

Abstract
Development of castration-resistant prostate cancer (CRPC) in a low androgen environment, arising from androgen deprivation therapy (ADT), is a major problem in patients with advanced prostate cancer (PCa). Several mechanisms have been hypothesized to explain the progression of PCa to CRPC during ADT, one of them is so called persistent intratumoral steroidogenesis. The existence of intratumoral steroidogenesis was hinted based on the residual levels of intraprostatic testosterone (T) and dihydrotestosterone (DHT) after ADT. Accumulating evidence has shown that the intraprostatic androgen levels after ADT are sufficient to induce cancer progression. Several studies now have demonstrated that PCa cells are able to produce T and DHT from different androgen precursors, such as cholesterol and the adrenal androgen, dehydroepiandrosterone (DHEA). Furthermore, up-regulation of genes encoding key steroidogenic enzymes in PCa cells seems to be an indicator for active intratumoral steroidogenesis in CRPC cells. Currently, several drugs are being developed targeting those steroidogenic enzymes, some of which are now in clinical trials or are being used as standard care for CRPC patients. In the future, novel agents that target steroidogenesis may add to the arsenal of drugs for CRPC therapy.
Keywords: Castration-resistant prostatic neoplasms, Enzyme inhibitors, Steroidogenesis, Molecular targeted therapy
INTRODUCTION
Prostate cancer (PCa) is rated to be the second most prevalent malignancy in males worldwide. According to GLOBOCAN, in 2008 there were about 903,500 new cases of PCa and 258,400 deaths of PCa worldwide [1]. The dependence of PCa on androgens, such as testosterone (T) and dihydrotestosterone (DHT), during early carcinogenesis and progression to metastatic disease has been broadly reported, and therefore androgen deprivation therapy (ADT) is given to reduce T and DHT levels [2]. During ADT, a reduction of serum T levels up to 90% is achieved after 2 to 4 weeks of treatment. However, most of the patients will develop a recurrence under low androgen levels, that is called castration-resistant prostate cancer (CRPC) [3].
Several mechanisms have been suggested to cause the progression of PCa to CRPC under ADT conditions, including hypersensitivity of the androgen receptor (AR) signaling pathway to androgens, enrichment or accumulation of androgen-insensitive stem cells, and activation of intratumoral steroidogenesis [4]. Among these, the intratumoral steroidogenesis pathway has recently gained much attention and support. Although serum T levels decrease during ADT, it was shown that the intraprostatic androgen levels in CRPC patients was equal before and after ADT [5–8]. Based on this fact, it was hypothesized, and later shown, that PCa cells are able to produce androgens itself via steroidogenesis [9], either by producing T and DHT from weak adrenal androgens (e.g., dehydroepiandrosterone [DHEA]) [10] or by de novo androgen synthesis starting from cholesterol [11]. It was also shown that several enzymes responsible for androgen synthesis are up regulated in CRPC tissue [12–14]. Due to the importance of intratumoral steroidogenesis to support the progression PCa to CRPC, new drugs are being developed that target the steroidogenic process, and hence may become new treatment options for CRPC. In this review, we will highlight the role of intratumoral steroidogenesis in CRPC and the status quo of developing novel targeted therapies for CRPC.
INTRATUMORAL STEROIDOGENESIS IN CRPC TISSUE
T and DHT are the main androgens for prostate cell differentiation and homeostasis [15]. T is synthesized in Leydig cells, while DHT is mainly produced in prostate tissue. In primary and metastatic PCa, the dependence of prostate cells on androgens persists, and androgens now directly support tumor cell proliferation, and hence tumor growth [16]. It was hypothesized that diminishing serum androgen levels should lead to inhibition of PCa cell growth, and thus ADT was recommended for advanced or metastatic PCa [17]. Unfortunately, the lower serum androgen levels obtained during ADT were not accompanied by a reduction of intraprostatic androgen levels within the tumor. In many studies, serum and intraprostatic T and DHT levels prior and after ADT have been measured (Table 1) [5–8,18,19].
Table 1.
Levels of T and DHT in serum and prostate tissue
| Conditions | Serum (ng/dL)
|
Prostate tissue (ng/g tissue)
|
||||
|---|---|---|---|---|---|---|
| T | DHT | DHEA | T | DHT | DHEA | |
| Before ADT | 410–465 | 43.5–55.68 | 90–203 | 0.07–1.34 | 4.6–6.4 | ~35 |
| After ADT | 11.5–13.4 | 3.48–3.98 | 60–211 | 0.74–1.44 | 1.0–1.9 | ~48 |
T, testosterone; DHT, dihydrotestosterone; DHEA, dehidroepiandroseterone; ADT, androgen deprivation therapy.
Serum T levels are reduced significantly from 410–465 ng/dL to 11.5–13.4 ng/dL after ADT [7]. In contrast, intraprostatic T levels after ADT (0.74–1.44 ng/g tissue) [6,8] were equal to that prior to ADT (0.07–1.3 ng/g tissue) [18,19]. A decline in both serum and prostatic DHT levels were reported after ADT [7]. Prior to ADT, serum DHT levels ranged from 43.5–55.68 ng/dL, and after ADT, the serum DHT levels were dropped to 3.48–3.98 ng/dL [7]. Prior to ADT, intraprostatic DHT levels ranged from 4.6–6.4 ng/g tissue [7], and after ADT, prostatic DHT levels were reduced approximately 75% (1.0–1.9 ng/g tissue) [7]. Still, in vitro and in vivo data indicate that these low intraprostatic DHT levels are sufficient to stimulate expression of androgen-regulated genes, and to support AR-mediated tumor-cell growth and survival [20]. In conclusion, current ADT strategies are not sufficient to reduce intraprostatic T and DHT to levels that can no longer activate AR signaling in prostate cancer cells [21].
