

Abstract
Background
A subset of patients with atopic dermatitis (AD) is prone to disseminated herpes simplex virus (HSV) infection, i.e. eczema herpeticum (ADEH+). Biomarkers that identify ADEH+ are lacking.
Objective
To search for novel ADEH+ gene signatures in peripheral blood mononuclear cells (PBMCs).
Methods
A RNA-sequencing (RNA-seq) approach was applied to evaluate global transcriptional changes using PBMCs from ADEH+ and AD without a history of EH (ADEH−). Candidate genes were confirmed by qPCR or ELISA.
RESULTS
ADEH+ PBMCs had distinct changes to the transcriptome when compared to ADEH− PBMCs following HSV-1 stimulation: 792 genes were differentially expressed at a false discovery rate (FDR) < 0.05 (ANOVA), and 15 type I and type III interferon (IFN) genes were among the top 20 most down-regulated genes in ADEH+. We further validated that IFN-α and IL-29 mRNA and protein levels were significantly decreased in HSV-1 stimulated PBMCs from ADEH+ compared to ADEH− and normal. Ingenuity pathway analysis (IPA) demonstrated that the up-stream regulators of type I and type III IFNs, IRF3 and IRF7, was significantly inhibited in ADEH+ based on the down-regulation of their target genes. Furthermore, we found that gene expression of IRF3 and IRF7 were significantly decreased in HSV-1 stimulated PBMC from ADEH+ subjects.
CONCLUSIONS
PBMCs from ADEH+ have a distinct immune response following HSV-1 exposure compared to ADEH−. Inhibition of the IRF3 and IRF7 innate immune pathways in ADEH+ may be important mechanism for increased susceptibility to disseminated viral infection.
Keywords: Atopic dermatitis, eczema herpeticum, Herpes simplex virus, IRF3, IRF7 Type I interferon, Type III interferon
INTRODUCTION
Atopic dermatitis (AD) is the most common chronic inflammatory skin disease worldwide affecting up to 30% of children and 3% of adults in some countries.1–2 A small subset (<3%) of patients with AD are susceptible to disseminated viral skin infections including eczema vaccinatum. This has led to the Centers for Disease Control recommendation that smallpox vaccination should be excluded in all patients with AD.3 Biomarkers that distinguish this subset of AD are therefore needed as the majority of AD would benefit from smallpox vaccination in the event of a bioterrorist attack.
Eczema herpeticum (EH) is a devastating complication occurring in AD patients, associated with disseminated herpes simplex virus (HSV) infection that can be life threatening without effective treatment.4 Previous studies have found that ADEH+ is associated with genetic variants in a number of genes, including filaggrin, Interferon (IFN) alpha receptor 1, IFN gamma receptor 1, IFN regulatory factor 2 and the TH2 promoting cytokine, thymic stromal lymphopoietin.5–8 In addition, ADEH+ patients often have increased Staphylococcus aureus (S. aureus) skin colonization,4 and S.aureus toxin, α-toxin, enhances viral loads in keratinocytes at sublytical concentrations.9 These previous studies suggest that ADEH+ is a complex disease determined by genetic predisposition and environmental factors. 10–12 However, the exact pathogenic mechanisms leading to disseminated HSV infections in AD patients remain elusive.
The primary goal of the current study was to determine whether subjects with ADEH+ have identifiable defects in their immune system that dampen their ability to control HSV-1 infections. Since peripheral blood mononuclear cells (PBMCs) contain all essential cells in the immune system to combat invading viruses, they are the most accessible sources for sampling to study human viral-infection diseases-related immune mechanisms. Indeed, previous studies have demonstrated that PBMCs are valuable sources for identification of abnormal immune pathways leading to serious HSV infections in humans.13–15 RNA sequencing (RNA-seq) technology is an emerging powerful global transcriptome analysis approach recently developed that applies the next generation sequencing technology.16–17 By deep sequencing the RNA of cells at a single nucleotide resolution, this approach provides unprecedented amounts of information of transcriptomes’ dynamics that prior microarray methods could not achieve. This method can not only quantitate the abundance of expressed transcripts, but also identify novel transcripts that are not yet in the public database. In addition, RNA-seq can also detect gene structure changes including different splicing forms, small insertions and deletions, as well as single nucleotide polymorphisms. In the current study, we applied this state-of-the-art technology to investigate global gene expression of PBMCs with or without HSV-1 exposure from ADEH+ and ADEH− subjects. We found that ADEH+ subjects showed distinct transcriptional responses to HSV-1 exposure when compared to the response of ADEH− subjects to the same virus.
METHODS
Subjects
Human subjects (age range from 6 years old to 65 years old) included 20 non-atopic healthy individuals, 20 ADEH−, and 20 ADEH+. Each ADEH+ subject was matched for age and gender of ADEH− and non-atopic subject to reduce confounding factors that might arise from age and gender. None of the ADEH+ subjects had acute HSV-1 infection at the time of participation which was confirmed by physical examination by the clinical investigator and the measurement of anti-HSV-1 IgM serum titers. All ADEH+ subjects were serological positive for anti-HSV-1 IgG. The detailed demographics information of human subjects is included in Supplemental Table I. The institutional review board at National Jewish Health approved this study. All subjects provided written informed consent to participate in the study. We sent total RNA from sham and HSV-1 stimulated PBMCs of 6 ADEH− and 6 ADEH+ subjects with the best RNA quality and quantity for RNA-seq assays. However, the sequence results from one ADEH+ subject had inadequate quality; so the data from this subject could not be used. The potential genes of interest were replicated by qPCR or ELISA assays in the larger group of 60 study subjects which included the 12 subjects used for RNA-seq assay.
