

To the Editor
Recombination activating genes (RAG) 1 and 2 are critical for V(D)J recombination and lymphocyte development.1 Mutations in RAG1/2 associated with <1% of wild-type recombination activity typically result in severe combined immunodeficiency (SCID) lacking T and B cells.2 Hypomorphic mutations with residual RAG activity can result in Omenn syndrome, characterized by erythroderma, lymphadenopathy, and the presence of oligoclonal T cells. More recently, studies have broadened the phenotypic variability associated with RAG1/2 mutations, which now includes granulomatous disease, γδ T-cell expansion, CD4+ lymphopenia with intact T cell function, and hyper-IgM syndrome.1, 3 We report a patient with a homozygous missense mutation in RAG1, who presented initially with agammaglobulinemia, absent B cells, and normal numbers of T cells, followed by evolution into a T-B-NK+ SCID phenotype with opportunistic infections and granulomas.
The patient was born to Kuwaiti first-degree cousin parents and presented at 6 months of age with pneumonia and failure to thrive The physical examination was notable for the absence of tonsils. Computerized tomography of the chest revealed a prominent thymus. He had undetectable IgG and IgA. IgM was 30 mg/dL. The absolute lymphocyte count was 5010 cells/μL with normal numbers of total CD3+ cells, CD4+ and CD8+ T cells, CD16+/CD56+ NK cells, but virtually no CD19+ B cells (Table 1). Genetic testing did not reveal any mutations in the genes encoding BTK, μ heavy chain, Igα, Igβ, λ5, and VpreB. He was given a diagnosis of agammaglobulinemia with absent B cells and was treated with IVIG.
Table 1.
Immune profiles
| 6 months | Proband 5 years | 11 years | |
|---|---|---|---|
| Lymphocytes, cells/μL (normal range)i | 5010 (3400-9000) | 1550 (2300-5400) | 1620 (1900-3700) |
|
| |||
| CD3+ | 4520 (1900-5900) | 1150 (1400-3700) | 541 (1200-2600) |
| CD3+CD4+ | 3300 (1400-4300) | 388 (700-2200) | 260 (650-1500) |
| CD3+CD8+ | 1220 (500-1700) | 679 (490-1300) | 279 (370-1100) |
| CD4+/CD8+ ratio | 2.7 (1.6-3.8) | 0.51 (0.9-2.6) | 0.93 (0.9 – 3.4) |
| CD4+CD45RA+ | ND | ND | 3.7 (46-77%) |
| CD4+CD45RO+ | ND | ND | 95 (13-30%) |
| CD4+CD45RA+CD31+ | ND | ND | 0.96 (45.7%)iv |
| CD19+ | 2 (610-2600) | 4 (390-1400) | 61 (270-860) |
| CD16+/CD56+ | 340 (160-950) | 349 (130-720) | 1004 (100-480) |
| Eosinophils, cells/μL (normal range)ii | 184 (200-300) | 95 (0-600) | 210 (0-600) |
|
| |||
| Immunoglobulins, mg/dL (normal range)iii | |||
|
| |||
| IgG | Undetectable (215-704) | ND | ND |
| IgA | Undetectable (8.1-68) | ND | ND |
| IgM | 30 (35-102) | ND | ND |
|
| |||
| Proliferation, cpm (normal control)iv | |||
|
| |||
| Phytohemagglutinin | ND | 1578 (16,485) | 541 (65,369) |
| Anti-CD3 | ND | ND | 232 (3,814) |
| Anti-CD3+CD28 | ND | ND | 6,797 (19,170) |
| Background | ND | 32 (192) | 131 (221.5) |
ND, Not Determined
Shearer WT, Rosenblatt HM, Gelman RS, Oyomopito R, Plaeger S, Stiehm ER, Wara DW, Douglas SD, Luzuriaga K, McFarland EJ, Yogev R, Rathore MH, Levy W, Graham BL, Spector SA; Pediatric AIDS Clinical Trial Group. Lymphocyte subsets in healthy children from birth through 18 years of age: the Pediatric AIDS Clinical Trials Group P1009 study. J Allergy Clin Immunol 2003; 112(5):973-80.
Tietz NW (Ed): Clinical Guide to Laboratory Tests, 3rd ed. W. B. Saunders, Philadelphia, PA, 1995
Jolliff CR Cost KM, Stivrins PC, Grossman PP, Nolte CR, Franco SM, Fijan KJ, Fletcher LL, Shriner HC. Clin Chem. Reference intervals for serum IgG, IgA, IgM, C3, and C4 as determined by rate nephelometry. 1982; 28:126-128.
Value in parenthesis for lymphocyte proliferation are derived from studies done on a healthy control the same day.