Although serum T and DHT levels are suppressed after ADT, serum levels of adrenal androgen precursors, such as DHEA (Table 1), remain constant after ADT (60–211 ng/dL vs. 90–203 ng/dL before ADT) and was found to be the most abundant adrenal androgen in PCa tissue [5,22,23]. Measurement of intraprostatic DHEA levels are ~35 ng/g tissue in untreated PCa patients, while in ADT treated patients, intraprostatic DHEA levels are even slightly increased to ~48 ng/g tissue [24]. In the later study, androstenedione (AD) and androstenediol levels in PCa tissue after ADT were also shown to be similar to those in untreated PCa. AD and androstenediol levels in untreated PCa versus after ADT are ~0.125 ng/g tissue versus ~0.06 ng/g tissue and ~2.5 ng/g tissue versus ~3.5 ng/g tissue, respectively [24]. In summary, after ADT, the total androgen pool in the circulation is reduced by only 59% [25]. The remaining 41% of androgens, including DHEA, are still available in the prostate for the synthesis of T and DHT, which can stimulate prostate cancer after castration.
THE MECHANISM OF INTRATUMORAL STEROIDOGENESIS IN CRPC
Many studies have unraveled that intratumoral steroidogenesis could be initiated from weak adrenal androgens, such as DHEA or even by de novo androgen synthesis starting from cholesterol [10,11]. These androgen precursors are then converted to androgens, T and DHT. In the next part, we will discuss the possible mechanism of CRPC cells to synthesize androgens.
Cholesterol is the natural precursor for androgen synthesis. It was reported that cholesterol levels could influence PCa progression. Xenograft tumors (derived from the LNCaP PCa cell line) in mice on a hypercholesterolemic diet were bigger and contained higher intratumoral T levels, compared to xenograft tumors in mice on a low fat/no cholesterol diet [11]. The enzymes required for de novo steroidogenesis from cholesterol, such as cytochrome P (CYP) 11A, CYP17A, and 3β-hydroxysteroid dehydrogenase (3βHSD) 1 were detected in LNCaP tumors in these mice fed on a hypercholesterol diet. A high expression of CYP17A, the key enzyme for de novo androgen synthesis, in tumor tissue was also correlated significantly with cholesterol levels [11]. In a study using patient tissue samples, metastatic CRPC exhibited significant increases in the expression levels of the FASN, CYP17A1, 3βHSD1, and 3βHSD2 genes when compared to primary PCa [8]. Also, immunohistochemical staining for the CYP11A1, CYP17A1, and 17β-hydroxysteroid dehydrogenase (17βHSD) 3 enzymes in lymph node metastasis showed a moderately higher staining intensity compared to primary PCa samples, indicating that all these enzymes are up-regulated in metastatic CRPC tissue [26]. The results above support the existence of intratumoral de novo steroidogenesis from cholesterol.
Subsequently, de novo synthesized androgen precursors need to be converted into active androgens. In PCa, the mechanism or pathway, by which androgen precursors are converted into T and DHT is under extensive investigation. Many studies have shown that in CRPC cells the androgens, T and DHT, can be synthesized via the classical and/or the backdoor pathways [27]. Fig. 1 illustrates the mechanism of intratumoral steroidogenesis in CRPC tissue and the steroidogenic enzymes that are involved in each particular pathway.
Fig. 1.
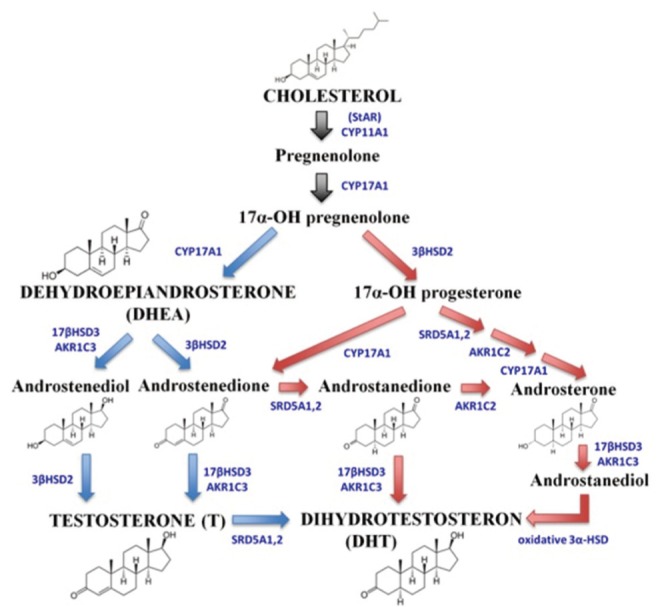
The illustration of intratumoral steroidogenesis pathway in castration-resistant prostate cancer tissue. T and DHT can be produced from cholesterol or DHEA via either the classical pathway, indicated by blue arrows or using the backdoor pathway, indicated by red arrows. The conversion of cholesterol into intermediate products is indicated by black arrows. Steroidogenic enzymes involved in each conversion are indicated in blue capital letters. T, testosterone; DHT, dihydrotestosterone; DHEA, dehidroepiandroseterone.
In the classical pathway, DHT, as a final androgen product, is produced by reduction of T, a reaction catalyzed by the 5α-reductase (SRD5A) enzyme (Fig. 1; blue arrow). The classical pathway plays an essential role in intratumoral steroidogenesis and can be initiated from DHEA. In one of our studies [10], we have shown that DuCaP cells, a CRPC cell line model, are able to use DHEA as an androgen precursor. DuCaP cells were able to proliferate in DHEA-supplemented medium, an environment that resembles ADT. Furthermore, T and DHT were detected in the medium soon after DHEA addition. These results suggested that these CRPC cells were able to convert DHEA into T and DHT, at levels sufficient to support cell growth.