Peripheral blood mononuclear cells isolation and HSV-1 stimulation
Human PBMCs were isolated using Ficoll-Hypaque® density gradient centrifugation of heparinized venous blood from donors. Briefly, heparinized blood (≤ 25 ml/tube) was transferred to ficoll tubes (Greiner® Leucosep tubes prepared with 15ml Ficoll-Hypaque) and centrifuged for 15 min at 2000 rpm. The white blood cells were washed three times with 1x phosphate buffered saline and the cells were re-suspended in RPMI 1640 containing 10% heat inactivated fetal bovine serum, 40 μmol/L L-glutamine, 100 U/mL penicillin, 100 U/mL streptomycin, and 20 mmol/L HEPES buffer solution (GIBCO BRL Life Technologies). One million PBMCs in 200μl culture media were stimulated with sham or HSV-1(VR-733, ATCC, Manassas, VA) at a multiplicity of infection (MOI) of 0.1 for 21 hours as our pilot experiments demonstrated that PBMCs stimulated with MOI of 0.1 of HSV-1 gave optimal cytokine induction (data not shown).
RNA isolation and sequencing
Total RNA was isolated from PBMCs using RNeasy Mini Kits (Qiagen, Valencia, CA) according to the manufacturer’s guidelines. RNA concentration and purity were determined using Nano Drop-1000 Spectrophotometer and Agilent 2100 Bioanalyzer RNA Nano Chip (Agilent Technol., Palo Alto, CA). Library preparation and HiSeq sequencing were performed at The University of Colorado Denver Genomics and Microarray Core facility. A total of 200–1000 ng of total RNA was used to prepare the Illumina HiSeq libraries according to manufacturer’s instructions for the TruSeq RNA kit. In brief, the poly-A containing mRNA molecules were purified using poly-T oligo-attached magnetic beads. Following purification, the mRNA was fragmented into small pieces using divalent cations under elevated temperature. The cleaved RNA fragments were converted into first strand cDNA using reverse transcriptase and random primers. This was followed by second strand cDNA synthesis using DNA polymerase I and RNase H. These cDNA fragments then went through an end repair process, the addition of a single “A” base, and then ligation of the adapters. The products were then purified and enriched with PCR to create the final cDNA library. The cDNA libraries were validated on the Agilent 2100 Bioanalyzer using DNA-1000 chip. After validation, the cDNA libraries were used for 2×100bp paired-end sequencing on the Illumina HiSeq2000 with a TruSeq SBS v3-HS Kit (Illumina). After sequencing, the samples were demultiplexed and the indexed adapter sequences were trimmed using the CASAVA v1.8.2 Software (Illumina).
Sequence Alignment and algorithm
The RNA-seq sequencing data were mapped and aligned using the computational pipeline of Bowtie and Tophat.18 Each read generated by each sample was mapped to the Homo sapiens genome by Bowtie. Subsequently Tophat was used to analyze these mapped locations and assign them their gene of origin, and detect splice variants therein. After mapping and aligning, the resulting. BAM files were imported into Partek Genomics Suite v6.6 (www.partek.com) and converted into gene transcript levels as reads per kilobase of exon per million mapped reads (RPKM) using a mixed-model approach (http://www.partek.com/Tutorials/microarray/User_Guides/RNASEQ.pdf). The RPKM reflects the molar concentration of a transcript in the starting sample by normalizing for gene length and for the total read number in the measurement. This allows for comparison of transcript levels both within and between experiments. The mapped and aligned files for this study, along with resulting transcript levels, have been deposited in the Gene Expression Omnibus (GEO) under the accession number [waiting for confirmation from NCBI for download, will update accession # once this goes through].
Quantitative real time RT-PCR
Total RNA was reverse transcribed into cDNA using SuperScript®III Reverse Transcriptase (Invitrogen, Grand Island, NY). Real time RT-PCR was conducted on an ABI Prism 7000 sequence detector (Applied Biosystems, Foster City, CA). Primers and probes for human IFNA4(Hs01681284_s1), IFNA5(Hs04186137_s1), IL-29(Hs00601677_g1), IRF3(Hs01547283_m1), IRF7(Hs01014809_g1)and 18S(Hs99999901_S1) were purchased from Applied Biosystems (Foster City, CA). The primer sequences for HSV-1 transcripts were prepared as previously described. 19 Quantities of all target genes in test samples were normalized to the corresponding 18S levels.
Measurement of IFN-α and IL-29
The concentration of IFN-α and IL-29 was measured by the enzyme-linked immunosorbent assay (ELISA) using The Human IFN-α multisubtype ELISA kit and the human IL-29 ELISA kit (PBL Biomedical Laboratories, Piscataway, NJ), respectively. The experiment procedures were carried out according to the manufacturer’s guidelines. The 96-well plates were analyzed on a DTX 880 Multimode detector (Beckman Coulter, Fullerton, CA).
