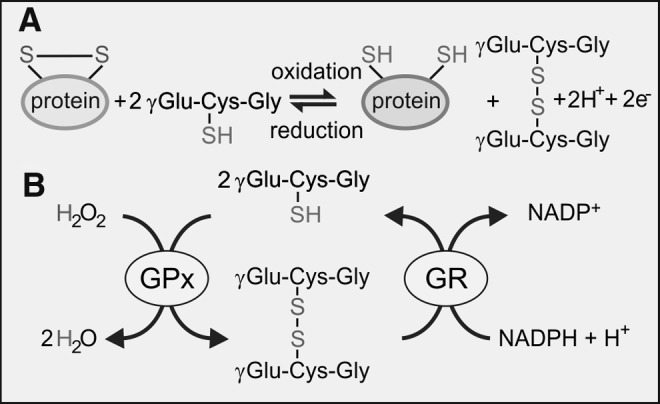
Oxidation of GSH by (A) non-enzymatic reduction of oxidized protein thiols and (B) GPx-catalyzed reduction of H2O2. Oxidized GSH is reduced by the NADPH-dependent enzyme, GR. GPx, glutathione peroxidase; GR, glutathione reductase; GSH, glutathione (γ-glutamyl-l-cysteinylglycine); NAD(P)H, nicotinamide adenine dinucleotide phosphate.
A. Glutathione metabolism
Synthesis of GSH takes place in the cytosol by two consecutive ATP-requiring reactions that form the first steps of the γ-glutamyl cycle, as illustrated in Figure 5. Loss of glutathione occurs as a result of its participation in redox or conjugation reactions, or due to export from the cell. During the GPx-catalyzed detoxification of ROS, GSH is oxidized to GSSG and is recycled back to GSH by glutathione reductase (GR), an enzyme that is highly expressed in microglia (104).
FIG. 5.

The γglutamyl cycle. Cystine enters the cytosol via the Xc− cystine-glutamate exchanger (1) and is reduced to cysteine. Glutamate cysteine ligase catalyzes the formation of γglutamyl cysteine (2), and GSH is formed by the addition of glycine by glutathione synthase (3). After the export of GSH, γGT catalyzes transfer of the γglutamyl moiety to an acceptor amino acid (4), which is transferred to the cytosol and recycled via 5-oxoproline to glutamate (5). Cysteinylglycine is hydrolyzed by a DP that liberates cysteine and glycine (6). GSSG: oxidized form of GSH. DP, dipeptidase; GSSG, glutathione disulfide; γGT, γ-glutamyl transpeptidase.
GSH synthesis is controlled by feedback inhibition of γ-glutamylcysteine synthetase (γ-GCL) by GSH and by availability of cysteine. As shown in Figure 5, cysteine is imported into microglia in its oxidized form, cystine, from the extracellular space using the plasma membrane Xc− cystine-glutamate exchanger. This is a two-subunit protein that operates as a sodium-independent, chloride-dependent electro-neutral antiporter, releasing glutamate in exchange for cystine in a 1:1 ratio (19, 184, 273). Glutamate released via the exchanger is recycled into microglia via high-affinity glutamate transporters [excitatory amino acid transporters (EAATs)], which is critical for avoiding the accumulation of extracellular glutamate that would pose a threat to neuronal survival (Fig. 6). Furthermore, glutamate uptake is necessary for providing the driving force to import more cystine via the exchanger. The capacity of microglia to uptake glutamate is∼10% that of astrocytes, but its value is closely coupled to GSH production (234, 235).
FIG. 6.

Impact of microglial activation on extracellular glutamate. Under physiological conditions, microglia accumulate cystine (CySS), via the Xc− cystine-glutamate exchanger, that is reduced to cysteine (Cys), the rate-limiting precursor for GSH synthesis. Glutamate released by the exchanger is taken up by high-affinity glutamate transporters on astrocytes. Activation of microglia by LPS acting through TLR4 causes up-regulation of the Xc− exchanger and increased release of glutamate. Simultaneously, activated cells release ROS and pro-inflammatory cytokines that block high-affinity glutamate transporters, leading to activation of neuronal glutamate receptors and risk of cell death by excitotoxic mechanisms. LPS, lipopolysaccharide; TLR, toll-like receptor.
B. Cystine-glutamate exchanger (Xc−) and glutamate homeostasis in microglia
Pro-inflammatory M1 activation of microglia causes up-regulation of the Xc− exchanger (240) through increased expression of its catalytic subunit (223). The glutamate transporter 1 (GLT-1) subtype of high-affinity glutamate transporters is similarly up-regulated, and it mediates a higher intracellular glutamate level to drive cystine import and GSH synthesis (234, 368). However, the rate of high-affinity uptake of glutamate in activated microglia never matches that of astrocytes and, curiously, astrocytic glutamate uptake is inhibited after microglial activation (368) (Fig. 6). Consequently, M1 microglia actively promote neuronal degeneration through failure of high-affinity glutamate uptake systems and remove extracellular glutamate effectively. The situation is exacerbated by enhanced release of glutamate from M1 microglia due to up-regulation of the Xc− exchanger (14, 15, 126, 368). Macrophage/microglial cells from patients with MS and mice with experimental autoimmune encephalomyelitis (EAE) have higher xCT (subunit of the Xc− exchanger) expression than controls (223) and may result in higher glutamate release, causing toxicity to oligodendrocytes. It has been proposed that the modulation of thiol redox balance in microglia, possibly by targeting the Xc− exchanger, could be an effective approach for attenuating injurious inflammatory cascades (126). Recent evidence shows that several flavonoid extracts from Rhus verniciflua were effective in preventing glutamate-mediated toxicity and oxidative stress in microglial cells in vitro (46).
C. Microglial phenotypes and the glutathione pool
Several studies report that microglial activation alters the GSH pool, although the response differs among cell lines. For instance, LPS/IFN-γ-induced activation of iNOS in both enriched primary microglial cultures and the N11 microglial cell line elicits a 40% decrease in GSH that can be blocked by the inhibition of iNOS (44, 200, 214). Similar observations have been recorded using N9 microglia, although it was noted that mitochondrial GSH was unaffected by the drop in total GSH after LPS/IFN-γ treatment (266). However, in BV2 microglial cells, the depletion in GSH in response to LPS/IFN-γ was independent of NO production (200). Furthermore, two independent studies have shown that TNF treatment of primary microglial cells in culture produced a significant increase in GSH that was accompanied by a significant reduction in ROS levels (75, 235). The opposite occurred in oligodendrocytes, and it was concluded that signals from TNF induce an antioxidant response in microglia which is absent in oligodendrocytes.
There are several instances in which changes in the GSH pool alter the extent of microglial activation. Thus, depletion of GSH in human microglial cells with the γ-GCL inhibitor buthionine-S-sulfoximine (BSO) induces oxidative stress and an inflammatory response that causes the cells to secrete TNF, IL-6, and nitrite ions which are toxic to neuroblastoma SH-SY5Y cells (145). A similar response occurs in astrocytes, which in both cell types is linked to calcium influx through TRPM2 channels. As expected, the stimulation of GSH synthesis in microglia has the opposite effect and reduces the release of pro-inflammatory factors that favorrs SH-SY5Y cell survival (146). Similarly, BV2 cells are less activated after exposure to Aβ peptide or LPS stimulation when γ-GCL is up-regulated, leading to augmentation of GSH (127, 329). Correspondingly, the expression of pro-inflammatory signals such as TNF, IL-1β and iNOS is reduced in N-acetyl cysteine-treated rats during experimental stroke compared with that in vehicle-treated animals (125). Taken together, these studies illustrate the direct link between oxidative signaling and the activation profile of microglial cells.
The GSH pool plays a critical role in chronic inflammation linked to neurodegenerative diseases and, thus, represents a potential target for treatment. This view arises from the results of numerous studies in which the depletion of brain GSH with BSO exacerbates the neurotoxic effects of a range of conditions that contribute to oxidative damage, such as ischemia (196), 1-methyl-4-phenylpyridinium (MPP+), or 6-OHDA [(290) and references therein]. In fact, the content of GSH is lowered in substantia nigra from PD patients, notwithstanding the fact that this area exhibits higher γGT expression (290). Similarly, microglial antioxidant pathways are being considered potential therapeutic targets in other neurological disorders. For instance, the phase II anti-Parkinson's drug, safinamide, may have potential as an anti-inflammatory agent in the treatment of MS. In activated microglia, safinamide blocked superoxide production and elevated GSH synthesis, thus providing protection against neuronal deficit and axonal degeneration in the EAE model of the disease (199). In other work, Pettit et al. (239) have recently shown that doecosahexaenoic acid (DHA), a naturally occurring anti-inflammatory agent, increases the GSH content of microglia in vitro, particularly when administered in combination with aspirin. In this context, it may be significant that DHA increases GLT-1-mediated uptake of glutamate (25). Finally, it has recently been discovered that brain tumors induced by chronic exposure to acrylonitrile are primarily of microglial origin and that the response of microglia to the chemical in vitro is different from that of astrocytes (34). Microglia display greater oxidative stress and a lower elevation in GSH compared with astrocytes that suggests a heightened sensitivity to the toxin in these cells.
V. Oxidized Lipid By-Products and Chronic Inflammation
Neural membranes are believed to be a Pandora's box with a wide spectrum of lipid mediators whose activities range from neuroprotective to neurotoxic effects (20). These mediators include free fatty acids such as arachidonic acid (AA), DHA, and lyso-glycerophospholipids (prostaglandins, leukotrienes, and platelet activating factor). While AA and lyso-glycerophospholipid metabolites are mostly pro-inflammatory, DHA metabolites (resolvins, lipoxins, and neuroprotectins) have primarily anti-inflammatory properties (85). The protective ability of resolvins and neuroprotectins lies in their capacity to inhibit IL-1β-induced NF-κB and COX-2 expression (176). In addition, DHA and eicosapentaenoic acid have been shown to reduce chronic inflammation, probably by decreasing IκB phosphorylation, thus attenuating NF-κB (65, 85).
A. Microglia can induce lipid peroxidation
As indicated in other sections of this review, NOX and iNOS activation in microglia lead to the generation of ROS and RNS, which combine together and form ONOO−. In fact, ONOO− production may be quantitatively more relevant than O2− and H2O2 production, at least in rodents (32). As shown in Figure 7, ONOO− further decomposes to highly reactive radicals—nitrogen dioxide radical (•NO2), •OH, and carbonate radical (•CO3). Each of these radicals has the potency to induce lipid peroxidation (96), a multistep autocatalytic process that results in the generation of oxysterols, hydroperoxides, and endoperoxides. The latter is further metabolized into a variety of reactive α,β aldehydes (4-hydroxy-2-nonenal [HNE], and acrolein [ACR]), dialdehydes such as malondialdehyde (MDA) and glyoxal, and keto-aldehydes (4-oxo-trans-2-nonenal, and isoketals) (93). Unlike ROS, reactive aldehydes are relatively stable and can diffuse from the original site of injury, thus spreading the initial oxidative damage (224). Notwithstanding the past two decades of extensive research in the field of reactive aldehydes, their role and function in cell biology remains largely unexplored. Of the three major aldehydes, HNE is the best characterized and participates in many signaling pathways that regulate cell physiology (236). Despite the fact that ACR is 100 times more reactive than HNE, its association with several brain pathologies, including neurodegenerative disease, is only beginning to be understood (60). MDA is, on the other hand, almost completely neglected from the viewpoint of its role as a signaling molecule.
To date, there has been little work done on analyzing the specific effects of lipids and their derivatives on microglial function. Nevertheless, several in vitro studies have highlighted their importance in the onset of neurodegeneration (Fig. 7). Thus, using an in vitro model of PD, Kim and colleagues (128) revealed that microglia are attracted by lipid components released from dying dopaminergic neurons. Similarly, lysophosphatidic acid was found to act as chemoattractant for microglial cells (276). The significance of lipid peroxidation products in neurodegeneration is further reinforced by the fact that both HNE and ACR are able to induce glutamate release from activated cultured microglia (15). Namely, as has been already discussed, an increase in extracellular glutamate arising from M1 microglia leads to neural degeneration mediated by HNE. On the other hand, DHA has displayed modulatory effects on the inflammatory properties of activated microglia through reduced secretion of pro-inflammatory cytokines and NO and an increase in the GSH pool (239). The only neurodegenerative disorder where lipid peroxidation products (HNE, 4-hydroxyhexenal and crotonaldehyde) were found present in microglia is ALS (287, 352). It seems that in the context of lipid peroxidation, microglia is explored predominantly as a source of highly reactive radicals which are able to initiate this process. Major oxidized lipid by-products related to chronic neuroinflammation are discussed next.
1. 4-Hydroxy-2-nonenal
As a product of ω-6 polyunsaturated fatty acid peroxidation, HNE is a second messenger of free radicals due to its involvement in cell signaling under normal and pathological conditions (82, 208, 354, 357, 358). The participation of HNE in brain pathologies is at the moment mostly explored in the context of AD, ranging from mild cognitive impairment (MCI), through early stage AD (EAD) and late AD (LAD). Reed et al. were the first to identify specific HNE-bound proteins in hippocampus and inferior parietal lobule from MCI patients, some of which were significantly elevated compared with the control (259). Throughout the course of AD, several significant categories of proteins were found to be HNE modified. While in MCI these categories include energy metabolism, mitochondrial dysfunction, cytoskeletal integrity, antioxidant defense, protein synthesis, stress response, neuronal communication, and excitoxicity, EAD is mainly characterized by HNE-modified proteins that are involved in energy and mitochondrial dysfunction. In the LAD, proteins involved in antioxidant defense and cytoskeletal integrity appear to be HNE modified (33). Increased levels of free HNE were observed in the amygdala, hippocampus, and parahippocampal gyrus of AD brain compared with age-matched controls. This increased alkenal concentration corresponded to the regions showing the most striking histopathologic alterations in AD (181). With regard to other neurodegenerative diseases, HNE was detected immunohistochemically in 58% of surviving neurons of substantia nigra from individuals affected by PD, as compared with 9% in control subjects (353). Besides this, HNE adducts were found in Lewy bodies in neocortical and brain stem neurons of PD patients (41). Moreover, HNE alters dopamine uptake after binding to sulfhydryl groups of the dopamine transporter and, therefore, may affect the onset and progression of PD (354). In another disease with a very strong inflammatory component, MS, a post-mortem analysis likewise revealed HNE in early and actively demyelinating plaques (210). Even though free HNE was not detected in MS, a large accumulation of both lysine and histidine HNE adducts was observed, implying a role of HNE in protein modifications (336). Finally, increased levels of HNE adducts were also found in Huntington disease (HD) (260). Together, these observations suggest a very significant production of HNE under inflammatory conditions as well as its participation in several proteinopathies, including AD, PD, and HD.
2. Acrolein
Unlike HNE, ACR has many sources of origin. It is not only formed during incomplete combustion of petrol, coal, wood, and plastic material but is also present in cigarette smoke and formed in overheated frying oils. ACR is a known metabolite of the widely used anticancer drug cyclophosphamide and can be formed intracellularly by enzymatic oxidation of polyamine metabolites. As a result, only a small part of ACR in the cell results directly from lipid peroxidation (82). Nevertheless, its role in numerous pathologies is becoming more pronounced. With regard to neurodegenerative diseases, ACR has been mostly studied in AD. Thus, more than half of neurofibrillary tangles in AD cases showed strong ACR immunoreactivity (39). A significant increase in extractable ACR in AD amygdala, hippocampus, and parahippocampal gyrus was observed compared with age-matched controls (162). ACR increases in a disease progression-dependent manner in AD. Significantly elevated ACR levels were found in superior and middle temporal gyrus during MCI with expansion to hippocampus and parahippocampal gyrus in EAD (337). Besides protein adducts, ACR can induce DNA modifications, resulting in ACR-deoxyguanosine adducts. An increase in these adducts was demonstrated in brain tissue from AD patients, implying their role in mutagenesis and carcinogenesis and thus contributing to the pathogenesis of neurodegenerative diseases (159). With regard to PD, it has been reported that in the substantia nigra from PD patients, ACR-modified α-synuclein accumulates mainly in the cytoplasm of the nigral melanized neurons (283).
3. Malondialdehyde
The main source of MDA in biological samples is the peroxidation of polyunsaturated fatty acids with two or more methylene-interrupted double bonds. However, MDA can be also generated in vivo by enzymatic conversion from various prostaglandins (69). Several post-mortem studies have shown elevated MDA levels in the hippocampus, pyriform cortex and amygdala, and temporal, frontal, parietal, and occipital cortices of the AD brain, as compared with young and age-matched controls (74). Serum levels of MDA have been suggested as markers of neurodegenerative processes. Indeed, increased MDA levels in serum of MCI, EAD, and LAD patients were observed (10, 220) and even erythrocyte MDA levels have been proposed as markers of cognitive deterioration (70). MDA immunoreactivity was seen in lipofuscin granules in neurons and macrophages in MS with additional staining predominantly present in myelin sheaths in active lesions (95). Increased MDA levels were also reported in the cerebrospinal fluid (CSF) of patients with PD (74).
4. Isoprostanes and neuroprostanes
Isoprostanes (IsoPs) are stable products of free radical reactions with AA, while neuroprostanes (NeuroPs) are oxidation products of DHA. NeuroPs provide a unique window for quantification of oxidative damage primarily in neuronal membranes in vivo (180). Total NeuroP, but not total IsoP, levels were found to be greater in AD patients than in controls, but only in those brain regions involved by AD (198). On the contrary, in MCI, increased IsoPs levels were detected in middle frontal gyrus, inferior parietal lobule, and occipital area compared with controls. In addition NeuroPs were elevated in the same areas, besides middle frontal gyrus, including hippocampus (180).
Taking into consideration the earlier mentioned implications of oxidized lipids in neurodegenerative disorders characterized by chronic inflammation, one can easily conclude that oxidized lipids have a far-reaching impact in the onset of these diseases. Affecting numerous proteins, most of which are still not identified, oxidized lipids interfere with the main signaling pathways that are responsible for the cell functioning. By elucidating the consequences that oxidized lipids and their metabolites have on the cell, we will be one step closer not only to determining the onset of the disease, but also to determining new therapeutic approaches.
VI. Redox Signaling Through Protein Sulfhydryl Groups
Traditionally, ROS/RNS have been viewed as molecules that compromise normal cellular physiology as a result of damage to DNA, proteins, and lipids. However, it is becoming increasingly clear that protein modification by ROS/RNS does not occur indiscriminately and that certain cysteine residues in proteins are more prone to oxidative modification than others. Indeed, it has been suggested that low levels of ROS/RNS may act as signaling molecules by causing reversible and covalent modification of redox-sensitive proteins via cysteine residue(s) which are exposed on the surface of the protein. Figure 8 depicts the main changes of proteins through reversible and irreversible modification of their cysteine residues.
FIG. 8.
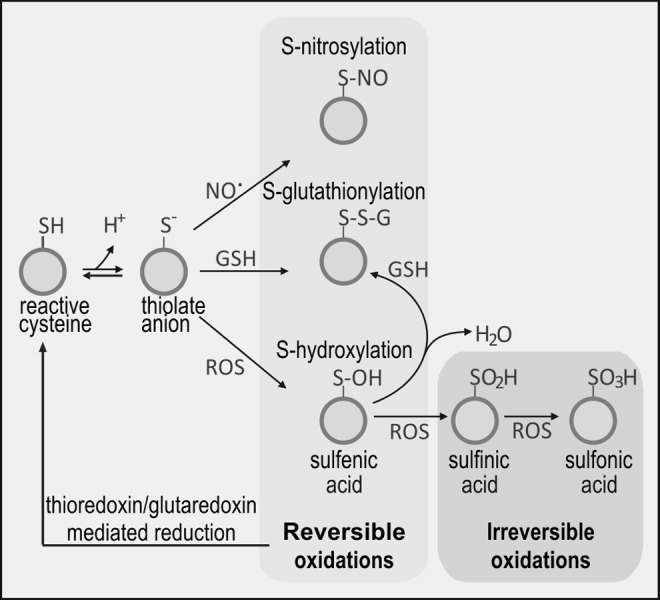
Protein thiolation. Oxidation of protein–SH groups generates thiolate anion that may react with NO (S-nitrosylation), GSH (S-glutathionylation), or ROS (S-hydroxylation). Exposure to high levels of ROS causes irreversible oxidation to sulfinic and sulfonic acids.
A. Protein-S-thiol modification
S-thiolation is an important means of protecting protein –SH groups from damage by oxidation, which, if not controlled, generates sulfenic (P–SOH), sulfinic (P–SO2H), or sulfonic (P–SO3H) acids. The extent of oxidation will depend on the cell oxidative level: high levels of ROS generate -sulfine and -sulfone derivatives, which are irreversible and associated with oxidative damage. Low levels of ROS/RNS generate reversible protein-cys-sulfene derivatives. However, these are intrinsically unstable and are prone to further oxidation to sulfinic or sulfonic acids. They may also bind to vicinal thiols, forming intramolecular disulfide bonds or be scavenged by GSH, yielding P–S–S–G. This process is known as S-glutathionylation (syn. S-glutathiolation) and depends on the local supply of GSH [(59) and references therein]. The mechanism of S-glutathionylation is not entirely clear, but at the concentration of GSH that pertains in most cells and particularly in microglia, spontaneous disulfide exchange is likely (318). In addition, there is evidence that enzymatically catalyzed S-glutathionylation may occur in conditions of severe oxidative stress. De-glutathionylation is commonly catalyzed by GR.