Several studies reported the up-regulation of key steroidogenic enzymes involved in DHT synthesis via the classical pathway in prostate cancer tissue. In an in vivo study using LNCaP xenograft, examination of tumor homogenate of castrated mice revealed an increased expression of SRD5A1, an enzyme involved in the conversion of T into DHT [28]. In clinical CRPC samples, expression of aldo-keto reductase family 1 (AKR1) C3 and 17βHSD3 was increased, but SRD5A2 expression was decreased [13]. Similar results were found in metastatic CRPC samples that displayed up-regulation of AKR1C3 transcript levels, and down-regulation of SRD5A2 expression [8]. In line with these findings in tissue samples, higher transcript expression levels of AKR1C3, SRD5A1, and SRD5A3 were found in circulating tumor cells (CTC) derived from primary PCa. In CTC derived from CRPC patients, up-regulation of AKR1C3 and SRD5A1 transcript was detected. However, in both CTC samples, SRD5A2 transcript levels were decreased [12]. Since the SRD5A isoforms all possess similar SRD5A activity, elevated SRD5A1 and/or SRD5A3 transcript levels suggest that PCa and CRPC cells actively convert T into DHT [12].
Beside the classical pathway, the backdoor pathway provides an alternative production of DHT that bypasses the need of T as an intermediate (Fig. 1; red arrow). The backdoor pathway is primarily active during organogenesis in the fetus to produce sufficient amounts of DHT for male sex development, whereas it is less active in the adult male [29]. Active intratumoral steroidogenesis via the backdoor pathway was demonstrated by the high conversion rate of androstenedione (AD) into DHT in CRPC cell lines and tissues from CRPC patients [30]. By treating CRPC cell lines with [3H]-AD and [3H]-T, followed by HPLC analysis, the conversion pathway, either AD → androstanedione (5α-dione) → DHT or AD → T → DHT, could be measured. The outcome of these studies showed that AD is more rapidly and uniformly converted into DHT via the backdoor pathway (i.e., via 5α-dione) than via the classical pathway (i.e., via T). Similarly, fresh CRPC metastases exhibited robust conversion of AD → 5α-dione → DHT [30]. In the same study, the essential role of SRD5A1 in the backdoor pathway synthesis of DHT was shown by knockdown of the SRD5A1 gene in LNCaP and LAPC4 cells. Accumulation of AD after SRD5A1 knockdown suggested that the conversion of AD into DHT was blocked [30]. In an in vivo study, high DHT levels up to 28 folds compared to control levels, were detected in tumors of CWR22R-bearing athymic mice after androstanediol dipropionate injection at the tumor site [31]. High conversion of androstanediol into DHT was correlated with increased mRNA and protein levels of 17βHSD6, an enzyme required for DHT synthesis. These studies suggest that the backdoor pathway is remarkably active in PCa, and may be responsible for PCa progression to CRPC [31].
In summary, multiple studies have shown that intratumoral steroidogenesis in CRPC cells is active. Both cholesterol and adrenal androgens, such as DHEA, are potential sources for the intratumoral synthesis of DHT. Albeit the possibility of using multiple routes for androgen synthesis, the conversion of DHEA via the backdoor pathway seems to be the major route to produce DHT. Regardless the pathway used for DHT synthesis, the same set of genes/enzymes are needed for the conversion of androgen precursors into DHT, and all of these genes, AKR1C3, 17βHSD3, and SRD5A1/3, were found up-regulated in PCa cells.
THERAPEUTIC IMPLICATION AND TARGETED THERAPY
The up-regulation of steroidogenic enzymes as shown in previous section is essential to maintain persistent intratumoral steroidogenesis and become attractive targets for therapy. Numerous compounds have been and are being developed to inhibit steroidogenic enzyme activity, yet only few of them accepted by the U.S. Food and Drug Administration (FDA) for clinical application. One of the approved steroidogenic enzyme inhibitors for the treatment of CRPC is the CYP17A inhibitor, abiraterone, while some other inhibitors are still under intensive development, such as inhibitors of AKR1C3, 17βHSD3, and SRD5A. The current status of development of these inhibitors is discussed below.
1. CYP17A inhibitors
The CYP17A enzyme plays an important role in androgen biosynthesis. It possesses 17α-hydroxylase and C17,20-lyase activities, with the C17,20-lyase being the key enzyme that involves in DHEA biosynthesis by the adrenal glands and the testes as well as several conversions during intratumoral steroidogenesis. Because of the central role of CYP17A in androgen synthesis (Fig. 1), it was hypothesized that specific inhibition of CYP17A enzyme activity may lead to clinical antitumor responses [32].
Abiraterone is the only CYP17A inhibitor approved by the FDA (April 2011) for the treatment of metastatic CRPC that previously received docetaxel-based chemotherapy [32]. It is formulated as the prodrug of abiraterone acetate, a nonsteroid, highly selective, and irreversible CYP17A inhibitor [33]. Strikingly, its nonspecific CYP17A inhibition leads to rise in mineralocorticoids [34]. Therefore, addition of prednisone or prednisolone as mineralocorticoid receptor antagonist during abiraterone therapy is needed to prevent mineralocorticoid excess syndrome (i.e., hypertension, hypokalemia, and lower-limb edema) [35].