The patient subsequently developed recurrent otitis media and cytomegalovirus-associated retinitis at 4 years of age. Immunologic evaluation performed at 5 years of age revealed virtually absent B cells, CD3+ and CD4+ T cell lymphopenia, and normal numbers of NK cells (Table I). Lymphocyte proliferation to PHA was severely decreased (Table I). NK cell cytotoxic function was normal (data not shown). At 8 years of age, he developed fever and diarrhea due to Epstein-Barr virus viremia and Salmonella enteritidis. At 10 years of age, the patient developed persistent colitis; endoscopy and colonoscopy revealed esophageal candidiasis and colonic granulomas. At that time, he had progressive T cell lymphopenia with a predominantly CD45RO+ activated phenotype, and nearly absent CD4+CD45RA+CD31+ recent thymic emigrants (Table I). HLA typing of the patient and his mother revealed no evidence of maternal engraftment. T cell proliferation to PHA and anti-CD3 stimulation was minimal. Proliferation to anti-CD3 stimulation increased with the addition of anti-CD28, suggesting residual signaling through the CD3 pathway. The combination of absent B cells, agammaglobulinemia, and progressive T cell lymphopenia with poor T cell function led to a diagnosis of combined immunodeficiency. The patient died at the age of 12 years from sepsis.
Whole genome sequencing was performed on DNA obtained from the patient prior to his death, his father, and his healthy sibling. Thirteen candidate genes had novel mutations that were homozygous in the patient, heterozygous in the father, and heterozygous or absent in the sibling (Table E1). Only the c.1211G>A mutation in RAG1 correlated with the patient’s phenotype of combined immunodeficiency. This mutation resulted in the substitution of arginine to glutamine at position 404 (p.R404Q) in the nonamer-binding region (NBR) of the protein. The NBR of RAG1 binds to recombination signal sequences that flank the Variable (V), Diversity (D) and Joining (J) elements of the immunoglobulin and T cell receptor genes, and is critical for V(D)J recombination activity.4 Using a flow cytometry-based assay that permits analysis of RAG1 recombination activity on an intrachromosomal substrate,5 we have demonstrated that the R404Q RAG1 mutant protein is normally expressed, but has markedly reduced recombination activity (1.18 ± 0.14% of wild-type) (Fig. 1). This mutation has been previously reported in patients with T-B- SCID or Omenn syndrome.6 In contrast to previously published patients with this mutation, our patient presented with clinical and laboratory features of agammaglobulinemia with absent B cells, a prominent thymus, normal numbers of circulating T cells, and no features of Omenn syndrome such as erythroderma or eosinophilia. Normal numbers of T cells with absence of B cells have been associated with another RAG1 mutation (c.T2686C, p.W896A) in a patient who presented with isolated culture-negative pneumonitis at 6 months of age.7 This patient, like ours, had a normal thymic shadow and normal T cell numbers. The W896A mutation resulted in impaired proliferation to mitogens and a highly restricted TCRβ repertoire that was not of maternal origin. We have previously demonstrated that the VDJ recombination activity of the W896A mutant is associated with minimally preserved RAG1 function (0.9 ± 0.1% of wild-type)5, similar to the R404Q mutation in our patient,
Figure 1.
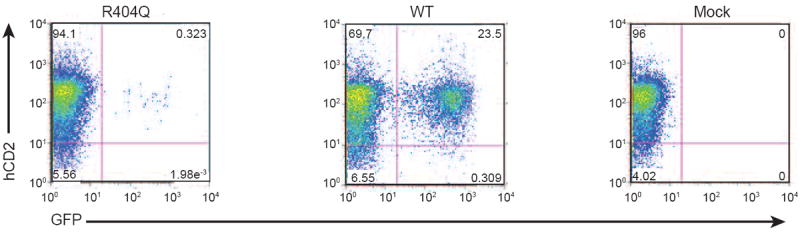
Recombination activity of the RAG1 R404Q mutant protein. Abelson (Abl) virus-transformed Rag1-/- mouse pro-B cells engineered to contain a single integrant of an inverted green fluorescent protein (GFP) cassette flanked by recombination signal sequences (RSS) were transduced with retroviral vectors allowing expression of wild-type (WT) human RAG1 or the mutant R404Q RAG1 protein, and human CD2 (hCD2) as a reporter. Mock-transduced cells served as a negative control. Following MACS-purification of hCD2-expressing cells and treatment in vitro with imatinib, recombination activity of the R404Q mutant protein was measured by normalizing GFP expression to levels observed in the presence of wild-type RAG1, as previously described.5 One representative experiment of three is shown.