Statistical and Bioinformatic Analysis
Partek Genomics Suite (www.partek.com) was used to calculate the normalized gene expression levels and run the statistical analysis on the RNA seq data. Partek’s transcript algorithm is similar to the one in Xing et al20 except Partek quantifies expression across the whole genome at the same time rather than each gene separately, and accounts for the fragmentation step in RNA-seq by normalizing by transcript length (http://www.partek.com/Tutorials/microarray/User_Guides/RNASEQ.pdf). A one-way ANOVA model was applied to the experimental groups and corrected for multiple testing at FDR<0.05. Genes that passed our statistical criteria were analyzed using the bioinformatics software Ingenuity Pathway Analysis (IPA) (www.ingenuity.com). IPA uses a Fisher’s exact test to identify over-represented connected biological units in a defined set of genes, which can include pathways, cellular functions or even known targets of regulatory genes. In some cases a confidence score, or z-score, can be made on the activity of the pathway or upstream regulator based on the expression pattern of the associated genes (http://pages.ingenuity.com/rs/ingenuity/images/0812%20upstream_regulator_analysis_whitepaper.pdf). The statistical analysis of real-time qPCR data was conducted using Graph Pad prism, version 5.03 (San Diego, CA). Comparisons of expression levels were performed using ANOVA techniques and independent sample t tests as appropriate. Differences were considered significant at P<0.05.
RESULTS
PBMCs from ADEH+ have distinct expression profiles from ADEH− in response to HSV
In order to more precisely investigate the global transcriptional changes of PBMCs’ early response to HSV-1 between ADEH+ versus ADEH−, we applied a RNA–seq approach to obtain gene expression profiles of PBMCs treated with and without HSV-1 for 21 hours. An average of 47 ± 11 million paired-end reads were obtained per sample, with 79% ± 4% of these sequences successfully aligned to the exon regions of the human genome (Supplemental Table II). A total of 19,463 transcripts were identified. We designated transcripts with RPKM greater than five and found in at least three samples as “present”. Using this designation, 8716 transcripts were considered as “present”. Only the “present” transcripts were subjected to subsequent analyses.
We plotted the transcriptional data in three-dimensional space using principal component analysis (PCA) as implemented by Partek Genomics Suite using a standard correlation method, observing clear separation of two main groups within the samples based on HSV-1 treatment (Figure 1B). As evidence of the difference in expression profiles, we found 5,837 and 5,520 transcripts (over half of all present genes) differentially expressed at a FDR<0.05 (ANOVA) between HSV-1 treated and sham treated PBMCs from ADEH+ and ADEH− patients, respectively (Figure 1A). The majority of transcripts that changed due to HSV-1 treatment were shared between the two groups (5,021 transcripts in common) (Figure 1A). We also observed spatial separation between the HSV-1 treated AD types in the PCA plot (Figure 1B), with 792 genes different at a FDR < 0.05 (ANOVA)(Supplemental Table III), while the sham-treated PBMCs from both types had similar expression profiles and tended to occupy the same three-dimensional spaces in the PCA plot, yielding only one gene, GSTM4, different between the two groups at a FDR < 0.05(ANOVA) (Figure 1A &Supplemental Table IV).
Figure 1. ADEH+ PBMCs expression profiles are distinct from ADEH− after HSV-1 exposure.

A) Venn diagram of genes significantly different (FDR<0.05, ANOVA) in RNA seq study between HSV-1 treatments and ADEH types. B) PCA analyses of the four experimental groups. HSV-1 stimulated ADEH+, red triangles; HSV-1 stimulated ADEH−, red squares; Sham-treated ADEH+, blue triangles; and sham-treated ADEH−, blue squares. A centroid connecting all associated members was added to the four different experimental groups in the PCA plot.
Type I and Type III interferons are significantly reduced in ADEH+ subjects
As shown in Figure 1B, the transcription profiles of HSV-1 –treated PBMCs from ADEH+ subjects were distinctly separated from ADEH− subjects. This suggested significantly different responses by these two types of AD to HSV-1 stimulation. We then examined the 792 genes different between the ADEH− and ADEH+ HSV-1 treated cells (Supplementary Table III), given the manageable number for bioinformatic interpretation, and the obvious functional implications these genes might have in the differential response to virus in ADEH+ subjects.
When we assessed the fold changes of gene expression levels between ADEH− and ADEH+, the most striking finding was that 15 different kinds of interferon (IFN) genes, including type I IFN and type III IFN, were among the top 20 most down-regulated genes in ADEH+ subjects as compared to ADEH− subjects (Table I). Since type I and type III IFNs are the most important cytokines that protect host cells from viral infection by inducing “anti-viral states”,21–22 these genes became our first group of candidate genes to validate. Using samples from 20 normal subjects, 20 ADEH− subjects, and 20 ADEH+ subjects, we validated by qPCR that IFNA4 and IFNA5 gene transcripts were significantly lower in HSV-1 stimulated ADEH+ PBMCs (Figure 2A&B). To further validate whether this decreased gene transcription leads to reduced protein expression, we used an ELISA kit that measured 13 different kinds of type I IFNs and confirmed that the protein levels of type I IFNs were indeed significantly decreased in HSV-1 stimulated ADEH+ PBMCs’ culture supernatants as compared to normal and ADEH− controls (Figure 2C). In addition, we also confirmed that type III IFN IL-29 gene transcription level by qPCR was significantly down-regulated in HSV-1 stimulated ADEH+ PBMCs as compared to ADEH− controls (Figure 3A), and its protein level in culture supernatants was also significantly lower in ADEH+ as compared to normal and ADEH− (Figure 3B).
