

Abstract
Background
Epithelial genes have previously been associated with asthma, but only explain a small fraction of heritability. In part, this may be due to epistasis that is often not considered.
Objective
To determine independent and epistatic associations between FLG, SPINK5 and TSLP gene variants and childhood asthma.
Methods
Using a candidate gene approach, we genotyped 29 variants in FLG, SPINK5 and TSLP in asthmatic, allergic, and non-allergic-non-asthmatic white and black children participating in the well-phenotyped Greater Cincinnati Pediatric Clinic Repository (GCPCR). Associations with asthma were also assessed in six replication populations.
Results
We observed independent associations of variants in SPINK5 (p=0.003) and TSLP (p=0.006) with childhood asthma; a SPINK5 SNP was replicated. In subjects with one or more SPINK5 risk alleles, the absence of the TSLP protective minor alleles was associated with a significant increase in asthma (67% vs. 53%, p=0.0017). In contrast, the presence or absence of TSLP minor alleles did not affect asthma risk in subjects without the SPINK5 risk alleles. The SPINK5 and TSLP epistasis was replicated in a black population (p=0.036) that did not display independent association with variants in these genes.
Conclusions
Our results support epistasis between SPINK5 and TSLP which contributes to childhood asthma. These findings emphasize the importance of utilizing biology to inform analyses to identify genetic susceptibility to complex diseases. The results from our study have clinical relevance and support that the therapeutic effects of anti-TSLP therapy in asthmatics may be dependent on SPINK5 genotype.
Keywords: childhood asthma, genetic association, epistasis, skin-related genes
Introduction
Asthma is the most common chronic illness in children in the US, affecting 7.1 million (9.5%) in 20111. Heritability estimates for asthma range between 0.36 and 0.772-5. Since 1989, >100 candidate genes have been described in >1000 publications on asthma or an associated disorder6. However, variants identified thus far confer small increments of risk, leading to questions about how the remaining genetic risk can be explained7. These studies have largely neglected genetic interactions including epistasis8 and the large sample sizes required for GWAS have often come at the expense of accurate, consistent phenotyping9.
Loss of function variation in the filaggrin (FLG) gene is the most replicated genetic risk factor for atopic dermatitis10, and the data is clear that FLG is associated with asthma in the presence of atopic features11-17. Thymic stromal lymphopoietin (TSLP), an epithelial-cell derived cytokine that promotes differentiation and activation of T-helper 2 (Th2) cells and their receptors, is a well-validated asthma susceptibility gene18. Murine studies have suggested that TSLP may share the same genetic pathway as serine protease inhibitor Kazal-type 5 (SPINK5)19, 20, which causes Netherton's Syndrome21 and has been associated with asthma, but studies are conflicting22-25.
To assess the effects of skin-barrier related genes and asthma, we evaluated SNPs in FLG, SPINK5 and TSLP in 973 white and 530 black children. To maximize the impact of the genetic associations in this study, cases and controls were rigorously and objectively phenotyped with regard to asthma and allergies (an important distinction for control selection that we recently reported26), our findings are replicated in six populations and we account for population substructure using ancestry informative markers. In addition, interactions of the candidate genes were evaluated given the mechanistic and biologic plausibility of epistasis.
Methods
Study Populations
The discovery population consisted of a subset of 4 to 17 year old Caucasian/white and African-American/black (the terms white and black will be used for simplicity) participants enrolled in either the Greater Cincinnati Pediatric Clinic Repository (GCPCR) or the Genomic Control Cohort (GCC), both described previously27. The GCPCR includes over 6,500 patients and the GCC has 1,020 children and DNA was available on all participants as previously described28, 29. Case-control definitions including those for asthma in the GCPCR have been previously described26. All asthmatics were rigorously phenotyped by a specialty physician (pediatric allergist or pulmonologist) according to ATS criteria30. Allergic controls (participants with allergic rhinitis, atopic dermatitis or environmental allergies) and non-allergic, non-asthmatic controls were available from both the GCPCR and the GCC. The protocols were approved by the CCHMC Institutional Review Board and participants gave written informed consent.
Among asthmatic children, asthma exacerbation was defined by previous hospitalizations for asthma. Results from skin prick testing (SPT) were available on 56% of asthmatic and allergic white children in the GCPCR. Children were defined as SPT positive if they had a positive test to a pollen (trees, weeds, grass), dust (dust mite, cockroach), animal (cat, dog) or mold at any time up to 6 months after their consent date.
Replication Cohorts
The replication populations were 1) 334 white trios (1002 individual samples) from the Childhood Asthma Management Program (CAMP)31; 2) 95 white trios (285 individual samples) from the Childhood Asthma Research and Education (CARE) Network32; 3) 382 white children (57 asthmatics, 184 non-asthmatic SPT-controls and 141 non-asthmatic SPT+ controls) participating in the Cincinnati Childhood Allergy and Air Pollution Study (CCAAPS)33; 4) 418 white individuals (207 GCPCR asthmatics enrolled after the discovery cohort and 211 non-asthmatic controls from the Cincinnati Control Cohort (CCC, described previously26)); 5) 347 white children (207 asthmatics and 140 allergic controls) and 6) 340 black children (272 asthmatics and 68 allergic controls) from the GCPCR enrolled after the discovery cohort. CAMP and CARE data were downloaded with permission from the NIH-based database of Genotypes and Phenotypes (dbGaP) (http://www.ncbi.nlm.nih.gov/gap). Phenotypic description and details about the CAMP CARE data can be found at http://www.ncbi.nlm.nih.gov/gap/?term=asthma. The ‘replication GCPCR’ were white and black asthmatics and allergic controls that were enrolled in the repository after the discovery GCPCR cohort. The CCC is a population-based cohort of white adults with no personal or family history of asthma (by self-report) representative of Greater Cincinnati.
Gene and SNP Selection and Genotyping
The methods for the gene and SNP selection for the discovery array have been previously described26, 34. FLG, SPINK5 and TSLP were chosen for these analyses based on their skin-related role and biologic relevance in the pathogenesis of asthma. To reduce the number of SNPs genotyped while generating the same amount of genetic information, tagging SNPs that maximized genomic coverage and captured the common genetic variation in these genes were selected using Haploview and Tagger (http://www.broad.mit.edu/mpg/haploview). Table 1 includes a description of the selected genes, their reported processes and functions as well as top associated disorders. A total of 40 SNPs, 1 truncation and 1 deletion in the FLG, SPINK5 and TSLP genes were considered for the discovery analyses.
Table 1.
Selected genes, functions and associated disorders.
| #SNPsa | Gene | Full Gene Name | Chr. | Reported Processes and Functionc | Reported Associated Disordersd |
|---|---|---|---|---|---|
| 8b | FLG | filaggrin | 1q21.3 | Keratinocyte differentiation, multicellular organismal development, cytoplasmic membrane bounded vesicle, intermediate filament, structural molecule activity | atopic dermatitis, eczema, ichthyosis vulgaris, asthma, rheumatoid arthritis, psoriasis, skin diseases, ichythyosis, atopy, inflammation |
|
| |||||
| 13 | SPINK 5 | Serine protease inhibitor Kazal-type 5 | 5q32 | Peptidase inhibitor activity, serine-type endopeptidase inhibitor activity | Netherton syndrome, atopic dermatitis, asthma, skin diseases, congenital ichthyosis, atopy, congenital erythroderma ichthyosiform, ichthyosis, psoriasis, growth retardation |
|
| |||||
| 8 | TSLP | Thymic stromal lymphopoietin | 5q22.1 | Extracellular region and space, cytokine activity, protein binding | inflammation, asthma, atopic dermatitis, necrosis, tumors, allergic asthma, skin lesions, mixed cryoglobulinemia, allergy, membranoproliferative glomerulonephritis |
Indicates the total number of tagging SNPs that entered the analyses.
This includes the 2282del4 deletion and R501X truncation.
