

Abstract
We previously reported that transglutaminase 2 (TG2) activity is markedly elevated in lungs of hypoxia-exposed rodent models of pulmonary hypertension (PH). Since vascular remodeling of pulmonary artery smooth muscle cells (PASMCs) is important in PH, we undertook the present study to determine whether TG2 activity is altered in PASMCs with exposure to hypoxia and whether that alteration participates in their proliferative response to hypoxia. Cultured distal bovine (b) and proximal human (h) PASMCs were exposed to hypoxia (3% O2) or normoxia (21% O2). mRNA and protein expression were determined by PCR and Western blot analyses. TG2 activity and function were visualized and determined by fluorescent labeled 5-pentylamine biotin incorporation and immunoblotting of serotonylated fibronectin. Cell proliferation was assessed by [3H]thymidine incorporation assay. At 24 h, both TG2 expression and activity were stimulated by hypoxia in bPASMCs. Activation of TG2 by hypoxia was blocked by inhibition of the extracellular calcium-sensing receptor or the transient receptor potential channel V4. In contrast, TG2 expression was blocked by inhibition of the transcription factor hypoxia-inducible factor-1α, supporting the presence of separate mechanisms for stimulation of activity and expression of TG2. Pulmonary arterial hypertension patient-derived hPASMCs were found to proliferate significantly more rapidly and respond to hypoxia more strongly than control-derived hPASMCs. Similar to bovine cells, hypoxia-induced proliferation of patient-derived cells was blocked by inhibition of TG2 activity. Our results suggest an important role for TG2, mediated by intracellular calcium fluxes and HIF-1α, in hypoxia-induced PASMC proliferation and possibly in vascular remodeling in PH.
Keywords: TG2, HIF-1α, CaSR, TRPV4, pulmonary artery smooth muscle cells
although it is well known that both humans and experimental animal models respond to hypoxia by the development of pulmonary hypertension (PH), the mechanism and related cellular events by which this occurs remain unclear. Transglutaminase 2 (TG2) is a calcium-dependent enzyme that is expressed ubiquitously in multiple cell types including smooth muscle cells and has been implicated in numerous physiological and pathological processes (23). We previously found that TG2 activity is markedly elevated in lungs of hypoxia-exposed mice and that inhibition of TG2 with ERW1041E blocks development of PH in these mice. (9). This is in agreement with our previous observations of increased serotonylated fibronectin (sFn), a product of increased TG2 activity, in the sera of humans with pulmonary arterial hypertension (PAH) and in sera and lungs of experimental rodent models of PH (36). In the animal model of hypoxia-induced PH, multiple physiological events are triggered concurrently. Our observations with intact animals therefore raise the question of whether hypoxia itself directly activates TG2 and/or whether it upregulates TG2 expression.
To address this question in the present study we have utilized an in vitro model of pulmonary artery smooth muscle cells (PASMCs) in culture where environmental oxygen tension can be controlled and cellular biochemical responses evaluated. We initially selected bovine (b) distal PASMCs for this model, because they grow well in culture and we have found their proliferation to be enhanced by a hypoxic environment (3% O2) (25). As a complementary in vitro model we explored the use of human (h) proximal PASMCs available from resected lungs of PAH patients at the time of transplantation. We found that available normal hPASMCs grow very slowly under ambient conditions, making it difficult to obtain reliable measurements of cellular proliferation. In contrast, PAH patient-derived smooth muscle cells from proximal pulmonary arteries proliferate more rapidly and we selected these cells for our studies.