B. Protein-thiol modification as a signaling mechanism in microglia
To date, there are a few reports of protein thiol modification specifically in microglial cells, yet evidence is accumulating, primarily from studies on macrophages/monocytes that this is undoubtedly an important aspect of the cellular response to, and protection from, oxidative stress. However, there is a fine balance between physiological and pathological function and, in conditions where the GSH pool is depleted, for example in neurodegenerative diseases, partially oxidized proteins may react with oxygen or other oxidants and produce irreversibly oxidized sulfines and sulfones, thus exacerbating oxidative damage.
In 1994, Ravichandran et al. (257) demonstrated that glyceraldehyde-3-phosphate was transiently S-thiolated (most commonly by glutathione, but also by cysteine) minutes after the respiratory burst in monocytes. Since then, there have been several other reports on the impact of S-thiolation or nitrosylation on cellular function that include, in the main, the following areas: (i) microglial activation and recruitment; (ii) cytoskeletal rearrangements and cellular trafficking; (iii) regulation of signaling pathways; and (iv) regulation of transcription. For example, oxidative S-glutathionylation is critical in the tyrosine kinase-dependent signaling pathway, leading to β2-integrin CD11b (Mac-1) activation required for cell migration (29) and for modification of β-actin assembly (59). In addition, the E1 and E2 components of the ubiquitin-proteasome system are reversibly inhibited by S-glutathionylation during oxidative stress (217), causing dysfunction of the protein elimination pathway. Furthermore, the Rpn1 and Rpn2 subunits of the 19S regulatory particle of the 26S proteasomal pathway are inhibited by S-glutathionylation after exposure to H2O2 and GSH in neutrophils in vitro (377). With regard to signaling pathways, NO, produced in response to IFN-γ treatment, negatively regulates c-Jun N-terminal kinase (JNK) by S-nitrosylation, thereby inhibiting interaction between JNK and c-JUN (227). TGF-β1 stimulates NOX4 and ROS production in murine fibroblasts via thiolation and inactivation of mitogen activated protein kinase (MAPK) phosphatase-1, a nuclear phosphatase (158), which leads to a sustained activation of JNK and p38 signaling cascades and is a prime example of cross-talk between regulation of protein phosphorylation and oxidative stress. Similarly, inactivation of calcium-calmodulin-dependent kinase 1 is achieved by S-glutathionylation of the active site cysteine (Cys179) (120). In macrophages, this kinase mediates the in vitro response to LPS and may also operate in vivo to regulate inflammation and organ dysfunction arising from sepsis (366). Similarly, most members of the PKC family of proteins contain several cysteine-rich regions in their catalytic and regulatory domains and, as such, are likely targets of modification by thiolation. Indeed, S-glutathionylation in vitro inhibits PKC isozymes, raising the possibility that GSH may be anti-tumourigenic via this mechanism (50). There are several examples of regulation of transcription by protein thiolation or nitrosylation. For example, redox-triggered reversible S-thiolation of transcription factor c-JUN regulates DNA binding (130). S-nitrosylation of karyopherin chromosomal region maintenance 1 (the major receptor for classical nuclear protein export) inhibits its interaction with nuclear export signals and blocks protein export from the nucleus (333). The human p53 tumor suppressor is inhibited by S-glutathionylation both in vitro and in human cancer cells as a result of oxidative stress (323). Other regulators of the inflammatory response, for example the transcription factor NRF2, are also influenced by S-thiol modification and are discussed in greater detail in Section VII.
An example of protein-thiolation in a pathological context comes from the observation that an oxidative burst in BV2 murine microglial cells leads to S-glutathionylation-mediated inhibition of insulin degrading enzyme and reduced degradation of Aβ peptide, with direct implications for progression of AD (254). Similarly, nitrosylation can also be associated with disease. For example, α-synuclein, the chief component of Lewy bodies released from damaged dopaminergic neurons in PD, is nitrosylated as a result of oxidative stress, and this initiates an inflammatory response in microglia (261).
In summary, S-glutathionylation and nitrosylation represent significant mechanisms for controlling the activity of microglia in a wide range of contexts and play an important role in determining cell fate and responsiveness to oxidative stress.
C. Thioredoxin reductase system
The thioredoxin reductase (TrxR) system comprises thioredoxin (Trx), TrxR, and NADPH, which, in mammalian cells, is derived from the pentose phosphate pathway and, to a lesser extent, by the malic enzyme. Trxs are a small family of redox-active proteins (10–12 kDa) that are critical for maintaining cellular redox balance and antioxidant function, including control of oxidative stress and cell death. While few, if any, studies have been performed with microglial cells, it is highly likely that this system plays an important role in the respiratory burst and protection of the cell from oxidative stress.
The redox activity of Trx is due to the presence of a highly conserved catalytic site with the sequence Trp-Cys-Gly-Pro-Cys-Lys. The negative redox potential of the two cysteine residues provides a strong reducing environment whose purpose is to protect protein-SH from oxidation and/or recycle S-thiolated proteins. Two forms of Trx exist in mammalian cells: Trx-1, which localizes to the cytoplasm, and Trx-2, which is found in mitochondria. In addition to re-cycling S-thiolated proteins, Trx has a number of additional roles, including acting as a co-factor in DNA synthesis by providing electrons for reduction of ribose by ribonucleotide reductase and inhibition of apoptosis (26). Once oxidized, Trx is itself recycled by the action of the flavoenzyme, TrxR which, similar to GPx, contains selenocysteine at its active site (8).
Hägglund et al. adopted a genomics-based approach in an effort to identify targets of Trx. Trx-reduced disulfides were discovered among a number of well-known proteins, including peroxiredoxin (Prx) and cyclophilin (94). In addition, a number of ribosomal proteins were identified, suggesting that Trx plays an active role in the regulation of translation. Dehydroascorbate reductase, the enzyme that re-generates vitamin C from dehydroascorbate, was also identified as an important target, implying that the Trx system is involved in the ascorbate-glutathione cycle. Additional Trx targets include oxidized protein tyrosine phosphatases, for example, PTP1B. In certain cell signaling cascades, the production of H2O2 inactivates PTP1B by oxidizing a cysteine sulfhydryl group (230). Trx has been shown to bind to apoptosis-signal-regulating kinase 1 (ASK1), thereby blocking its activity and preventing both stress- and cytokine-mediated apoptosis. As Trx becomes oxidized, it disassociates from the kinase and apoptosis is stimulated. Furthermore, the binding of Trx by its inhibitor, Trx-interacting protein (TXNIP) also contributes to apoptosis by removing Trx from ASK1 (185). Glutaredoxin 2 has also been identified as a TrxR target (164). Other substrates for TrxR include cytochrome C (205) and heme oxygenase-1 (HO-1) (315). Trx-1 induces p53-dependent cell p21 transactivation, leading to cell-cycle arrest and DNA repair, which suggests a link between a response to oxidative stress and DNA repair mechanisms (317). In this context, it is worth noting that up-regulation of p53 has been detected in glial cells of neurodegenerative disease patients (62, 115). Furthermore, there are several reports of an increase in Trx in response to conditions that are known to cause activation of microglia, such as LPS (334), Aβ peptide (363), and IL-1β (284). Recent work has shown that ROS removal by brain mitochondria requires Trx (77).
In summary, it is becoming increasingly clear that the Trx system, as in the case of GSH, plays a pivotal role in the protection of microglial cells from oxidative stress. Future determinations will, doubtless, lead to a greater understanding of the interplay between these two key redox systems in controlling microglial cell function and stability.
VII. Microglial Polarization Is Controlled by Transcriptional Regulation
The eventual fate of microglial cells is tightly controlled by their gene expression profile and two ROS-regulated transcription factors, namely NF-κB and NRF2, are particularly important in this regard.
A. NF-κB transcription factor
The transcription factor NF-κB is expressed in neurons and microglia and exhibits a dual role in neurodegenerative diseases (183). Thus, activation of NF-κB in neurons promotes their survival; whereas activation in microglia may lead to pathological neuroinflammation. It is likely that in the brain, the pro-survival effect of NF-κB in neurons is more relevant than its pro-inflammatory role in microglia. Nevertheless, NF-κB is considered a master regulator of the inflammatory responses to brain infections and to environmental and cellular damage (Fig. 9).
FIG. 9.

NF-κB is the master regulator of the inflammatory response. NF-κB modulates the expression of genes containing κB regulatory elements in their gene promoters. These genes code for enzymes, cytokines, chemokines, survival factors, and adhesion molecules: iNOS, COX-2, manganese SOD2, chemokine (C-X-C motif ) ligand 1, 2, and 10 (CXCL1, CXCL2, and CXCL10), Bcl2, VCAM-1, and ICAM-1. Bcl2, B-cell lymphoma-2; COX2, cyclooxygenase-2; ICAM-1, intercellular adhesion molecule 1; SOD, superoxide dismutase; VCAM-1, vascular cell adhesion molecule 1.
The NF-κB family comprises five structural homologs: NF-κB1 (p50), NF-κB2 (p52), RelA (p65), RelB, and c-Rel. These proteins share a highly conserved 300-amino-acid N-terminal Rel homology domain (RHD) that is responsible for DNA binding, dimerization, and association with the ankyrin-containing protein IκB. The combination of the different dimers confers selectivity on the signal mediated through NF-κB in diverse cell types and under different conditions. The p65/p50 dimer is the best-characterized inducer of pro-inflammatory genes and is fully functional in microglia. Probably, since it happens in macrophages, p50 dimers participate in acquisition of the M2 phenotype. Thus, p50 null mice show exacerbated M1-driven inflammation and a defective capacity to induce M2-polarized reaction (245).
1. Redox levels regulate regulatory kinases upstream of NF-κB
NOX-derived ROS has been implicated in the activation of NF-κB and in the release of pro-inflammatory mediators by microglia. For instance, NOX mediate LPS-induced expression of TNF in M1 microglia, leading to loss of dopaminergic neurons and this effect is attenuated in the gp91−/− mice (251). Simultaneously, NF-κB inhibitors attenuate microglial activation (237). Several lines of evidence support the regulation of NF-κB by ROS at the level of upstream kinases. Most of the following studies correspond to in vitro cultures of macrophages and to a lesser extent of microglia and will be presented here on the assumption that NF-κB regulation is similar in these two monocytic cell types (Figs. 10 and 11). In addition, some of these general mechanisms may take place in neurons.
FIG. 10.

Redox regulation of NF-κB. ROS may activate or inactivate the IKK complex through phosphorylation or cysteine oxidation, respectively, leading to modulation of downstream targets. On the other hand, ROS influence the DNA-binding properties of the NF-κB proteins, (i) phosphorylation of p65 on two serine residues mediated by ROS leads to greater NF-κB activation; (ii) oxidation of p50 prevents its DNA binding, and this oxidation is reversed in the nucleus by a Trx1-dependent process. Trx, thioredoxin.
FIG. 11.
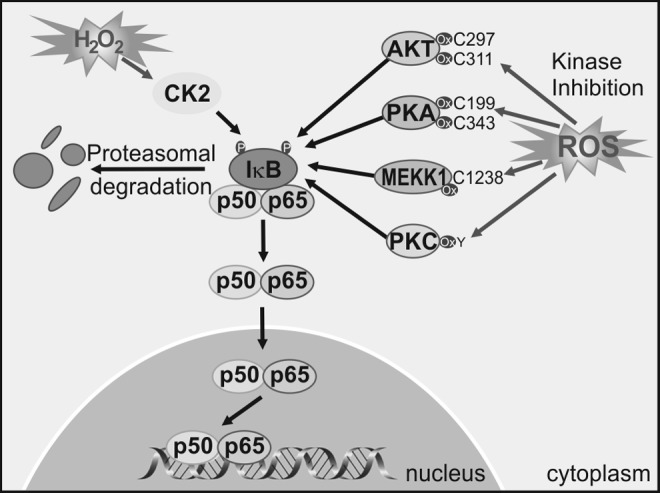
Redox regulation of NF-κB-upstream kinases. ROS may activate and inactivate NF-κB upstream kinases. Cysteine oxidation of residues in AKT, PKA, MEKK1, and PKC leads to kinase inactivation. On the other hand, ROS may activate CK2 that phosphorylates IκB, leading to activation of the NF-κB pathway. IκB, inhibitor of NF-κB; MEKK, mitogen-activated protein kinase/ERK kinase; PKA, protein kinase A; PKC, protein kinase C.
ROS activates upstream kinases that lead to NF-κB activation. Among those are IKKβ and probably IKKɛ, which create phosphorylate-specific serines in the NF-κB inhibitor IκBα. This phosphorylation targets IκB for degradation through the ubiquitin/proteasome pathway, thus relieving NF-κB from this constrain, and enabling NF-κB stabilization and nuclear translocation (362). Other kinases that participate indirectly in phosphorylation of NF-κB isoforms or regulate NF-κB indirectly through the IKK pathway include the Mitogen-activated protein kinase/ERK kinase-1 (209), AKT (121), PKC (73, 137, 149), CK2 (122), and PKA (370). The phosphorylation of the p65 isoform of NF-κB on Ser276 is necessary for the interaction of p65 with co-activators such as CBP/p300s required for the expression of a subset of NF-κB-dependent genes (216). PKA mediates phosphorylation of Ser276 (369, 371), and this event is considered dependent on ROS, as antioxidants inhibit Ser276 phosphorylation and CBP/p300 binding (114).
2. Redox state also controls NF-κB nuclear levels
ROS can influence the DNA-binding activity of NF-κB in the nucleus. Post-translational modifications of NF-κB are required for complex activation of NF-κB-dependent genes (91). Chromatin relaxation and remodeling are necessary for NF-κB binding to DNA, and transcriptional control is tightly regulated by acetylation/deacetylation of lysine residues in histone N-terminal tails. This regulation has been widely analyzed in lung macrophages. In resting monocyte cells, histone deacetylases (HDAC)1/p50 complexes bind to the DNA and repress transcription by local histone H3 deacetylation, which prevents RNA Pol II recruitment. On stimulation, p65 enters the nucleus, and associates with CBP/p300, leading to the displacement of p50/HDAC1 repressive complexes and enabling transcription (369). ROS levels profoundly alter HDAC1-3 function in activated macrophages, enabling NF-κB induction of pro-inflammatory genes (189, 347). The reduced HDAC2 activity is due to ROS-induced post-translational modifications such as nitration and nitrosylation (189, 347).
In addition to control of NF-κB levels, IKKα also regulates NF-κB nuclear stage in response to ROS. Macrophages stimulated with ROS inducers exhibited association of IKKα to the promoter of pro-inflammatory genes, leading to phosphorylation of Ser10 and acetylation of Lys9 on histone H3, enabling NF-κB interaction with DNA (348). Direct phosphorylation of p65 by PKA up-regulates the expression of a subset of NF-κB-dependent genes, including TNF, macrophage inflammatory protein 2, and monocyte chemotactic protein-1 (216). Phosphorylation of p65 in Ser276 by PKA enables an interaction of nuclear p65 with the co-activator CBP/p300 and, therefore, stimulates pro-inflammatory gene transactivation (369). This step is also regulated by the redox state, because antioxidant compounds impair PKA-induced phosphorylation of p65 at Ser276 and subsequent gene activation (114). Another post-translational modification that can occur is tyrosine nitration of p65 at two different tyrosines (Tyr66 and Tyr152) located in the RHD domain. Nitration of p65 results in the replacement of p65/p50 with the p50/p50 complexes or the association of p65 with IkBα that export NF-κB back to the cytosol (91). Cys62 of p50 provides another brilliant example of how cells use redox and S-thiol protein modification to control gene expression. Cys62 within the RHD domain of p50 should be reduced for efficient NF-κB DNA binding (311). In the cytoplasm of resting cells, Cys62-p50 is highly oxidized, but once NF-κB enters the nucleus, Cys62 is reduced by Trx (242) or AP endonuclease/redox factor 1 (6, 103). Interestingly, the same example illustrates how S-thiol modification by NO is used as a negative feedback mechanism to switch off pro-inflammatory signaling induced by NF-κB. NO production on cytokine stimulation is associated with S-nytrosylation of Cys62-p50 and Cys38-p65 (123) that could be prevented with antioxidant compounds.
A corollary of the previous data is that redox state governs microglial activation toward a pro-inflammatory or alternative phenotype. In the presence of ROS signaling molecules, more p65/p50 dimers should be formed, leading to pro-inflammatory gene transcription. On the contrary, under homeostatic redox conditions, p65 will not be available and other NF-κB dimers such as p50/p50 will be formed, leading to increased expression of anti-inflammatory genes.
B. NRF2 transcription factor
Redox homeostasis is tightly controlled by the transcription factor NRF2, which is a crucial player in the antioxidant protection of microglial cells, thus avoiding pernicious effects of oxidative stress and over-activation of the M1 phenotype.
NRF2, a member of the Cap'n'Collar family of basic region-leucine zipper transcription factors, was cloned by virtue of its binding to the promoter of the β-globin coding gene (HBB) that was necessary for erythropoiesis and platelet development (197). However, its function is not related to erythropoiesis, but to control of redox homeostasis and it is currently viewed as the master regulator of the antioxidant response. NRF2 binds to a cis-acting element termed the ARE and modulates transcription of its target genes (268). In addition, NRF2 requires a member of the small MAF proteins (F, G or K in vertebrates) in order to form a functional ARE-binding transcriptional complex (345). Over the past decade, information on NRF2 antioxidant function has been obtained from studies with NRF2-deficient mice. NRF2 deficiency substantially increases susceptibility to a broad range of chemical toxins and disease conditions associated with oxidative pathology, whereas pharmacological activation of NRF2 protects against oxidative damage (305).
Although NRF2 is ubiquitously expressed, it has a very significant role in the context of microglial redox homeostasis. ARE-regulated genes include (i) enzymes involved in ROS clearance such as SOD3, GPx, and Prx; (ii) enzymes that synthetize reducing factors, such as GSH (catalytic and regulatory subunits γ-GCL), NADPH (glucose-6-phosphate dehydrogenase, and phosphogluconate dehydrogenase); (iii) enzymes related with the regeneration of reduced cofactors and proteins, for example, reduction of GSSG by GR; (iv) reduction of oxidized Trx by TrxR and Prx-SO2H by sulfiredoxin; (v) redox transporters, such as Xc−; (vi) metallothioneins (MT1 and 2) and ferritin, responsible for clearing metals by chelation reactions; and (vii) enzymes directly related with production of antioxidant molecules such as HO-1, NAD(P)H:quinone oxidoreductase 1 (NQO1), and Trx as well as inhibition of expression of Trx inhibitor (TXNIP). In addition, NRF2 also participates in the regulation of proteasomal protein degradation, cell proliferation, autophagy, and metabolic reprogramming (136, 166, 195, 301). The functions of NRF2 are summarized in Figure 12, and Figure 13 highlights the mechanisms that are known to regulate NRF2 activity at the levels of protein stability, phosphorylation, and epigenetic modifications.
FIG. 12.

NRF2 is the master regulator of the redox homeostasis. NRF2 modulates redox homeostasis through the control of a wide battery of cytoprotective genes that share in common ARE sequences in their promoter. SOD3, GPx, Prx, glutamate-cysteine ligase catalytic and modulator subunits (GCLC and GCLM), G6PDH, 6PGD, GSR, TrxR, Srx, cystine/glutamate transport, metallothioneins (MT1 and 2), FT, HO-1, NQO1, γGT, γGS, Trx, and Trx inhibitor (TXNIP). 6PGD, phosphogluconate dehydrogenase; FT, ferritin; G6PDH, glucose-6-phosphate dehydrogenase; GCLC, glutamate-cysteine ligase catalytic subunit; GCLM, glutamate-cysteine ligase modulatory subunit; GSR, glutathione reductase; HO-1, heme oxygenase-1; Prx, peroxiredoxin; Srx, sulfiredoxin; TrxR, thioredoxin reductase; γGS, γ-glutamate cysteine synthetase.
FIG. 13.

Regulatory mechanisms of NRF2. Under surveillance mode, microglia maintains low NRF2 protein levels by its inhibition by the E3 ligase adapter KEAP1. (A), two NRF2-motifs, ETGE and DLG, interact with a separate Kelch-repeat domain present in the KEAP1 homodimer. KEAP1 and NRF2 interaction is controlled by the redox status of at least three critical cysteine residues of KEAP1 (Cys151, Cys273, and Cys288). High levels of ROS/RNS alter these cysteine residues, enabling NRF2 activation. (B) NRF2 is controlled by signaling cascades that are also profoundly influenced by ROS/RNS concentration. For instance, GSK-3β phosphorylates NRF2 and activates a KEAP1-independent degradation pathway that is dependent of a different E3-ligase named β-TrCP. (C) In addition, NRF2 is modulated by epigenetic mechanisms, including acetylation, CpG island methylation of its promoter region, and miRNA generation. β-TrCP, β-transducin repeat containing E3 ubiquitin protein ligase; GSK-3, glycogen synthase kinase-3; KEAP1, Kelch-like ECH-associated protein 1.
1. ROS control the NRF2 through redox modification of Kelch-like ECH-associated protein 1
In surveillance mode, microglia maintain low NRF2 protein levels by a well-established mechanism that requires the participation of the ubiquitin E3 ligase adapter Kelch-like ECH-associated protein 1 (KEAP1). KEAP1 associates with the N-terminal region of Cullin3 (CUL3), a subunit of the ubiquitin E3 ligase complex, and promotes the ubiquitination of NRF2 in cooperation with the CUL3-ROC1 complex, resulting in proteasomal degradation (57, 87, 131, 361).