Obtained from Gene Ontology website (www.geneontology.org).
Reported disease associations were obtained from the top 10 Novoseek disease relationships hits (number of articles in which both the gene's symbol or description and the disease appear) from GeneCards® (www.genecards.org).
Subjects from the GCPCR were selected for genotyping if they had 1) a pulmonary function test or completed asthma symptom questionnaire, 2) a SPT or completed Children's Health Survey for Asthma35 and 3) ≥250ng DNA with an optical density of 1.6-2.2. Allergic and non-asthmatic, non-allergic controls were selected from the GCC that had available DNA. Genomic DNA was isolated from GCPCR buccal swabs with either the Zymo Research Genomic DNA II Kit (Zymo Research Corp., Orange, CA) or the Purgene DNA Purification System (Gentra Systems Minneapolis, MN), and from Oragene saliva samples per the kit's instructions. Genomic DNA was extracted from GCC blood samples using Manual PerfectPure DNA Blood Kit (Invitrogen, Carlsbad, CA). Genotyping of GCPCR and GCC samples was performed using a custom Illumina Golden Gate assay according to the manufacturer's protocol (http://www.illumina.com; San Diego, CA)26, 34. Genotypes were assigned using BeadStudio's genotyping module (BeadStudio v3.2, San Diego, CA). Genotyping of FLG R501X truncation and 2282del4 deletion was performed by PCR and restriction fragment length polymorphism in both CCAAPS and the GCPCR as previously described36, 37. Genotyping of the CCAAPS population for SPINK5 (29 SNPs) and TSLP (10 SNPs) was performed on banked saliva samples using a second custom Illumina Golden Gate assay. The publicly available data downloaded from dbGaP for both the CAMP and CARE replication cohorts were generated using the Affymetrix 6.0 SNP chip. Genotyping data from the Affymetrix 6.0 SNP chip was also available for the CCC. For the replications, all SNPs available in SPINK5 or TSLP for each population were included; only those SNPs that were on the discovery SNP array and/or those that had a p<0.05 are displayed.
Statistical Analyses
SNPs failing Hardy Weinberg Equilibrium in the non-allergic control group (p<0.0001, having minor allele frequencies below 10% in the combined controls; or missing call rates greater than 10%) were excluded (n=11). In addition, individuals with more than 20% of their total SNPs missing were excluded. Principal component (PC) analyses were performed using the 30 included ancestry-informative markers (AIMs) in EIGENSTRAT38, 39 to account for potential population stratification. Sets of AIMS ranging from 24-128 are useful tools for ascertaining the origin of subjects from particular continents, and to correct for population stratification in admixed population sample sets40. After PCs were included in the model the genomic inflation factor was 1.0, suggesting minimal impact of population stratification for these AIMs, which is not unexpected since the general Cincinnati population does not show any North/South or East/West stratification based on larger panels of AIMS used for the larger GCC population (data not shown). Using PLINK41, associations with asthma were tested adjusting for age, sex and PCs (four PCs used in white and no PCs needed in black analyses) using the additive logistic regression model stratified by race. To address multiple testing, we determined the average pairwise LD (as measured by r2) for all SNP combinations and calculated the Bonferroni correction using Simple Interactive Statistical Analyses Software (http://www.quantitativeskills.com/sisa/).
For the GCPCR whites and blacks, associations with the primary phenotype of asthma were therefore considered significant at 0.0077 and 0.0039, respectively. For the CAMP and CARE cohorts, association analysis was performed using the transmission disequilibrium test (TDT)42. The case-control analyses for the GCPCR/CCC replication was accomplished by fitting a logistic regression model adjusted for sex. We used the strictest definition of genetic replication provided by Sullivan et al, which requires that the replication be the same SNP, phenotype and direction of association resulting in a mean false replication rate of 2%43.
Permutation tests provide a computationally intensive approach to generating significance levels empirically. The purpose of the permutation test is to show that it is very unlikely that a permuted dataset will achieve the same significance and the observed effect is not due to chance. It also provides a framework for correction for multiple testing by keeping the patterns of LD between SNPs the same under the observed and permuted samples. We permuted the binary outcome (asthma vs. non-allergic control, or asthma vs. allergic control) 10,000 times and estimated the probability distribution of the statistic under the null hypothesis. The empirical p value was defined as the probability of observing permuted statistics larger than the observed statistics.
After the primary analyses, secondary analyses were performed to ensure that the observed genetic associations with asthma were not due to other co-morbid allergic conditions such as atopic dermatitis (AD) and sensitization. The genetic associations with asthma excluding all cases with known co-morbid AD were performed by fitting a logistic regression model adjusted for age, sex and PCs. Genetic associations with SPT were evaluated for any SPT+ as well as positivity to molds, pollens, animals or dust. Subjects that were SPT+ were compared to non-asthmatic, non-allergic controls as well as SPT+/SPT- restricted to asthmatics. The SPT analyses was carried out in PLINK41 adjusted for age, sex and PCs using the additive logistic regression model in whites only due to sample size limitations for the blacks.
To evaluate epistasis between SPINK5 and TSLP, we conducted analyses using a gene collapsing technique since the significant SNPs had low minor allele frequencies (∼10%). SNPs included in the analysis were identified by the magnitude of effect (OR) and significance (p-value). Four SNPs in SPINK5 had an OR>1.3 and p<0.01 (rs2303064, rs7445392, rs2303063 or rs9325071) and 3 SNPs in TSLP had an OR<0.70 and p<0.01 (rs10062929, rs11466749, and rs11466750). The children were then identified as having any (SPINK5+) or none (SPINK5-) of the minor alleles in the four risk SNPs from SPINK5, as well as having any (TSLP+) or none (TSLP-) of the minor alleles in the three protective SNPs in TSLP. For FLG, children having one or more copies of the R501X truncation or 2282del4 deletion were defined as FLG+ and those with neither of these two polymoprphisms were defined as FLG-.
Results
Demographics of Discovery Subjects
A total of 1503 children from the GCPCR and GCC were eligible for genotyping on the custom SNP chip after inclusion criteria were applied: age (4-17), race (Caucasian or African-American), diagnoses (asthma, AR or AD), DNA quantity (≥250ng), DNA quality (optical density 1.6-2.2) and available pulmonary function test, SPT or questionnaire data (asthmatics only). After excluding 37 genotyped children due to missing SNP call rates >20%, the discovery GCPCR/GCC population consisted of 1,466 asthmatic (402 and 308), allergic (257 and 152) and non-allergic (297 and 50) white and black children respectively (Supplementary Table 1). The non-allergic control children were older than the asthmatics of similar race (p<0.05), and there were more male black asthmatics than black non-allergic controls (p<0.05).
Genetic Associations with Asthma in the Discovery GCPCR Population
The associations of SNPs in FLG, SPINK5 and TSLP were evaluated independently for whites and blacks, adjusted for age, sex and population stratification. After consideration for multiple comparisons, a non-synonymous SNP in SPINK5, rs2303064, was significantly associated with asthma in white subjects compared to non-allergic controls (p=0.003) and a tagging SNP in TSLP (rs11466750) was significantly associated with asthma in whites compared to allergic children (p=0.0056, Table 2). Multiple SNPs in both SPINK5 and TSLP were nominally associated with asthma (p<0.05). No SNPs or deletion variants in FLG achieved significance.
Table 2.