For the measurement of TG2 activity in these studies we used an exogenous reporter 5(biotinamido)pentylamine (5-BP) and also quantified the endogenous formation of sFn. We have evaluated the influence of a small molecule TG2 inhibitor, ERW1041E (9), and TG2 mutants on hypoxia-induced PASMC proliferation. Cellular calcium fluxes, and thereby intracellular Ca2+ concentrations, are known to regulate PASMC proliferation and contractility (18, 22, 31, 38, 44, 47). Intracellular Ca2+ is also known to induce TG2 activity (41, 46) and we previously showed that calcium chelator EGTA significantly reduced TG2 activity in bPASMCs (19). However, there is no information on specific pathways that are responsible for Ca2+ regulation in this process. In the present study we assessed the role of the extracellular calcium-sensing receptor (CaSR) and the transient receptor potential (TRP) vanilloid 4 channel (TRPV4) in the hypoxia-induced TG2 responses with the use of pharmacological inhibitors. Furthermore, we evaluated the role of hypoxia-inducible factor (HIF)-1α, a transcription factor that is upregulated by hypoxia, on TG2 expression through the use of cobalt chloride as a hypoxia mimic and a HIF-1α inhibitor, PX-478.
We report here that hypoxia stimulates both TG2 expression and activity along with proliferation of PASMCs. Hypoxia-enhanced cell proliferation occurs in both bovine and human PAH PASMCs and is blocked by inhibition of TG2 activity. Furthermore, effects on TG2 by inhibitors of the CaSR and the TRPV4 suggest that Ca2+ fluxes may initiate posttranslational TG2 activation in PASMCs and participate in the cellular response to hypoxia. HIF-1α responds to hypoxia and cobalt chloride, and we found that HIF-1α plays an integral role in induction of TG2 expression. Our observations constitute the first report of a direct influence of hypoxia on TG2 of PASMCs in culture and with addition of our previous studies (9) suggest that TG2 may be a therapeutic target for hypoxia-induced pulmonary vascular remodeling in PH.
MATERIALS AND METHODS
Reagents.
ERW1041E (2-[(3-bromo-4,5-dihydro-isoxazol-5-ylmethyl)-carbamoyl]-pyrrolidine-1-carboxylic acid quinolin-3-ylmethyl ester) and 5-BP [5-(biotinamido)pentylamine] were synthesized as described previously (9). NPS2390 (N-tricyclo[3.3.1.13,7]dec-1-yl-2-quinoxalinecarboxamide) was purchased from Tocris (Minneapolis, MN). HC-067047 (2-methyl-1-[3-(4-morpholinyl)propyl]-5-phenyl-N-[3-(trifluoromethyl)phenyl]-1H-pyrrole-3-carboxamide) and cobalt chloride were purchased from Sigma (St. Louis, MO). PX-478 [(S)-4-(2-amino-2-carboxyethyl)-N,N-bis(2-chloroethyl)aniline oxide dihydrochloride] was purchased from MedKoo Biosciences (Chapel Hill, NC).
Cell culture.
Two-day-old calf lungs were purchased from a local slaughterhouse and smooth muscle cells from distal pulmonary arteries (bPASMCs) measuring ∼0.5–1.5 mm in diameter were isolated and cultured as previously described (33). Human (h) PASMCs were isolated from lungs of idiopathic PAH patients and unused control donor subjects (Cleveland Clinic, Cleveland, OH). bPASMCs were grown in DMEM media (Life Technologies, Grand Island, NY) and hPASMCs were grown in DMEM: F-12 (1:1) media (Life Technologies) supplemented with 10% fetal bovine serum (FBS; Thermo Scientific, Waltham, MA), pen-strep antibiotic, and antimycotic (Life Technologies). Cell passages 3–5 (bPASMCs) and 5–7 (hPASMCs) were used and cellular purity was assessed by morphological appearance under phase-contrast microscopy and by immunofluorescence staining for smooth muscle α-actin.
PASMC treatment and cell proliferation assay.
Cells were grown to ∼80% confluence and serum starved for 24 h before the experiments. Cells were pretreated for 1 h with inhibitors or vehicle and then exposed to hypoxia (3% O2, 5% CO2, and balance N2) or normoxia (95% room air and 5% CO2) in tightly sealed humidified modular incubator chambers (Billups-Rothenberg, Del Mar, CA) at 37°C as previously described (25). The cells were immediately processed for downstream analysis to avoid loss of hypoxia effects as described. Cell proliferation was assessed by a [3H]thymidine incorporation assay (PerkinElmer, Waltham, MA) as previously described (25).