Figure 2. The induction of IFN-α by HSV-1 is significantly decreased in ADEH+.

A) IFNA4 and B) IFNA5 mRNA levels were evaluated by real-time qPCR in sham and HSV-1 treated PBMCs from normal (n=20), ADEH− (n=20) and ADEH+ (n=20). C) IFN-α protein levels were evaluated by ELISA in the culture supernatants from HSV-1 stimulated PBMCs from normal subjects (n=20), ADEH− (n=20) and ADEH+ (n=20).
Figure 3. The induction of type III IFN IL-29 by HSV-1 is significantly decreased in ADEH+.
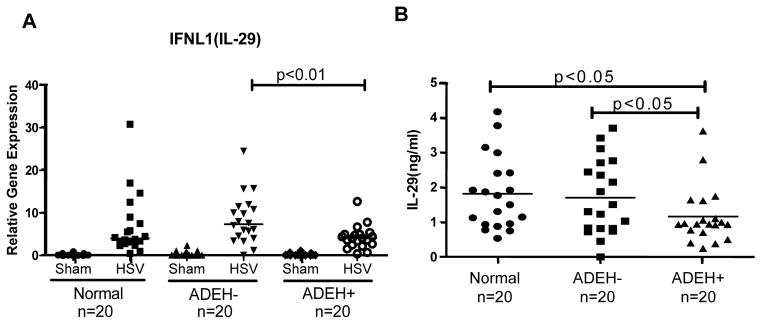
A) IL-29 mRNA level was evaluated by real-time qPCR in sham and HSV-1 treated PBMCs from normal (n=20), ADEH− (n=20) and ADEH+ (n=20); B) IL-29 protein levels in HSV-1 stimulated PBMCs from normal (n=20), ADEH− (n=20) and ADEH+ (n=20) were evaluated by ELISA.
We also examined the top up-regulated genes revealed by RNA-seq assay. We found that IFNγ is the most up-regulated gene (Supplemental Table V) in this short term culture. However, up-regulation of IFNγ was not replicated when we included the remainder of subjects in our study cohort (supplemental Figure 1).
Since part of our normal controls and ADEH− subjects are not serological anti-HSV-1 IgG positive, in order to exclude the confounding effect of anti-HSV-1 IgG antibody, we re-analyzed the data that only included subjects with anti-HSV-1 IgG positive. The results demonstrated that the gene expression of type I and type III IFNs were still significantly down-regulated in ADEH+ subjects as compared to ADEH− subjects or normal controls (Supplemental Figure 2). We further confirmed the sources of type I and type III IFNs were predominantly produced by innate immune cells or antigen-presenting cells, but not T cells and NK cells (Supplemental Figure 3). These data demonstrated that innate immune responses were significantly impaired in ADEH+ subjects.
Up-stream regulators of type I and type III IFNs were down-regulated in ADEH+
In order to identify the important regulators that are involved in type I and type III IFN expression, we employed IPA software to compute the probability of obtaining a set of target genes for a given protein, miRNA, chemical or drug. Since much is known about the effect these master regulators have on their target molecules, IPA also gives a confidence score, or z-score, on the activity of these molecules based on the direction of expression of the target genes. This analysis revealed that five interferon regulatory (IRF) genes (Figure 4A), including IRF1, IRF3, IRF7, IRF8 and IRF9, were predicted to be inhibited in ADEH+ PBMCs upon HSV-1 stimulation. Among them, IRF3, IRF7 and IRF9 had statistical significance with z-score less than −1.0. The gene with the most statistically over-represented list of target genes (Fisher’s Exact Test, P = 1.2 × 10−16) and the lowest inhibition score (z-score = −3.6) was IRF7, with 30 target genes included in our list of 792 genes different between ADEH+ and ADEH− samples after exposure to virus, with all but 6 of these target molecules suggesting decreased activity of IRF7 (Figure 4B). IRF3 stands as the second regulator in terms of affected target genes.
Figure 4. Up-stream regulators of type I and type III IFNs were inhibited in ADEH+.

A) IPA analysis demonstrated that 5 IRFs are predicted to be inhibited in ADEH+. Each IRF is connected with its target genes; dashed and solid connecting lines indicate an indirect or direct relationship with the master regulator respectively. The colors of lines connecting IRFs and their corresponding targets indicate the predicted activation status of the IRF gene based on expression patterns of its target genes. B) Statistical parameters of 5 IRFs.
Both IRF3 and IRF7 are important transcription factors involved in many viral recognition signaling pathways,23–25 such as Toll like receptors TLR3, TLR7/8, and TLR9, cytoplasmic RNA sensor RIG1 and MDA5 as well as cytoplasmic DNA sensors26–27, their activation lead to the production of type I and type III IFNs. Using qPCR, we investigated IRF3 and IRF7 gene expression in PBMCs from ADEH+ as compared to ADEH− and normal subjects. We found that the expression of IRF3 was significantly lower in ADEH+ subjects upon HSV-1 stimulation as compared to normal and ADEH− subjects. IRF7 was significantly induced by HSV-1 stimulation in all three diagnostic groups; however, the induction of IRF7 in ADEH+ was significantly reduced as compared to normals and ADEH− (Figure 5A&B).