SNP associations between asthmatics and allergic and non-allergic controls in the GCPCR.
| Asthma versus Non-allergic Controls | Asthma versus Allergic Controls | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||
| Population | whitea | blacka | whitea | blacka | ||||||||||
|
| ||||||||||||||
| # cases/ # controls | 402/297 | 308/50 | 402/257 | 308/152 | ||||||||||
|
| ||||||||||||||
| Gene | SNP | Major/ Minor Allele | MAFb | OR | P-value | MAFb | OR | P-value | MAFb | OR | P-value | MAFb | OR | P-value |
| FLG | rs2184951 | T/G | 14%/14% | 1.00 | 0.99 | - | - | - | 14%/15% | 0.93 | 0.65 | - | - | - |
| rs11204976 | C/T | 15%/14% | 0.99 | 0.96 | - | - | - | 15%/14% | 1.04 | 0.82 | - | - | - | |
| rs12730241 | G/A | 15%/14% | 1.03 | 0.85 | - | - | - | 15%/14% | 1.06 | 0.72 | - | - | - | |
| rs2065956 | C/T | 13%/12% | 1.06 | 0.74 | - | - | - | 13%/11% | 1.09 | 0.62 | - | - | - | |
| rs2184953 | A/Gc | 16%/14% | 1.12 | 0.47 | 34%/31% | 1.14 | 0.61 | 16%/14% | 1.10 | 0.56 | 34%/35% | 0.95 | 0.74 | |
| rs11204980 | G/C | 15%/15% | 0.95 | 0.76 | 14%/16% | 0.84 | 0.57 | 15%/14% | 1.05 | 0.78 | 14%/14% | 0.93 | 0.73 | |
| R501X | na | na | 2.01 | 0.18d | - | - | - | na | 0.75 | 0.57e | - | - | - | |
| 2282del4 | na | na | 1.20 | 0.57d | - | - | - | na | 1.01 | 0.99e | - | - | - | |
|
| ||||||||||||||
| SPINK5 | rs3822744 | C/T | 36%/34% | 1.09 | 0.45 | 9%/15% | 0.62 | 0.12 | 36%/35% | 1.06 | 0.64 | 9%/11% | 0.87 | 0.56 |
| rs1423007 | A/G | 37%/34% | 1.13 | 0.32 | 25%/26% | 0.94 | 0.80 | 37%/35% | 1.10 | 0.42 | 25%/26% | 0.97 | 0.86 | |
| rs7445392 | T/Cc | 51%/44% | 1.33 | 0.01 | 33%/29% | 1.21 | 0.45 | 51%/47% | 1.17 | 0.18 | 33%/29% | 1.20 | 0.25 | |
| rs6892205 | G/Ac | 49%/43% | 1.25 | 0.05 | 47%/37% | 1.59 | 0.05 | 49%/46% | 1.08 | 0.49 | 47%/45% | 1.13 | 0.41 | |
| rs2303063 | A/Gc | 49%/43% | 1.32 | 0.01 | 25%/22% | 1.14 | 0.63 | 49%/46% | 1.12 | 0.33 | 25%/23% | 1.13 | 0.51 | |
| rs2303064 | G/A | 14%/9% | 1.70 | 0.003f | 48%/48% | 1.03 | 0.90 | 14%/12% | 1.21 | 0.26 | 48%/48% | 0.98 | 0.91 | |
| rs2287768 | A/G | 20%/19% | 1.03 | 0.81 | - | - | - | 20%/22% | 0.90 | 0.44 | - | - | - | |
| rs9325071 | A/G | 15%/11% | 1.58 | 0.01 | 32%/41% | 0.65 | 0.07 | 15%/12% | 1.17 | 0.35 | 32%/32% | 0.95 | 0.76 | |
| rs3777134 | T/Cc | 38%/35% | 1.20 | 0.13 | 36%/37% | 0.92 | 0.70 | 38%/36% | 1.11 | 0.40 | 36%/35% | 1.01 | 0.96 | |
| rs2112767 | T/C | 39%/35% | 1.23 | 0.09 | 44%/43% | 1.03 | 0.88 | 39%/36% | 1.12 | 0.36 | 44%/44% | 1.04 | 0.79 | |
| rs4263489 | C/G | 11%/14% | 0.80 | 0.19 | - | - | - | 11%/12% | 0.89 | 0.51 | - | - | - | |
| rs3815741 | A/G | - | - | - | 26%/27% | 0.89 | 0.67 | - | - | - | 26%/27% | 0.93 | 0.69 | |
| rs28408445 | C/G | - | - | - | 14%/18% | 0.76 | 0.33 | - | - | - | 14%/12% | 1.11 | 0.63 | |
|
| ||||||||||||||
| TSLP | rs3806933 | C/T | 40%/41% | 0.93 | 0.53 | 25%/26% | 0.96 | 0.87 | 40%/42% | 0.92 | 0.46 | 25%/29% | 0.83 | 0.26 |
| rs2289276 | C/T | 28%/28% | 0.93 | 0.57 | 15%/14% | 1.06 | 0.86 | 28%/25% | 1.13 | 0.34 | 15%/17% | 0.87 | 0.45 | |
| rs1898671 | C/T | 37%/34% | 1.10 | 0.40 | - | - | - | 37%/32% | 1.19 | 0.15 | - | - | - | |
| rs10062929 | C/A | 12%/14% | 0.93 | 0.68 | 10%/12% | 0.86 | 0.67 | 12%/18% | 0.64 | 0.009 | 10%/12% | 0.85 | 0.47 | |
| rs2289277 | C/G | 39%/42% | 0.90 | 0.33 | 39%/40% | 1.01 | 0.98 | 39%/43% | 0.87 | 0.24 | 39%/44% | 0.83 | 0.20 | |
| rs11466749 | A/G | 15%/16% | 0.95 | 0.73 | 12%/14% | 0.83 | 0.59 | 15%/20% | 0.67 | 0.009 | 12%/14% | 0.86 | 0.50 | |
| rs11466750 | G/A | 12%/14% | 0.92 | 0.60 | 26%/27% | 0.94 | 0.81 | 12%/18% | 0.63 | 0.0056f | 26%/25% | 1.06 | 0.73 | |
| rs2289278 | C/G | - | - | - | 11%/8% | 1.60 | 0.24 | - | - | - | 11%/12% | 1.03 | 0.89 | |
White and black asthmatic children were from the GCPCR and non-asthmatic/non-allergic controls and allergic controls were from the GCPCR and GCC. Associations were tested using an additive model. Odds ratios (OR) were determined using logistic regression based on the minor allele adjusted for age, gender and population stratification. Bold indicates association reached linkage disequilibrium-corrected bonferroni cut-off of 0.0077 for whites and 0.0039 for blacks.
MAF (minor allele frequency) in cases/controls.
Major/Minor allele are reverse for blacks.
Sample size was 114/221 for R501X and 401/256 for 2282del4.
Sample size was 114/72 for R501X and 401/216 for 2282del4.
These associations remained significant after asthmatics with known co-morbid eczema were excluded (p=0.003 and 0.004), indicating an asthma specific association.
To further substantiate our results and ensure that the family wise error rate was appropriately controlled, we performed 10,000 permutations. The point-wise empirical p-value was consistent with the observed p-value (0.0026 for rs2303064 and 0.0046 for rs11466750; results not shown) supporting its validity. The family-wise corrected p-values were 0.039 and 0.069 for rs2303064 and rs11466750, respectively.
In order to ensure that the genetic associations with asthma were not attributed to other co-morbid allergic conditions such as atopic dermatitis (AD), we performed secondary analyses excluding asthmatic cases with AD and evaluated SPT+ as an outcome. These analyses were performed in whites only due to sample size limitations for the blacks. The associations with asthma remained significant after asthmatics with known co-morbid AD were excluded (p=0.003 for rs2303064 and 0.004 for rs11466750; data not shown), indicating an asthma specific association. We did not observe any significant associations with SPT+ (data not shown).
Genetic Associations with Asthma in Replication Cohorts
We evaluated SNP associations in the SPINK5 and TSLP genes with asthma in six additional populations: CAMP, CARE, CCAAPS, replication GCPCR/CCC, and replication GCPCR (whites and blacks). Based on our stringent replication definition (same SNP, phenotype and direction of association), we achieved replication of the SPINK5 rs2303064 variant in the CARE population (p=0.046; Table 3). In addition, we observed additional SNPs in SPINK5 and TSLP to be nominally (p<0.05) associated with asthma in five of the six populations (Table 3).