RNA isolation and PCR.
Total RNA was extracted from cells by use of TRIzol reagent and reverse transcription was performed by using a High-Capacity cDNA Reverse Transcription Kit according to manufacturer's instructions (Life Technologies). For mRNA detection, cDNA probes and Platinum PCR SuperMix with bovine specific assay by design primer sets (OligoPerfect Designer; Life Technologies) were used. The primer F: 5′-GGAGAAGAGCGAAGGGACTT-3′ and primer R: 5′-GACTTCGGCAAAGAGAAAG-3′ were used to amplify the cDNA of bovine TG2. The primer F: 5′-GGGTCATCATCTCTGCACCT-3′ and primer R: 5′-GGTCATAAGTCCCTCCACGA-3′ were used to amplify the cDNA of bovine GAPDH. PCR products were separated by 3% agarose gel electrophoresis in Tri-acetate-EDTA buffer and stained with ethidium bromide as previously described (24).
Cell lysis and protein extraction.
Cells were rinsed briefly with ice-cold Dulbecco's phosphate-buffered saline (DPBS; Life Technologies) and scraped into ice-cold Nonidet P-40 lysis buffer (Boston BioProducts, Ashland, MA) containing protease inhibitor cocktail (Sigma) and phosphatase inhibitor cocktail set I (Calbiochem, Billerica, MA). After incubating for 30 min at 4°C, lysates were centrifuged at 14,000 g for 15 min at 4°C. Supernatants were collected and the protein concentration was determined by use of a Bradford assay kit (Bio-Rad, Hercules, CA). Equal amounts of protein lysate were denatured at 96°C for 6 min (Laemmli sample buffer; Boston BioProducts) and resolved by SDS-PAGE (Bio-Rad).
Western blot analysis.
Cell lysates were electrophoresed and transferred to a polyvinylidene difluoride membrane (Millipore, Billerica, MA). The membrane was blocked with 5% milk in Tris-buffered saline (TBS) and incubated with primary antibody diluted in 5% bovine serum albumin (BSA; Sigma) in TBS. Serotonylated fibronectin was detected by anti-5-HT-BSA conjugate antibody (1:2,000; Sigma). Fibronectin was measured by anti-fibronectin (H-300) antibody (1:2,000; Santa Cruz Biotechnology, Dallas, TX). TG2 was detected by use of anti-TG2 (H-237) polyclonal antibody (1:1,000; Santa Cruz Biotechnology). For detecting HIF-1α, anti-HIF-1α (H-206) polyclonal antibody (1:1,000; Santa Cruz Biotechnology) was used. The respective protein bands were then detected by use of horseradish peroxidase (HRP)-tagged secondary antibodies (1:5,000; Santa Cruz Biotechnology) and the ECL System (Thermo Scientific). Densitometry analysis was performed as previously described (19) with Un-Scan-It gel analysis software (Silk Scientific, Orem, UT).
5-BP immunofluorescence assay.
For measurement of TG2 activity, 5-BP incorporation was visualized with fluorochrome-conjugated streptavidin HRP. PASMCs were grown to ∼80% confluence on glass coverslips (BD Bioscience, San Jose, CA). After 24 h of serum starvation, cells were incubated with 5-BP for 1 h prior to hypoxia/normoxia exposure. For negative control, 5-BP incubation was omitted. After a brief wash with PBS, cells were fixed with 4% formaldehyde (Tousimis, Rockville, MD) in PBS. Fixed cells were then blocked for nonspecific background with 5% milk in TBS and incubated with Streptavidin AlexaFluor 555 HRP conjugate (Life Technologies) for 1 h in 5% BSA in TBS. The coverslips were mounted on to the slides by using Vectashield mounting medium with DAPI (Vector Laboratories, Burlingame, CA) and sealed with nail polish. The stained cells were imaged under an Axio light microscope (Carl Zeiss, Thornwood, NY) using Volocity software (PerkinElmer). The TG2 activity was quantitatively assessed by measuring the intensity per cell by use of ImageJ analysis software (NIH).