Figure 5.
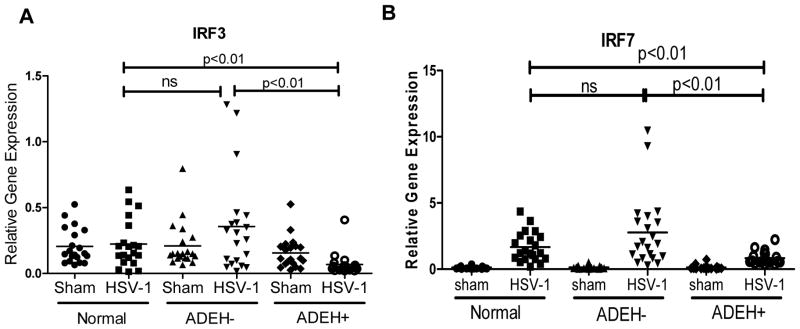
IRF3 and IRF7 are significantly decreased in HSV-1 stimulated ADEH+ PBMCs. IRF3 (A) and IRF7 (B) mRNA levels were evaluated by real-time qPCR.
DISCUSSION
Diseases resulting from HSV infections are serious public health concerns. In the United States, it is estimated that over 100 million individuals are infected by HSV-1, and at least 40–60 million individuals are infected with HSV-2.28 While most infected individuals are asymptomatic or have mild disease, others can develop serious diseases include encephalitis (HSE), keratoconjunctivitis, and the focus of our current study, ADEH+ disease. Despite acyclovir treatment, the fatality rate of HSE remains around 10%, and about 70% of HSE patients recovered with neurological sequelae.29 HSV ocular infections are the leading cause for the corneal blindness. 30 ADEH+ can be severely disfiguring and life-threatening.31 Therefore, studies leading to a better understanding of human host immune defense mechanisms against HSV are needed for the development of more effective approaches to prevent and treat herpetic diseases.
The current study applied next generation sequencing technology, RNA-seq, to explore changes in global transcriptome of PBMCs from ADEH− and ADEH+ with or without HSV-1 exposure ex vivo, with the goals of determining whether ADEH+ subjects have identifiable abnormalities in their immune systems as well as finding candidate genes that can serve as biomarkers of AD patients at risk of disseminated HSV infection. The most striking finding from the RNA-seq data is that multiple type I and type III IFN genes are significantly decreased in ADEH+ PBMCs after HSV-1 exposure as compared to ADEH− (Table I). Using qPCR and ELISA as well as additional subjects, we further confirmed at the transcription level and protein level that IFN-α and IL-29 were indeed significantly decreased in ADEH+ as compared to ADEH− and normal controls (Fig 2 & Fig 3). Since type I and type III IFNs signaling are the hallmarks for antiviral innate immune responses,32–33 these data demonstrate that innate immune responses are impaired in ADEH+ subjects. All ADEH+ subjects were serological positive of anti-HSV-1 IgG (Supplemental Table I), indicating that ADEH+ patients are capable to establish specific adaptive immune response to HSV-1 infection. These data support the findings from other herpetic diseases and classic HSV vaccine studies that specific cell-mediated and humoral immune responses are not sufficient to protect humans from HSV infection diseases.34–35
Previous studies from mice and humans have demonstrated that type I and/or type III IFNs are the first line of host defense combating HSV infection. Murine infection models demonstrated that production of type I IFN(IFNα/β) during the first several hours of virus exposure is critical in control of HSV-1 infections.36 Type I IFN can inhibit the onset of HSV immediate early gene expression,37 limit viral spread into the nervous system,38 and activate the NK cell defense system.39 Type III IFNs, IFN-λ1(IL-29), IFN-λ2(IL-28A) and IFN-λ3(IL-28B), discovered in 2003, was shown to protect human cells from HSV infection as well.40 The studies of human HSV encephylitis (HSE) further highlight the importance of type I and type III IFN in the control of severe human HSV diseases. Loss-of –function mutations in STAT1 and TYK2, the two important signal components in the down-stream signaling pathway of type I IFN receptors, predisposes to the development of HSE.41–42 Inborn gene mutations in the up-stream signaling pathway of type I and/or type III IFNs have also been found in HSE patients. The reported genes’ mutations include TLR3, UNC93B, TRIF and TRAF3.43–46 TLR3 recognizes double-strained RNA from HSV-1 intermediates, binds UNC93B and TRIF the down-stream adaptors, and subsequently activates IRF3 and IRF7 to transcribe type I and/or type III IFNs. Cells isolated from autosomal recessive (AR) UNC-93B-deficient patients, autosomal dominant (AD) TLR3 deficient patients, AD TRIF and AD TRAF3 deficient patients have been reported to be more susceptible to HSV-1 infection. The loss-of -function mutations in these genes lead to either completely abolished or reduced type I and type III IFNs production in innate immune cells following TLR3 agonist Poly(I:C) stimulation. Based on the significance of type I/type III IFNs in combating HSV-1 infections in both mouse and human, our findings of the decreased type I and type III IFNs’ production in ADEH+ subjects suggest that the major innate anti-viral defense mechanism in ADEH+ patients is compromised. This could be the mechanism underlining increased propensity to disseminated HSV-1 in ADEH+. Recently, several case reports demonstrated that application of synthesized type I IFN can effectively cure human severe HSV-1 infection manifestation.47–48 The current study has found that the two master regulators of innate immune responses IRF3 and IRF7 are suppressed in ADEH+, suggesting that abnormalities of ADEH+ exist up-stream of IRF3 and IRF7 signal pathways. These data open new research avenues for future investigation into the etiology of ADEH+.