Table 3.
SNP associations with asthma in replication cohorts.
| Population | CAMPa | CAREa | CCAAPSb | replication GCPCR/CCCd | replication GCPCRe | replication GCPCRe | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||||
| # cases/ # controls | 334 trios | 95 trios | 57/184c | 206/211 | 206/132 | 263/67 | ||||||||
|
| ||||||||||||||
| Race= | white | white | white | white | white | black | ||||||||
|
| ||||||||||||||
| Gene | SNP | Major/ Minor Allele | OR | P-value | OR | P-value | OR | P-value | OR | P-value | OR | P-value | OR | P-value |
| SPINK5 | rs2303064f | G/A | 0.82 | 0.26 | 2.00 | 0.046 | 1.34 | 0.35 | 1.22 | 0.36 | - | - | 0.93 | 0.71 |
| rs7725861 | A/T | 0.68 | 0.03 | 1.53 | 0.19 | - | - | - | - | - | - | - | - | |
| rs9791022 | T/C | 0.45 | 0.004 | 1.55 | 0.26 | - | - | - | - | - | - | - | - | |
| rs3777138 | A/G | 1.00 | 1.00 | 7.00 | 0.03 | - | - | - | - | - | - | - | - | |
| rs3777142 | A/T | - | - | - | - | 2.14 | 0.04 | - | - | - | - | - | - | |
| rs17704764 | A/G | - | - | - | - | 1.75 | 0.04 | - | - | - | - | - | - | |
| rs6864920 | A/G | - | - | - | - | 0.62 | 0.04 | - | - | - | - | - | - | |
| rs2287772 | C/T | - | - | - | - | 0.63 | 0.049 | - | - | - | - | - | - | |
| rs9325073 | G/C | - | - | - | - | - | - | 1.53 | 0.003 | - | - | - | - | |
| rs7445392f | T/C | - | - | - | - | - | - | - | - | - | - | 0.98 | 0.92 | |
| rs2303063f | G/A | - | - | - | - | - | - | - | - | - | - | 1.07 | 0.76 | |
| rs6892205f | A/G | - | - | - | - | - | - | - | - | - | - | 1.23 | 0.27 | |
|
| ||||||||||||||
| TSLP | rs11466750f | G/A | - | - | - | - | 1.18 | 0.65 | - | - | 0.98 | 0.93 | 0.85 | 0.47 |
| rs11466749f | T/C | 0.92 | 0.59 | 0.48 | 0.02 | 1.47 | 0.24 | - | - | 0.98 | 0.94 | 0.82 | 0.55 | |
| rs11466744 | G/T | 0.00 | 0.03 | - | - | - | - | - | - | - | - | - | - | |
| rs2289276f | C/T | - | - | - | - | 1.08 | 0.77 | - | - | 1.36 | 0.10 | 0.64 | 0.08 | |
| rs3806933f | C/T | - | - | - | - | 1.13 | 0.57 | - | - | 1.24 | 0.21 | 0.64 | 0.04 | |
The Childhood Asthma Management Program (CAMP) and Childhood Research and Education (CARE) are clinical trials included in the SNP Health Association Resource (SHARe) Asthma Resource project (SHARP) and were used as replication cohorts in this analysis. Whole genome genotyping data from the Affymetrix 6.0 chip was available on 334 CAMP and 95 CARE family trios of asthmatic children. Transmission disequilibrium tests were used for SNP associations.
The Cincinnati Childhood Allergy and Air Pollution (CCAAPS) birth cohort included 57 white asthmatics and 184 white non-asthmatic, SPT negative control children.
For TSLP analysis, controls were 141 non-asthmatic SPT positive children. Asthma was diagnosed at age 7 using pulmonary function and methacholine challenge testing.
New GCPCR cases were asthmatic individuals that were recruited into the GCPCR after the discovery cohort and genotyped on a separate custom Illumina chip. The CCC consisted of white adults with no history of asthma. A logistic model was fitted adjusted for sex.
New GCPCR controls were recruited after the discovery cohort and were non-asthmatic children with allergic rhinitis (both with and without atopic dermatitis). Shading indicates replication defined by same SNP, phenotype and direction of effect. Bolding indicates gene-level replication at p<0.05.
Indicates SNP was included on discovery array.
Epistasis between SPINK5 and TSLP
The literature suggests that SPINK5 and TSLP occupy a shared pathway where LEKTI deficiency ultimately leads to TSLP production, increasing the Th2 response and causing allergic lung inflammation21, 44-48. Therefore, we sought to determine if there were epistatic effects of variation in the SPINK5 and TSLP genes on asthma prevalence. Formal gene:gene analyses did not show an interaction between SPINK5 and TSLP (p=0.29). Further analyses were performed stratified by either SPINK5 or TSLP genotype by conducting gene collapsing analyses. We evaluated the effects of having one or more of the 3 protective TSLP alleles (rs10062929, rs11466749, and rs11466750) among subjects with and without one or more of the risk SPINK5 alleles (rs2303064, rs7445392, rs2303063 or rs9325071). There was modest linkage disequilibrium among the SPINK5 SNPs as well as the TSLP SNPs (Figure 1). In subjects with at least one of the SPINK5 risk alleles (SPINK5+), the absence of the TSLP protective minor alleles (TSLP-) was associated with a significant increase in asthma risk (67% vs. 53%, p=0.0017, Figure 2A). In contrast, the presence (TSLP+) or absence of TSLP minor alleles did not affect asthma risk in subjects without any of the SPINK5 risk alleles (SPINK5-, p=0.71).
Figure 1.
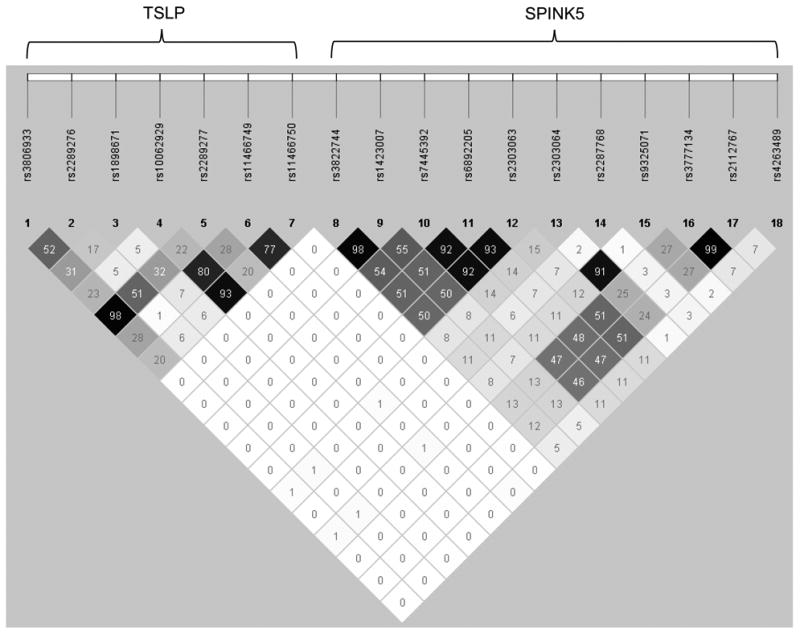
Linkage disequilibrium (r2) among SNPs included in the gene collapsing analyses for 407 asthmatics compared to 257 allergic controls in the white discovery GCPCR populations.
Figure 2.