TG2 plasmid transfections.
pcDNA3 vector constructs encoding the Myc-tagged forms of transglutaminase-defective TG2 mutant C277V and GTP-binding defective TG2 mutant R580L (gifts from Dr. Richard Cerione, Cornell University, NY) were transfected into cells by using Lipofectamine 2000 (Life Technologies) according to manufacturer's instructions.
Statistical analysis.
All experiments were independently replicated at least three times. Data were expressed as means ± SE. Statistical analysis was performed by Student's t-test for comparing two treatment groups and one-way analysis of variance (Holm-Sidak method) for multiple comparisons using SigmaPlot 12.5 software (Systat Software, San Jose, CA). A P value of <0.05 is considered statistically significant.
RESULTS
Hypoxia stimulates activity, mRNA, and protein expression of TG2 in bPASMCs.
To determine the effect of hypoxia on TG2 transcription, expression, and activity in bPASMCs, we measured these changes after incubating the cells for up to 24 h in either normoxia (room air) or hypoxia (3% O2). As noted in Fig. 1, TG2 mRNA, protein expression and TG2 activity are all markedly increased in response to hypoxia at 24 h compared with cells incubated under normoxic conditions. Similarly, serotonylation of fibronectin, a product of TG2 activity, is elevated at 24 h exposure to hypoxia (Fig. 2A), reflecting enhanced TG2 activity. In addition, the ratio of sFn to fibronectin (Fn) levels is significantly increased in response to hypoxia (Fig. 2B). ERW1041E is an irreversible inhibitor of TG2 that has been shown to selectively suppress TG2 activity in rodents (9). ERW1041E significantly inhibited the formation of hypoxia-induced sFn (Fig. 2, C and D).
Fig. 1.

Transglutaminase 2 (TG2) mRNA, protein expression, and activity are increased in bovine (b) pulmonary artery smooth muscle cells (PASMCs) exposed to hypoxia. A: RT-PCR analysis showing TG2 mRNA in serum-starved bPASMCs exposed to 24-h normoxia (21% O2) or hypoxia (3% O2) (representative images of PCR products shown in duplicates). B: representative Western blots of cell extracts demonstrating TG2 protein expression under normoxia and hypoxia conditions for 2–24 h. C: bar graph demonstrating the effect of hypoxia on TG2 protein expression measured by densitometry analysis (n = 3 blots/condition). TG2 protein expression (78 kDa) was detected with anti-TG2 antibody. Equal amounts of RNA and protein were loaded as indicated by the loading control GAPDH and smooth muscle α-actin (42 kDa). D: representative images showing intracellular TG2 activity detected by 5-(biotinamido)pentylamine (5-BP) assay and imaged on a microscope with an oil immersion ×40 objective lens. Serum starved bPASMCs cultured on coverslips were incubated with 5-BP (400 μM) under normoxia or hypoxia conditions for 2 and 24 h. The 5-BP incubation step was omitted in control cells. Streptavidin AlexaFluor555-conjugate and DAPI counterstain were used to stain intracellular TG2 activity (red) and nucleus (blue), respectively. E: bar graph showing changes in TG2 activity assessed by measuring the relative staining intensity per cell with ImageJ software. At least 2 slides/condition were used (n = 10 images/condition). *P < 0.05, significantly different from normoxia control. #P < 0.05, significantly different from hypoxia control. ns, Not significant.
Fig. 2.