Supplementary Material
Table 1.
The top 20 most down-regulated genes in ADEH+ as compared to ADEH− in response to HSV-1 stimulation
| Refseq Symbol | Entrez Gene Name | Gene symbol | p-value | ADEH+ vs ADEH− (Fold Changes) |
|---|---|---|---|---|
| KCNE1L | KCNE1-like | KCNE1L | 4.08E-03 | −7.813 |
| ANKRD1 | ankyrin repeat domain 1 | ANKRD1 | 4.93E-05 | −5.907 |
| IFNA4 | interferon, alpha 4 | IFNA4 | 8.46E-04 | −4.333 |
| IFNA5 | interferon, alpha 5 | IFNA5 | 1.12E-03 | −4.042 |
| IFNA6 | interferon, alpha 6 | IFNA6 | 1.92E-03 | −3.846 |
| IFNA17 | interferon, alpha 17 | IFNA17 | 7.78E-04 | −3.797 |
| IFNA16 | interferon, alpha 16 | IFNA16 | 1.15E-03 | −3.651 |
| IFNA8 | interferon, alpha 8 | IFNA8 | 1.08E-03 | −3.482 |
| IFNA21 | interferon, alpha 21 | IFNA21 | 7.49E-04 | −3.417 |
| IFNA7 | interferon, alpha 7 | IFNA7 | 1.32E-03 | −3.383 |
| IFNA2 | interferon, alpha 2 | IFNA2 | 5.35E-04 | −3.202 |
| IFNA14 | interferon, alpha 14 | IFNA14 | 6.70E-04 | −3.188 |
| IFNA1/IFNA13 | interferon, alpha 1 | IFNA13 | 1.37E-03 | −3.133 |
| IFNA10 | interferon, alpha 10 | IFNA10 | 2.74E-03 | −3.065 |
| IFNW1 | interferon, omega 1 | IFNW1 | 6.74E-04 | −3.036 |
| IFNL1 | interferon, lambda 1 | IL29 | 7.03E-04 | −2.949 |
| LRRN3 | leucine rich repeat neuronal 3 | LRRN3 | 1.43E-03 | −2.939 |
| GSTM4 | glutathione S-transferase mu 4 | GSTM4 | 1.00E-03 | −2.886 |
| IFNB1 | interferon, beta 1, fibroblast | IFNB1 | 5.87E-04 | −2.823 |
| NMT2 | N-myristoyltransferase 2 | NMT2 | 4.13E-03 | −2.033 |
Key messages.
ADEH+ PBMCs demonstrated distinct gene expression profiles after HSV-1 stimulation.
Type I and type III IFN responses are impaired in ADEH+.
IRF3 and IRF7 gene expression are reduced in ADEH+.
Acknowledgments
This work was funded by NIH/NIAID Atopic Dermatitis Research Network contract HHSN272201000020C. The authors acknowledge Patricia Taylor, Gayle Spears, and The CTRC nurses for their hard work in recruiting human subjects for this study. CTRC is supported in part by the Colorado Clinical and Translational Science Award/Colorado Clinical &Translational Sciences Institute grant UL1 RR025780 from National Center for Research Resources/NIH and UL1 TR000154 from NIH/National Center for Advancing Translational Sciences. Additionally, the authors wish to acknowledge The Edelstein Family Foundation for their generous support of our work. The authors thank Shih-Yun Lyman for her assistance in preparation of this manuscript.
ABBREVIATIONS
- AD
Atopic dermatitis
- ADEH+
Atopic dermatitis with a history of eczema herpeticum
- ADEH−
Atopic dermatitis without a history of eczema herpeticum
- ANOVA
Analysis of variance
- CTRC
Clinical and Translational Research Center
- ELISA
Enzyme-linked immunosorbent assay
- FDR
False discovery rate
- HSV-1
Herpes simplex virus 1
- IPA
Ingenuity Pathway Analysis
- IFN
Interferon
- MOI
Multiplicity of infection
- GSTM4
Glutathione S-transferase mu 4
- NIH
National Institutes of Health
- NIAID
National Institute of Allergy and Infectious Diseases
- PBMC
Peripheral blood mononuclear cells
- qPCR
Quantitative polymerase chain reaction
- RNA-seq
RNA-sequencing
- RPKM
Reads Per Kilobase per Million mapped reads
Footnotes
The authors have no financial conflict of interest related to this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bieber T. Atopic dermatitis. N Engl J Med. 2008;358:1483–94. doi: 10.1056/NEJMra074081. [DOI] [PubMed] [Google Scholar]
- 2.Leung DY. New insights into atopic dermatitis: role of skin barrier and immune dysregulation. Allergol Int. 2013;62:151–61. doi: 10.2332/allergolint.13-RAI-0564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bicknell WJ. The case for voluntary smallpox vaccination. N Engl J Med. 2002;346:1323–5. doi: 10.1056/NEJM200204253461713. [DOI] [PubMed] [Google Scholar]
- 4.Beck LA, Boguniewicz M, Hata T, Schneider LC, Hanifin J, Gallo R, et al. Phenotype of atopic dermatitis subjects with a history of eczema herpeticum. J Allergy Clin Immunol. 2009;124:260–9. 9 e1–7. doi: 10.1016/j.jaci.2009.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao PS, Leung DY, Rafaels NM, Boguniewicz M, Hand T, Gao L, et al. Genetic variants in interferon regulatory factor 2 (IRF2) are associated with atopic dermatitis and eczema herpeticum. J Invest Dermatol. 2012;132:650–7. doi: 10.1038/jid.2011.374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao PS, Rafaels NM, Hand T, Murray T, Boguniewicz M, Hata T, et al. Filaggrin mutations that confer risk of atopic dermatitis confer greater risk for eczema herpeticum. J Allergy Clin Immunol. 2009;124:507–13. 13 e1–7. doi: 10.1016/j.jaci.2009.