This case broadens the spectrum of disease associated with a RAG1 mutation that nearly abolishes recombination activity. This phenomenon has been seen in families where the same mutation gives rise to different clinical and immunological phenotypes, suggesting that the manifestations of RAG1/2 mutations may reflect interactions with other uncharacterized genetic modifiers, epigenetic factors, or environmental exposures. The progression of the patient’s disease also underscores how a single mutation can have variable time-dependent effects in the same individual. Although he had normal numbers of T cells at 6 months of age, T cell function may have been abnormal at that time as suggested by his history of pneumonia and failure to thrive. He developed progressively worsening T cell numbers and function, opportunistic infections, granulomas, and ultimately died of sepsis, which, in retrospect, are consistent with an underlying RAG1/2 mutation. The diagnostic delay in this case underscores the importance of SCID newborn screening, particularly in areas of the world where lymphocyte proliferation or TCR repertoire studies are not available. Quantification of T cell receptor excision circles (TRECs) can identify poor thymic output, even in the case of normal T cell numbers due to maternal engraftment or oligoclonality.8 Additionally, cases of SCID which present with absent B cell numbers, as in our patient and others7, support the inclusion of quantification of κ-deleting recombination excision circles, which can detect absent B cells, as part of newborn screening for SCID for improved early detection.9
Supplementary Material
Table E1. Candidate genes are defined as genes with novel mutations in coding regions or canonical splice sites that were homozygous in the patient, heterozygous in his father, and either heterozygous or absent in his healthy brother. Novel mutations are defined as those absent from public polymorphism databases (dbSNP and 1000 Genomes) and our in-house database of 73 Middle Eastern exomes/genomes.
Acknowledgments
Supported by: NIH P01AI-076210, AI-094017, a grant from the Dubai Harvard Foundation for Medical Research (RSG), a Manton Foundation Pilot Award (JC), and Kuwait Foundation for Advancement of Sciences grant 2010-1302-05 (WA).
Abbreviations
- RAG
Recombination activating genes
- SCID
severe combined immunodeficiency
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Niehues T, Perez-Becker R, Schuetz C. More than just SCID--the phenotypic range of combined immunodeficiencies associated with mutations in the recombinase activating genes (RAG) 1 and 2. Clinical Immunology. 2010;135:183–92. doi: 10.1016/j.clim.2010.01.013. [DOI] [PubMed] [Google Scholar]
- 2.Notarangelo LD, Villa A, Schwarz K. RAG and RAG defects. Current Opinion in Immunology. 1999;11:435–42. doi: 10.1016/S0952-7915(99)80073-9. [DOI] [PubMed] [Google Scholar]
- 3.Chou J, Hanna-Wakim R, Tirosh I, Kane J, Fraulino D, Lee YN, et al. A novel homozygous mutation in recombination activating gene 2 in 2 relatives with different clinical phenotypes: Omenn syndrome and hyper-IgM syndrome. Journal of Allergy and Clinical Immunology. 2012;130:1414–6. doi: 10.1016/j.jaci.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kirch SA, Sudarsanam P, Oettinger MA. Regions of RAG1 protein critical for V(D)J recombination. European Journal of Immunology. 1996;26:886–91. doi: 10.1002/eji.1830260425. [DOI] [PubMed] [Google Scholar]
- 5.Lee Y, Frugoni F, Dobbs K, Walter JE, Giliani S, Gennery AR, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human RAG1-deficiency. Journal of Allergy and Clinical Immunology. 2013 doi: 10.1016/j.jaci.2013.10.007. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sobacchi C, Marrella V, Rucci F, Vezzoni P, Villa A. RAG-dependent primary immunodeficiencies. Human Mutation. 2006;27:1174–84. doi: 10.1002/humu.20408. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, Quintal L, Atkinson A, Williams B, Grunebaum E, Roifman CM. Novel RAG1 mutation in a case of severe combined immunodeficiency. Pediatrics. 2005;116:e445–9. doi: 10.1542/peds.2005-0369. [DOI] [PubMed] [Google Scholar]
- 8.Puck JM. Laboratory technology for population-based screening for severe combined immunodeficiency in neonates: the winner is T-cell receptor excision circles. Journal of Allergy and Clinical Immunology. 2012;129:607–16. doi: 10.1016/j.jaci.2012.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borte S, von Dobeln U, Fasth A, Wang N, Janzi M, Winiarski J, et al. Neonatal screening for severe primary immunodeficiency diseases using high-throughput triplex real-time PCR. Blood. 2012;119:2552–5. doi: 10.1182/blood-2011-08-371021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table E1. Candidate genes are defined as genes with novel mutations in coding regions or canonical splice sites that were homozygous in the patient, heterozygous in his father, and either heterozygous or absent in his healthy brother. Novel mutations are defined as those absent from public polymorphism databases (dbSNP and 1000 Genomes) and our in-house database of 73 Middle Eastern exomes/genomes.