The interaction between NRF2 and KEAP1 occurs via a “two-site tethering” mechanism, otherwise known as the “hinge and latch” mechanism. In this model, two NRF2-motifs, ETGE and DLG, interact with a separate Kelch-repeat domain present in the KEAP1 homodimer. It is thought that a single NRF2 polypeptide first binds one KEAP1 subunit through its high-affinity ETGE motif, and that this, in turn, enables the second low-affinity DLG motif to engage with the other Kelch-repeat domain of the remaining KEAP1 subunit (186, 313). The interaction between KEAP1 and NRF2 is controlled by the redox status of at least three critical cysteine residues of KEAP1 (Cys151, Cys273, and Cys288). When these cysteines are oxidized or participate in adduct formation with electrophilic compounds, the NRF2/KEAP1 interaction is disrupted in a way that NRF2 escapes degradation, enters the nucleus, dimerizes with small MAF proteins, and binds AREs in its target genes (132, 153, 346, 360). Microglial activation of NOX results in increased ROS levels that lead to modification of KEAP1 cysteines and further activation of NRF2.
Pharmacological regulation of the KEAP1-NRF2 complex has been crucial for understanding the physiological role of NRF2. For instance, sulforaphane (SFN) has been successfully used to increase NRF2 levels in the brain. SFN is an isothiocyanate derived from broccoli that crosses the BBB efficiently enough to induce NRF2 and its target genes in the brain parenchyma (111). Intraperitoneal administration of SFN in wild-type mice, but not in NRF2-null mice, protects against MPTP-induced death of nigral dopaminergic neurons. Interestingly, the neuroprotective effects are accompanied by a decrease in M1 markers of reactive microglia (116). A large number of other plant antioxidants and plant-derived dietary supplements have been reported to target the KEAP1/NRF2 complex and might have a therapeutic value in the modulation of microglial activation (150, 274).
2. NRF2 regulation by signaling kinases
NRF2 is controlled by signaling kinases that are closely regulated by ROS/RNS levels. For instance, PERK and PKA kinases, activated by misfolded proteins or phorbol esters, respectively, as well as PKC phosphorylate NRF2 and disrupt the association of NRF2 with KEAP1 (58, 107). Other kinases such as CK2 (7), Fyn (113), or MAPKs (124, 286, 299, 344, 375) phosphorylate NRF2 without altering its interaction with KEAP1.
In recent years, Cuadrado's group has reported that glycogen synthase kinase-3 (GSK-3)β, a kinase abnormally active in AD, inhibits NRF2 by nuclear exclusion (270). Additional studies have shown that GSK-3β phosphorylates NRF2 and activates an alternative degradation pathway that is dependent on a different E3-ligase named β-transducin repeat containing E3 ubiquitin protein ligase (51, 252, 253). The physiological relevance of the inhibition of NRF2 by GSK-3 is reflected in two studies where GSK-3β decreased NRF2 activity, thereby sensitizing neurons against oxidative damage (111, 263).
GSK-3β activity profoundly influences microglial physiology. Thus, inhibition of GSK-3β by genetic or pharmacological approaches reduces the induction of M1 markers in response to different challenges, including LPS, traumatic injury, or 6-OHDA, and this is accompanied by a reduction in brain inflammation. These inflammatory challenges fail to increase typical M1-markers when GSK-3 is inactive and, therefore, NRF2 is active (108, 134, 331).
3. Epigenetic modulation of NRF2
A growing body of evidence supports the existence of novel mechanisms to modulate NRF2 other than proteasomal degradation. In 2009, Zhang's group demonstrated that NRF2 is directly acetylated by CBP/p300 within the DNA-binding domain that augments binding of NRF2 to the AREs (298). In addition, H2O2 released by M1 microglia can up-regulate the protein p300/CBP and down-regulate HDACs. These events lead to increased binding of NRF2 to AREs (190). Astrocyte-rich cultures treated with conditioned medium from M1 microglia showed a time-dependent increase in astroglial HDAC activity that correlated with reduced acetylation of histones H3 and H4 and a further decrease in NRF2-related transcription (55). Accordingly, HDAC inhibition increased the expression of NRF2-target genes, Hmox1 (encoding HO-1), Nqo1, and Gclm. In contrast to wild-type animals, NRF2-deficient mice are not protected by pharmacological inhibition of HDAC after cerebral ischemia (330).
Methylation of cytosine-guanine (CG) islands within the NRF2 promoter has been shown to suppress its expression in mice with prostate cancer. Interestingly, NRF2 activity is restored by phytochemicals that reduce methylation of CG islands (297). With regard to oxidative stress, AD patients display high homocysteine and low vitamin B12 and folate levels in blood, suggesting a dysregulation of the S-adenosyl methionine cycle that is required for DNA methylation (275). Considering that the NRF2/HO-1 axis is up-regulated in AD microglia, the correlation between high NRF2 levels and lower methylation of NRF2 promoter sequences is reasonable.
NRF2 regulation by specific miRNAs is just beginning to be explored. Of note, miR153, miR27a, miR142-5p, and miR144 reduce NRF2 activation (206). Interestingly, miRNA metabolism is sensitive to oxidative stress and, therefore, may play a role in controlling NRF2 levels in microglia. It has recently been shown that DICER, a key enzyme for the synthesis of mature functional miRNA, is down-regulated by aging and as a result of oxidative stress elicited by H2O2. Interestingly, in the same study, the authors show that the recovery of redox balance restored DICER expression (319).
It is noteworthy that all mechanisms which are known to regulate NRF2 are sensitive to redox status. It is speculated that the increase in ROS/RNS levels during execution of the M1 program increases pro-inflammatory gene expression through the NF-κB pathway, as well as anti-oxidant gene expression by NRF2. This dual mechanism enables microglia to be primed to shut down the immune response and to return to a resting or surveillance state.
4. NRF2 and its target genes modulate microglial phenotypes
The first evidence for an immunomodulatory role for NRF2 was reported in models of lung inflammation. Genetic deficiency in NRF2 enhanced susceptibility to emphysema (45, 112, 255), diesel exhaust chemicals (152), and asthma (256). Accordingly, NRF2-null mice are more susceptible to the inflammatory response elicited by LPS and sepsis (307, 308). Considering this background and that microglia belong to the monocyte lineage, it follows that NRF2 should also participate in the final acquisition of microglial phenotype. This was demonstrated by comparing parameters of inflammation in the hippocampus of NRF2-deficient and wild-type mice after systemic treatment with LPS. NRF2-knockout mice were hypersensitive to neuroinflammation induced by LPS, as determined by an increase in M1 typical markers, including F4/80, iNOS, IL-6, and TNF, compared with wild-type littermates. In line with these observations, NRF2 activation by SFN reduced the abundance of microglial cells in the hippocampus and attenuated the production of M1 markers in response to LPS (111).
With regard to neurodegenerative disorders, it has been reported that, as with LPS, chronic administration of MPTP i.p. resulted in enhanced microglial release of proinflammatory cytokines. This observation correlated with enhanced generation of M1-mediators in primary microglial cultures of NRF2-null mice compared with wild-type littermates. Interestingly, SFN administration prevented chronic inflammation only in the wild-type animals, providing formal proof that oxidative stress and NRF2 are definitely associated with the etiopathology of several neurodegenerative diseases. At the same time that M1 inflammation markers were increased in response to MPTP, M2 markers were decreased in NRF2-null mice compared with wild-type animals. These results have been confirmed in microglial cultures that are stimulated with conditioned medium from dopaminergic cells treated with MPP+, further demonstrating a role of NRF2 in maintaining a balance between classical and alternative microglial activation (262).
Among the NRF2 target genes, numerous data strongly implicate HO-1 in immunomodulation. HO-1 catalyzes the degradation of heme to generate carbon monoxide, biliverdin, and free iron (167). Biliverdin is transformed by biliverdin reductase into bilirubin, which is considered one of the most powerful antioxidant molecules (168, 169). Carbon monoxide released by glial cells after induction HO-1 under stress conditions, or by its constitutive isoform HO-2, is associated with neurosecretory functions of the hypothalamus (172, 173). Initial observations on the immunomodulatory role of HO-1 were derived from experiments using peripheral monocytes/macrophages. HO-1 end-products reduce classic monocyte activation, because carbon monoxide and biliverdin inhibit NOX (138, 302) and TLR-4 (204) signaling. Furthermore, HO-1 might regulate NOX via the reduction of heme availability (303). In the brain, astrocytes secrete factors that increase microglial HO-1 protein levels and lead to a reduction of microglial ROS in response to IFN-γ (194). Moreover, heme oxygenase activation increases prostaglandin production in rat astrocytes through the activation of COX enzymes (171), which is one of the earliest events during neuroinflammation (243). On the other hand, cyclopentanone prostaglandins induced HO-1 in both astroglia and microglia, thus amplifying the inflammatory response (129, 133).
Polarization of monocytes by viral IL-10 results in up-regulation of HO-1 and acquisition of M2c. HO-1 inhibition results in a loss of capacity of IL-10 to suppress TNF and IL-1 production reducing alternative M2c activation (9). Both heme regulatory molecules, CD163 and HO-1, are preferentially expressed by M2-macrophages (288). This interesting observation is also supported in microglia, where CD163- and HO-1-positive cells preferentially activate the M2 program after traumatic brain injury (367). It is interesting that recent evidence indicated that HO-1 and biliverdin reductase undergo oxidative, nitrosative, and phosphorylative post-traductional modifications in the brain of subjects with AD and MCI (17, 18). Although those studies did not identify the cell type where these post-translational modifications occur, they suggest that the anti-oxidant and anti-inflammatory roles of these two enzymes may be lost, at least partially, in the AD brain.
Emerging evidence indicate a strong relationship between redox homeostasis and the vitagene system. Vitagenes constitute a network genes participating in longevity assurance processes (36). The vitagene family includes several NRF2 regulated genes such as Hmox1 and Trx, as heat shock protein Hsp70 and sirtuin (Sirt1). Its participation in modulation of neuroinflammation is being now characterized. For instance, Hsp70 induces IL-6 and TNF production, phagocytosis, and clearance of Aβ peptides by microglia. Activation of SIRT1 decreases Aβ-induced neuronal death and transgenic over-expression of SIRT1 in AD models increases processing of APP through the non-amyloidogenic route (54). On the contrary, SIRT2 overexpression inhibited microglial activation and brain inflammation in response to LPS through NF-κB deacetylation (222). The vitagene system can be activated by nutritional antioxidants, including curcumin, resveratrol, acetyl-l-carnitine, or carnosine (35, 37).
On the other hand, NF-κB, the master regulator of inflammation, is hyperactivated under oxidative stress conditions and, therefore, is sensitive to modulation by NRF2. We and others have reported the hyperactivation of p65-NF-κB in NRF2-deficient microglia/macrophages as well as the down-regulation of p65-NF-κB by NRF2 inducers such as SFN. This functional interference has not yet been convincingly characterized at the molecular level. Thus, while NF-κB is the master regulator of the M1 phenotype, NRF2 may be a master regulator of the M2 phenotype.
VIII. BBB Permeability Is Altered by ROS, Enabling Homing of Peripheral Immune Cells
For many years, the brain was one of the several organs described as immunologically privileged on the basis that allo- and xeno-grafts transplanted into the CNS are “in some way partially or fully exempted from the normal rigors imposed by their histoincompatible status” (16). In the early years of the twentieth century, Murphy and Sturm (202) proposed two possible explanations: (i) mechanical impediment of cells to migrate beyond the perivascular spaces; (ii) lymphoid cells find the brain tissue an uncongenial environment. These authors were the first to define BBB as an “anatomic barrier that restricts diapedesis” and ensures the immune privileged state of the brain. The lack of constitutive MHC class I and II expression on CNS parenchymal cells (molecules required by T cells to recognize their antigen), the inability of microglial and astroglial cells to sustain immune responses, the paucity of dendritic cells in the parenchyma, and the lack of lymphatic vessels within the CNS further contributed to the concept of an immune privileged tissue, inaccessible to circulating immune cells (79, 201). However, this view was questioned as early as 1948 by Medawar (188), who concluded that T cells activated outside the CNS found a way across the brain barriers to provoke an immune response within the CNS (81). These days, it is known that leucocytes, monocytes, and macrophages can enter the CNS, not only during inflammation and disease (79, 279), but also under homeostatic conditions. In such non-disease states, leucocytes accumulate on the abluminal side of CNS microvessels (219), where they encounter perivascular antigen-presenting cells, collect CNS antigens, and patrol the CNS barriers (92). Infiltrated cells then act together with resident microglia (63) and even in some cases, adopt a microglial phenotype (88, 101).
There are several routes for leukocytes to enter the CNS: (i) migration from the microvessels into parenchymal perivascular space; (ii) migration via the choroid plexus into the cerebro-spinal fluid; and (iii) migration through post-capillary venules at the pial surface into subarachnoid and Virchow–Robin perivascular spaces. A fourth route has been recently suggested that involves migration from subependymal vessels via the ependyma into the ventricles (304). These routes involve crossing the endothelial BBB, the epithelial blood–CSF barrier, and the blood–spinal cord barrier.
A. Blood–brain barrier
Localized at the interface between the blood and the cerebral tissue and formed by the endothelial cells (ECs) of cerebral blood vessels, the BBB is, by far, the largest surface area for blood–brain exchange (12–18 m2 in the adult human) (203). Moreover, the maximal distance between brain cells and a capillary is about 25 μm, meaning that once the BBB is crossed, the distance for diffusion of solutes and drugs to neurons and glial cell bodies is short (1). The integrity of the BBB is controlled by functional interactions of several cell types residing in close proximity to brain ECs (pericytes, astrocytes, neurons, and microglia). All these cells, along with the basal lamina (endothelial basement membrane), constitute the neurovascular unit (335). Figure 14 shows the elements that constitute the BBB and its alteration by ROS.
FIG. 14.

The BBB is made of highly specialized ECs that secrete ECM (basal lamina) where many pericytes can be found. Astrocytes, on the other hand, secrete ECM of a different composition, which along with astrocytic endfeet, that embed EC at the capillaries, establishes the glia limitans. At the post-capillary venules, basal lamina and glia limitans are separated by the perivascular (Robin-Virchov) space in which antigen-presenting cells can be found. During inflammation, P-selectin and/or VCAM-1 are up-regulated, providing enough receptors for leukocytes expressing PSGL-1 or α4β1 integrin recognition and thus enabling the beginning of the diapedesis. In the BBB, diapedesis occurs preferentially transcellular, leaving TJs intact. However, activated microglia produce high amounts of ROS and RNS through the NOX system. These reactive species impair TJs, causing thr disappearance of occludin and claudin-5 and causing an increase in BBB permeability. In addition, ROS and RNS initiate lipid peroxidation of cell membranes, resulting in HNE formation. HNE not only disrupts TJs, thereby increasing BBB permeability, but can also spread from the site of origin, thus affecting cell functioning far from the initial injury. Increased levels of HNE, ACR, and MDA-modified proteins can be found in neurodegenerative disorders that are associated with chronic inflammation. BBB, blood–brain barrier; EC, endothelial cell; ECM, extracellular matrix; TJ, tight junction. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
The cerebral ECs differ significantly from ECs of other organs by having specialized transport systems, uniform thickness with no fenestrations, low pinocytic activity, continuous intercellular tight junctions (TJs) and adherens junctions, and greater number and volume of mitochondria (64). Astrocytes secrete and provide important factors to ECs, including TGF-β, glial-derived neurotropic factor, and fibroblast growth factor, which modulate the BBB to control the passage of most molecules (2, 304). On the other hand, the regulation of BBB formation in embryogenesis, its maintenance, vascular stability, regulation of capillary blood flow, angiogenesis, and clearance of toxic cellular products are under the control of pericytes (338).
B. Oxidative stress affects BBB permeability
Under non-pathological conditions, the BBB maintains the ionic composition of the brain interstitium that is necessary for normal neuronal function. However, almost every CNS pathology (MS, hypoxia and ischemia, edema, stroke, PD, AD, epilepsy, tumors, glaucoma, and lysosomal storage diseases) includes an element of BBB dysfunction ranging from mild and transient TJ opening to chronic barrier breakdown or even changes in transport systems and enzymes (1, 72, 376).
One of the earliest steps preceding clinical diagnosis of neurodegenerative disorders seems to be disruption of BBB caused by oxidative stress (97, 161, 280). ROS have been shown to modulate BBB integrity by transient activation of PI3K/AKT pathway via RhoA, which acutely disrupts the integrity of the TJs, as indicated by the disappearance of occludin and claudin-5 (280). The inhibition of Rho kinase completely prevented the ROS-induced increase in permeability and the ROS-induced polymerization of the actin cytoskeleton (119). The increase in BBB permeability correlates with the down-regulation of occludin expression (135), alterations in the oligomeric assembly of the occludin (185), and its movement away from the TJs (161). In fact, occludin is degraded in response to H2O2-induced impairment of BBB function (156). Accordingly, IL-17-induced NOX- or xanthine oxidase-dependent ROS production activates the BBB-endothelial contractile machinery, which is accompanied by a down-regulation of occludin (109). The main enzymes responsible for TJ degradation are the matrix metalloproteinases (MMPs), which are also activated by ROS in a later phase of BBB disruption (97, 349). MMPs as well degrade collagen and laminin in basal lamina, causing the disruption of basement membrane and contributing to an increase in BBB permeability. In addition, increased ROS causes recruitment and migration of leukocytes to the CNS whose released products further degrade basal lamina and increase BBB permeability (76).
C. Activated microglia and BBB impairment
As discussed earlier, ROS produced from activated microglia can induce lipid peroxidation, thereby modulating the integrity of the BBB. For example, HNE was shown to affect functional integrity of the BBB, causing its permeability within 20 minutes in vitro (191). The mechanism was explained by Usatyuk et al. (320), who revealed that HNE modulates cell-cell adhesion through modifications of focal adhesion, adherens, and TJ proteins as well as integrin. These findings, along with HNE abundance in the BBB found under various pathophysiological circumstances (277, 356) and as shown in Figure 15, suggest that HNE is associated with several regulatory functions, including post-traumatic recovery (355, 359).
FIG. 15.

Immunohistochemical detection of HNE in the BBB (courtesy of Prof. K. Zarkovic, Medical Faculty of the University of Zagreb). The photo shows abundance of HNE, as detected by genuine monoclonal antibodies that are specific for the HNE-His adducts (courtesy of Prof. Georg Waeg, University of Graz) in the specimen of the baboon brain after hypovolemic shock. Dark brown color indicates strong immunopositivity for HNE, in particular, in glial cells surrounding the blood vessel in the center. HNE, 4-hydroxy-2-nonenal. To see this illustration in color, the reader is referred to the web version of this article at www.liebertpub.com/ars
Studies on CD200 deficient mice have revealed that fine regulation of microglial activation occurs through an interaction of CD200 (mainly expressed on neurons) and its receptor CD200R (mainly expressed on microglia). Under inflammatory conditions, microglial activation in these mice is exacerbated, due to increases in TLR2 and TLR4 expression (56). This model has also shown that an increase in BBB permeability precedes microglial activation (71). Evidence suggests that microglial activation inevitably causes BBB impairment. Thus, Kacimi et al. (118) showed that activated microglia promote BBB disruption through injury of ECs by releasing high amounts of ROS. This mechanism was further explored in EAE, where fibrinogen leaking from the CNS was found to trigger microglial activation (61). This condition takes place before the onset of the neurological symptoms of EAE; whereas, at the peak of the disease, microglia start clustering around the vasculature in areas of fibrin deposition and release ROS, leading to axonal damage (61). These findings are in accordance with immunohistochemical and post-mortem magnetic resonance imaging analyses of human MS lesions where microglial activation and BBB disruption were identified as the earliest pathological signs (178, 325).
In the case of impaired BBB and microglial activation, there is a thin line between what happens first. Nonetheless, the microglial NOX system seems to be the major source of O2− (28, 119). This species appears to be the most important ROS in cerebral diseases, causing not only BBB permeability but also enhancing monocyte adhesion and migration (322, 351).
D. Leukocyte trafficking across the BBB
The BBB might be considered well regulated but a delicate boundary between humoral and cellular components of “systemic integrative tissue of the organism” (blood) and the most sophisticated neuronal system that controls most of the major body functions and should, therefore, be well protected from potentially dangerous humoral and cellular components present in the blood. Even though leukocytes interact with ECs of the BBB within small capillaries, their extravasation into the brain parenchyma occurs at the level of post-capillary venules (79). Specifically, in pre- and post-capillary vessels, astrocytic endfeet of the glia limitans are separated from the endothelial layer by pericytes and smooth muscle cells and by the perivascular (Virchow–Robin) space (22). This perivascular space seems to serve as a specific “checkpoint” for immune competent cells before the delicate CNS milieu is put at risk for the benefit of successfully fighting a potential infection (79). During various local and systemic stress conditions (hypovolemia, hypoxia, shock, sepsis, traumatic injury, etc.), humoral and cellular components of the blood collide with BBB and the neural tissue produces cytokines and soluble factors that act on microglia, astrocytes, or neurons, causing further immune system activation, in particular, affecting leukocytes [reviewed in (139)]. ROS, originating from microglia and leukocytes after their interaction with ECs, seem to be the key players in promoting leukocyte trafficking across the BBB through activation of redox signaling pathways. Namely, triggered inflammatory responses to altered BBB by both leukocytes and microglia lead to TNF production and expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and platelet/endothelial cell adhesion molecule 1 (PECAM 1) in ECs (139). TNF appears to be the major signaling mediator for endothelium activation and leukocyte recruitment as observed by a decreased number of rolling and adherent leukocytes (approximately 80%) after minocycline (effective inhibitor for microglial activation) treatment (372). In addition, sustained overexpression of pro-inflammatory cytokine IL-1β is also capable of inducing persistent leukocyte recruitment to the brain (282). As shown in the same study, BBB leakage is not reduced when neutrophil infiltration is abolished, indicating high importance of ROS and their second messengers (such as HNE), humoral factors (such as cytokines), and glial cells in BBB malfunction under stress (282). These data verify the fact that myriad of factors mainly induced by ROS affect not only BBB permeability but also immune cells crossing it.