Hypoxia increases serotonylation of fibronectin that is blocked by TG2 inhibitor ERW1041E. A: serum-starved bPASMCs were exposed to normoxia (21% O2) or hypoxia (3% O2) for 24 h. Western blots of cell extracts showing the effect of normoxia and hypoxia on serotonylated fibronectin (sFn) and fibronectin (Fn) levels. B: bar graph demonstrating changes in TG2 activity assessed by measuring sFn normalized to Fn levels by densitometry analysis of blots in A (n = 5 blots/condition). C: serum-starved bPASMCs were pretreated for 1 h with vehicle or ERW1041E and exposed to hypoxia for 24 h. Representative Western blots showing the effect of ERW1041E on TG2 expression and activity (sFn). D: bar graph demonstrating the effect of ERW1041E on sFn/Fn ratio measured by densitometry analysis (n = 3 blots/treatment group). sFN (220 kDa) was detected with anti-5-HT antiserum. Fn (220 kDa) was detected by use of anti-Fn antibody. TG2 protein expression (78 kDa) was detected with anti-TG2 antibody. Smooth muscle α-actin (42 kDa) was blotted on the stripped membrane as loading control. *P < 0.05, significantly different from normoxia control. #P < 0.05, significantly different from vehicle-treated hypoxia control.
Hypoxia stimulates bPASMC proliferation that is blocked by the TG2 inhibitor ERW1041E and expression of the catalytically inactive C277V TG2 mutant.
Hypoxia is known to induce increased proliferation of bPASMCs (25). An illustration of this stimulation at 24-h hypoxia exposure is shown in Fig. 3. Hypoxia-induced bPASMC proliferation is blocked by ERW1041E (Fig. 3A). Furthermore, to determine the role of TG2 cross-linking activity and the GTP-binding activity on proliferative responses to hypoxia, empty vector (pcDNA) and TG2 mutant transfected bPASMCs were used. The control cells received only the transfection reagent. The dominant-negative mutant that lacks cross-linking activity of TG2, C277V, significantly blocked hypoxia-induced cellular proliferation (Fig. 3B). However, there was no significant effect on hypoxia-induced cell proliferation with either transfection of pcDNA or the GTP-binding defective TG2 mutant R580L. These results suggest that TG2 cross-linking activity is important for TG2-mediated cell proliferation.
Fig. 3.

Hypoxia-induced bPASMC proliferation is blocked by TG2 inhibitor ERW1041E and TG2 cross-linking defective mutant C277V. A: serum-starved bPASMCs were pretreated with TG2 inhibitor ERW1041E for 1 h and exposed to normoxia (21% O2) or hypoxia (3% O2) for 24 h. B: bPASMCs were either mock transfected or transfected with pcDNA vector alone or with TG2 point mutants, the GTP-binding defective R580L, and the cross-linking defective C277V. Control cells were treated with transfection reagent alone. The next day bPASMCs were serum starved and then exposed to normoxia or hypoxia for 24 h. Cell proliferation was quantified by [3H]thymidine incorporation assay and expressed as cell counts (n = 12 wells/treatment group). *P < 0.05, significantly different from vehicle-treated or mock-transfected normoxia control. #P < 0.05, significantly different from vehicle-treated or mock-transfected hypoxia control.
Influence of CaSR and TRPV4 inhibitors on TG2 activity and expression.
To test the hypothesis that intracellular Ca2+ fluxes are required for hypoxia-induced TG2 activity in bPASMCs, we used pharmacological inhibitors of the CaSR (NPS2390) and the TRPV4 (HC-067047). When tested for their influence on TG2 activity after hypoxia exposure for 24 h, both NPS2390 (1–10 μM) and HC-067047 (0.5–10 μM) inhibited serotonylation of fibronectin compared with vehicle control (Fig. 4, A and C). The ratio of sFn to Fn levels was significantly reduced in both NPS2390- (Fig. 4B) and HC-067047 (Fig. 4D)-treated cells. However, there was no change in TG2 expression as demonstrated in Fig. 4, A and C. These findings suggest a difference in the mechanism of regulation of TG2 activity compared with its expression.
Fig. 4.