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao PS, Rafaels NM, Mu D, Hand T, Murray T, Boguniewicz M, et al. Genetic variants in thymic stromal lymphopoietin are associated with atopic dermatitis and eczema herpeticum. J Allergy Clin Immunol. 2010;125:1403–7. e4. doi: 10.1016/j.jaci.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Leung DY, Gao PS, Grigoryev DN, Rafaels NM, Streib JE, Howell MD, et al. Human atopic dermatitis complicated by eczema herpeticum is associated with abnormalities in IFN-gamma response. J Allergy Clin Immunol. 2011;127:965–73. e1–5. doi: 10.1016/j.jaci.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bin L, Kim BE, Brauweiler A, Goleva E, Streib J, Ji Y, et al. Staphylococcus aureus alpha-toxin modulates skin host response to viral infection. J Allergy Clin Immunol. 2012;130:683–91. e2. doi: 10.1016/j.jaci.2012.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barnes KC. An update on the genetics of atopic dermatitis: scratching the surface in 2009. J Allergy Clin Immunol. 2010;125:16–29. e1–11. doi: 10.1016/j.jaci.2009.11.008. quiz 30–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kuo IH, Carpenter-Mendini A, Yoshida T, McGirt LY, Ivanov AI, Barnes KC, et al. Activation of epidermal toll-like receptor 2 enhances tight junction function: implications for atopic dermatitis and skin barrier repair. J Invest Dermatol. 2013;133:988–98. doi: 10.1038/jid.2012.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schlievert PM, Strandberg KL, Lin YC, Peterson ML, Leung DY. Secreted virulence factor comparison between methicillin-resistant and methicillin-sensitive Staphylococcus aureus, and its relevance to atopic dermatitis. J Allergy Clin Immunol. 2010;125:39–49. doi: 10.1016/j.jaci.2009.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Casrouge A, Zhang SY, Eidenschenk C, Jouanguy E, Puel A, Yang K, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314:308–12. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 14.Jabara HH, McDonald DR, Janssen E, Massaad MJ, Ramesh N, Borzutzky A, et al. DOCK8 functions as an adaptor that links TLR-MyD88 signaling to B cell activation. Nat Immunol. 2012;13:612–20. doi: 10.1038/ni.2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dupuis S, Jouanguy E, Al-Hajjar S, Fieschi C, Al-Mohsen IZ, Al-Jumaah S, et al. Impaired response to interferon-alpha/beta and lethal viral disease in human STAT1 deficiency. Nat Genet. 2003;33:388–91. doi: 10.1038/ng1097. [DOI] [PubMed] [Google Scholar]
- 16.Nagalakshmi U, Waern K, Snyder M. RNA-Seq: a method for comprehensive transcriptome analysis. Curr Protoc Mol Biol. 2010;Chapter 4(Unit 4 11):1–3. doi: 10.1002/0471142727.mb0411s89. [DOI] [PubMed] [Google Scholar]
- 17.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics 448. Nat Rev Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bin L, Howell MD, Kim BE, Streib JE, Hall CF, Leung DY. Specificity protein 1 is pivotal in the skin’s antiviral response. J Allergy Clin Immunol. 2011;127:430–8. e1–2. doi: 10.1016/j.jaci.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xing Y, Yu T, Wu YN, Roy M, Kim J, Lee C. An expectation-maximization algorithm for probabilistic reconstructions of full-length isoforms from splice graphs. Nucleic Acids Res. 2006;34:3150–60. doi: 10.1093/nar/gkl396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ank N, Paludan SR. Type III IFNs: new layers of complexity in innate antiviral immunity. Biofactors. 2009;35:82–7. doi: 10.1002/biof.19. [DOI] [PubMed] [Google Scholar]
- 22.Fensterl V, Sen GC. Interferons and viral infections. Biofactors. 2009;35:14–20. doi: 10.1002/biof.6. [DOI] [PubMed] [Google Scholar]
- 23.Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J Biol Chem. 2007;282:15325–9. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- 24.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, et al. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–7. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 25.Seth RB, Sun L, Chen ZJ. Antiviral innate immunity pathways. Cell Res. 2006;16:141–7. doi: 10.1038/sj.cr.7310019. [DOI] [PubMed] [Google Scholar]
- 26.Nie Y, Wang YY. Innate immune responses to DNA viruses. Protein Cell. 2013;4:1–7. doi: 10.1007/s13238-012-2122-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xiao TS, Fitzgerald KA. The cGAS-STING pathway for DNA sensing. Mol Cell. 2013;51:135–9. doi: 10.1016/j.molcel.2013.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Whitley RJ, Roizman B. Herpes simplex virus infections. Lancet. 2001;357:1513–8. doi: 10.1016/S0140-6736(00)04638-9. [DOI] [PubMed] [Google Scholar]