Intravital fluorescence video microscopy has provided direct in vivo evidence of the unique interaction of T lymphocytes with the BBB in the spinal cord. Unlike the diapedesis across postcapillary venules in lymph nodes, diapedesis across the BBB lacks T-cell rolling and lasts approximately 6 h, rather than minutes (321, 339). Thus, instead of rolling, T cells were captured by the post-capillary venule wall, followed by firm adhesion to the venule. These events (capture and adhesion) are mediated through the interaction of α4-integrins on T cells with endothelial VCAM-1, whereas diapedesis of T cells across the venule wall is supported by the αLβ2-integrin (leukocyte function-associated antigen) (81, 140).
Constitutively expressed VCAM-1 mediates the G-protein-independent prompt arrest (capture) of circulating encephalitogenic T-cell blasts via α4-integrin to the endothelium of the healthy BBB (321). Transient capture is followed by G-protein-dependent α4-integrin/VCAM-1-mediated adhesion strengthening (321). Thus, leukocytes cross the brain endothelium, irrespective of the TJ presence, suggesting that BBB disruption and cell migration are not necessarily connected (339). After their diapedesis across the inflamed BBB, T cells need to make their way across the ECM components of the endothelial basement membrane. This membrane consists of a number of glycoproteins such as laminins, type IV collagen, nidogens, and heparan sulfate proteoglycans (80). Just recently, T-cell extravasation sites were shown to be preferentially the endothelium basement membrane sites containing laminin α4 (340). After breaching the endothelial basement membrane, T cells reach the perivascular space where they have to re-encounter their cognate antigen presented by antigen-presenting cells, in order to pursue their route into the neuropil (CNS parenchyma) (80). The final obstacle in this long journey is glia limitans composed of parenchymal basement membrane laid down by astrocytes (219). Activities of MMPs, especially MMP-2 and MMP-9, are essential for penetration of glia limitans (3).
After reaching the CNS parenchyma, astrocytes are the first cells encountered by leukocytes. Among many functions, astrocytes even take part in the regulation of leukocyte trafficking primarily by forming the scar-like barriers. These barriers serve to restrict leukocytes to perivascular clusters and limit their infiltration into adjacent CNS parenchyma, thus restricting the inflammatory process (326). The next step of leukocyte trafficking is contact with neurons that occurs through ICAM-5. This binding enables neurons to present T cells to neighboring glial cells and to help them induce T-cell apoptosis (309). Besides, neurons can induce proliferation of encephalitogenic T cells through up-regulation of TGF-β1 signaling pathway. Eventually, an interaction of neurons with T cells results in the conversion of encephalitogenic T cells into T-regulatory cells (Treg), which suppress encephalitogenic T cells showing bifunctional roles of neurons in the regulation of T-cell function and CNS inflammation (160).
Moreover, in vitro data indicate that astrocytes can prevent microglial cell cytotoxicity by mechanisms mediated by TGFβ1-Smad3 pathway in an age-dependent (310), suggesting that activation of the TGFβ1-Smad3 pathway is impaired in aging. If so, age-related impairment of TGFβ1-Smad3 could reduce protective activation and enhance cytotoxic activation of microglia, enhancing microglia and ROS-mediated age and stress-associated neurodegeneration.
Therefore, a multistep process of leukocytes crossing the BBB and a consequential reaction of neuronal and glial cells still need to be further clarified. Latest findings reveal that immune cell trafficking across the BBB appears to be independent on BBB integrity due to transcellular diapedesis stressing the relevance of the humoral components, mainly cytokine and ROS production which activates numerous signaling pathways in order to control the inflammation reaction.
IX. Concluding Remarks
The role of ROS as second messengers that control microglial dynamics is gaining increasing attention. Oxidant modifications on lipids and proteins reviewed here indicate the profound relevance that redox changes have in many cell types, and in microglia in particular, as well as in other aspects of neuroinflammation, including the integrity of the BBB and peripheral leukocyte infiltration. Transcription factor NF-κB has long been considered a master regulator of inflammation, but it is clear that transcription factor NRF2 is also closely associated with microglial activation. We have provided evidence that it is through maintenance of redox homeostasis that this protein opposes uncontrolled effects of ROS which might polarize microglía toward an exacerbated M1 pro-inflammatory phenotype. It is anticipated that, in the near future, the most prevalent diseases will be those affecting the elderly. Considering that ROS levels increase during aging and that ROS deregulate normal neuroinflammatory responses, we expect to see a growing interest in the redox pathophysiology of microglia-derived neuroinflammation which will uncover new drug targets to modulate the low-grade chronic neuroinflammation that characterizes neurodegenerative diseases.
Abbreviations Used
- β-TrCP
β-transducin repeat containing E3 ubiquitin protein ligase
- 6-OHDA
6-hydroxydopamine
- 6PGD
phosphogluconate dehydrogenase
- Aβ
amyloid beta
- AA
arachidonic acid
- ACR
acrolein
- AD
Alzheimer's disease
- ALS
amyotrophic lateral sclerosis
- ARE
antioxidant response element
- ARG1
arginase-1
- ASK1
apoptosis-signal-regulating kinase 1
- BBB
blood–brain barrier
- Bcl2
B-cell lymphoma-2
- BH4
tetrahydrobiopterin
- BSO
buthionine-S-sulfoximine
- CAT
cationic amino acid transporter
- CCL-18
chemokine 18
- CD172a
receptor for CD47
- CD200R1
receptor for CD200
- CG islands
cytosine guanine islands
- CNS
central nervous system
- COX2
cyclooxygenase-2
- CSF
cerebrospinal fluid
- CXCR2
receptor for CXC3-chemokine ligand-1
- DC-SIGN
dendritic cell-specific intracellular adhesion molecule 3-grabbing integrin
- DHA
doecosahexaenoic acid
- DUOX
dual oxygenase
- EAD
early stage Alzheimer's disease
- EAE
experimental autoimmune encephalomyelitis
- EC
endothelial cell
- ECM
extracellular matrix
- ET
endotoxin tolerance
- FIZZ1
found in inflammatory zone
- FT
ferritin
- G6PDH
glucose-6-phosphate dehydrogenase
- GCLC
glutamate-cysteine ligase catalytic subunit
- GCLM
glutamate-cysteine ligase modulatory subunit
- GLT-1
glutamate transporter 1
- GPx
glutathione peroxidase
- GR, GSR
glutathione reductase
- GSH
glutathione (γ-glutamyl-l-cysteinylglycine)
- GSK-3
glycogen synthase kinase-3
- GSSG
glutathione disulfide
- GST
glutathione S-transferase
- H2O2
hydrogen peroxide
- HDAC
histone deacetylase
- HNE
4-hydroxy-2-nonenal
- •HO
hydroxyl radical
- HO-1
heme oxygenase-1
- IBA-1
ionized calcium-binding adapter molecule 1
- ICAM-1
intercellular adhesion molecule 1
- IFN-γ
interferon-γ
- IL
interleukin
- IL-12p40
interleukin 12 p40 subunit
- IL-1Ra
interleukin 1 receptor antagonist
- IL-4Rα
interleukin 4 receptor α
- iNOS
inducible isoform of nitric oxide synthase
- IsoPs
Isoprostanes
- IκB
inhibitor of NF-κB
- JNK
c-Jun N-terminal kinase
- KEAP1
Kelch-like ECH-associated protein 1
- LAD
late-stage Alzheimer's disease
- LCN2
lipocalin 2
- LPS
lipopolysaccharide
- LTP
long-term potentiation
- MAPK
mitogen-activated protein kinase
- MARCO
macrophage receptor with collagenous structure
- MCI
mild cognitive impairment
- MDA
malondialdehyde
- MEKK
mitogen-activated protein kinase/ERK kinase
- MHCII
major histocompatibility antigen II
- MMP
matrix metalloproteinase
- MPP+
1-methyl-4-phenylpyridinium
- MPTP
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- MR
manose receptor
- MS
multiple sclerosis
- NAD(P)H
nicotinamide adenine dinucleotide phosphate
- NeuroPs
neuroprostanes
- NF-κB
nuclear factor kappa-light-chain-enhancer of activated B cells
- NO
nitric oxide
- NOS
nitric oxide synthase
- nNOS
neuronal nitric oxide synthase
- NOX
NADPH oxidase
- NQO1
NAD(P)H:quinone oxidoreductase 1
- NRF2
nuclear factor (erythroid-derived 2)-like 2
- O2−
superoxide anion
- ONOO−
peroxynitrite
- PD
Parkinson's disease
- PI3K
phosphatidyl inositol-3 kinase
- PKA
protein kinase A
- PKC
protein kinase C
- Prx
peroxiredoxin
- RAGE
receptor for advanced glycation endproducts
- RHD
Rel homology domain
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SFN
sulforaphane
- SOCS
suppressors of cytokine signaling
- SOCS3
suppressor of cytokine signaling 3
- SOD
superoxide dismutase
- SOD1
Cu/Zn-superoxide dismutase 1
- sphk
sphingosine kinase
- Srx
sulfiredoxin
- TGF-β
transforming growth factor-β
- TGF-b
tumor growth factor b
- TJ
tight junctions
- TLR
toll-like receptor
- TNF
tumor necrosis factor
- Trx
thioredoxin
- TrxR
thioredoxin reductase
- TXNIP
Trx-interacting protein
- VCAM-1
vascular cell adhesion molecule 1
- YM-1
mouse homolog chitinase 3 like 1 (CHI3L1)
- γ-GCL
γ-glutamylcysteine synthetase
- γGS
γ-glutamate cysteine synthetase
- γGT
γ-glutamyl transpeptidase
Acknowledgments
This review was supported by the Spanish Ministry of Science and Innovation Refs. SAF2012-32222 (M.G.L.) and SAF2010-17822 (A.C.), the Spanish Ministry of Health (Instituto de Salud Carlos III) RETICS-RD06/0026 (M.G.L.), Pathfinder phase II of the Network of Centres of Excellence on Neurodegeneration (COEN) (A.C.), the Science Foundation, Ireland (G.M.) and the European Cooperation in Science and Research (COST Action BM1203/EU-ROS).
References
- 1.Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, and Begley DJ. Structure and function of the blood–brain barrier. Neurobiol Dis 37: 13–25, 2010 [DOI] [PubMed] [Google Scholar]
- 2.Abbott NJ, Ronnback L, and Hansson E. Astrocyte-endothelial interactions at the blood–brain barrier. Nat Rev Neurosci 7: 41–53, 2006 [DOI] [PubMed] [Google Scholar]
- 3.Agrawal S, Anderson P, Durbeej M, van Rooijen N, Ivars F, Opdenakker G, and Sorokin LM. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J Exp Med 203: 1007–1019, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ajami B, Bennett JL, Krieger C, Tetzlaff W, and Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10: 1538–1543, 2007 [DOI] [PubMed] [Google Scholar]
- 5.Altenhofer S, Kleikers PW, Radermacher KA, Scheurer P, Rob Hermans JJ, Schiffers P, Ho H, Wingler K, and Schmidt HH. The NOX toolbox: validating the role of NADPH oxidases in physiology and disease. Cell Mol Life Sci 69: 2327–2343, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ando K, Hirao S, Kabe Y, Ogura Y, Sato I, Yamaguchi Y, Wada T, and Handa H. A new APE1/Ref-1-dependent pathway leading to reduction of NF-kappaB and AP-1, and activation of their DNA-binding activity. Nucleic Acids Res 36: 4327–4336, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Apopa PL, He X, and Ma Q. Phosphorylation of Nrf2 in the transcription activation domain by casein kinase 2 (CK2) is critical for the nuclear translocation and transcription activation function of Nrf2 in IMR-32 neuroblastoma cells. J Biochem Mol Toxicol 22: 63–76, 2008 [DOI] [PubMed] [Google Scholar]
- 8.Arnér ES. Focus on mammalian thioredoxin reductases - importante selenoproteins with versatile functions. Biochim Biophys Acta 1790: 495–526, 2009 [DOI] [PubMed] [Google Scholar]
- 9.Avdic S, Cao JZ, McSharry BP, Clancy LE, Brown R, Steain M, Gottlieb DJ, Abendroth A, and Slobedman B. Human cytomegalovirus interleukin-10 polarizes monocytes toward a deactivated M2c phenotype to repress host immune responses. J Virol 87: 10273–10282, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aybek H, Ercan F, Aslan D, and Sahiner T. Determination of malondialdehyde, reduced glutathione levels and APOE4 allele frequency in late-onset Alzheimer's disease in Denizli, Turkey. Clin Biochem 40: 172–176, 2007 [DOI] [PubMed] [Google Scholar]
- 11.Baek KJ, Thiel BA, Lucas S, and Stuehr DJ. Macrophage nitric oxide synthase subunits. Purification, characterization, and role of prosthetic groups and substrate in regulating their association into a dimeric enzyme. J Biol Chem 268: 21120–21129, 1993 [PubMed] [Google Scholar]
- 12.Baker BJ, Akhtar LN, and Benveniste EN. SOCS1 and SOCS3 in the control of CNS immunity. Trends Immunol 30: 392–400, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bal-Price A, Matthias A, and Brown GC. Stimulation of the NADPH oxidase in activated rat microglia removes nitric oxide but induces peroxynitrite production. J Neurochem 80: 73–80, 2002 [DOI] [PubMed] [Google Scholar]
- 14.Barger SW. and Basile AS. Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem 76: 846–854, 2001 [DOI] [PubMed] [Google Scholar]
- 15.Barger SW, Goodwin ME, Porter MM, and Beggs ML. Glutamate release from activated microglia requires oxidative burst and lipid peroxidation. J Neurochem 101: 1205–1213, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Barker CF. and Billingham RE. Immunologically privileged sites. Adv Immunol 25: 1–54, 1977 [PubMed] [Google Scholar]
- 17.Barone E, Di Domenico F, Cenini G, Sultana R, Coccia R, Preziosi P, Perluigi M, Mancuso C, and Butterfield DA. Oxidative and nitrosative modifications of biliverdin reductase-A in the brain of subjects with Alzheimer's disease and amnestic mild cognitive impairment. J Alzheimers Dis 25: 623–633, 2011 [DOI] [PubMed] [Google Scholar]
- 18.Barone E, Di Domenico F, Sultana R, Coccia R, Mancuso C, Perluigi M, and Butterfield DA. Heme oxygenase-1 posttranslational modifications in the brain of subjects with Alzheimer disease and mild cognitive impairment. Free Radic Biol Med 52: 2292–2301, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bassi MT, Gasol E, Manzoni M, Pineda M, Riboni M, Martin R, Zorzano A, Borsani G, and Palacin M. Identification and characterisation of human xCT that co-expresses, with 4F2 heavy chain, the amino acid transport activity system xc. Pflugers Arch 442: 286–296, 2001 [DOI] [PubMed] [Google Scholar]
- 20.Bazan NG. and Flower RJ. Medicine: lipid signals in pain control. Nature 420: 135–138, 2002 [DOI] [PubMed] [Google Scholar]
- 21.Beckman JS. and Crow JP. Pathological implications of nitric oxide, superoxide and peroxynitrite formation. Biochem Soc Trans 21: 330–334, 1993 [DOI] [PubMed] [Google Scholar]
- 22.Bechmann I, Galea I, and Perry VH. What is the blood–brain barrier (not)? Trends Immunol 28: 5–11, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Bedard K. and Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 87: 245–313, 2007 [DOI] [PubMed] [Google Scholar]
- 24.Beloosesky Y, Salman H, Bergman M, Bessler H, and Djaldetti M. Cytokine levels and phagocytic activity in patients with Alzheimer's disease. Gerontology 48: 128–132, 2002 [DOI] [PubMed] [Google Scholar]
- 25.Berry CB, Hayes D, Murphy A, Wießner M, Rauen T, and McBean GJ. Differential modulation of the glutamate transporters GLT1, GLAST and EAAC1 by docosahexaenoic acid. Brain Res 1037: 123–133, 2005 [DOI] [PubMed] [Google Scholar]
- 26.Biaglow JE. and Miller RA. The thioredoxin reductase/thioredoxin system. Novel redox targets for cancer therapy. Cancer Biol Ther 4: 6–13, 2005 [DOI] [PubMed] [Google Scholar]
- 27.Biswas SK. and Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol 11: 889–896, 2010 [DOI] [PubMed] [Google Scholar]
- 28.Block ML. and Hong JS. Microglia and inflammation-mediated neurodegeneration: multiple triggers with a common mechanism. Prog Neurobiol 76: 77–98, 2005 [DOI] [PubMed] [Google Scholar]
- 29.Blouin E, Halbwachs-Mecarelli L, and Rieu P. Redox regulation of beta2-integrin CD11b/CD18 activation. Eur J Immunol 29: 3419–3431, 1999 [DOI] [PubMed] [Google Scholar]
- 30.Boveris A. and Chance B. The mitochondrial generation of hydrogen peroxide. General properties and effect of hyperbaric oxygen. Biochem J 134: 707–716, 1973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brown GC. and Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett 356: 295–298, 1994 [DOI] [PubMed] [Google Scholar]
- 32.Brown GC. and Neher JJ. Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol Neurobiol 41: 242–247, 2010 [DOI] [PubMed] [Google Scholar]
- 33.Butterfield DA, Reed T, and Sultana R. Roles of 3-nitrotyrosine- and 4-hydroxynonenal-modified brain proteins in the progression and pathogenesis of Alzheimer's disease. Free Radic Res 45: 59–72, 2011 [DOI] [PubMed] [Google Scholar]
- 34.Caito S, Yu Y, and Aschner M. Differential response to acrylonitrile toxicity in rat primary astrocytes and microglia. Neurotoxicology 37: 93–99, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Calabrese V, Cornelius C, Cuzzocrea S, Iavicoli I, Rizzarelli E, and Calabrese EJ. Hormesis, cellular stress response and vitagenes as critical determinants in aging and longevity. Mol Aspects Med 32: 279–304, 2011 [DOI] [PubMed] [Google Scholar]
- 36.Calabrese V, Cornelius C, Dinkova-Kostova AT, Calabrese EJ, and Mattson MP. Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid Redox Signal 13: 1763–1811, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Calabrese V, Cornelius C, Dinkova-Kostova AT, Iavicoli I, Di Paola R, Koverech A, Cuzzocrea S, Rizzarelli E, and Calabrese EJ. Cellular stress responses, hormetic phytochemicals and vitagenes in aging and longevity. Biochim Biophys Acta 1822: 753–783, 2012 [DOI] [PubMed] [Google Scholar]
- 38.Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, and Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci 8: 766–775, 2007 [DOI] [PubMed] [Google Scholar]
- 39.Calingasan NY, Uchida K, and Gibson GE. Protein-bound acrolein: a novel marker of oxidative stress in Alzheimer's disease. J Neurochem 72: 751–756, 1999 [DOI] [PubMed] [Google Scholar]
- 40.Cand F. and Verdetti J. Superoxide dismutase, glutathione peroxidase, catalase, and lipid peroxidation in the major organs of the aging rats. Free Radic Biol Med 7: 59–63, 1989 [DOI] [PubMed] [Google Scholar]
- 41.Castellani RJ, Perry G, Siedlak SL, Nunomura A, Shimohama S, Zhang J, Montine T, Sayre LM, and Smith MA. Hydroxynonenal adducts indicate a role for lipid peroxidation in neocortical and brainstem Lewy bodies in humans. Neurosci Lett 319: 25–28, 2002 [DOI] [PubMed] [Google Scholar]
- 42.Chan AC, Tran K, Raynor T, Ganz PR, and Chow CK. Regeneration of vitamin E in human platelets. J Biol Chem 266: 17290–17295, 1991 [PubMed] [Google Scholar]
- 43.Chatterjee S, Noack H, Possel H, Keilhoff G, and Wolf G. Glutathione levels in primary glial cultures: monochlorobimane provides exidence of cell type-specific distribution. Glia 27: 152–161, 1999 [PubMed] [Google Scholar]
- 44.Chatterjee S, Noack H, Possel H, and Wolf G. Induction of nitric oxide synthesis lowers intracellular glutathione in microglia of primary microglial cultures. Glia 29: 98–101, 2000 [PubMed] [Google Scholar]
- 45.Cho HY, Reddy SP, Yamamoto M, and Kleeberger SR. The transcription factor NRF2 protects against pulmonary fibrosis. FASEB J 18: 1258–1260, 2004 [DOI] [PubMed] [Google Scholar]
- 46.Cho N, Choi JH, Yang H, Jeong EJ, Lee KY, Kim YC, and Sung SH. Neuroprotective and anti-inflammatory effects of flavonoids isolated from Rhus verniciflua in neuronal HT22 and microglial BV2 cell lines. Food Chem Toxicol 50: 1940–1945, 2012 [DOI] [PubMed] [Google Scholar]