Influence of CaSR and TRPV4 inhibitors on hypoxia-induced TG2 activity and expression. Serum-starved bPASMCs were pretreated for 1 h with vehicle or CaSR inhibitor (NPS2390) or TRPV4 inhibitor (HC-067047) and exposed to hypoxia (3% O2) conditions for 24 h. Representative Western blots showing the effect of NPS2390 (A) and HC-067047 (C) on hypoxia-induced TG2 activity (sFn) and expression of Fn and TG2. Bar graph demonstrating the effect of NPS2390 (B) and HC-067047 (D) on sFn/Fn ratio measured by densitometry analysis (n = 4 blots/treatment group). sFN (220 kDa) was detected with anti-5-HT antiserum. Fn (220 kDa) was detected by using anti-Fn antibody. TG2 protein expression (78 kDa) was detected with anti-TG2 antibody. Smooth muscle α-actin (42 kDa) was blotted on the stripped membrane as loading control. #P < 0.05, significantly different from vehicle-treated hypoxia control.
Role of HIF-1α in expression and activation of TG2 by hypoxia.
Experiments were performed to test the hypothesis that TG2 expression is regulated by HIF-1α in bPASMCs exposed to hypoxia or hypoxia-mimicking agent cobalt chloride. PX-478 is a selective inhibitor of constitutive and inducible HIF-1α (14). As noted in Fig. 5, A and B, both hypoxia exposure and cobalt chloride treatment significantly induced HIF-1α and TG2 expression compared with normoxia controls at 24 h. Accumulation of HIF-1α by cobalt chloride was attenuated when cells were incubated with PX-478 (Fig. 5D). Furthermore, PX-478 treatment also significantly inhibited cobalt chloride- and hypoxia-induced TG2 expression at 24 h (Fig. 5, E and G). Similar to hypoxia exposure, cobalt chloride treatment induced serotonylation of fibronectin in bPASMCs (Fig. 6B). However, PX-478 did not prevent the cobalt chloride-induced serotonylation of fibronectin, meaning that it inhibited TG2 expression but not its activity. In addition, PX-478 treatment significantly inhibited total fibronectin expression (Fig. 6C). The sFn-to-Fn ratio was significantly increased with PX-478 treatment compared with vehicle-treated normoxia control and cobalt chloride-treated control (Fig. 6B). Conversely, when testing the influence of TG2 activity on HIF-1α expression we observed that TG2 inhibitor ERW1041E did not significantly alter HIF-1α expression in bPASMCs (Fig. 6D).
Fig. 5.

TG2 expression is blocked by HIF-1α inhibitor, PX-478. A: representative Western blots demonstrating the effect of cobalt chloride (CoCl2; 300 μM) and vehicle on HIF-1α and TG2 expression under normoxia (21% O2) or hypoxia (3% O2) conditions at 24 h. B: bar graph demonstrating the effect of CoCl2 and hypoxia on HIF-1α and TG2 expression measured by densitometry analysis (n = 4 blots/treatment group). C: representative Western blots demonstrating the effect of vehicle and PX-478 in presence of CoCl2 for 24 h. Bar graph demonstrating the effect of PX-478 on CoCl2-induced expression of HIF-1α (D) and TG2 (E) measured by densitometry analysis (n = 4 blots/treatment group). F: representative Western blots demonstrating the effect of vehicle or PX-478 under hypoxia for 24 h. G: bar graph demonstrating the effect of PX-478 on hypoxia-induced TG2 expression measured by densitometry analysis (n = 4 blots/treatment group). HIF-1α (120 kDa) was detected by immunoblotting with anti-HIF-1α antibody. TG2 protein expression (78 kDa) was detected with anti-TG2 antibody. Smooth muscle α-actin (42 kDa) was blotted on the stripped membrane as loading control. *P < 0.05, significantly different from vehicle-treated normoxia control. #P < 0.05, significantly different from CoCl2-treated control or vehicle-treated hypoxia control.
Fig. 6.