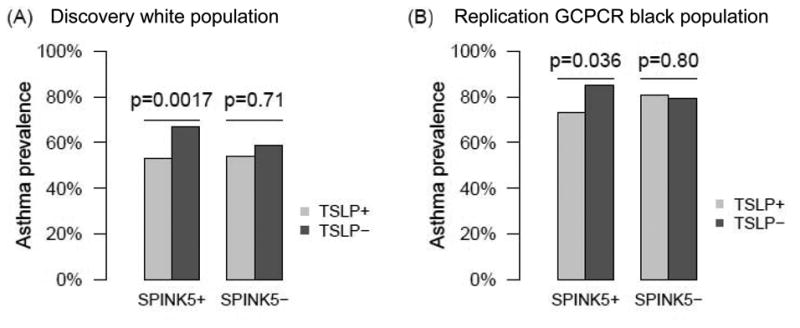
Epistasis of SPINK5 and TSLP. (A) Discovery white GCPCR/GCC children. (B) Replication of epistasis of SPINK5 and TSLP in replication GCPCR black children.
Replication of Epistasis between SPINK5 and TSLP
Using biologic plausibility of a shared pathway from the published literature and the results from above, we pursued replication of the observed epistasis of SPINK5 and TSLP in the discovery GCPCR black, replication GCPCR white and replication GCPCR black populations. Notably, there were no independent genetic associations at our LD-adjusted Bonferroni level with any SNPs in SPINK5 or TSLP in these three populations. Therefore, we included SNPs with p<0.10 and OR >1.2 for SPINK5 (rs6892205 and rs2303063) and p<0.10 and OR <0.7 for TSLP (rs3806933 and rs2289276) in the analyses (Table 3). We replicated the epistasis between SPINK5 and TSLP in the replication GCPCR black population (Figure 2B); the other two populations did not replicate. As in the discovery GCPCR white population, SPINK5+ black subjects that were SPINK5+ TSLP+ had a significantly decreased asthma risk compared to subjects that were SPINK5+ TSLP- (73% versus 85%, p=0.036; Figure 2B), whereas there was no effect of TSLP minor alleles among subjects that were SPINK5- (p=0.80).
Discussion
While the role of skin-related genes in asthma has been previously reported, our data support that asthma risk is, in part, a result of epistasis between skin-related genes (TSLP and SPINK5). Using a well characterized cohort of white asthmatic cases and controls, we identified SNPs in these genes to be significantly associated with asthma in children. The finding with SPINK5 was replicated according to the strictest replication definition43. Additionally, we discovered epistasis between TSLP and SPINK5 in whites. This was replicated using a black cohort where neither gene was independently associated. These findings highlight the importance of evaluating epistasis in genetic association studies. Further, our results emphasize the need to utilize biology to inform analyses in order to identify genetic susceptibility to complex diseases, substantiate the value of smaller well-characterized cohorts in genetic association studies, and demonstrate the importance and value of looking at functionally related genes, rather than one at a time as in GWAS, to identify genes with clinical relevance.
This study further implicates skin-related genes in the pathogenesis of childhood asthma. Interestingly, the associations of the skin genes with asthma were sustained when children with co-morbid AD were excluded from the analysis, suggesting an independent asthma effect. Associations between asthma and SPINK5 have been reported in the Chinese24 and German23 populations. Previous studies have also found that TSLP SNPs are associated with asthma49, including a meta-analysis of GWASs in subjects of multiple ancestries50. This is the first report of an epistatic effect of SNPs in the SPINK5 and TSLP genes with asthma. We discovered epistasis between TSLP and SPINK5 whereby children that had one or more SPINK5 risk alleles and were wild-type for all TSLP SNPs had the highest risk of asthma. The addition of TSLP minor alleles conferred protection from asthma in the presence of a SPINK5 risk allele, and this protection was stronger than the association between TSLP and asthma alone. Importantly, our primary analyses did not detect independent significant associations with SPINK5 and TSLP in the black population. Only after stratified analyses were we able to uncover the epistatic effect of SPINK5 and TSLP in the black population. Thus, ignoring epistatic effects may mask important genetic susceptibility. While other studies have utilized biologic plausibility51, 52, results from linkage analyses53, 54 and bioinformatics54, 55 to inform analyses of genetic interactions in asthma, our study is the first to apply this approach in an independent population and replicate findings where no significant independent effects were observed (blacks).
Observations from animal models provide valuable insights into the possible mechanistic basis for the observed epistasis between SPINK5 and TSLP. Mice that lack Spink5 overexpress TSLP in the skin in the absence of lympho-epithelial kazal-type-related inhibitor (LEKTI)20. Overexpression in the skin leads to high systemic availability of TSLP driving susceptibility to allergic inflammation in the lung, even in the absence of any skin pathology19. TSLP transgenic mice develop severe asthma in the absence of a skin phenotype and express low levels of the TSLP transgene in the trachea but not in the lung19. Further, the protein is absent in the bronchoalveolar fluid suggesting systemic skin-derived TSLP is sufficient to predispose mice to allergic lung inflammation19. Models using lung-specific expression of the TSLP transgene or intranasal administration of TSLP show that increased lung TSLP drives an inflammatory response in the presence of antigen, suggesting that TSLP modifies the response to aero-antigen, promoting inflammation20, 56. Murine models of asthma treated with anti-TSLP antibody reversed airway inflammation, prevented structural alterations and decreased airway hyperresponsiveness57, while suppression of signaling with anti-TSLP receptor reduces eosinophilic airway inflammation, goblet cell hyperplasia and TH2 cytokine production58.
These animal data, along with our results, support a shared pathway for SPINK5 and TSLP (and possibly FLG) leading to airway inflammation and asthma development (Figure 3). SPINK5 overexpression in human epithelial cells leads to increased inflammatory cytokines including IL6, IL8 and RANTES, indicating it may have a role in the pathogenesis of asthma that is independent of protease inhibition59 (Figure 3A). This suggests that SPINK5 stimulates the involvement of B and T lymphocytes, neutrophils and eosinophils active in the inflammatory process through its effects on pro-inflammatory cytokines and chemokines. In humans, LEKTI deficiency results in a lack of kallikrein-related peptidase 5 (KLK5) and KLK7 inhibition21, 60 and unopposed KLK14 activity21, activating through protease-activated receptor-2 (PAR-2) which is found at the surface of keratinocytes. PAR-2 then activates nuclear factor kappa B (NF-κB) leading to the production of the TSLP cytokine21, 60 causing the differentiation of Th0 cells to Th2 which produce IL-4, IL-13 and IL-5, amplifying the Th2 response and promoting inflammation, allergy and asthma (Figure 3B). LEKTI deficiency also promotes increased profilaggrin cleavage44 leading to a compromised barrier45 promoting sensitization and inflammation (Figure 3C).
Figure 3.
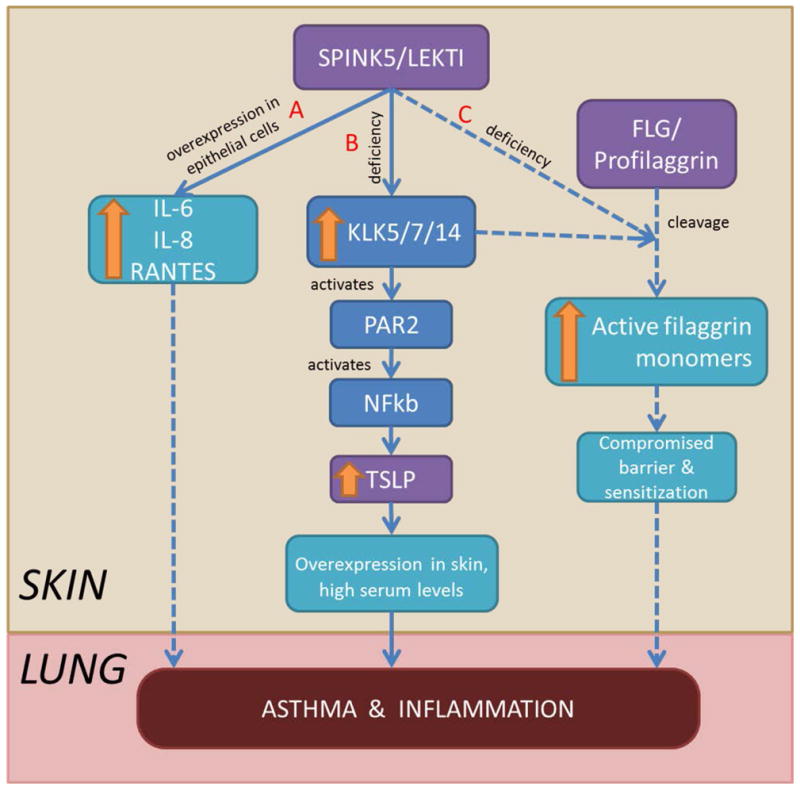
Hypothesized shared pathways leading to asthma and inflammation. (A) SPINK5 overexpression increases inflammatory cytokines. (B) LEKTI deficiency increases KLK5/7/14, activating through PAR-2, and NF-κB leading to TSLP production, causing allergy and lung inflammation. (C) LEKTI deficiency increases profillagrin cleavage compromising the skin barrier and contributing to sensitization and subsequent asthma. Solid lines represent published pathways, dashed lines represent hypothesized pathways.