In preclinical studies, intraperitoneal administration of abiraterone acetate in a rodent model resulted in inhibition of CYP17A activity as shown by reduced weight of the ventral prostate [36]. Approval of abiraterone by the FDA was based on the outcome of a multinational phase III clinical trial that included 1,195 CRPC patients who previously received chemotherapy. In this study, oral administration of 1,000 mg abiraterone with 5-mg prednisone prolonged the patient overall survival by an average of 14.8 months and increased the prostate-specific antigen (PSA) response rate by 29% [37]. Nowadays, oral abiraterone acetate (Zytiga, Janssen Biotech Inc., Horsham, PA, USA) is used in combination with prednisone or prednisolone in Europe and the United States for metastatic CRPC previously treated with docetaxel-containing chemotherapy [38].
Recently, a novel selective CYP17A inhibitor with more potent inhibition of C17,20-lyase over 17α-hydroxylase activities was developed, namely TAK-700 (Orteronel, Takeda Ltd., Osaka, Japan) [39]. It is a nonsteroidal, reversible 17,20-lyase inhibitor that is five-fold more selective to inhibit C17,20-lyase activity than 17α-hydroxylase activity [40]. By selectively inhibiting C17,20-lyase activity, the need of prednisone supplementation would be reduced because of less influence on mineralocorticoid synthesis [39]. Administration of TAK-700 to intact male rats resulted in a reduction of serum T levels and prostate weight [41]. In a recent phases 1 and 2 trial in metastatic CRPC patients, dose-escalating toxicities were observed. In the phase 2 efficacy study, treatment with 400-mg TAK-700, supplemented with 5-mg prednisone, significantly reduced PSA, T, and circulating DHEA-sulfate levels. A phase 3 study in chemotherapy naïve and postdocetaxel metastatic CRPC patients is ongoing [42]. In summary, inhibition of the CYP17A enzyme is an approved treatment for metastatic CRPC, and further improvements of inhibitory compounds and treatment schedules are ongoing.
2. AKR1C3 inhibitors
The AKR1C enzyme family is involved in normal androgen metabolism and in intratumoral steroidogenesis. There are three AKR1C isoforms, namely AKR1C1, AKR1C2, and AKR1C3. Both AKR1C1 and AKR1C2 are involved in the inactivation of DHT, whereas AKR1C3 converts AD and androstanedione into active T and DHT, respectively (Fig. 1) [43]. A recent study reported that high AKR1C3 levels were detected in a subset of CRPC patients and related to tumor progression. Therefore, AKR1C3 could be used as a biomarker to monitor active intratumoral steroidogenesis in CRPC and considered as a potential therapeutic target [10].
However, development of selective AKR1C3 inhibitors is challenging due to its >86% sequence identity with the other human members of the AKR1C subfamily, AKR1C1 and AKR1C2. Inhibition of AKR1C1 and 2 activities would inhibit DHT turnover, and hence promote proliferative signaling in the prostate. The first AKR1C3 inhibitors were developed in 2005 from nonsteroidal anti-inflammatory drugs analogs, particularly indomethacin [44]. Later, additional selective AKR1C3 compound were developed based on N-phenylanthranilic acid, such as 3-((4-(trifluoromethyl)phenyl)amino)benzoic acid and 3-((4-nitronaphthalen-1-yl)amino)benzoic acid [45–47]. These compounds showed AKR1C3 selective inhibition in biochemical assays [46,47], but, so far, none of the developed compounds has been tested in CRPC cell line and tumor models. Therefore, the effect of AKR1C3 inhibitors on CRPC still remains to be elucidated and awaits the development of highly selective AKR1C3 inhibitors and extensive pre-clinical testing.
3. 17βHSD3 inhibitors
The 17βHSD3 enzyme converts AD and androstanedione into T and DHT, respectively during androgen synthesis (Fig. 1) [43]. In PCa, involvement of 17βHSD3 in intratumoral steroidogenesis was proven using LNCaP cell line transfected with pCEP4.17βHSD3. Incubation of stable LNCaP[HSD3] clone with AD efficiently stimulated cell proliferation, suggesting the active conversion of available AD by the cells [48]. Besides, high levels of the 17βHSD3 expression in prostatic tissue derived from primary PCa and lymph node metastasis were observed, indicating its role in PCa and advance disease [26]. Therefore, development of 17βHSD3 inhibitors may be beneficial in reducing disease progression and promoting survival of CRPC patients.
17βHSD3 inhibitors could be developed from different compound structures. Coumarin compounds, such as oxazolidinediones and thiazolidinediones, were reported to exhibit 17βHSD3 inhibitory activity at low nanomolar concentrations, with acceptable selectivity over other 17β-HSD isoenzymes [49,50]. The STX2171 compound is another novel selective nonsteroidal 17βHSD3 inhibitor that has an IC50 of ~200 nM in a whole-cell assay in vitro [48,51]. In a preclinical study, STX2171 was able to significantly reduce plasma T levels and xenografted tumor growth in castrated male MF-1 mice supplemented with the androgen precursor AD [51]. Thus, the development of selective inhibitors of 17βHSD3 show promising results, but the specificity and efficacy of these new compounds needs to be validated in preclinical studies before going into clinical trials.
4. SRD5A inhibitors
Other essential enzymes involved in intratumoral steroidogenesis are SRD5A1, SRD5A2, and SRD5A3, all 3 belonging to the SRD5A family. The SRD5A1 and SRD5A2 are able to catalyze the conversion of T into DHT, whilst recombinant SRD5A3 protein is also able to do so in in vitro assays [43,52,53]. Inhibition of DHT synthesis by SRD5A inhibitors offers novel therapeutic options in CRPC therapy, because most of the SRD5A enzyme isoforms are expressed at higher levels in PCa cells compared to normal prostate cells [30]. Two SRD5A inhibitors are available in the clinic, namely finasteride and dutasteride that were approved by the FDA for the treatment of benign prostatic hyperplasia (BPH) [54,55]. Finasteride inhibits SRD5A type 2, while dutasteride selectively inhibits SRD5A type 1 and 2 enzymes [56,57]. Dutasteride is a 45 times more potent in inhibiting SRD5A1 and two times more potent in inhibiting SRD5A2 compared to Finasteride [57].