- 47.Choi SH, Aid S, and Bosetti F. The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: implications for translational research. Trends Pharmacol Sci 30: 174–181, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Choi SH, Aid S, Kim HW, Jackson SH, and Bosetti F. Inhibition of NADPH oxidase promotes alternative and anti-inflammatory microglial activation during neuroinflammation. J Neurochem 120: 292–301, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Choi SH, Lee DY, Kim SU, and Jin BK. Thrombin-induced oxidative stress contributes to the death of hippocampal neurons in vivo: role of microglial NADPH oxidase. J Neurosci 25: 4082–4090, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chu F, Ward NE, and O'Brian CA. Potent activation of representative members of each PKC isozyme subfamily and PKD via S-thiolation by the tumor-promotion/progression antagonist glutathione but not by its precursor, cysteine. Carcinogenesis 22: 1221–1229, 2001 [DOI] [PubMed] [Google Scholar]
- 51.Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, and Hayes JD. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 32: 3765–3781, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Colton CA, Mott RT, Sharpe H, Xu Q, Van Nostrand WE, and Vitek MP. Expression profiles for macrophage alternative activation genes in AD and in mouse models of AD. J Neuroinflammation 3: 27, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cordle A. and Landreth G. 3-Hydroxy-3-methylglutaryl-coenzyme A reductase inhibitors attenuate beta-amyloid-induced microglial inflammatory responses. J Neurosci 25: 299–307, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cornelius C, Perrotta R, Graziano A, Calabrese EJ, and Calabrese V. Stress responses, vitagenes and hormesis as critical determinants in aging and longevity: Mitochondria as a “chi”. Immun Ageing 10: 15, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Correa F, Mallard C, Nilsson M, and Sandberg M. Activated microglia decrease histone acetylation and Nrf2-inducible anti-oxidant defence in astrocytes: restoring effects of inhibitors of HDACs, p38 MAPK and GSK3beta. Neurobiol Dis 44: 142–151, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Costello DA, Lyons A, Denieffe S, Browne TC, Cox FF, and Lynch MA. Long term potentiation is impaired in membrane glycoprotein CD200-deficient mice: a role for Toll-like receptor activation. J Biol Chem 286: 34722–34732, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cullinan SB, Gordan JD, Jin J, Harper JW, and Diehl JA. The Keap1-BTB protein is an adaptor that bridges Nrf2 to a Cul3-based E3 ligase: oxidative stress sensing by a Cul3-Keap1 ligase. Mol Cell Biol 24: 8477–8486, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cullinan SB, Zhang D, Hannink M, Arvisais E, Kaufman RJ, and Diehl JA. Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol Cell Biol 23: 7198–7209, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dalle-Donne I, Rossi R, Colombo G, Giustarini D, and Milzani A. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem Sci 34: 85–96, 2009 [DOI] [PubMed] [Google Scholar]
- 60.Dang TN, Arseneault M, Murthy V, and Ramassamy C. Potential role of acrolein in neurodegeneration and in Alzheimer's disease. Curr Mol Pharmacol 3: 66–78, 2010 [PubMed] [Google Scholar]
- 61.Davalos D, Ryu JK, Merlini M, Baeten KM, Le Moan N, Petersen MA, Deerinck TJ, Smirnoff DS, Bedard C, Hakozaki H, Gonias Murray S, Ling JB, Lassmann H, Degen JL, Ellisman MH, and Akassoglou K. Fibrinogen-induced perivascular microglial clustering is required for the development of axonal damage in neuroinflammation. Nat Commun 3: 1227, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davenport CM, Sevastou IG, Hooper C, and Pocock JM. Inhibiting p53 pathways in microglia attenuates microglial-evoked neurotoxicity following exposure to Alzheimer peptides. J Neurochem 112: 552–563, 2010 [DOI] [PubMed] [Google Scholar]
- 63.Davoust N, Vuaillat C, Androdias G, and Nataf S. From bone marrow to microglia: barriers and avenues. Trends Immunol 29: 227–234, 2008 [DOI] [PubMed] [Google Scholar]
- 64.de Boer AG. and Gaillard PJ. Blood–brain barrier dysfunction and recovery. J Neural Transm 113: 455–462, 2006 [DOI] [PubMed] [Google Scholar]
- 65.De Caterina R. and Massaro M. Omega-3 fatty acids and the regulation of expression of endothelial pro-atherogenic and pro-inflammatory genes. J Membr Biol 206: 103–116, 2005 [DOI] [PubMed] [Google Scholar]
- 66.DeChatelet LR, McPhail LC, Mullikin D, and McCall CE. An isotopic assay for NADPH oxidase activity and some characteristics of the enzyme from human polymorphonuclear leukocytes. J Clin Invest 55: 714–721, 1975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.del Rio-Hortega P. El “tercer elemento” de los centros nerviosos. Bol Soc Esp Biol 9: 69–120, 1919 [Google Scholar]
- 68.del Rio Hortega P. Microglia. In: Cytology and Cellular Pathology of the Nervous System, edited by Penfield W, vol. 2 New York: PB Hoeber, 1032, pp. 481–534, [Google Scholar]
- 69.Del Rio D, Stewart AJ, and Pellegrini N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr Metab Cardiovasc Dis 15: 316–328, 2005 [DOI] [PubMed] [Google Scholar]
- 70.Delibas N, Ozcankaya R, and Altuntas I. Clinical importance of erythrocyte malondialdehyde levels as a marker for cognitive deterioration in patients with dementia of Alzheimer type: a repeated study in 5-year interval. Clin Biochem 35: 137–141, 2002 [DOI] [PubMed] [Google Scholar]
- 71.Denieffe S, Kelly RJ, McDonald C, Lyons A, and Lynch MA. Classical activation of microglia in CD200-deficient mice is a consequence of blood brain barrier permeability and infiltration of peripheral cells. Brain Behav Immun 34: 86–97, 2013 [DOI] [PubMed] [Google Scholar]
- 72.Desai BS, Monahan AJ, Carvey PM, and Hendey B. Blood–brain barrier pathology in Alzheimer's and Parkinson's disease: implications for drug therapy. Cell Transplant 16: 285–299, 2007 [DOI] [PubMed] [Google Scholar]
- 73.Diaz-Meco MT, Berra E, Municio MM, Sanz L, Lozano J, Dominguez I, Diaz-Golpe V, Lain de Lera MT, Alcami J, Paya CV, et al. A dominant negative protein kinase C zeta subspecies blocks NF-kappa B activation. Mol Cell Biol 13: 4770–4775, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dib M, Garrel C, Favier A, Robin V, and Desnuelle C. Can malondialdehyde be used as a biological marker of progression in neurodegenerative disease? J Neurol 249: 367–374, 2002 [DOI] [PubMed] [Google Scholar]
- 75.Dopp JM, Sarafian TA, Spinella FM, Kahn MA, Shau H, and de Vellis J. Expression of the p75 TNF receptor is linked to TNF-induced NFKappaB translocation and oxyradical neutralization in glial cells. Neurochem Res 27: 1535–1542, 2002 [DOI] [PubMed] [Google Scholar]
- 76.Doyle KP, Simon RP, and Stenzel-Poore MP. Mechanisms of ischemic brain damage. Neuropharmacology 55: 310–318, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Drechsel DA. and Patel M. Respiration-dependent H2O2 removal in brain mitochondria via the thioredoxin/peroxiredoxin system. J Biol Chem 285: 27850–27858, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Egwuagu CE. and Larkin Iii J. Therapeutic targeting of STAT pathways in CNS autoimmune diseases. JAKSTAT 2: e24134, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Engelhardt B. Regulation of immune cell entry into the central nervous system. Results Probl Cell Differ 43: 259–280, 2006 [DOI] [PubMed] [Google Scholar]
- 80.Engelhardt B. Immune cell entry into the central nervous system: involvement of adhesion molecules and chemokines. J Neurol Sci 274: 23–26, 2008 [DOI] [PubMed] [Google Scholar]
- 81.Engelhardt B. and Coisne C. Fluids and barriers of the CNS establish immune privilege by confining immune surveillance to a two-walled castle moat surrounding the CNS castle. Fluids Barriers CNS 8: 4, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Esterbauer H, Schaur RJ, and Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 11: 81–128, 1991 [DOI] [PubMed] [Google Scholar]
- 83.Eugenin EA, King JE, Hazleton JE, Major EO, Bennett MV, Zukin RS, and Berman JW. Differences in NMDA receptor expression during human development determine the response of neurons to HIV-tat-mediated neurotoxicity. Neurotox Res 19: 138–148, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Eyo UB. and Wu LJ. Bidirectional microglia-neuron communication in the healthy brain. Neural Plast 2013: 456857, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Farooqui AA, Horrocks LA, and Farooqui T. Modulation of inflammation in brain: a matter of fat. J Neurochem 101: 577–599, 2007 [DOI] [PubMed] [Google Scholar]
- 86.Fischer MT, Sharma R, Lim JL, Haider L, Frischer JM, Drexhage J, Mahad D, Bradl M, van Horssen J, and Lassmann H. NADPH oxidase expression in active multiple sclerosis lesions in relation to oxidative tissue damage and mitochondrial injury. Brain 135: 886–899, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Furukawa M. and Xiong Y. BTB protein Keap1 targets antioxidant transcription factor Nrf2 for ubiquitination by the Cullin 3-Roc1 ligase. Mol Cell Biol 25: 162–171, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gehrmann J, Matsumoto Y, and Kreutzberg GW. Microglia: intrinsic immuneffector cell of the brain. Brain Res Brain Res Rev 20: 269–287, 1995 [DOI] [PubMed] [Google Scholar]
- 89.Ghosh A. and Pahan K. Gemfibrozil, a lipid-lowering drug, induces suppressor of cytokine signaling 3 in glial cells: implications for neurodegenerative disorders. J Biol Chem 287: 27189–27203, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, and Merad M. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330: 841–845, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gloire G. and Piette J. Redox regulation of nuclear post-translational modifications during NF-kappaB activation. Antioxid Redox Signal 11: 2209–2222, 2009 [DOI] [PubMed] [Google Scholar]
- 92.Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, and Becher B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med 11: 328–334, 2005 [DOI] [PubMed] [Google Scholar]
- 93.Gueraud F, Atalay M, Bresgen N, Cipak A, Eckl PM, Huc L, Jouanin I, Siems W, and Uchida K. Chemistry and biochemistry of lipid peroxidation products. Free Radic Res 44: 1098–1124, 2010 [DOI] [PubMed] [Google Scholar]
- 94.Hägglund P, Bunkenborg J, Maeda K, and Svensson B. Identification of thioredoxin disulfide targets using a quantitative proteomics approach based on isotope-coded affinity tags. J Proteome Res 7: 5270–5276, 2008 [DOI] [PubMed] [Google Scholar]
- 95.Haider L, Fischer MT, Frischer JM, Bauer J, Hoftberger R, Botond G, Esterbauer H, Binder CJ, Witztum JL, and Lassmann H. Oxidative damage in multiple sclerosis lesions. Brain 134: 1914–1924, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hall ED, Detloff MR, Johnson K, and Kupina NC. Peroxynitrite-mediated protein nitration and lipid peroxidation in a mouse model of traumatic brain injury. J Neurotrauma 21: 9–20, 2004 [DOI] [PubMed] [Google Scholar]
- 97.Haorah J, Ramirez SH, Schall K, Smith D, Pandya R, and Persidsky Y. Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood–brain barrier dysfunction. J Neurochem 101: 566–576, 2007 [DOI] [PubMed] [Google Scholar]
- 98.Harraz MM, Marden JJ, Zhou W, Zhang Y, Williams A, Sharov VS, Nelson K, Luo M, Paulson H, Schoneich C, and Engelhardt JF. SOD1 mutations disrupt redox-sensitive Rac regulation of NADPH oxidase in a familial ALS model. J Clin Invest 118: 659–670, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Harrigan TJ, Abdullaev IF, Jourd'heuil D, and Mongin AA. Activation of microglia with zymosan promotes excitatory amino acid release via volume-regulated anion channels: the role of NADPH oxidases. J Neurochem 106: 2449–2462, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hernandes MS, Santos GD, Cafe-Mendes CC, Lima LS, Scavone C, Munhoz CD, and Britto LR. Microglial cells are involved in the susceptibility of NADPH oxidase knockout mice to 6-hydroxy-dopamine-induced neurodegeneration. PLoS One 8: e75532, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hess DC, Abe T, Hill WD, Studdard AM, Carothers J, Masuya M, Fleming PA, Drake CJ, and Ogawa M. Hematopoietic origin of microglial and perivascular cells in brain. Exp Neurol 186: 134–144, 2004 [DOI] [PubMed] [Google Scholar]
- 102.Heyes MP, Achim CL, Wiley CA, Major EO, Saito K, and Markey SP. Human microglia convert l-tryptophan into the neurotoxin quinolinic acid. Biochem J 320 (Pt 2): 595–597, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hirota K, Matsui M, Iwata S, Nishiyama A, Mori K, and Yodoi J. AP-1 transcriptional activity is regulated by a direct association between thioredoxin and Ref-1. Proc Natl Acad Sci U S A 94: 3633–3638, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hirrlinger J, Gutterer JM, Kussmaul L, Hamprecht B, and Dringen R. Microglial cells in culture express a prominent glutathione system for defense against reactive oxygen species. Dev Neuroscience 22: 384–392, 2000 [DOI] [PubMed] [Google Scholar]
- 105.Hoetzenecker W, Echtenacher B, Guenova E, Hoetzenecker K, Woelbing F, Bruck J, Teske A, Valtcheva N, Fuchs K, Kneilling M, Park JH, Kim KH, Kim KW, Hoffmann P, Krenn C, Hai T, Ghoreschi K, Biedermann T, and Rocken M. ROS-induced ATF3 causes susceptibility to secondary infections during sepsis-associated immunosuppression. Nat Med 18: 128–134, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hollensworth SB, Shen C, Sim JE, Spitz DR, Wilson GL, and LeDoux SP. Glial cell type-specific responses to menadione-induced oxidative stress. Free Radic Biol Med 15: 1161–1174, 2000 [DOI] [PubMed] [Google Scholar]
- 107.Huang HC, Nguyen T, and Pickett CB. Phosphorylation of Nrf2 at Ser-40 by protein kinase C regulates antioxidant response element-mediated transcription. J Biol Chem 277: 42769–42774, 2002 [DOI] [PubMed] [Google Scholar]
- 108.Huang Z, Li J, Zhang S, and Zhang X. Inorganic arsenic modulates the expression of selenoproteins in mouse embryonic stem cell. Toxicol Lett 187: 69–76, 2009 [DOI] [PubMed] [Google Scholar]
- 109.Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, Bechmann I, Becher B, Luhmann HJ, Waisman A, and Kuhlmann CR. Cellular mechanisms of IL-17-induced blood–brain barrier disruption. FASEB J 24: 1023–1034, 2010 [DOI] [PubMed] [Google Scholar]
- 110.Iantomasi T, Favilli F, Marraccini P, Stio M, Treves C, and Quattrone A. Age and GSH metabolism in rat cerebral cortex, as related to oxidative and energy parameters. Mech Ageing Dev 70: 65–82, 1993 [DOI] [PubMed] [Google Scholar]
- 111.Innamorato NG, Rojo AI, Garcia-Yague AJ, Yamamoto M, de Ceballos ML, and Cuadrado A. The transcription factor Nrf2 is a therapeutic target against brain inflammation. J Immunol 181: 680–689, 2008 [DOI] [PubMed] [Google Scholar]
- 112.Ishii Y, Itoh K, Morishima Y, Kimura T, Kiwamoto T, Iizuka T, Hegab AE, Hosoya T, Nomura A, Sakamoto T, Yamamoto M, and Sekizawa K. Transcription factor Nrf2 plays a pivotal role in protection against elastase-induced pulmonary inflammation and emphysema. J Immunol 175: 6968–6975, 2005 [DOI] [PubMed] [Google Scholar]
- 113.Jain AK. and Jaiswal AK. GSK-3beta acts upstream of Fyn kinase in regulation of nuclear export and degradation of NF-E2 related factor 2. J Biol Chem 282: 16502–16510, 2007 [DOI] [PubMed] [Google Scholar]
- 114.Jamaluddin M, Wang S, Boldogh I, Tian B, and Brasier AR. TNF-alpha-induced NF-kappaB/RelA Ser(276) phosphorylation and enhanceosome formation is mediated by an ROS-dependent PKAc pathway. Cell Signal 19: 1419–1433, 2007 [DOI] [PubMed] [Google Scholar]
- 115.Jayadev S, Yun B, Nguyen H, Yokoo H, Morrison RS, and Garden GA. The glial response to CNS HIV infection includes p53 activation and increased expression of p53 target genes. J Neuroimmune Pharmacol 2: 359–370, 2007 [DOI] [PubMed] [Google Scholar]
- 116.Jazwa A, Rojo AI, Innamorato NG, Hesse M, Fernandez-Ruiz J, and Cuadrado A. Pharmacological targeting of the transcription factor Nrf2 at the basal ganglia provides disease modifying therapy for experimental parkinsonism. Antioxid Redox Signal 14: 2347–2360, 2011 [DOI] [PubMed] [Google Scholar]
- 117.Jekabsone A, Mander PK, Tickler A, Sharpe M, and Brown GC. Fibrillar beta-amyloid peptide Abeta1-40 activates microglial proliferation via stimulating TNF-alpha release and H2O2 derived from NADPH oxidase: a cell culture study. J Neuroinflammation 3: 24, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kacimi R, Giffard RG, and Yenari MA. Endotoxin-activated microglia injure brain derived endothelial cells via NF-kappaB, JAK-STAT and JNK stress kinase pathways. J Inflamm (Lond) 8: 7, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kahles T, Luedike P, Endres M, Galla HJ, Steinmetz H, Busse R, Neumann-Haefelin T, and Brandes RP. NADPH oxidase plays a central role in blood–brain barrier damage in experimental stroke. Stroke 38: 3000–3006, 2007 [DOI] [PubMed] [Google Scholar]
- 120.Kambe T, Song T, Takata T, Hatano N, Miyamoto Y, Nozaki N, Naito Y, Tokumitsu H, and Watanabe Y. Inactivation of Ca2+/calmodulin-dependent protein kinase 1 by S-glutathionylation of the active site cysteine residue. FEBS Lett 584: 2478–2484, 2010 [DOI] [PubMed] [Google Scholar]
- 121.Kang CH, Jayasooriya RG, Dilshara MG, Choi YH, Jeong YK, Kim ND, and Kim GY. Caffeine suppresses lipopolysaccharide-stimulated BV2 microglial cells by suppressing Akt-mediated NF-kappaB activation and ERK phosphorylation. Food Chem Toxicol 50: 4270–4276, 2012 [DOI] [PubMed] [Google Scholar]
- 122.Kato T, Jr., Delhase M, Hoffmann A, and Karin M. CK2 is a C-terminal IkappaB kinase responsible for NF-kappaB activation during the UV response. Mol Cell 12: 829–839, 2003 [DOI] [PubMed] [Google Scholar]
- 123.Kelleher ZT, Matsumoto A, Stamler JS, and Marshall HE. NOS2 regulation of NF-kappaB by S-nitrosylation of p65. J Biol Chem 282: 30667–30672, 2007 [DOI] [PubMed] [Google Scholar]
- 124.Keum YS, Owuor ED, Kim BR, Hu R, and Kong AN. Involvement of Nrf2 and JNK1 in the activation of antioxidant responsive element (ARE) by chemopreventive agent phenethyl isothiocyanate (PEITC). Pharm Res 20: 1351–1356, 2003 [DOI] [PubMed] [Google Scholar]
- 125.Khan M, Sekhon B, Jatana M, Giri S, Gilg AG, Sekhon C, Singh I, and Singh AK. Administration of N-acetylcysteine after focal cerebral ischemia protects brain and reduces inflammation in a rat model of experimental stroke. J Neurosci Res 76: 519–527, 2004 [DOI] [PubMed] [Google Scholar]
- 126.Kigerl KA, Ankeny DA, Garg SK, Wei P, Guan Z, Wenmin L, McTigue DM, Banerjee R, and Popovich PG. System xc- regulated microglia and macrophage glutamate excitotoxicity in vivo. Exp Neurol 233: 333–341, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kim CY, Lee C, Park GH, and Jang JH. Neuroprotective effect of epigallocatechin-3-gallate against beta-amyloid-induced oxidative and nitrosative cell death via augmentation of antioxidant defence capacity. Arch Pharm Res 32: 869–881, 2009 [DOI] [PubMed] [Google Scholar]
- 128.Kim H, Parka JH, Kim K. Lipid-like components released from degenerating dopaminergic neurons trigger the dynamic migration of microglia. Biochem Biophys Res Commun 426: 18–25, 2012 [DOI] [PubMed] [Google Scholar]
- 129.Kitamura Y, Kakimura J, Koike H, Umeki M, Gebicke-Haerter PJ, Nomura Y, and Taniguchi T. Effects of 15-deoxy-delta(12,14) prostaglandin J(2) and interleukin-4 in Toll-like receptor-4-mutant glial cells. Eur J Pharmacol 411: 223–230, 2001 [DOI] [PubMed] [Google Scholar]
- 130.Klatt P, Molina EP, De Lacoba MG, Padilla CA, Martinez-Galesteo E, Barcena JA, and Lamas S. Redox regulation of c-Jun DNA binding by reversible S-glutathiolation. FASEB J 13: 1481–1490, 1999 [DOI] [PubMed] [Google Scholar]
- 131.Kobayashi A, Kang MI, Okawa H, Ohtsuji M, Zenke Y, Chiba T, Igarashi K, and Yamamoto M. Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol Cell Biol 24: 7130–7139, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kobayashi M, Li L, Iwamoto N, Nakajima-Takagi Y, Kaneko H, Nakayama Y, Eguchi M, Wada Y, Kumagai Y, and Yamamoto M. The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol Cell Biol 29: 493–502, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Koppal T, Petrova TV, and Van Eldik LJ. Cyclopentenone prostaglandin 15-deoxy-Delta(12,14)-prostaglandin J(2) acts as a general inhibitor of inflammatory responses in activated BV-2 microglial cells. Brain Res 867: 115–121, 2000 [DOI] [PubMed] [Google Scholar]