While we were successful in identifying both main and epistatic effects with TSLP and SPINK5, no associations with FLG were identified. This is not surprising given the low frequency of the functional FLG variants in our population resulting in inadequate power to detect an association. However, our data does support that common variation in FLG, represented by our 6 tagging SNPs, is not associated with asthma in these populations. A meta-analysis of asthma regardless of eczema revealed a significant association with the combined R501X and 2282del4 genotype (pooled OR 1.48, 95%CI 1.32-1.66)61, indicating that FLG deficiency may predispose to asthma. However, the asthma association was driven by the asthma plus eczema phenotype, supporting that FLG is only associated with asthma in the presence of atopic features.
The identification of epistasis between SPINK5 and TSLP is has immediate clinical implications; there are clinical trials underway testing AMG157, a human monoclonal antibody that inhibits the action of TSLP. Phase 1 escalation studies to evaluate the safety, tolerability and pharmacokinetics in both healthy subjects and subjects with atopic dermatitis have been completed62, 63 and another study evaluating the effects of AMG157 on FEV1 after allergen inhalation challenge in mild atopic asthmatics is currently recruiting64. The results from our study suggest that the therapeutic effects of anti-TSLP therapy in asthmatics may be dependent on SPINK5 genotype.
In conclusion, our results support a novel epistasis between SPINK5 and TSLP, which contributes to childhood asthma. These findings emphasize the importance of utilizing biology to inform analyses to identify genetic susceptibility to complex diseases, especially in populations that do not exhibit independent genetic associations. The results from our study are clinically relevant with respect to anti-TSLP therapy and suggest that therapeutic effects in asthmatics may be dependent on SPINK5 genotype.
Supplementary Material
Key Messages.
Our study supports epistasis between SPINK5 and TSLP which contributes to childhood asthma.
Genetic association studies should use biologic plausibility to inform analyses to identify genetic susceptibility to complex disease.
The therapeutic effects of anti-TSLP therapy in asthmatics may be dependent on SPINK5 genotype.
Acknowledgments
This research was supported in part by the Cincinnati Children's Research Foundation and its Cincinnati Genomic Control Cohort. We thank the physicians, nurses and staff of Cincinnati Children's Hospital Medical Center Allergy and Immunology, Pulmonary, Dermatology, Dental, and Orthopedic clinics, Headache Center, and Emergency Department for their contributions to the Greater Cincinnati Pediatric Clinic Repository. We also thank all the patients and their families who participated in the GCPCR, GCC, CCC and CCAAPS studies. The CAMP/CARE datasets used for the replication analyses described in this manuscript were obtained through dbGaP accession number phs000166.v2.p1.
Funding: This work was supported by National Institutes of Health grants U19AI070235 (GKKH), R01ES011170 (GKL) and R21ES016830 (MBK)
Abbreviations
- FLG
filaggrin
- TSLP
thymic stromal lymphopoietin
- Th2
T-helper 2
- SPINK5
serine protease inhibitor Kazal-type 5
- GCPCR
Greater Cincinnati Pediatric Clinic Repository
- GCC
Genomic Control Cohort
- SPT
skin prick testing
- CAMP
Childhood Asthma Management Program
- CARE
Childhood Asthma Research and Education
- CCAAPS
Cincinnati Childhood Allergy and Air Pollution Study
- CCC
Cincinnati Control Cohort
- dbGaP
database of Genotypes and Phenotypes
- AIMs
ancestry-informative markers
- PCs
principal components
- TDT
transmission disequilibrium test
- OR
odds ratio
- LEKTI
lympho-epithelial kazal-type-related inhibitor
- KLK5
kallikrein-related peptidase 5
- KLK7
kallikrein-related peptidase 7
- KLK14
kallikrein-related peptidase 14
- PAR-2
protease-activated receptor-2
- NF-κB
nuclear factor kappa B
Footnotes
Author Contributions: G.K.K.H. conceived the project, oversaw its design, implementation, interpretation, and analysis, gave critical review of the manuscript and provided the funding for the project. J.M.B.M. wrote the manuscript and L.J.M, M.B.K. and T.M.B. provided conceptual advice and critical review. J.M.B.M. and M.B.K. oversaw subject recruitment, data collection and storage. J.M.B.M. and L.J.M. oversaw the statistical analysis. M.A.L. oversaw the data management and prepared the data for analysis and M.B.E. oversaw preparation of the DNA samples from the GCPCR and the GCC for genotyping. H.H. and V.P. performed the data analyses and helped prepare tables and figures. J.E.L, G.K.L and D.I.B. provided the CCAAPS data and samples as well as conceptual advice and critical review.