Finasteride was shown to be ineffective in PCa and CRPC treatment with no difference in local recurrence or distant metastasis [58]. Currently, studies focus on the use of dutasteride, marketed under the trade name Avodart (GlaxoSmithKlinex, Brentford, UK), to reduce intratumoral androgen levels [59,60]. By inhibiting both SRD5A1 and SRD5A2, a reduction of intratumoral DHT is expected and may lead to tumor suppression [52]. In a double-blinded, randomized, parallel-group trial, dutasteride lowered intraprostatic DHT levels by 93% after 4 months of treatment. The reduced DHT level with dutasteride was accompanied by a reciprocal increase in serum and intraprostatic T levels, suggesting that dutasteride provides near-maximal suppression of intraprostatic DHT levels in PCa patients [61]. On the other hand, in CRPC patient, a phase II study of dutasteride reported that 14 out of 25 evaluable men had disease progression by 2 months, 9 had stable disease, and 2 had partial response, indicating that dutasteride had restricted biochemical response in CRPC [62].
The limited effect of dutasteride, that only inhibits SRD5A1 and 2, could be caused by high expression of SRD5A3 observed in CRPC tissue samples [53]. The existence of SRD5A3 may actively convert androgen precursors into DHT as was shown in an in vivo study using castration-resistant CWR22 xenograft bearing mice. Incubation of tumor lysates with higher concentrations of dutasteride than clinically achieved demonstrated persistent biosynthesis of DHT most likely by uninhibited SRD5A3 activity [53]. In summary, inhibition of SRD5A enzyme activity, so far, has shown limited therapeutic effects on CRPC. Novel SRD5A3 inhibitors are required to be combined with dutasteride to completely block SRD activity in CRPC cells.
FUTURE DIRECTIONS
The progression of primary PCa into CRPC during ADT remains a major hurdle to improve overall survival of PCa patients. Intratumoral steroidogenesis is an essential mechanism leading to disease progression (i.e., resistance to ADT). Intratumoral steroidogenesis fuels PCa cells with sufficient amounts of active androgens, T and DHT, in the low androgenic environment during ADT. The key steroidogenic enzymes that are up-regulated in CRPC become novel attractive targets for therapy to lower intraprostatic androgen levels.
In the last decade, several enzyme inhibitors have been developed that inhibit specific enzymes involved in intratumoral steroidogenesis. Clinical trials showed promising results, and the CYP17A inhibitor, abiraterone, now is approved for the treatment of chemotherapy resistant CRPC. However, targeting single enzyme activity may be of limited use, due to the tumor’s ability to use different androgen precursor sources and steroidogenesis pathways to maintain T and DHT levels. Therefore, combination therapies, using inhibitors of different enzymes may be the most optimal strategy to reduce intratumoral T and DHT levels, and hence to combat CRPC [63]. Moreover, combination therapy might allow the use of lower doses of enzyme inhibitors with subsequent reductions in side and/or toxic effects. At the end, preclinical and clinical studies are still needed to investigate individual and combinations of enzyme inhibitors for reducing intratumoral androgen levels in CRPC patients.
ACKNOWLEDGMENTS
Agus Rizal Hamid was supported by the EU - Marie Curie ITN project “PRO-NEST” (contract# 238278).
Footnotes
CONFLICT OF INTEREST
No potential conflict of interest relevant to this article was reported.
REFERENCES
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Cannata DH, Kirschenbaum A, Levine AC. Androgen deprivation therapy as primary treatment for prostate cancer. J Clin Endocrinol Metab. 2012;97:360–5. doi: 10.1210/jc.2011-2353. [DOI] [PubMed] [Google Scholar]
- 3.Amaral TM, Macedo D, Fernandes I, Costa L. Castration-resistant prostate cancer: mechanisms, targets, and treatment. Prostate Cancer. 2012;2012:327253. doi: 10.1155/2012/327253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azzouni F, Mohler J. Biology of castration-recurrent prostate cancer. Urol Clin North Am. 2012;39:435–52. doi: 10.1016/j.ucl.2012.07.002. [DOI] [PubMed] [Google Scholar]
- 5.Nishiyama T, Hashimoto Y, Takahashi K. The influence of androgen deprivation therapy on dihydrotestosterone levels in the prostatic tissue of patients with prostate cancer. Clin Cancer Res. 2004;10:7121–6. doi: 10.1158/1078-0432.CCR-04-0913. [DOI] [PubMed] [Google Scholar]
- 6.Titus MA, Schell MJ, Lih FB, Tomer KB, Mohler JL. Testosterone and dihydrotestosterone tissue levels in recurrent prostate cancer. Clin Cancer Res. 2005;11:4653–7. doi: 10.1158/1078-0432.CCR-05-0525. [DOI] [PubMed] [Google Scholar]
- 7.Nishiyama T, Ikarashi T, Hashimoto Y, Wako K, Takahashi K. The change in the dihydrotestosterone level in the prostate before and after androgen deprivation therapy in connection with prostate cancer aggressiveness using the Gleason score. J Urol. 2007;178(4 Pt 1):1282–8. doi: 10.1016/j.juro.2007.05.138. [DOI] [PubMed] [Google Scholar]