- 134.Koriyama Y, Nakayama Y, Matsugo S, and Kato S. Protective effect of lipoic acid against oxidative stress is mediated by Keap1/Nrf2-dependent heme oxygenase-1 induction in the RGC-5 cellline. Brain Res 1499: 145–157, 2013 [DOI] [PubMed] [Google Scholar]
- 135.Krizbai IA, Bauer H, Bresgen N, Eckl PM, Farkas A, Szatmari E, Traweger A, Wejksza K, and Bauer HC. Effect of oxidative stress on the junctional proteins of cultured cerebral endothelial cells. Cell Mol Neurobiol 25: 129–139, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kwak MK, Wakabayashi N, Greenlaw JL, Yamamoto M, and Kensler TW. Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway. Mol Cell Biol 23: 8786–8794, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Lallena MJ, Diaz-Meco MT, Bren G, Paya CV, and Moscat J. Activation of IkappaB kinase beta by protein kinase C isoforms. Mol Cell Biol 19: 2180–2188, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lanone S, Bloc S, Foresti R, Almolki A, Taille C, Callebert J, Conti M, Goven D, Aubier M, Dureuil B, El-Benna J, Motterlini R, and Boczkowski J. Bilirubin decreases nos2 expression via inhibition of NAD(P)H oxidase: implications for protection against endotoxic shock in rats. FASEB J 19: 1890–1892, 2005 [DOI] [PubMed] [Google Scholar]
- 139.Larochelle C, Alvarez JI, and Prat A. How do immune cells overcome the blood–brain barrier in multiple sclerosis? FEBS Lett 585: 3770–3780, 2011 [DOI] [PubMed] [Google Scholar]
- 140.Laschinger M, Vajkoczy P, and Engelhardt B. Encephalitogenic T cells use LFA-1 for transendothelial migration but not during capture and initial adhesion strengthening in healthy spinal cord microvessels in vivo. Eur J Immunol 32: 3598–3606, 2002 [DOI] [PubMed] [Google Scholar]
- 141.Lassmann H, Schmied M, Vass K, and Hickey WF. Bone marrow derived elements and resident microglia in brain inflammation. Glia 7: 19–24, 1993 [DOI] [PubMed] [Google Scholar]
- 142.Lastres-Becker I, Innamorato NG, Jaworski T, Rabano A, Kugler S, Van Leuven F, and Cuadrado A. Fractalkine activates NRF2/NFE2L2 and heme oxygenase 1 to restrain tauopathy-induced microgliosis. Brain 137: 78–91, 2014 [DOI] [PubMed] [Google Scholar]
- 143.Lawson LJ, Perry VH, Dri P, and Gordon S. Heterogeneity in the distribution and morphology of microglia in the normal adult mouse brain. Neuroscience 39: 151–170, 1990 [DOI] [PubMed] [Google Scholar]
- 144.Lee H, Lee S, Cho IH, and Lee SJ. Toll-like receptors: sensor molecules for detecting damage to the nervous system. Curr Protein Pept Sci 14: 33–42, 2013 [DOI] [PubMed] [Google Scholar]
- 145.Lee M, Cho T, Jantaratnotai N, Wang YT, McGeer E, and McGeer PL. Depletion of GSH in glial cells induces neurotoxicity: relevance to aging and degeneration in neurological diseases. FASEB J 24: 2533–2545, 2010 [DOI] [PubMed] [Google Scholar]
- 146.Lee M, Tazzari V, Giustarini D, Rossi R, Sparatore A, Del Soldato P, McGeer E, and McGeer PL. Effects of hydrogen sulfide-releasing L-DOPA derivatives on glial activation: potential for treating Parkinson's disease. J Biol Chem 285: 17318–17328, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Lee S, Lee J, Kim S, Park JY, Lee WH, Mori K, Kim SH, Kim IK, and Suk K. A dual role of lipocalin 2 in the apoptosis and deramification of activated microglia. J Immunol 179: 3231–3241, 2007 [DOI] [PubMed] [Google Scholar]
- 148.Lee S, Park JY, Lee WH, Kim H, Park HC, Mori K, and Suk K. Lipocalin-2 is an autocrine mediator of reactive astrocytosis. J Neurosci 29: 234–249, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Leitges M, Sanz L, Martin P, Duran A, Braun U, Garcia JF, Camacho F, Diaz-Meco MT, Rennert PD, and Moscat J. Targeted disruption of the zetaPKC gene results in the impairment of the NF-kappaB pathway. Mol Cell 8: 771–780, 2001 [DOI] [PubMed] [Google Scholar]
- 150.Leonardo CC. and Dore S. Dietary flavonoids are neuroprotective through Nrf2-coordinated induction of endogenous cytoprotective proteins. Nutr Neurosci 14: 226–236, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Li B, Bedard K, Sorce S, Hinz B, Dubois-Dauphin M, and Krause KH. NOX4 expression in human microglia leads to constitutive generation of reactive oxygen species and to constitutive IL-6 expression. J Innate Immun 1: 570–581, 2009 [DOI] [PubMed] [Google Scholar]
- 152.Li N, Alam J, Venkatesan MI, Eiguren-Fernandez A, Schmitz D, Di Stefano E, Slaughter N, Killeen E, Wang X, Huang A, Wang M, Miguel AH, Cho A, Sioutas C, and Nel AE. Nrf2 is a key transcription factor that regulates antioxidant defense in macrophages and epithelial cells: protecting against the proinflammatory and oxidizing effects of diesel exhaust chemicals. J Immunol 173: 3467–3481, 2004 [DOI] [PubMed] [Google Scholar]
- 153.Li W. and Kong AN. Molecular mechanisms of Nrf2-mediated antioxidant response. Mol Carcinog 48: 91–104, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liao B, Zhao W, Beers DR, Henkel JS, and Appel SH. Transformation from a neuroprotective to a neurotoxic microglial phenotype in a mouse model of ALS. Exp Neurol 237: 147–152, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lindenau J, Noack H, Asayama K, and Wolf G. Enhanced cellular glutathione peroxidase imunoreactivity in activated astrocytes and in microglia during excitotoxin-induced neurodegeneration. Glia 24: 252–256, 1998 [PubMed] [Google Scholar]
- 156.Lischper M, Beuck S, Thanabalasundaram G, Pieper C, and Galla HJ. Metalloproteinase mediated occludin cleavage in the cerebral microcapillary endothelium under pathological conditions. Brain Res 1326: 114–127, 2010 [DOI] [PubMed] [Google Scholar]
- 157.Liu R. and Choi J. Age-associated decline in gamma-glutamylcysteine synthetase gene expression in rats. Free Radic Biol Med 28: 566–574, 2000 [DOI] [PubMed] [Google Scholar]
- 158.Liu RM, Choi J, Wu JH, Gaston Pravia KA, Lewis KM, Brand JD, Mochel NS, Krzywanski DM, Lambeth JD, Hagood JS, Forman HJ, Thannickal VJ, and Postlethwait EM. Oxidative modification of nuclear mitogen-activated protein kinase phosphatase 1 is involved in transforming growth factor beta1-induced expression of plasminogen activator inhibitor 1 in fibroblasts. J Biol Chem 285: 16239–16247, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Liu X, Lovell MA, and Lynn BC. Development of a method for quantification of acrolein-deoxyguanosine adducts in DNA using isotope dilution-capillary LC/MS/MS and its application to human brain tissue. Anal Chem 77: 5982–5989, 2005 [DOI] [PubMed] [Google Scholar]
- 160.Liu Y, Teige I, Birnir B, and Issazadeh-Navikas S. Neuron-mediated generation of regulatory T cells from encephalitogenic T cells suppresses EAE. Nat Med 12: 518–525, 2006 [DOI] [PubMed] [Google Scholar]
- 161.Lochhead JJ, McCaffrey G, Quigley CE, Finch J, DeMarco KM, Nametz N, and Davis TP. Oxidative stress increases blood–brain barrier permeability and induces alterations in occludin during hypoxia-reoxygenation. J Cereb Blood Flow Metab 30: 1625–1636, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lovell MA, Xie C, and Markesbery WR. Acrolein is increased in Alzheimer's disease brain and is toxic to primary hippocampal cultures. Neurobiol Aging 22: 187–194, 2001 [DOI] [PubMed] [Google Scholar]
- 163.Lu DY, Huang BR, Yeh WL, Lin HY, Huang SS, Liu YS, and Kuo YH. Anti-neuroinflammatory effect of a novel caffeamide derivative, KS370G, in microglial cells. Mol Neurobiol 48: 863–874, 2013 [DOI] [PubMed] [Google Scholar]
- 164.Lu J. and Holmgren A. Thioredoxin system in cell death progression. Antioxid Redox Signal 17: 1738–1747, 2012 [DOI] [PubMed] [Google Scholar]
- 165.Lyons A, Downer EJ, Crotty S, Nolan YM, Mills KH, and Lynch MA. CD200 ligand receptor interaction modulates microglial activation in vivo and in vitro: a role for IL-4. J Neurosci 27: 8309–8313, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Malhotra D, Portales-Casamar E, Singh A, Srivastava S, Arenillas D, Happel C, Shyr C, Wakabayashi N, Kensler TW, Wasserman WW, and Biswal S. Global mapping of binding sites for Nrf2 identifies novel targets in cell survival response through ChIP-Seq profiling and network analysis. Nucleic Acids Res 38: 5718–5734, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mancuso C. and Barone E. The heme oxygenase/biliverdin reductase pathway in drug research and development. Curr Drug Metab 10: 579–594, 2009 [DOI] [PubMed] [Google Scholar]
- 168.Mancuso C, Barone E, Guido P, Miceli F, Di Domenico F, Perluigi M, Santangelo R, and Preziosi P. Inhibition of lipid peroxidation and protein oxidation by endogenous and exogenous antioxidants in rat brain microsomes in vitro. Neurosci Lett 518: 101–105, 2012 [DOI] [PubMed] [Google Scholar]
- 169.Mancuso C, Capone C, Ranieri SC, Fusco S, Calabrese V, Eboli ML, Preziosi P, Galeotti T, and Pani G. Bilirubin as an endogenous modulator of neurotrophin redox signaling. J Neurosci Res 86: 2235–2249, 2008 [DOI] [PubMed] [Google Scholar]
- 170.Mancuso C, Navarra P, and Preziosi P. Roles of nitric oxide, carbon monoxide, and hydrogen sulfide in the regulation of the hypothalamic-pituitary-adrenal axis. J Neurochem 113: 563–575, 2010 [DOI] [PubMed] [Google Scholar]
- 171.Mancuso C, Pistritto G, Tringali G, Grossman AB, Preziosi P, and Navarra P. Evidence that carbon monoxide stimulates prostaglandin endoperoxide synthase activity in rat hypothalamic explants and in primary cultures of rat hypothalamic astrocytes. Brain Res Mol Brain Res 45: 294–300, 1997 [DOI] [PubMed] [Google Scholar]
- 172.Mancuso C, Preziosi P, Grossman AB, and Navarra P. The role of carbon monoxide in the regulation of neuroendocrine function. Neuroimmunomodulation 4: 225–229, 1997 [DOI] [PubMed] [Google Scholar]
- 173.Mancuso C, Ragazzoni E, Tringali G, Liberale I, Preziosi P, Grossman A, and Navarra P. Inhibition of heme oxygenase in the central nervous system potentiates endotoxin-induced vasopressin release in the rat. J Neuroimmunol 99: 189–194, 1999 [DOI] [PubMed] [Google Scholar]
- 174.Mander P. and Brown GC. Activation of microglial NADPH oxidase is synergistic with glial iNOS expression in inducing neuronal death: a dual-key mechanism of inflammatory neurodegeneration. J Neuroinflammation 2: 20, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mantovani A, Sozzani S, Locati M, Allavena P, and Sica A. Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23: 549–555, 2002 [DOI] [PubMed] [Google Scholar]
- 176.Marcheselli VL, Hong S, Lukiw WJ, Tian XH, Gronert K, Musto A, Hardy M, Gimenez JM, Chiang N, Serhan CN, and Bazan NG. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem 278: 43807–43817, 2003 [DOI] [PubMed] [Google Scholar]
- 177.Marden JJ, Harraz MM, Williams AJ, Nelson K, Luo M, Paulson H, and Engelhardt JF. Redox modifier genes in amyotrophic lateral sclerosis in mice. J Clin Invest 117: 2913–2919, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Marik C, Felts PA, Bauer J, Lassmann H, and Smith KJ. Lesion genesis in a subset of patients with multiple sclerosis: a role for innate immunity? Brain 130: 2800–2815, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Marin-Teva JL, Dusart I, Colin C, Gervais A, van Rooijen N, and Mallat M. Microglia promote the death of developing Purkinje cells. Neuron 41: 535–547, 2004 [DOI] [PubMed] [Google Scholar]
- 180.Markesbery WR, Kryscio RJ, Lovell MA, and Morrow JD. Lipid peroxidation is an early event in the brain in amnestic mild cognitive impairment. Ann Neurol 58: 730–735, 2005 [DOI] [PubMed] [Google Scholar]
- 181.Markesbery WR. and Lovell MA. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer's disease. Neurobiol Aging 19: 33–36, 1998 [DOI] [PubMed] [Google Scholar]
- 182.Martinez FO, Sica A, Mantovani A, and Locati M. Macrophage activation and polarization. Front Biosci 13: 453–461, 2008 [DOI] [PubMed] [Google Scholar]
- 183.Mattson MP. NF-kappaB in the survival and plasticity of neurons. Neurochem Res 30: 883–893, 2005 [DOI] [PubMed] [Google Scholar]
- 184.McBean GJ. Cerebral cystine uptake: a tale of two transporters. Trends Pharmacol Sci 23: 299–302, 2002 [DOI] [PubMed] [Google Scholar]
- 185.McCaffrey G, Willis CL, Staatz WD, Nametz N, Quigley CA, Hom S, Lochhead JJ, and Davis TP. Occludin oligomeric assemblies at tight junctions of the blood–brain barrier are altered by hypoxia and reoxygenation stress. J Neurochem 110: 58–71, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.McMahon M, Thomas N, Itoh K, Yamamoto M, and Hayes JD. Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J Biol Chem 279: 31556–31567, 2004 [DOI] [PubMed] [Google Scholar]
- 187.McNeill E. and Channon KM. The role of tetrahydrobiopterin in inflammation and cardiovascular disease. Thromb Haemost 108: 832–839, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Medawar PB. Immunity to homologous grafted skin; the fate of skin homografts transplanted to the brain, to subcutaneous tissue, and to the anterior chamber of the eye. Br J Exp Pathol 29: 58–69, 1948 [PMC free article] [PubMed] [Google Scholar]
- 189.Meja KK, Rajendrasozhan S, Adenuga D, Biswas SK, Sundar IK, Spooner G, Marwick JA, Chakravarty P, Fletcher D, Whittaker P, Megson IL, Kirkham PA, and Rahman I. Curcumin restores corticosteroid function in monocytes exposed to oxidants by maintaining HDAC2. Am J Respir Cell Mol Biol 39: 312–323, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Mercado N, Thimmulappa R, Thomas CM, Fenwick PS, Chana KK, Donnelly LE, Biswal S, Ito K, and Barnes PJ. Decreased histone deacetylase 2 impairs Nrf2 activation by oxidative stress. Biochem Biophys Res Commun 406: 292–298, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Mertsch K, Blasig I, and Grune T. 4-Hydroxynonenal impairs the permeability of an in vitro rat blood–brain barrier. Neurosci Lett 314: 135–138, 2001 [DOI] [PubMed] [Google Scholar]
- 192.Mildner A, Schmidt H, Nitsche M, Merkler D, Hanisch UK, Mack M, Heikenwalder M, Bruck W, Priller J, and Prinz M. Microglia in the adult brain arise from Ly-6ChiCCR2+monocytes only under defined host conditions. Nat Neurosci 10: 1544–1553, 2007 [DOI] [PubMed] [Google Scholar]
- 193.Min KJ, Pyo HK, Yang MS, Ji KA, Jou I, and Joe EH. Gangliosides activate microglia via protein kinase C and NADPH oxidase. Glia 48: 197–206, 2004 [DOI] [PubMed] [Google Scholar]
- 194.Min KJ, Yang MS, Kim SU, Jou I, and Joe EH. Astrocytes induce hemeoxygenase-1 expression in microglia: a feasible mechanism for preventing excessive brain inflammation. J Neurosci 26: 1880–1887, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, and Motohashi H. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22: 66–79, 2012 [DOI] [PubMed] [Google Scholar]
- 196.Miziu T, Kinouchi H, and Chan PH. Depletion of brain glutathione by buthionine sulfoximine enhances cerebral ischaemic injury in rats. Am J Physiol 262: H313–H317, 1992 [DOI] [PubMed] [Google Scholar]
- 197.Moi P, Chan K, Asunis I, Cao A, and Kan YW. Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region. Proc Natl Acad Sci U S A 91: 9926–9930, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Montine KS, Quinn JF, Zhang J, Fessel JP, Roberts LJ, 2nd, Morrow JD, and Montine TJ. Isoprostanes and related products of lipid peroxidation in neurodegenerative diseases. Chem Phys Lipids 128: 117–124, 2004 [DOI] [PubMed] [Google Scholar]
- 199.Morsali D, Bechtold D, Lee W, Chauhdry S, Palchaudhuri U, Hassoon P, Snell DM, Malpass K, Piers T, Pocock J, Roach A, and Smith KJ. Safinamide and flecainide protect axons and reduce microglial activation in models of multiple sclerosis. Brain 136: 1067–1082, 2013 [DOI] [PubMed] [Google Scholar]
- 200.Moss DW. and Bates TE. Activation of murine microglial cell lines by lipopolysaccharide and interferon-gamma causes NO-mediated decreases in mitochondrial and cellular function. Eur J Neurosci 13: 529–538, 2001 [DOI] [PubMed] [Google Scholar]
- 201.Muldoon LL, Alvarez JI, Begley DJ, Boado RJ, Del Zoppo GJ, Doolittle ND, Engelhardt B, Hallenbeck JM, Lonser RR, Ohlfest JR, Prat A, Scarpa M, Smeyne RJ, Drewes LR, and Neuwelt EA. Immunologic privilege in the central nervous system and the blood–brain barrier. J Cereb Blood Flow Metab 33: 13–21, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Murphy JB. and Sturm E. Conditions determining the transplantability of tissues in the brain. J Exp Med 38: 183–197, 1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Nag S. and Begley DJ. Blood–brain barrier, exchange of metabolites and gases. In: Pathology and Genetics. Cerebrovascular Diseases. edited by Basel Kalimo H., Switzerland: ISN Neuropath. Press, 2005, pp. 22–29 [Google Scholar]
- 204.Nakahira K, Kim HP, Geng XH, Nakao A, Wang X, Murase N, Drain PF, Wang X, Sasidhar M, Nabel EG, Takahashi T, Lukacs NW, Ryter SW, Morita K, and Choi AM. Carbon monoxide differentially inhibits TLR signaling pathways by regulating ROS-induced trafficking of TLRs to lipid rafts. J Exp Med 203: 2377–2389, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Nalvarte I, Damdimopolous AE, and Spyrou G. Human mitochondrial thioredoxin reductase reduces cytochrome c and confers resistance to complex III inhibition. Free Radic Biol Med 36: 1270–1278, 2004 [DOI] [PubMed] [Google Scholar]
- 206.Narasimhan M, Patel D, Vedpathak D, Rathinam M, Henderson G, and Mahimainathan L. Identification of novel microRNAs in post-transcriptional control of Nrf2 expression and redox homeostasis in neuronal, SH-SY5Y cells. PLoS One 7: e51111, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Nayernia Z, Jaquet V, and Krause K.-H.New insights on NOX enzymes in the central nervous system. Antioxid Redox Signal 20: 2815–2837, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Negre-Salvayre A, Auge N, Ayala V, Basaga H, Boada J, Brenke R, Chapple S, Cohen G, Feher J, Grune T, Lengyel G, Mann GE, Pamplona R, Poli G, Portero-Otin M, Riahi Y, Salvayre R, Sasson S, Serrano J, Shamni O, Siems W, Siow RC, Wiswedel I, Zarkovic K, and Zarkovic N. Pathological aspects of lipid peroxidation. Free Radic Res 44: 1125–1171, 2010 [DOI] [PubMed] [Google Scholar]
- 209.Nemoto S, DiDonato JA, and Lin A. Coordinate regulation of IkappaB kinases by mitogen-activated protein kinase kinase kinase 1 and NF-kappaB-inducing kinase. Mol Cell Biol 18: 7336–7343, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Newcombe J, Li H, and Cuzner ML. Low density lipoprotein uptake by macrophages in multiple sclerosis plaques: implications for pathogenesis. Neuropathol Appl Neurobiol 20: 152–162, 1994 [DOI] [PubMed] [Google Scholar]
- 211.Nimmerjahn A, Kirchhoff F, and Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308: 1314–1318, 2005 [DOI] [PubMed] [Google Scholar]
- 212.Nisimoto Y, Jackson HM, Ogawa H, Kawahara T, and Lambeth JD. Constitutive NADPH-dependent electron transferase activity of the Nox4 dehydrogenase domain. Biochemistry 49: 2433–2442, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Njie EG, Boelen E, Stassen FR, Steinbusch HW, Borchelt DR, and Streit WJ. Ex vivo cultures of microglia from young and aged rodent brain reveal age-related changes in microglial function. Neurobiol Aging 33: 195 e1–e12, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Noack H, Possel H, Chatterjee S, Keilhoff G, and Wolf G. Nitrosative stress in primary glial cultures after induction of the inducible isoform of nitric oxide synthase (i-NOS). Toxicology 148: 133–142, 2000 [DOI] [PubMed] [Google Scholar]