Literature Cited
- 1.Bloom B, Cohen R, Freeman G Summary health statistics for U.S. children: National Health Interview Survey 2011. Vital Health Stat. 2012;10:254. [PubMed] [Google Scholar]
- 2.Duffy DL, Martin NG, Battistutta D, Hopper JL, Mathews JD. Genetics of asthma and hay fever in Australian twins. Am Rev Respir Dis. 1990;142:1351–8. doi: 10.1164/ajrccm/142.6_Pt_1.1351. [DOI] [PubMed] [Google Scholar]
- 3.Harris JR, Magnus P, Samuelsen SO, Tambs K. No evidence for effects of family environment on asthma. A retrospective study of Norwegian twins. Am J Respir Crit Care Med. 1997;156:43–9. doi: 10.1164/ajrccm.156.1.9609094. [DOI] [PubMed] [Google Scholar]
- 4.Koppelman GH, Los H, Postma DS. Genetic and environment in asthma: the answer of twin studies. Eur Respir J. 1999;13:2–4. doi: 10.1183/09031936.99.13100299. [DOI] [PubMed] [Google Scholar]
- 5.Nieminen MM, Kaprio J, Koskenvuo M. A population-based study of bronchial asthma in adult twin pairs. Chest. 1991;100:70–5. doi: 10.1378/chest.100.1.70. [DOI] [PubMed] [Google Scholar]
- 6.March ME, Sleiman PM, Hakonarson H. The genetics of asthma and allergic disorders. Discov Med. 2011;11:35–45. [PubMed] [Google Scholar]
- 7.Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, Hunter DJ, et al. Finding the missing heritability of complex diseases. Nature. 2009;461:747–53. doi: 10.1038/nature08494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zuk O, Hechter E, Sunyaev SR, Lander ES. The mystery of missing heritability: Genetic interactions create phantom heritability. Proc Natl Acad Sci U S A. 2012;109:1193–8. doi: 10.1073/pnas.1119675109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weiss ST, Raby BA, Rogers A. Asthma genetics and genomics 2009. Curr Opin Genet Dev. 2009;19:279–82. doi: 10.1016/j.gde.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Palmer CN, Irvine AD, Terron-Kwiatkowski A, Zhao Y, Liao H, Lee SP, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis. Nat Genet. 2006;38:441–6. doi: 10.1038/ng1767. [DOI] [PubMed] [Google Scholar]
- 11.Henderson J, Northstone K, Lee SP, Liao H, Zhao Y, Pembrey M, et al. The burden of disease associated with filaggrin mutations: a population-based, longitudinal birth cohort study. J Allergy Clin Immunol. 2008;121:872–7. doi: 10.1016/j.jaci.2008.01.026. e9. [DOI] [PubMed] [Google Scholar]
- 12.Marenholz I, Nickel R, Ruschendorf F, Schulz F, Esparza-Gordillo J, Kerscher T, et al. Filaggrin loss-of-function mutations predispose to phenotypes involved in the atopic march. J Allergy Clin Immunol. 2006;118:866–71. doi: 10.1016/j.jaci.2006.07.026. [DOI] [PubMed] [Google Scholar]
- 13.Morar N, Cookson WO, Harper JI, Moffatt MF. Filaggrin mutations in children with severe atopic dermatitis. J Invest Dermatol. 2007;127:1667–72. doi: 10.1038/sj.jid.5700739. [DOI] [PubMed] [Google Scholar]
- 14.Palmer CN, Ismail T, Lee SP, Terron-Kwiatkowski A, Zhao Y, Liao H, et al. Filaggrin null mutations are associated with increased asthma severity in children and young adults. J Allergy Clin Immunol. 2007;120:64–8. doi: 10.1016/j.jaci.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 15.Poninska J, Samolinski B, Tomaszewska A, Raciborski F, Samel-Kowalik P, Walkiewicz A, et al. Filaggrin gene defects are independent risk factors for atopic asthma in a Polish population: a study in ECAP cohort. PLoS One. 2011;6:e16933. doi: 10.1371/journal.pone.0016933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rogers AJ, Celedon JC, Lasky-Su JA, Weiss ST, Raby BA. Filaggrin mutations confer susceptibility to atopic dermatitis but not to asthma. J Allergy Clin Immunol. 2007;120:1332–7. doi: 10.1016/j.jaci.2007.09.037. [DOI] [PubMed] [Google Scholar]
- 17.Weidinger S, O'Sullivan M, Illig T, Baurecht H, Depner M, Rodriguez E, et al. Filaggrin mutations, atopic eczema, hay fever, and asthma in children. J Allergy Clin Immunol. 2008;121:1203–9. doi: 10.1016/j.jaci.2008.02.014. e1. [DOI] [PubMed] [Google Scholar]
- 18.Ober C, Yao TC. The genetics of asthma and allergic disease: a 21st century perspective. Immunol Rev. 2011;242:10–30. doi: 10.1111/j.1600-065X.2011.01029.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Demehri S, Morimoto M, Holtzman MJ, Kopan R. Skin-derived TSLP triggers progression from epidermal-barrier defects to asthma. PLoS Biol. 2009;7:e1000067. doi: 10.1371/journal.pbio.1000067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ziegler SF. Thymic stromal lymphopoietin and allergic disease. J Allergy Clin Immunol. 2012;130:845–52. doi: 10.1016/j.jaci.2012.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hovnanian A. Netherton syndrome: skin inflammation and allergy by loss of protease inhibition. Cell Tissue Res. 2013;351:289–300. doi: 10.1007/s00441-013-1558-1. [DOI] [PubMed] [Google Scholar]
- 22.Genuneit J, Cantelmo JL, Weinmayr G, Wong GW, Cooper PJ, Riikjarv MA, et al. A multi-centre study of candidate genes for wheeze and allergy: the International Study of Asthma and Allergies in Childhood Phase 2. Clin Exp Allergy. 2009;39:1875–88. doi: 10.1111/j.1365-2222.2009.03364.x. [DOI] [PubMed] [Google Scholar]
- 23.Kabesch M, Carr D, Weiland SK, von Mutius E. Association between polymorphisms in serine protease inhibitor, kazal type 5 and asthma phenotypes in a large German population sample. Clin Exp Allergy. 2004;34:340–5. doi: 10.1111/j.1365-2222.2004.01860.x. [DOI] [PubMed] [Google Scholar]
- 24.Liu Q, Xia Y, Zhang W, Li J, Wang P, Li H, et al. A functional polymorphism in the SPINK5 gene is associated with asthma in a Chinese Han Population. BMC Med Genet. 2009;10:59. doi: 10.1186/1471-2350-10-59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walley AJ, Chavanas S, Moffatt MF, Esnouf RM, Ubhi B, Lawrence R, et al. Gene polymorphism in Netherton and common atopic disease. Nat Genet. 2001;29:175–8. doi: 10.1038/ng728. [DOI] [PubMed] [Google Scholar]
- 26.Kovacic MB, Myers JM, Wang N, Martin LJ, Lindsey M, Ericksen MB, et al. Identification of KIF3A as a novel candidate gene for childhood asthma using RNA expression and population allelic frequencies differences. PLoS One. 2011;6:e23714. doi: 10.1371/journal.pone.0023714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Butsch Kovacic M, Biagini Myers JM, Lindsey M, Patterson T, Sauter S, Ericksen MB, et al. The Greater Cincinnati Pediatric Clinic Repository: A Novel Framework for Childhood Asthma and Allergy Research. Pediatr Allergy Immunol Pulmonol. 2012;25:104–13. doi: 10.1089/ped.2011.0116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Butsch Kovacic M, Biagini Myers JM, Wang N, Martin LJ, Lindsey M, Ericksen MB, et al. A novel selection approach identifies six candidate epithelial genes for childhood asthma including KIF3A. PLoS One. 2011;6:e23714. doi: 10.1371/journal.pone.0023714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martin LJ, Gupta J, Jyothula SS, Butsch Kovacic M, Biagini Myers JM, Patterson TL, et al. Functional variant in the autophagy-related 5 gene promotor is associated with childhood asthma. PLoS One. 2012;7:e33454. doi: 10.1371/journal.pone.0033454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.ATS. Standardization of Spirometry. Am J Respir Crit Care Med. 1994;152:1107–36. doi: 10.1164/ajrccm.152.3.7663792. [DOI] [PubMed] [Google Scholar]
- 31.The Childhood Asthma Management Program (CAMP): design, rationale, and methods. Childhood Asthma Management Program Research Group. Control Clin Trials. 1999;20:91–120. [PubMed] [Google Scholar]
- 32.Guilbert TW, Morgan WJ, Krawiec M, Lemanske RF, Jr, Sorkness C, Szefler SJ, et al. The Prevention of Early Asthma in Kids study: design, rationale and methods for the Childhood Asthma Research and Education network. Control Clin Trials. 2004;25:286–310. doi: 10.1016/j.cct.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 33.LeMasters GK, Wilson K, Levin L, Biagini J, Ryan P, Lockey JE, et al. High prevalence of aeroallergen sensitization among infants of atopic parents. J Pediatr. 2006;149:505–11. doi: 10.1016/j.jpeds.2006.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baye TM, Butsch Kovacic M, Biagini Myers JM, Martin LJ, Lindsey M, Patterson TP, et al. Differences in candidate gene association between European ancestry and African American asthmatic children. PLoS One. 2011;6:e16522. doi: 10.1371/journal.pone.0016522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asmussen L, Olson LM, Grant EN, Fagan J, Weiss KB. Reliability and validity of the Children's Health Survey for Asthma. Pediatrics. 1999;104:e71. doi: 10.1542/peds.104.6.e71. [DOI] [PubMed] [Google Scholar]