- 8.Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–54. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mostaghel EA, Nelson PS. Intracrine androgen metabolism in prostate cancer progression: mechanisms of castration resistance and therapeutic implications. Best Pract Res Clin Endocrinol Metab. 2008;22:243–58. doi: 10.1016/j.beem.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamid AR, Pfeiffer MJ, Verhaegh GW, Schaafsma E, Brandt A, Sweep FC, et al. Aldo-keto reductase family 1 member C3 (AKR1C3) is a biomarker and therapeutic target for castration-resistant prostate cancer. Mol Med. 2013;18:1449–55. doi: 10.2119/molmed.2012.00296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mostaghel EA, Solomon KR, Pelton K, Freeman MR, Montgomery RB. Impact of circulating cholesterol levels on growth and intratumoral androgen concentration of prostate tumors. PLoS One. 2012;7:e30062. doi: 10.1371/journal.pone.0030062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mitsiades N, Sung CC, Schultz N, Danila DC, He B, Eedunuri VK, et al. Distinct patterns of dysregulated expression of enzymes involved in androgen synthesis and metabolism in metastatic prostate cancer tumors. Cancer Res. 2012;72:6142–52. doi: 10.1158/0008-5472.CAN-12-1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pfeiffer MJ, Smit FP, Sedelaar JP, Schalken JA. Steroidogenic enzymes and stem cell markers are upregulated during androgen deprivation in prostate cancer. Mol Med. 2011;17:657–64. doi: 10.2119/molmed.2010.00143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–25. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- 15.van der Sluis TM, Vis AN, van Moorselaar RJ, Bui HN, Blankenstein MA, Meuleman EJ, et al. Intraprostatic testosterone and dihydrotestosterone. Part I: concentrations and methods of determination in men with benign prostatic hyperplasia and prostate cancer. BJU Int. 2012;109:176–82. doi: 10.1111/j.1464-410X.2011.10651.x. [DOI] [PubMed] [Google Scholar]
- 16.Imamoto T, Suzuki H, Yano M, Kawamura K, Kamiya N, Araki K, et al. The role of testosterone in the pathogenesis of prostate cancer. Int J Urol. 2008;15:472–80. doi: 10.1111/j.1442-2042.2008.02074.x. [DOI] [PubMed] [Google Scholar]
- 17.Mottet N, Bellmunt J, Bolla M, Joniau S, Mason M, Matveev V, et al. EAU guidelines on prostate cancer. Part II: Treatment of advanced, relapsing, and castration-resistant prostate cancer. Eur Urol. 2011;59:572–83. doi: 10.1016/j.eururo.2011.01.025. [DOI] [PubMed] [Google Scholar]
- 18.Gleave M, Qian J, Andreou C, Pommerville P, Chin J, Casey R, et al. The effects of the dual 5alpha-reductase inhibitor dutasteride on localized prostate cancer: results from a 4-month pre-radical prostatectomy study. Prostate. 2006;66:1674–85. doi: 10.1002/pros.20499. [DOI] [PubMed] [Google Scholar]
- 19.Heracek J, Hampl R, Hill M, Starka L, Sachova J, Kuncova J, et al. Tissue and serum levels of principal androgens in benign prostatic hyperplasia and prostate cancer. Steroids. 2007;72:375–80. doi: 10.1016/j.steroids.2007.01.004. [DOI] [PubMed] [Google Scholar]
- 20.Marks LS, Mostaghel EA, Nelson PS. Prostate tissue androgens: history and current clinical relevance. Urology. 2008;72:247–54. doi: 10.1016/j.urology.2008.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishizaki F, Nishiyama T, Kawasaki T, Miyashiro Y, Hara N, Takizawa I, et al. Androgen deprivation promotes intratumoral synthesis of dihydrotestosterone from androgen metabolites in prostate cancer. Sci Rep. 2013;3:1528. doi: 10.1038/srep01528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Narimoto K, Mizokami A, Izumi K, Mihara S, Sawada K, Sugata T, et al. Adrenal androgen levels as predictors of outcome in castration-resistant prostate cancer patients treated with combined androgen blockade using flutamide as a second-line anti-androgen. Int J Urol. 2010;17:337–45. doi: 10.1111/j.1442-2042.2010.02473.x. [DOI] [PubMed] [Google Scholar]
- 23.Labrie F. DHEA, important source of sex steroids in men and even more in women. Prog Brain Res. 2010;182:97–148. doi: 10.1016/S0079-6123(10)82004-7. [DOI] [PubMed] [Google Scholar]
- 24.Arai S, Miyashiro Y, Shibata Y, Tomaru Y, Kobayashi M, Honma S, et al. Effect of castration monotherapy on the levels of adrenal androgens in cancerous prostatic tissues. Steroids. 2011;76:301–8. doi: 10.1016/j.steroids.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 25.Labrie F. Blockade of testicular and adrenal androgens in prostate cancer treatment. Nat Rev Urol. 2011;8:73–85. doi: 10.1038/nrurol.2010.231. [DOI] [PubMed] [Google Scholar]
- 26.Bennett NC, Hooper JD, Lambie D, Lee CS, Yang T, Vesey DA, et al. Evidence for steroidogenic potential in human prostate cell lines and tissues. Am J Pathol. 2012;181:1078–87. doi: 10.1016/j.ajpath.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 27.Auchus ML, Auchus RJ. Human steroid biosynthesis for the oncologist. J Investig Med. 2012;60:495–503. doi: 10.231/JIM.0b013e3182408567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Locke JA, Guns ES, Lubik AA, Adomat HH, Hendy SC, Wood CA, et al. Androgen levels increase by intratumoral de novo steroidogenesis during progression of castration-resistant prostate cancer. Cancer Res. 2008;68:6407–15. doi: 10.1158/0008-5472.CAN-07-5997. [DOI] [PubMed] [Google Scholar]