- 215.Norden DM. and Godbout JP. Review: microglia of the aged brain: primed to be activated and resistant to regulation. Neuropathol Appl Neurobiol 39: 19–34, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Nowak DE, Tian B, Jamaluddin M, Boldogh I, Vergara LA, Choudhary S, and Brasier AR. RelA Ser276 phosphorylation is required for activation of a subset of NF-kappaB-dependent genes by recruiting cyclin-dependent kinase 9/cyclin T1 complexes. Mol Cell Biol 28: 3623–3638, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Obin M, Shang F, Gong X, Handelman G, Blumberg J, and Taylor A. Redox regulation of ubiquitin-conjugating enzymes: mechanistic insights using the thiol-specific oxidant diamide. FASEB J 12: 561–569, 1998 [DOI] [PubMed] [Google Scholar]
- 218.Oh J, Hur MW, and Lee CE. SOCS1 protects protein tyrosine phosphatases by thioredoxin upregulation and attenuates Jaks to suppress ROS-mediated apoptosis. Oncogene 28: 3145–3156, 2009 [DOI] [PubMed] [Google Scholar]
- 219.Owens T, Bechmann I, and Engelhardt B. Perivascular spaces and the two steps to neuroinflammation. J Neuropathol Exp Neurol 67: 1113–1121, 2008 [DOI] [PubMed] [Google Scholar]
- 220.Padurariu M, Ciobica A, Hritcu L, Stoica B, Bild W, and Stefanescu C. Changes of some oxidative stress markers in the serum of patients with mild cognitive impairment and Alzheimer's disease. Neurosci Lett 469: 6–10, 2010 [DOI] [PubMed] [Google Scholar]
- 221.Pahan K, Sheikh FG, Namboodiri AM, and Singh I. N-acetyl cysteine inhibits induction of no production by endotoxin or cytokine stimulated rat peritoneal macrophages, C6 glial cells and astrocytes. Free Radic Biol Med 24: 39–48, 1998 [DOI] [PubMed] [Google Scholar]
- 222.Pais TF, Szego EM, Marques O, Miller-Fleming L, Antas P, Guerreiro P, de Oliveira RM, Kasapoglu B, and Outeiro TF. The NAD-dependent deacetylase sirtuin 2 is a suppressor of microglial activation and brain inflammation. EMBO J 32: 2603–2616, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Pampliega O, Domercq M, Soria FN, Villoslada P, Rodríguez-Antigüedad A, and Matute C. Increased expression of cystine/glutamate antiporter in multiple sclerosis. J Neuroinflammation 8: 63–75, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Pamplona R. Advanced lipoxidation end-products. Chem Biol Interact 192: 14–20, 2011 [DOI] [PubMed] [Google Scholar]
- 225.Pannu R, Won JS, Khan M, Singh AK, and Singh I. A novel role of lactosylceramide in the regulation of lipopolysaccharide/interferon-gamma-mediated inducible nitric oxide synthase gene expression: implications for neuroinflammatory diseases. J Neurosci 24: 5942–5954, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Paolicelli RC, Bolasco G, Pagani F, Maggi L, Scianni M, Panzanelli P, Giustetto M, Ferreira TA, Guiducci E, Dumas L, Ragozzino D, and Gross CT. Synaptic pruning by microglia is necessary for normal brain development. Science 333: 1456–1458, 2011 [DOI] [PubMed] [Google Scholar]
- 227.Park HS, Mo JS, and Choi EJ. Nitric oxide inhibits an interaction between JNK1 and c-Jun through nitrosylation. Biochem Biophys Res Commun 351: 281–286, 2006 [DOI] [PubMed] [Google Scholar]
- 228.Park L, Anrather J, Zhou P, Frys K, Pitstick R, Younkin S, Carlson GA, and Iadecola C. NADPH-oxidase-derived reactive oxygen species mediate the cerebrovascular dysfunction induced by the amyloid beta peptide. J Neurosci 25: 1769–1777, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Park L, Zhou P, Pitstick R, Capone C, Anrather J, Norris EH, Younkin L, Younkin S, Carlson G, McEwen BS, and Iadecola C. Nox2-derived radicals contribute to neurovascular and behavioral dysfunction in mice overexpressing the amyloid precursor protein. Proc Natl Acad Sci U S A 105: 1347–1352, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Parsons ZD. and Gates KS. Thiol-dependent recovery of catalytic activity from oxidized protein tyrosine phosphatases. Biochemistry 37: 6412–6423, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Pawate S, Shen Q, Fan F, and Bhat NR. Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J Neurosci Res 77: 540–551, 2004 [DOI] [PubMed] [Google Scholar]
- 232.Pena OM, Pistolic J, Raj D, Fjell CD, and Hancock RE. Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. J Immunol 186: 7243–7254, 2011 [DOI] [PubMed] [Google Scholar]
- 233.Perry VH, Nicoll JA, and Holmes C. Microglia in neurodegenerative disease. Nat Rev Neurol 6: 193–201, 2010 [DOI] [PubMed] [Google Scholar]
- 234.Persson M. and Rönnbäck L. Microglial self-defence mediated through GLT-1 and gluathione. Amino Acids 42: 207–219, 2012 [DOI] [PubMed] [Google Scholar]
- 235.Persson M, Sandberg M, Hansson E, and Rönnbäck L. Microglial glutamate uptake is coupled to glutathione synthesis and glutamate release. Eur J Neurosci 24: 1063–1070, 2006 [DOI] [PubMed] [Google Scholar]
- 236.Petersen DR. and Doorn JA. Reactions of 4-hydroxynonenal with proteins and cellular targets. Free Radic Biol Med 37: 937–945, 2004 [DOI] [PubMed] [Google Scholar]
- 237.Peterson LJ. and Flood PM. Oxidative stress and microglial cells in Parkinson's disease. Mediators Inflamm 2012: 401264, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Petry A, Weitnauer M, and Gorlach A. Receptor activation of NADPH oxidases. Antioxid Redox Signal 13: 467–487, 2010 [DOI] [PubMed] [Google Scholar]
- 239.Pettit LK, Varsanyi C, Tadros J, and Vassiliou E. Modulating the inflammatory properties of activated microglia with docosahexaenoic acid and aspirin. Lipids Health Dis 12: 16, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Piani D. and Fontana A. Involvement of the cystine transport system xc- in the macrophage-induced glutamate-dependent cytotoxicity to neurons. J Immunol 152: 3578–3585, 1994 [PubMed] [Google Scholar]
- 241.Piani D, Spranger M, Frei K, Schaffner A, and Fontana A. Macrophage-induced cytotoxicity of N-methyl-d-aspartate receptor positive neurons involves excitatory amino acids rather than reactive oxygen intermediates and cytokines. Eur J Immunol 22: 2429–2436, 1992 [DOI] [PubMed] [Google Scholar]
- 242.Pineda-Molina E, Klatt P, Vazquez J, Marina A, Garcia de Lacoba M, Perez-Sala D, and Lamas S. Glutathionylation of the p50 subunit of NF-kappaB: a mechanism for redox-induced inhibition of DNA binding. Biochemistry 40: 14134–14142, 2001 [DOI] [PubMed] [Google Scholar]
- 243.Pistritto G, Mancuso C, Tringali G, Perretti M, Preziosi P, and Navarra P. The relative contribution of constitutive and inducible cyclooxygenase activity to lipopolysaccharide-induced prostaglandin production by primary cultures of rat hypothalamic astrocytes. Neurosci Lett 246: 45–48, 1998 [DOI] [PubMed] [Google Scholar]
- 244.Ponomarev ED, Maresz K, Tan Y, and Dittel BN. CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J Neurosci 27: 10714–10721, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Porta C, Rimoldi M, Raes G, Brys L, Ghezzi P, Di Liberto D, Dieli F, Ghisletti S, Natoli G, De Baetselier P, Mantovani A, and Sica A. Tolerance and M2 (alternative) macrophage polarization are related processes orchestrated by p50 nuclear factor kappaB. Proc Natl Acad Sci U S A 106: 14978–14983, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Powell CS. and Jackson RM. Mitochondrial complex I, aconitase, and succinate dehydrogenase during hypoxia-reoxygenation: modulation of enzyme activities by MnSOD. Am J Physiol Lung Cell Mol Physiol 285: L189–L198, 2003 [DOI] [PubMed] [Google Scholar]
- 247.Power JH. and Blumbergs PC. Cellular glutathione peroxidase in human brain: celluar distribution and its potential role in the degradation of Lewy bodies in Parkinson's disease and dementia with Lewy bodies. Acta Neuropathol 117: 63–73, 2009 [DOI] [PubMed] [Google Scholar]
- 248.Puntener U, Booth SG, Perry VH, and Teeling JL. Long-term impact of systemic bacterial infection on the cerebral vasculature and microglia. J Neuroinflammation 9: 146, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Qin H, Holdbrooks AT, Liu Y, Reynolds SL, Yanagisawa LL, and Benveniste EN. SOCS3 deficiency promotes M1 macrophage polarization and inflammation. J Immunol 189: 3439–3448, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Qin L. and Crews FT. NADPH oxidase and reactive oxygen species contribute to alcohol-induced microglial activation and neurodegeneration. J Neuroinflammation 9: 5, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Qin L, Liu Y, Wang T, Wei SJ, Block ML, Wilson B, Liu B, and Hong JS. NADPH oxidase mediates lipopolysaccharide-induced neurotoxicity and proinflammatory gene expression in activated microglia. J Biol Chem 279: 1415–1421, 2004 [DOI] [PubMed] [Google Scholar]
- 252.Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, and Cuadrado A. SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol 31: 1121–1133, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, Tobon-Velasco JC, Devijver H, Garcia-Mayoral MF, Van Leuven F, Hayes JD, Bertho G, and Cuadrado A. Structural and functional characterization of Nrf2 degradation by the glycogen synthase kinase 3/beta-TrCP axis. Mol Cell Biol 32: 3486–3499, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Ralat LA, Ren M, Schilling AB, and Tang WJ. Protective role of Cys-178 against the inactivation and oligomerization of human insulin-degrading enzyme by oxidation and nitrosylation. J Biol Chem 284: 34005–34018, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Rangasamy T, Cho CY, Thimmulappa RK, Zhen L, Srisuma SS, Kensler TW, Yamamoto M, Petrache I, Tuder RM, and Biswal S. Genetic ablation of Nrf2 enhances susceptibility to cigarette smoke-induced emphysema in mice. J Clin Invest 114: 1248–1259, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Rangasamy T, Guo J, Mitzner WA, Roman J, Singh A, Fryer AD, Yamamoto M, Kensler TW, Tuder RM, Georas SN, and Biswal S. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J Exp Med 202: 47–59, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Ravichandran V, Seres T, Moriguchi T, Thomas JA, and Johnston RBJ. S-thiolation of glyceraldehyde-3-phosphate dehydrogenase induced by the phagocytosis-associated respiratory burst in blood monocytes. J Biol Chem 269: 25010–25015, 1994 [PubMed] [Google Scholar]
- 258.Rebrin I. and Sohal RS. Pro-oxidant shift in glutathione redox state during aging. Adv Drug Deliv Rev 60: 1545–1552, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Reed T, Perluigi M, Sultana R, Pierce WM, Klein JB, Turner DM, Coccia R, Markesbery WR, and Butterfield DA. Redox proteomic identification of 4-hydroxy-2-nonenal-modified brain proteins in amnestic mild cognitive impairment: insight into the role of lipid peroxidation in the progression and pathogenesis of Alzheimer's disease. Neurobiol Dis 30: 107–120, 2008 [DOI] [PubMed] [Google Scholar]
- 260.Reed TT. Lipid peroxidation and neurodegenerative disease. Free Radic Biol Med 51: 1302–1319, 2011 [DOI] [PubMed] [Google Scholar]
- 261.Reynolds AD, Kadiu I, Garg SK, Glanzer JG, Nordgren T, Ciborowski P, Banerjee R, and Gendelman HE. Nitrated alpha-synuclein and microglial neuroregulatory activities. J Neuroimmune Pharmacol 3: 59–74, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Rojo AI, Innamorato NG, Martin-Moreno AM, De Ceballos ML, Yamamoto M, and Cuadrado A. Nrf2 regulates microglial dynamics and neuroinflammation in experimental Parkinson's disease. Glia 58: 588–598, 2010 [DOI] [PubMed] [Google Scholar]
- 263.Rojo AI, Sagarra MR, and Cuadrado A. GSK-3beta down-regulates the transcription factor Nrf2 after oxidant damage: relevance to exposure of neuronal cells to oxidative stress. J Neurochem 105: 192–202, 2008 [DOI] [PubMed] [Google Scholar]
- 264.Rossi-George A, Guo CJ, Oakes BL, and Gow AJ. Copper modulates the phenotypic response of activated BV2 microglia through the release of nitric oxide. Nitric Oxide 27: 201–209, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Roy A, Jana A, Yatish K, Freidt MB, Fung YK, Martinson JA, and Pahan K. Reactive oxygen species up-regulate CD11b in microglia via nitric oxide: implications for neurodegenerative diseases. Free Radic Biol Med 45: 686–699, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Roychowdhury S, Wolf G, Keilhoff G, and Horn TF. Cytosolic and mitochondrial glutathione in microglial cells are differentially affected by oxidative/nitrosative stress. Nitric Oxide 8: 39–47, 2002 [DOI] [PubMed] [Google Scholar]
- 267.Ruoppolo M, Freedman RB, Pucci P, and Marino G. Glutathione-dependent pathways of refolding of RNase T1 by oxidation and disulfide isomerization: catalysis by protein disulfide isomerase. Biochemistry 35: 13636–13646, 1996 [DOI] [PubMed] [Google Scholar]
- 268.Rushmore TH, King RG, Paulson KE, and Pickett CB. Regulation of glutathione S-transferase Ya subunit gene expression: identification of a unique xenobiotic-responsive element controlling inducible expression by planar aromatic compounds. Proc Natl Acad Sci U S A 87: 3826–3830, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Saha RN. and Pahan K. Regulation of inducible nitric oxide synthase gene in glial cells. Antioxid Redox Signal 8: 929–947, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Salazar M, Rojo AI, Velasco D, de Sagarra RM, and Cuadrado A. Glycogen synthase kinase-3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem 281: 14841–14851, 2006 [DOI] [PubMed] [Google Scholar]
- 271.Sanchez-Guajardo V, Barnum CJ, Tansey MG, and Romero-Ramos M. Neuroimmunological processes in Parkinson's disease and their relation to alpha-synuclein: microglia as the referee between neuronal processes and peripheral immunity. ASN Neuro 5: 113–139, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Sanson M, Distel E, and Fisher EA. HDL induces the expression of the M2 macrophage markers arginase 1 and fizz-1 in a STAT6-dependent process. PLoS One 8: e74676, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Sato H, Tamba M, Ishii T, and Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem 274: 11455–11458, 1999 [DOI] [PubMed] [Google Scholar]
- 274.Scapagnini G, Vasto S, Abraham NG, Caruso C, Zella D, and Fabio G. Modulation of Nrf2/ARE pathway by food polyphenols: a nutritional neuroprotective strategy for cognitive and neurodegenerative disorders. Mol Neurobiol 44: 192–201, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Scarpa S, Cavallaro RA, D'Anselmi F, and Fuso A. Gene silencing through methylation: an epigenetic intervention on Alzheimer disease. J Alzheimers Dis 9: 407–414, 2006 [DOI] [PubMed] [Google Scholar]
- 276.Schilling T, Stock C, Schwab A, and Eder C. Functional importance of Ca2+-activated K+channels for lysophosphatidic acid-induced microglial migration. Eur J Neurosci 19: 1469–1474, 2004 [DOI] [PubMed] [Google Scholar]
- 277.Schlag GŽK, Redl H, Žarković N, Waeg G. Brain damage secondary to hemorrhagic traumatic shock in baboons. 5th Wiggers Bernard Conference “Shock, sepsis, and organ failure - brain damage secondary to hemorrhagic - traumatic shock, sepsis, and traumatic brain injury” Schlag G, Redl H, Traber D. (ur.). Heildelberg: Springer-Verlag, 1997, pp. 3–19 [Google Scholar]
- 278.Schlegelmilch T, Henke K, and Peri F. Microglia in the developing brain: from immunity to behaviour. Curr Opin Neurobiol 21: 5–10, 2011 [DOI] [PubMed] [Google Scholar]
- 279.Scholz M, Cinatl J, Schadel-Hopfner M, and Windolf J. Neutrophils and the blood–brain barrier dysfunction after trauma. Med Res Rev 27: 401–416, 2007 [DOI] [PubMed] [Google Scholar]
- 280.Schreibelt G, Kooij G, Reijerkerk A, van Doorn R, Gringhuis SI, van der Pol S, Weksler BB, Romero IA, Couraud PO, Piontek J, Blasig IE, Dijkstra CD, Ronken E, and de Vries HE. Reactive oxygen species alter brain endothelial tight junction dynamics via RhoA, PI3 kinase, and PKB signaling. FASEB J 21: 3666–3676, 2007 [DOI] [PubMed] [Google Scholar]
- 281.Serrander L, Cartier L, Bedard K, Banfi B, Lardy B, Plastre O, Sienkiewicz A, Forro L, Schlegel W, and Krause KH. NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem J 406: 105–114, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Shaftel SS, Carlson TJ, Olschowka JA, Kyrkanides S, Matousek SB, and O'Banion MK. Chronic interleukin-1beta expression in mouse brain leads to leukocyte infiltration and neutrophil-independent blood brain barrier permeability without overt neurodegeneration. J Neurosci 27: 9301–9309, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Shamoto-Nagai M, Maruyama W, Hashizume Y, Yoshida M, Osawa T, Riederer P, and Naoi M. In parkinsonian substantia nigra, alpha-synuclein is modified by acrolein, a lipid-peroxidation product, and accumulates in the dopamine neurons with inhibition of proteasome activity. J Neural Transm 114: 1559–1567, 2007 [DOI] [PubMed] [Google Scholar]
- 284.Sharma V, Mishra M, Ghosh S, Tewari R, Basu A, Seth P, and Sen E. Modulation of interleukin-1beta mediated inflammatory response in human astrocytes by flavonoids: implications in neuroprotection. Brain Res Bull 73: 55–63, 2007 [DOI] [PubMed] [Google Scholar]
- 285.Sheldon KE, Shandilya H, Kepka-Lenhart D, Poljakovic M, Ghosh A, and Morris SM., Jr.Shaping the murine macrophage phenotype: IL-4 and cyclic AMP synergistically activate the arginase I promoter. J Immunol 191: 2290–2298, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Shen G, Hebbar V, Nair S, Xu C, Li W, Lin W, Keum YS, Han J, Gallo MA, and Kong AN. Regulation of Nrf2 transactivation domain activity. The differential effects of mitogen-activated protein kinase cascades and synergistic stimulatory effect of Raf and CREB-binding protein. J Biol Chem 279: 23052–23060, 2004 [DOI] [PubMed] [Google Scholar]
- 287.Shibata N, Nagai R, Uchida K, Horiuchi S, Yamada S, Hirano A, Kawaguchi M, Yamamoto T, Sasaki S, and Kobayashi M. Morphological evidence for lipid peroxidation and protein glycoxidation in spinal cords from sporadic amyotrophic lateral sclerosis patients. Brain Res 917: 97–104, 2001 [DOI] [PubMed] [Google Scholar]
- 288.Sierra-Filardi E, Vega MA, Sanchez-Mateos P, Corbi AL, and Puig-Kroger A. Heme oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology 215: 788–795, 2010 [DOI] [PubMed] [Google Scholar]
- 289.Sierra A, Encinas JM, Deudero JJ, Chancey JH, Enikolopov G, Overstreet-Wadiche LS, Tsirka SE, and Maletic-Savatic M. Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7: 483–495, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Smeyne M. and Smeyne RJ. Glutathione metabolism and Parkinson's disease. Free Radic Biol Med [Epub ahead of print]; DOI: 10.1016/j.freeradbiomed.2013.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Sorce S. and Krause KH. NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal 11: 2481–2504, 2009 [DOI] [PubMed] [Google Scholar]
- 292.Srinivasan G, Aitken JD, Zhang B, Carvalho FA, Chassaing B, Shashidharamurthy R, Borregaard N, Jones DP, Gewirtz AT, and Vijay-Kumar M. Lipocalin 2 deficiency dysregulates iron homeostasis and exacerbates endotoxin-induced sepsis. J Immunol 189: 1911–1919, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Stewart VC, Heslegrave AJ, Brown GC, Clark JB, and Heales SJ. Nitric oxide-dependent damage to neuronal mitochondria involves the NMDA receptor. Eur J Neurosci 15: 458–464, 2002 [DOI] [PubMed] [Google Scholar]
- 294.Streit WJ. Microglia and neuroprotection: implications for Alzheimer's disease. Brain Res Brain Res Rev 48: 234–239, 2005 [DOI] [PubMed] [Google Scholar]
- 295.Streit WJ, Braak H, Xue QS, and Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol 118: 475–485, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Streit WJ, Sammons NW, Kuhns AJ, and Sparks DL. Dystrophic microglia in the aging human brain. Glia 45: 208–212, 2004 [DOI] [PubMed] [Google Scholar]