- 36.Enomoto H, Hirata K, Otsuka K, Kawai T, Takahashi T, Hirota T, et al. Filaggrin null mutations are associated with atopic dermatitis and elevated levels of IgE in the Japanese population: a family and case-control study. J Hum Genet. 2008;53:615–21. doi: 10.1007/s10038-008-0293-z. [DOI] [PubMed] [Google Scholar]
- 37.Epstein TG, LeMasters GK, Bernstein DI, Ericksen MB, Martin LJ, Ryan PH, et al. Genetic variation in small proline rich protein 2B as a predictor for asthma among children with eczema. Ann Allergy Asthma Immunol. 2012;108:145–50. doi: 10.1016/j.anai.2012.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Narayanaswamy CR, Raghavarao D. Principal Component Analysis of Large Dispersion Matrices. Applied Statistics-Journal of the Royal Statistical Society Series C. 1991;40:309–16. [Google Scholar]
- 39.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–9. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 40.Kosoy R, Nassir R, Tian C, White PA, Butler LM, Silva G, et al. Ancestry informative marker sets for determining continental origin and admixture proportions in common populations in America. Hum Mutat. 2009;30:69–78. doi: 10.1002/humu.20822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–75. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spielman RS, Ewens WJ. The TDT and other family-based tests for linkage disequilibrium and association. Am J Hum Genet. 1996;59:983–9. [PMC free article] [PubMed] [Google Scholar]
- 43.Sullivan PF. Spurious genetic associations. Biol Psychiatry. 2007;61:1121–6. doi: 10.1016/j.biopsych.2006.11.010. [DOI] [PubMed] [Google Scholar]
- 44.Descargues P, Deraison C, Bonnart C, Kreft M, Kishibe M, Ishida-Yamamoto A, et al. Spink5-deficient mice mimic Netherton syndrome through degradation of desmoglein 1 by epidermal protease hyperactivity. Nat Genet. 2005;37:56–65. doi: 10.1038/ng1493. [DOI] [PubMed] [Google Scholar]
- 45.Hewett DR, Simons AL, Mangan NE, Jolin HE, Green SM, Fallon PG, et al. Lethal, neonatal ichthyosis with increased proteolytic processing of filaggrin in a mouse model of Netherton syndrome. Hum Mol Genet. 2005;14:335–46. doi: 10.1093/hmg/ddi030. [DOI] [PubMed] [Google Scholar]
- 46.Sakabe JI, Yamamoto M, Hirakawa S, Motoyama A, Ohta I, Tatsuno K, et al. Kallikrein-related peptidase 5 functions in proteolytic processing of profilaggrin in cultured human keratinocytes. J Biol Chem. 2013 doi: 10.1074/jbc.M113.476820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sandilands A, Sutherland C, Irvine AD, McLean WH. Filaggrin in the frontline: role in skin barrier function and disease. J Cell Sci. 2009;122:1285–94. doi: 10.1242/jcs.033969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weidinger S, Baurecht H, Wagenpfeil S, Henderson J, Novak N, Sandilands A, et al. Analysis of the individual and aggregate genetic contributions of previously identified serine peptidase inhibitor Kazal type 5 (SPINK5), kallikrein-related peptidase 7 (KLK7), and filaggrin (FLG) polymorphisms to eczema risk. J Allergy Clin Immunol. 2008;122:560–8. doi: 10.1016/j.jaci.2008.05.050. e4. [DOI] [PubMed] [Google Scholar]
- 49.Harada M, Hirota T, Jodo AI, Hitomi Y, Sakashita M, Tsunoda T, et al. Thymic stromal lymphopoietin gene promoter polymorphisms are associated with susceptibility to bronchial asthma. Am J Respir Cell Mol Biol. 2011;44:787–93. doi: 10.1165/rcmb.2009-0418OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Torgerson DG, Ampleford EJ, Chiu GY, Gauderman WJ, Gignoux CR, Graves PE, et al. Meta-analysis of genome-wide association studies of asthma in ethnically diverse North American populations. Nat Genet. 2011;43:887–92. doi: 10.1038/ng.888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Acevedo N, Saaf A, Soderhall C, Melen E, Mandelin J, Pietras CO, et al. Interaction between Retinoid Acid Receptor-Related Orphan Receptor Alpha (RORA) and Neuropeptide S Receptor 1 (NPSR1) in Asthma. PLoS One. 2013;8:e60111. doi: 10.1371/journal.pone.0060111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Orsmark-Pietras C, Melen E, Vendelin J, Bruce S, Laitinen A, Laitinen LA, et al. Biological and genetic interaction between tenascin C and neuropeptide S receptor 1 in allergic diseases. Hum Mol Genet. 2008;17:1673–82. doi: 10.1093/hmg/ddn058. [DOI] [PubMed] [Google Scholar]
- 53.Ferreira MA, Zhao ZZ, Thomsen SF, James M, Evans DM, Postmus PE, et al. Association and interaction analyses of eight genes under asthma linkage peaks. Allergy. 2009;64:1623–8. doi: 10.1111/j.1398-9995.2009.02091.x. [DOI] [PubMed] [Google Scholar]
- 54.Ungvari I, Hullam G, Antal P, Kiszel PS, Gezsi A, Hadadi E, et al. Evaluation of a partial genome screening of two asthma susceptibility regions using bayesian network based bayesian multilevel analysis of relevance. PLoS One. 2012;7:e33573. doi: 10.1371/journal.pone.0033573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Han B, Chen XW, Talebizadeh Z. FEPI-MB: identifying SNPs-disease association using a Markov Blanket-based approach. BMC Bioinformatics. 2011;12(Suppl 12):S3. doi: 10.1186/1471-2105-12-S12-S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhou B, Comeau MR, De Smedt T, Liggitt HD, Dahl ME, Lewis DB, et al. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat Immunol. 2005;6:1047–53. doi: 10.1038/ni1247. [DOI] [PubMed] [Google Scholar]
- 57.Chen ZG, Zhang TT, Li HT, Chen FH, Zou XL, Ji JZ, et al. Neutralization of TSLP inhibits airway remodeling in a murine model of allergic asthma induced by chronic exposure to house dust mite. PLoS One. 2013;8:e51268. doi: 10.1371/journal.pone.0051268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shi L, Leu SW, Xu F, Zhou X, Yin H, Cai L, et al. Local blockade of TSLP receptor alleviated allergic disease by regulating airway dendritic cells. Clin Immunol. 2008;129:202–10. doi: 10.1016/j.clim.2008.07.015. [DOI] [PubMed] [Google Scholar]
- 59.Birben E, Sackesen C, Turgutoglu N, Kalayci O. The role of SPINK5 in asthma related physiological events in the airway epithelium. Respir Med. 2012;106:349–55. doi: 10.1016/j.rmed.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 60.Briot A, Deraison C, Lacroix M, Bonnart C, Robin A, Besson C, et al. Kallikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J Exp Med. 2009;206:1135–47. doi: 10.1084/jem.20082242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rodriguez E, Baurecht H, Herberich E, Wagenpfeil S, Brown SJ, Cordell HJ, et al. Meta-analysis of filaggrin polymorphisms in eczema and asthma: robust risk factors in atopic disease. J Allergy Clin Immunol. 2009;123:1361–70. doi: 10.1016/j.jaci.2009.03.036. e7. [DOI] [PubMed] [Google Scholar]
- 62.ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine (US), National Institutes of Health. Bethesda, MD.]; A Randomized, Double-Blind, Placebo-Controlled, Ascending Multiple Dose Study to Evaluate the Safety, Tolerability and Pharmacokinetics of AMG 157 in Healthy Subjects. Available from http://clinicaltrials.gov/ct2/show/NCT00972179?term=amg157&rank=1. [Google Scholar]
- 63.ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine (US), National Institutes of Health.]; A Randomized, Double-Blind, Placebo-Controlled, Ascending Single Dose Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of AMG 157 in Healthy Subjects and Subjects With Moderate to Severe Atopic Dermatitis. [Google Scholar]
- 64.ClinicalTrials.gov. Bethesda (MD): U.S. National Library of Medicine (US), National Institutes of Health.]; A Randomized, Double-Blind, Placebo-Controlled, Parallel Design, Multiple-Dose Study to Evaluate the Safety, Tolerability, Pharmacokinetics and Pharmacodynamics of AMG 157 in Subjects With Mild Atopic Asthma. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.