- 29.Fukami M, Homma K, Hasegawa T, Ogata T. Backdoor pathway for dihydrotestosterone biosynthesis: implications for normal and abnormal human sex development. Dev Dyn. 2013;242:320–9. doi: 10.1002/dvdy.23892. [DOI] [PubMed] [Google Scholar]
- 30.Chang KH, Li R, Papari-Zareei M, Watumull L, Zhao YD, Auchus RJ, et al. Dihydrotestosterone synthesis bypasses testosterone to drive castration-resistant prostate cancer. Proc Natl Acad Sci U S A. 2011;108:13728–33. doi: 10.1073/pnas.1107898108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mohler JL, Titus MA, Bai S, Kennerley BJ, Lih FB, Tomer KB, et al. Activation of the androgen receptor by intratumoral bioconversion of androstanediol to dihydrotestosterone in prostate cancer. Cancer Res. 2011;71:1486–96. doi: 10.1158/0008-5472.CAN-10-1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vasaitis TS, Bruno RD, Njar VC. CYP17 inhibitors for prostate cancer therapy. J Steroid Biochem Mol Biol. 2011;125:23–31. doi: 10.1016/j.jsbmb.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rowlands MG, Barrie SE, Chan F, Houghton J, Jarman M, McCague R, et al. Esters of 3-pyridylacetic acid that combine potent inhibition of 17 alpha-hydroxylase/C17,20-lyase (cytochrome P45017 alpha) with resistance to esterase hydrolysis. J Med Chem. 1995;38:4191–7. doi: 10.1021/jm00021a008. [DOI] [PubMed] [Google Scholar]
- 34.Potter GA, Barrie SE, Jarman M, Rowlands MG. Novel steroidal inhibitors of human cytochrome P45017 alpha (17 alpha-hydroxylase-C17,20-lyase): potential agents for the treatment of prostatic cancer. J Med Chem. 1995;38:2463–71. doi: 10.1021/jm00013a022. [DOI] [PubMed] [Google Scholar]
- 35.Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, Settatree S, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563–71. doi: 10.1200/JCO.2007.15.9749. [DOI] [PubMed] [Google Scholar]
- 36.Barrie SE, Potter GA, Goddard PM, Haynes BP, Dowsett M, Jarman M. Pharmacology of novel steroidal inhibitors of cytochrome P450(17) alpha (17 alpha-hydroxylase/C17-20 lyase) J Steroid Biochem Mol Biol. 1994;50:267–73. doi: 10.1016/0960-0760(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 37.de Bono JS, Logothetis CJ, Molina A, Fizazi K, North S, Chu L, et al. Abiraterone and increased survival in metastatic prostate cancer. N Engl J Med. 2011;364:1995–2005. doi: 10.1056/NEJMoa1014618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Scott LJ, Yang LP, Lyseng-Williamson KA. Abiraterone acetate: a guide to its use in metastatic castration-resistant prostate cancer. Drugs Aging. 2012;29:243–8. doi: 10.2165/11209160-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 39.Kaku T, Hitaka T, Ojida A, Matsunaga N, Adachi M, Tanaka T, et al. Discovery of orteronel (TAK-700), a naphthylmethylimidazole derivative, as a highly selective 17,20-lyase inhibitor with potential utility in the treatment of prostate cancer. Bioorg Med Chem. 2011;19:6383–99. doi: 10.1016/j.bmc.2011.08.066. [DOI] [PubMed] [Google Scholar]
- 40.Yamaoka M, Hara T, Hitaka T, Kaku T, Takeuchi T, Takahashi J, et al. Orteronel (TAK-700), a novel non-steroidal 17,20-lyase inhibitor: effects on steroid synthesis in human and monkey adrenal cells and serum steroid levels in cynomolgus monkeys. J Steroid Biochem Mol Biol. 2012;129:115–28. doi: 10.1016/j.jsbmb.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 41.Hara T, Kouno J, Kaku T, Takeuchi T, Kusaka M, Tasaka A, et al. Effect of a novel 17,20-lyase inhibitor, orteronel (TAK-700), on androgen synthesis in male rats. J Steroid Biochem Mol Biol. 2013;134:80–91. doi: 10.1016/j.jsbmb.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 42.Dreicer R, MacLean D, Suri A, Stadler WM, Shevrin D, Hart L, et al. Phase I/II trial of orteronel (TAK-700)–an investigational 17,20-lyase inhibitor–in patients with metastatic castration-resistant prostate cancer. Clin Cancer Res. 2014;20:1335–44. doi: 10.1158/1078-0432.CCR-13-2436. [DOI] [PubMed] [Google Scholar]
- 43.Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011;32:81–151. doi: 10.1210/er.2010-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Byrns MC, Steckelbroeck S, Penning TM. An indomethacin analogue, N-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3alpha-HSD, type 5 17beta-HSD, and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochem Pharmacol. 2008;75:484–93. doi: 10.1016/j.bcp.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bauman DR, Rudnick SI, Szewczuk LM, Jin Y, Gopishetty S, Penning TM. Development of nonsteroidal anti-inflammatory drug analogs and steroid carboxylates selective for human aldo-keto reductase isoforms: potential antineoplastic agents that work independently of cyclooxygenase isozymes. Mol Pharmacol. 2005;67:60–8. doi: 10.1124/mol.104.006569. [DOI] [PubMed] [Google Scholar]