- 297.Su ZY, Shu L, Khor TO, Lee JH, Fuentes F, and Kong AN. A perspective on dietary phytochemicals and cancer chemoprevention: oxidative stress, nrf2, and epigenomics. Top Curr Chem 329: 133–162, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Sun Z, Chin YE, and Zhang DD. Acetylation of Nrf2 by p300/CBP augments promoter-specific DNA binding of Nrf2 during the antioxidant response. Mol Cell Biol 29: 2658–2672, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Sun Z, Huang Z, and Zhang DD. Phosphorylation of Nrf2 at multiple sites by MAP kinases has a limited contribution in modulating the Nrf2-dependent antioxidant response. PLoS One 4: e6588, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Surai PF, Speake BK, Noble RC, and Sparks NH. Tissue-specific antioxidant profiles and susceptibility to lipid peroxidation of the newly hatched chick. Biol Trace Elem Res 68: 63–78, 1999 [DOI] [PubMed] [Google Scholar]
- 301.Taguchi K, Fujikawa N, Komatsu M, Ishii T, Unno M, Akaike T, Motohashi H, and Yamamoto M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc Natl Acad Sci U S A 109: 13561–13566, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Taille C, El-Benna J, Lanone S, Boczkowski J, and Motterlini R. Mitochondrial respiratory chain and NAD(P)H oxidase are targets for the antiproliferative effect of carbon monoxide in human airway smooth muscle. J Biol Chem 280: 25350–25360, 2005 [DOI] [PubMed] [Google Scholar]
- 303.Taille C, El-Benna J, Lanone S, Dang MC, Ogier-Denis E, Aubier M, and Boczkowski J. Induction of heme oxygenase-1 inhibits NAD(P)H oxidase activity by down-regulating cytochrome b558 expression via the reduction of heme availability. J Biol Chem 279: 28681–28688, 2004 [DOI] [PubMed] [Google Scholar]
- 304.Takeshita Y. and Ransohoff RM. Inflammatory cell trafficking across the blood–brain barrier: chemokine regulation and in vitro models. Immunol Rev 248: 228–239, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Talalay P, Dinkova-Kostova AT, and Holtzclaw WD. Importance of phase 2 gene regulation in protection against electrophile and reactive oxygen toxicity and carcinogenesis. Adv Enzyme Regul 43: 121–134, 2003 [DOI] [PubMed] [Google Scholar]
- 306.Tchaikovskaya T, Fraifeld V, Urphanishvili T, Andorfer JH, Davies P, and Listowsky I. Glutathione S-transferase hGSTM3 and ageing-associated neurodegeneration: relationship to Alzheimer's disease. Mech Ageing Dev 126: 309–315, 2005 [DOI] [PubMed] [Google Scholar]
- 307.Thimmulappa RK, Lee H, Rangasamy T, Reddy SP, Yamamoto M, Kensler TW, and Biswal S. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J Clin Invest 116: 984–995, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Thimmulappa RK, Scollick C, Traore K, Yates M, Trush MA, Liby KT, Sporn MB, Yamamoto M, Kensler TW, and Biswal S. Nrf2-dependent protection from LPS induced inflammatory response and mortality by CDDO-Imidazolide. Biochem Biophys Res Commun 351: 883–889, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Tian L, Kilgannon P, Yoshihara Y, Mori K, Gallatin WM, Carpen O, and Gahmberg CG. Binding of T lymphocytes to hippocampal neurons through ICAM-5 (telencephalin) and characterization of its interaction with the leukocyte integrin CD11a/CD18. Eur J Immunol 30: 810–818, 2000 [DOI] [PubMed] [Google Scholar]
- 310.Tichauer JE, Flores B, Soler B, Eugenin-von Bernhardi L, Ramirez G, and von Bernhardi R. Age-dependent changes on TGFbeta1 Smad3 pathway modify the pattern of microglial cell activation. Brain Behav Immun 37: 187–196, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Toledano MB, Ghosh D, Trinh F, and Leonard WJ. N-terminal DNA-binding domains contribute to differential DNA-binding specificities of NF-kappa B p50 and p65. Mol Cell Biol 13: 852–860, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Tomimoto H, Shibata M, Ihara M, Akiguchi I, Ohtani R, and Budka H. A comparative study on the expression of cyclooxygenase and 5-lipoxygenase during cerebral ischemia in humans. Acta Neuropathol 104: 601–607, 2002 [DOI] [PubMed] [Google Scholar]
- 313.Tong KI, Katoh Y, Kusunoki H, Itoh K, Tanaka T, and Yamamoto M. Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol Cell Biol 26: 2887–2900, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Tremblay ME, Lowery RL, and Majewska AK. Microglial interactions with synapses are modulated by visual experience. PLoS Biol 8: e1000527, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Trigona WL, Mullarky IK, Cao Y, and Sordillo LM. Thioredoxin reductase regulates the induction of haem-oxygenase-1 expression in aortic endothelial cells. Biochem J 394: 207–216, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Tringali G, Mancuso C, Mirtella A, Pozzoli G, Parente L, Preziosi P, and Navarra P. Evidence for the neuronal origin of immunoreactive interleukin-1 beta released by rat hypothalamic explants. Neurosci Lett 219: 143–146, 1996 [DOI] [PubMed] [Google Scholar]
- 317.Ueno M, Masutani H, Arai RJ, Yamauchi A, Hirota K, Sakai T, Inamoto T, Tamaoka Y, Yodoi J, and Nikaido T. Thioredoxin-dependent redox regulation of p53-mediated p21 activation. J Biol Chem 274: 35809–35815, 1999 [DOI] [PubMed] [Google Scholar]
- 318.Ulleveig S, Kim HS, and Asmis R. S-glutathionylation in monocyte and macrophage (dys)function. Int J Mol Sci 14: 15212–15232, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Ungvari Z, Tucsek Z, Sosnowska D, Toth P, Gautam T, Podlutsky A, Csiszar A, Losonczy G, Valcarcel-Ares MN, Sonntag WE, and Csiszar A. Aging-induced dysregulation of dicer1-dependent microRNA expression impairs angiogenic capacity of rat cerebromicrovascular endothelial cells. J Gerontol A Biol Sci Med Sci 68: 877–891, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Usatyuk PV, Parinandi NL, and Natarajan V. Redox regulation of 4-hydroxy-2-nonenal-mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. J Biol Chem 281: 35554–35566, 2006 [DOI] [PubMed] [Google Scholar]
- 321.Vajkoczy P, Laschinger M, and Engelhardt B. Alpha4-integrin-VCAM-1 binding mediates G protein-independent capture of encephalitogenic T cell blasts to CNS white matter microvessels. J Clin Invest 108: 557–565, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Van der Goes A, Wouters D, Van Der Pol SM, Huizinga R, Ronken E, Adamson P, Greenwood J, Dijkstra CD, and De Vries HE. Reactive oxygen species enhance the migration of monocytes across the blood–brain barrier in vitro. FASEB J 15: 1852–1854, 2001 [DOI] [PubMed] [Google Scholar]
- 323.Velu CS, Niture SK, Doneanu CE, Pattabiraman N, and Srivenugopal KS. Human p53 is inhibited by glutahionylation of cysteines present in the proximal DNA-binding domain during oxidative stress. Biochemistry 46: 7765–7780, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Venema RC, Ju H, Zou R, Ryan JW, and Venema VJ. Subunit interactions of endothelial nitric-oxide synthase. Comparisons to the neuronal and inducible nitric-oxide synthase isoforms. J Biol Chem 272: 1276–1282, 1997 [DOI] [PubMed] [Google Scholar]
- 325.Vos CM, Geurts JJ, Montagne L, van Haastert ES, Bo L, van der Valk P, Barkhof F, and de Vries HE. Blood–brain barrier alterations in both focal and diffuse abnormalities on postmortem MRI in multiple sclerosis. Neurobiol Dis 20: 953–960, 2005 [DOI] [PubMed] [Google Scholar]
- 326.Voskuhl RR, Peterson RS, Song B, Ao Y, Morales LB, Tiwari-Woodruff S, and Sofroniew MV. Reactive astrocytes form scar-like perivascular barriers to leukocytes during adaptive immune inflammation of the CNS. J Neurosci 29: 11511–11522, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Waetzig V, Czeloth K, Hidding U, Mielke K, Kanzow M, Brecht S, Goetz M, Lucius R, Herdegen T, and Hanisch UK. c-Jun N-terminal kinases (JNKs) mediate pro-inflammatory actions of microglia. Glia 50: 235–246, 2005 [DOI] [PubMed] [Google Scholar]
- 328.Wake H, Moorhouse AJ, Jinno S, Kohsaka S, and Nabekura J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J Neurosci 29: 3974–3980, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Wang B, Navath RS, Romero R, Kannan S, and Kannan R. Anti-inflamatory and anti-oxidant activity of anionic dendrimer-N-acetyl cysteine conjugates in activated microglial cells. Int J Pharm 377: 159–168, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Wang B, Zhu X, Kim Y, Li J, Huang S, Saleem S, Li RC, Xu Y, Dore S, and Cao W. Histone deacetylase inhibition activates transcription factor Nrf2 and protects against cerebral ischemic damage. Free Radic Biol Med 52: 928–936, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Wang H, Khor TO, Saw CL, Lin W, Wu T, Huang Y, and Kong AN. Role of Nrf2 in suppressing LPS-induced inflammation in mouse peritoneal macrophages by polyunsaturated fatty acids docosahexaenoic acid and eicosapentaenoic acid. Mol Pharm 7: 2185–2193, 2010 [DOI] [PubMed] [Google Scholar]
- 332.Wang H, Liu H, and Liu RM. Gender difference in glutathione metabolism during aging in mice. Exp Gerontol 38: 507–517, 2003 [DOI] [PubMed] [Google Scholar]
- 333.Wang P, Liu GH, Wu K, Qu J, Huang B, Zhang X, Zhou X, Gerace L, and Chen C. Repression of classical nuclear export by S-nitrosylation of CRM1. J Cell Sci 122: 3772–3779, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Wang X, Svedin P, Nie C, Lapatto R, Zhu C, Gustavsson M, Sandberg M, Karlsson JO, Romero R, Hagberg H, and Mallard C. N-acetylcysteine reduces lipopolysaccharide-sensitized hypoxic-ischemic brain injury. Ann Neurol 61: 263–271, 2007 [DOI] [PubMed] [Google Scholar]
- 335.Weiss N, Miller F, Cazaubon S, and Couraud PO. The blood–brain barrier in brain homeostasis and neurological diseases. Biochim Biophys Acta 1788: 842–857, 2009 [DOI] [PubMed] [Google Scholar]
- 336.Wheeler D, Bandaru VV, Calabresi PA, Nath A, and Haughey NJ. A defect of sphingolipid metabolism modifies the properties of normal appearing white matter in multiple sclerosis. Brain 131: 3092–3102, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Williams TI, Lynn BC, Markesbery WR, and Lovell MA. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer's disease. Neurobiol Aging 27: 1094–1099, 2006 [DOI] [PubMed] [Google Scholar]
- 338.Winkler EA, Bell RD, and Zlokovic BV. Central nervous system pericytes in health and disease. Nat Neurosci 14: 1398–1405, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Wolburg H, Wolburg-Buchholz K, and Engelhardt B. Diapedesis of mononuclear cells across cerebral venules during experimental autoimmune encephalomyelitis leaves tight junctions intact. Acta Neuropathol 109: 181–190, 2005 [DOI] [PubMed] [Google Scholar]
- 340.Wu C, Ivars F, Anderson P, Hallmann R, Vestweber D, Nilsson P, Robenek H, Tryggvason K, Song J, Korpos E, Loser K, Beissert S, Georges-Labouesse E, and Sorokin LM. Endothelial basement membrane laminin alpha5 selectively inhibits T lymphocyte extravasation into the brain. Nat Med 15: 519–527, 2009 [DOI] [PubMed] [Google Scholar]
- 341.Wu DC, Re DB, Nagai M, Ischiropoulos H, and Przedborski S. The inflammatory NADPH oxidase enzyme modulates motor neuron degeneration in amyotrophic lateral sclerosis mice. Proc Natl Acad Sci U S A 103: 12132–12137, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Wu DC, Teismann P, Tieu K, Vila M, Jackson-Lewis V, Ischiropoulos H, and Przedborski S. NADPH oxidase mediates oxidative stress in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of Parkinson's disease. Proc Natl Acad Sci U S A 100: 6145–6150, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Wu SZ, Bodles AM, Porter MM, Griffin WS, Basile AS, and Barger SW. Induction of serine racemase expression and D-serine release from microglia by amyloid beta-peptide. J Neuroinflammation 1: 2, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Xu C, Yuan X, Pan Z, Shen G, Kim JH, Yu S, Khor TO, Li W, Ma J, and Kong AN. Mechanism of action of isothiocyanates: the induction of ARE-regulated genes is associated with activation of ERK and JNK and the phosphorylation and nuclear translocation of Nrf2. Mol Cancer Ther 5: 1918–1926, 2006 [DOI] [PubMed] [Google Scholar]
- 345.Yamamoto T, Kyo M, Kamiya T, Tanaka T, Engel JD, Motohashi H, and Yamamoto M. Predictive base substitution rules that determine the binding and transcriptional specificity of Maf recognition elements. Genes Cells 11: 575–591, 2006 [DOI] [PubMed] [Google Scholar]
- 346.Yamamoto T, Suzuki T, Kobayashi A, Wakabayashi J, Maher J, Motohashi H, and Yamamoto M. Physiological significance of reactive cysteine residues of Keap1 in determining Nrf2 activity. Mol Cell Biol 28: 2758–2770, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Yang SR, Chida AS, Bauter MR, Shafiq N, Seweryniak K, Maggirwar SB, Kilty I, and Rahman I. Cigarette smoke induces proinflammatory cytokine release by activation of NF-kappaB and posttranslational modifications of histone deacetylase in macrophages. Am J Physiol Lung Cell Mol Physiol 291: L46–L57, 2006 [DOI] [PubMed] [Google Scholar]
- 348.Yang SR, Valvo S, Yao H, Kode A, Rajendrasozhan S, Edirisinghe I, Caito S, Adenuga D, Henry R, Fromm G, Maggirwar S, Li JD, Bulger M, and Rahman I. IKK alpha causes chromatin modification on pro-inflammatory genes by cigarette smoke in mouse lung. Am J Respir Cell Mol Biol 38: 689–698, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Yang Y. and Rosenberg GA. Blood–brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 42: 3323–3328, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Yasukawa H, Misawa H, Sakamoto H, Masuhara M, Sasaki A, Wakioka T, Ohtsuka S, Imaizumi T, Matsuda T, Ihle JN, and Yoshimura A. The JAK-binding protein JAB inhibits Janus tyrosine kinase activity through binding in the activation loop. EMBO J 18: 1309–1320, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Yenari MA, Xu L, Tang XN, Qiao Y, and Giffard RG. Microglia potentiate damage to blood–brain barrier constituents: improvement by minocycline in vivo and in vitro. Stroke 37: 1087–1093, 2006 [DOI] [PubMed] [Google Scholar]
- 352.Yip PK, Pizzasegola C, Gladman S, Biggio ML, Marino M, Jayasinghe M, Ullah F, Dyall SC, Malaspina A, Bendotti C, and Michael-Titus A. The omega-3 fatty acid eicosapentaenoic acid accelerates disease progression in a model of amyotrophic lateral sclerosis. PLoS One 8: e61626, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, and Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc Natl Acad Sci U S A 93: 2696–2701, 1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 354.Zarkovic K. 4-hydroxynonenal and neurodegenerative diseases. Mol Aspects Med 24: 293–303, 2003 [DOI] [PubMed] [Google Scholar]
- 355.Zarkovic KCM, Kolenc D, and Zarkovic N. 4-Hydroxynoneal as second messenger of free radicals in the blood–brain barrier. Conference Abstract: Pharmacology and Toxicology of the Blood–Brain Barrier: State of the Art, Needs for Future Research and Expected Benefits for the EU 2010 [Google Scholar]
- 356.Zarkovic KZN, Schlag G, Redl H, and Waeg G. Histological aspects of sepsis induced brain changes in a baboon model. In: Shock, Sepsis and Organ Failure, 5th Wiggers Bernard Conference, edited by Schlag G, Redl H, and Traber DL. Heidelberg: Springer-Verlag, 1997, pp. 146–160 [Google Scholar]
- 357.Zarkovic N. 4-hydroxynonenal as a bioactive marker of pathophysiological processes. Mol Aspects Med 24: 281–291, 2003 [DOI] [PubMed] [Google Scholar]
- 358.Zarkovic N, Cipak A, Jaganjac M, Borovic S, and Zarkovic K. Pathophysiological relevance of aldehydic protein modifications. J Proteomics 92: 239–247, 2013 [DOI] [PubMed] [Google Scholar]
- 359.Zarkovic N, Zarkovic K, Schaur RJ, Stolc S, Schlag G, Redl H, Waeg G, Borovic S, Loncaric I, Juric G, and Hlavka V. 4-Hydroxynonenal as a second messenger of free radicals and growth modifying factor. Life Sci 65: 1901–1904, 1999 [DOI] [PubMed] [Google Scholar]
- 360.Zhang DD. and Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol 23: 8137–8151, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Zhang DD, Lo SC, Cross JV, Templeton DJ, and Hannink M. Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol Cell Biol 24: 10941–10953, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Zhang F, Qian L, Flood PM, Shi JS, Hong JS, and Gao HM. Inhibition of IkappaB kinase-beta protects dopamine neurons against lipopolysaccharide-induced neurotoxicity. J Pharmacol Exp Ther 333: 822–833, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Zhang L, Yu H, Zhao X, Lin X, Tan C, Cao G, and Wang Z. Neuroprotective effects of salidroside against beta-amyloid-induced oxidative stress in SH-SY5Y human neuroblastoma cells. Neurochem Int 57: 547–555, 2010 [DOI] [PubMed] [Google Scholar]
- 364.Zhang W, Wang T, Pei Z, Miller DS, Wu X, Block ML, Wilson B, Zhou Y, Hong JS, and Zhang J. Aggregated alpha-synuclein activates microglia: a process leading to disease progression in Parkinson's disease. FASEB J 19: 533–542, 2005 [DOI] [PubMed] [Google Scholar]
- 365.Zhang W, Wang T, Qin L, Gao HM, Wilson B, Ali SF, Hong JS, and Liu B. Neuroprotective effect of dextromethorphan in the MPTP Parkinson's disease model: role of NADPH oxidase. FASEB J 18: 589–591, 2004 [DOI] [PubMed] [Google Scholar]
- 366.Zhang X, Guo L, Collage RD, Stripay JL, Tsung A, Lee JS, and Rosengart MR. Calcium/calmodulin-dependent protein kinase (CaMK) 1 alpha mediates the macrophage inflammatory response to sepsis. J Leukoc Biol 90: 249–261, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Zhang Z, Zhang ZY, Wu Y, and Schluesener HJ. Lesional accumulation of CD163+macrophages/microglia in rat traumatic brain injury. Brain Res 1461: 102–110, 2012 [DOI] [PubMed] [Google Scholar]
- 368.Zhao W, Xie W, Le W, Beers DR, He Y, Henkel JS, Simpson EP, Yes AA, Xiao Q, and Appel SH. Activated microglia initiate motor neuron injury by a nitric oxide and glutamate-mediated mechanism. J Neuropathol Exp Neurol 63: 964–977, 2004 [DOI] [PubMed] [Google Scholar]
- 369.Zhong H, May MJ, Jimi E, and Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell 9: 625–636, 2002 [DOI] [PubMed] [Google Scholar]
- 370.Zhong H, SuYang H, Erdjument-Bromage H, Tempst P, and Ghosh S. The transcriptional activity of NF-kappaB is regulated by the IkappaB-associated PKAc subunit through a cyclic AMP-independent mechanism. Cell 89: 413–424, 1997 [DOI] [PubMed] [Google Scholar]
- 371.Zhong H, Voll RE, and Ghosh S. Phosphorylation of NF-kappa B p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol Cell 1: 661–671, 1998 [DOI] [PubMed] [Google Scholar]
- 372.Zhou H, Lapointe BM, Clark SR, Zbytnuik L, and Kubes P. A requirement for microglial TLR4 in leukocyte recruitment into brain in response to lipopolysaccharide. J Immunol 177: 8103–8110, 2006 [DOI] [PubMed] [Google Scholar]
- 373.Zhou Y, Ling EA, and Dheen ST. Dexamethasone suppresses monocyte chemoattractant protein-1 production via mitogen activated protein kinase phosphatase-1 dependent inhibition of Jun N-terminal kinase and p38 mitogen-activated protein kinase in activated rat microglia. J Neurochem 102: 667–678, 2007 [DOI] [PubMed] [Google Scholar]
- 374.Zhu Y, Carvey PM, and Ling Z. Age-related changes in glutathione and glutathione-related enzymes in rat brain. Brain Res 1090: 35–44, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Zipper LM. and Mulcahy RT. The Keap1 BTB/POZ dimerization function is required to sequester Nrf2 in cytoplasm. J Biol Chem 277: 36544–36552, 2002 [DOI] [PubMed] [Google Scholar]
- 376.Zlokovic BV. The blood–brain barrier in health and chronic neurodegenerative disorders. Neuron 57: 178–201, 2008 [DOI] [PubMed] [Google Scholar]
- 377.Zmijewski JW, Banerjee S, and Abraham E. S-glutathionylation of the Rpn2 regulatory subunit inhibits 26 S proteasomal function. J Biol Chem 284: 22213–22221, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]