- 29.Sili U, Kaya A, Mert A. Herpes simplex virus encephalitis: Clinical manifestations, diagnosis and outcome in 106 adult patients. J Clin Virol. 2014;60:112–8. doi: 10.1016/j.jcv.2014.03.010. [DOI] [PubMed] [Google Scholar]
- 30.Liesegang TJ. Classification of herpes simplex virus keratitis and anterior uveitis. Cornea. 1999;18:127–43. doi: 10.1097/00003226-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 31.Leung DY. Why is eczema herpeticum unexpectedly rare? Antiviral Res. 2013;98:153–7. doi: 10.1016/j.antiviral.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stetson DB, Medzhitov R. Type I interferons in host defense. Immunity. 2006;25:373–81. doi: 10.1016/j.immuni.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 33.Lopusna K, Rezuchova I, Betakova T, Skovranova L, Tomaskova J, Lukacikova L, et al. Interferons lambda, new cytokines with antiviral activity. Acta Virol. 2013;57:171–9. doi: 10.4149/av_2013_02_171. [DOI] [PubMed] [Google Scholar]
- 34.Whitley RJ, Roizman B. Herpes simplex viruses: is a vaccine tenable? J Clin Invest. 2002;110:145–51. doi: 10.1172/JCI16126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Coleman JL, Shukla D. Recent advances in vaccine development for herpes simplex virus types I and II. Hum Vaccin Immunother. 2013;9:729–35. doi: 10.4161/hv.23289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vollstedt S, Arnold S, Schwerdel C, Franchini M, Alber G, Di Santo JP, et al. Interplay between alpha/beta and gamma interferons with B, T, and natural killer cells in the defense against herpes simplex virus type 1. J Virol. 2004;78:3846–50. doi: 10.1128/JVI.78.8.3846-3850.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mittnacht S, Straub P, Kirchner H, Jacobsen H. Interferon treatment inhibits onset of herpes simplex virus immediate-early transcription. Virology. 1988;164:201–10. doi: 10.1016/0042-6822(88)90637-x. [DOI] [PubMed] [Google Scholar]
- 38.Halford WP, Veress LA, Gebhardt BM, Carr DJ. Innate and acquired immunity to herpes simplex virus type 1. Virology. 1997;236:328–37. doi: 10.1006/viro.1997.8738. [DOI] [PubMed] [Google Scholar]
- 39.Leib DA, Harrison TE, Laslo KM, Machalek MA, Moorman NJ, Virgin HW. Interferons regulate the phenotype of wild-type and mutant herpes simplex viruses in vivo. J Exp Med. 1999;189:663–72. doi: 10.1084/jem.189.4.663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou L, Li J, Wang X, Ye L, Hou W, Ho J, et al. IL-29/IL-28A suppress HSV-1 infection of human NT2-N neurons. J Neurovirol. 2011;17:212–9. doi: 10.1007/s13365-011-0031-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Averbuch D, Chapgier A, Boisson-Dupuis S, Casanova JL, Engelhard D. The clinical spectrum of patients with deficiency of Signal Transducer and Activator of Transcription-1. Pediatr Infect Dis J. 2011;30:352–5. doi: 10.1097/INF.0b013e3181fdff4a. [DOI] [PubMed] [Google Scholar]
- 42.Minegishi Y, Saito M, Morio T, Watanabe K, Agematsu K, Tsuchiya S, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006;25:745–55. doi: 10.1016/j.immuni.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 43.Perez de Diego R, Sancho-Shimizu V, Lorenzo L, Puel A, Plancoulaine S, Picard C, et al. Human TRAF3 adaptor molecule deficiency leads to impaired Toll-like receptor 3 response and susceptibility to herpes simplex encephalitis. Immunity. 2010;33:400–11. doi: 10.1016/j.immuni.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sancho-Shimizu V, Perez de Diego R, Lorenzo L, Halwani R, Alangari A, Israelsson E, et al. Herpes simplex encephalitis in children with autosomal recessive and dominant TRIF deficiency. J Clin Invest. 2011;121:4889–902. doi: 10.1172/JCI59259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tabeta K, Hoebe K, Janssen EM, Du X, Georgel P, Crozat K, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol. 2006;7:156–64. doi: 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]
- 46.Zhang SY, Jouanguy E, Ugolini S, Smahi A, Elain G, Romero P, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–7. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 47.Keles S, Jabara HH, Reisli I, McDonald DR, Barlan I, Hanna-Wakim R, et al. Plasmacytoid dendritic cell depletion in DOCK8 deficiency: Rescue of severe herpetic infections with IFN-alpha 2b therapy. J Allergy Clin Immunol. 2014 doi: 10.1016/j.jaci.2014.03.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Papan C, Hagl B, Heinz V, Albert MH, Ehrt O, Sawalle-Belohradsky J, et al. Beneficial IFN-alpha treatment of tumorous herpes simplex blepharoconjunctivitis in dedicator of cytokinesis 8 deficiency. J Allergy Clin Immunol. 2014;133:1456–8. doi: 10.1016/j.jaci.2014.02.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.