- 46.Adeniji AO, Twenter BM, Byrns MC, Jin Y, Winkler JD, Penning TM. Discovery of substituted 3-(phenylamino)benzoic acids as potent and selective inhibitors of type 5 17β-hydroxysteroid dehydrogenase (AKR1C3) Bioorg Med Chem Lett. 2011;21:1464–8. doi: 10.1016/j.bmcl.2011.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chen M, Adeniji AO, Twenter BM, Winkler JD, Christianson DW, Penning TM. Crystal structures of AKR1C3 containing an N-(aryl)amino-benzoate inhibitor and a bifunctional AKR1C3 inhibitor and androgen receptor antagonist. Therapeutic leads for castrate resistant prostate cancer. Bioorg Med Chem Lett. 2012;22:3492–7. doi: 10.1016/j.bmcl.2012.03.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Day JM, Tutill HJ, Foster PA, Bailey HV, Heaton WB, Sharland CM, et al. Development of hormone-dependent prostate cancer models for the evaluation of inhibitors of 17beta-hydroxysteroid dehydrogenase type 3. Mol Cell Endocrinol. 2009;301:251–8. doi: 10.1016/j.mce.2008.08.014. [DOI] [PubMed] [Google Scholar]
- 49.Harada K, Kubo H, Tomigahara Y, Nishioka K, Takahashi J, Momose M, et al. Coumarins as novel 17beta-hydroxysteroid dehydrogenase type 3 inhibitors for potential treatment of prostate cancer. Bioorg Med Chem Lett. 2010;20:272–5. doi: 10.1016/j.bmcl.2009.10.111. [DOI] [PubMed] [Google Scholar]
- 50.Harada K, Kubo H, Tanaka A, Nishioka K. Identification of oxazolidinediones and thiazolidinediones as potent 17β-hydroxysteroid dehydrogenase type 3 inhibitors. Bioorg Med Chem Lett. 2012;22:504–7. doi: 10.1016/j.bmcl.2011.10.095. [DOI] [PubMed] [Google Scholar]
- 51.Day JM, Foster PA, Tutill HJ, Schmidlin F, Sharland CM, Hargrave JD, et al. STX2171, a 17β-hydroxysteroid dehydrogenase type 3 inhibitor, is efficacious in vivo in a novel hormone-dependent prostate cancer model. Endocr Relat Cancer. 2013;20:53–64. doi: 10.1530/ERC-12-0231. [DOI] [PubMed] [Google Scholar]
- 52.Vis AN, Schroder FH. Key targets of hormonal treatment of prostate cancer. Part 2: the androgen receptor and 5alpha-reductase. BJU Int. 2009;104:1191–7. doi: 10.1111/j.1464-410X.2009.08743.x. [DOI] [PubMed] [Google Scholar]
- 53.Titus MA, Li Y, Kozyreva OG, Maher V, Godoy A, Smith GJ, et al. 5α-reductase type 3 enzyme in benign and malignant prostate. Prostate. 2014;74:235–49. doi: 10.1002/pros.22745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McConnell JD, Roehrborn CG, Bautista OM, Andriole GL, Jr, Dixon CM, Kusek JW, et al. The long-term effect of doxazosin, finasteride, and combination therapy on the clinical progression of benign prostatic hyperplasia. N Engl J Med. 2003;349:2387–98. doi: 10.1056/NEJMoa030656. [DOI] [PubMed] [Google Scholar]
- 55.Roehrborn CG, Barkin J, Siami P, Tubaro A, Wilson TH, Morrill BB, et al. Clinical outcomes after combined therapy with dutasteride plus tamsulosin or either monotherapy in men with benign prostatic hyperplasia (BPH) by baseline characteristics: 4-year results from the randomized, double-blind Combination of Avodart and Tamsulosin (CombAT) trial. BJU Int. 2011;107:946–54. doi: 10.1111/j.1464-410X.2011.10124.x. [DOI] [PubMed] [Google Scholar]
- 56.Rasmusson GH, Reynolds GF, Steinberg NG, Walton E, Patel GF, Liang T, et al. Zasteroids: structure-activity relationships for inhibition of 5 alpha-reductase and of androgen receptor binding. J Med Chem. 1986;29:2298–315. doi: 10.1021/jm00161a028. [DOI] [PubMed] [Google Scholar]
- 57.Frye SV. Discovery and clinical development of dutasteride, a potent dual 5alpha-reductase inhibitor. Curr Top Med Chem. 2006;6:405–21. doi: 10.2174/156802606776743101. [DOI] [PubMed] [Google Scholar]
- 58.Andriole G, Lieber M, Smith J, Soloway M, Schroeder F, Kadmon D, et al. Treatment with finasteride following radical prostatectomy for prostate cancer. Urology. 1995;45:491–7. doi: 10.1016/s0090-4295(99)80021-1. [DOI] [PubMed] [Google Scholar]
- 59.Nacusi LP, Tindall DJ. Targeting 5α-reductase for prostate cancer prevention and treatment. Nat Rev Urol. 2011;8:378–84. doi: 10.1038/nrurol.2011.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thompson IM, Goodman PJ, Tangen CM, Lucia MS, Miller GJ, Ford LG, et al. The influence of finasteride on the development of prostate cancer. N Engl J Med. 2003;349:215–24. doi: 10.1056/NEJMoa030660. [DOI] [PubMed] [Google Scholar]
- 61.Rittmaster R, Hahn RG, Ray P, Shannon JB, Wurzel R. Effect of dutasteride on intraprostatic androgen levels in men with benign prostatic hyperplasia or prostate cancer. Urology. 2008;72:808–12. doi: 10.1016/j.urology.2008.06.032. [DOI] [PubMed] [Google Scholar]
- 62.Shah SK, Trump DL, Sartor O, Tan W, Wilding GE, Mohler JL. Phase II study of Dutasteride for recurrent prostate cancer during androgen deprivation therapy. J Urol. 2009;181:621–6. doi: 10.1016/j.juro.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Agarwal N, Sonpavde G, Sternberg CN. Novel molecular targets for the therapy of castration-resistant prostate cancer. Eur Urol. 2012;61:950–60. doi: 10.1016/j.eururo.2011.12.028. [DOI] [PubMed] [Google Scholar]