Effect of PX-478 and ERW1041E on TG2 and HIF-1α activity. A: representative Western blots shows the effect of vehicle and HIF-1α inhibitor PX-478 in presence of CoCl2 for 24 h. Bar graph demonstrating the effect of PX-478 on sFn (B) and Fn (C) expression measured by densitometry analysis (n = 3 blots/treatment group). D: representative Western blots shows the effect of vehicle or TG2 inhibitor ERW1041E in presence of CoCl2 for 24 h. E: bar graph demonstrating the effect of ERW1041E on HIF-1α expression measured by densitometry analysis (n = 4 blots/treatment group). HIF-1α (120 kDa) was detected by immunoblotting with anti-HIF-1α antibody. sFN (220 kDa) was detected with anti-5-HT antiserum. Fn (220 kDa) was detected with anti-Fn antibody. Smooth muscle α-actin (42 kDa) was blotted on the stripped membrane as loading control. *P < 0.05, significantly different from vehicle-treated normoxia control. #P < 0.05, significantly different from CoCl2-treated control.
Participation of TG2 in hypoxia-stimulated human PASMC proliferation.
To determine whether human PASMCs respond to hypoxia similarly to bovine ones we carried out cell proliferation studies with human cells. We found that hPASMCs from normal control subjects grew very slowly and showed low thymidine counts, making results difficult to interpret (Fig. 7A). While in the process of carrying out these studies we found that hPASMCs obtained from pulmonary arteries of lungs of transplanted patients with PAH grew much more rapidly than those from control subjects and were more consistent in measurable responses to hypoxia (e.g., Fig. 7A). Therefore, we used these PAH cells to assess for similarities to bPASMCs in their response to hypoxia. As shown in Fig. 7, A and B, there was a marked stimulation in growth response of these cells to hypoxia, and the growth responses of cells under both normoxic and hypoxic conditions were abrogated by ERW1041E at a 100–200 μM concentration (Fig. 7B). Thus these cells like the bovine ones are dependent on TG2 for their growth under both normoxic and hypoxic conditions.
Fig. 7.

Hypoxia-induced pulmonary arterial hypertension (PAH) human (h)PASMC proliferation is reduced with TG2 inhibitor ERW1041E. A: control subject and PAH patient-derived hPASMCs were exposed to normoxia (21% O2) or hypoxia (3% O2) for 24 h. B: PAH hPASMCs were pretreated with ERW1041E or vehicle for 1 h and exposed to normoxia or hypoxia for 24 h. Cell proliferation was quantified by [3H]thymidine incorporation assay and expressed as cell counts (n = 8 wells/treatment group). *P < 0.05, significantly different from vehicle-treated normoxia control. #P < 0.05, significantly different from vehicle-treated hypoxia control.
DISCUSSION
It is well known that PH occurs with exposure to altitude and is present in a variety of clinical conditions such as chronic obstructive pulmonary disease and sleep apnea that are associated with alveolar hypoxia. The biological mechanism for the relationship between hypoxia and PH has been under investigation for several decades. Both animal models and cell culture systems have been utilized in this exploration. With the discovery of HIF-1α, efforts were focused on this transcription factor as a central responder to low oxygen tensions during the pathogenesis of hypoxic pulmonary hypertension (3, 30). Several studies have targeted cellular Ca2+ fluxes as an important regulator of smooth muscle growth and reactivity, a part of the “vascular remodeling” that occurs with pulmonary hypertension (21, 27, 35, 40). A variety of vasoactive substances including serotonin, endothelin-1, and histamine have been broadly implicated in the process of vascular remodeling (31). In recent years TG2, an enzyme that catalyzes the formation of covalent bonds between lysine and glutamine residues of proteins or of proteins with primary monoamines such as serotonin, has become recognized to participate in smooth muscle function (19, 23). Furthermore, our data suggest that serotonin may be synthesized in PASMCs since we observed that serotonylation of fibronectin is induced with hypoxia exposure even without adding exogenous serotonin to the medium (Fig. 2A).
Although TG2 belongs to a cross-linking family of enzymes, prior studies suggest that the TG2 induction does not always result in increased cross-linking activity (20). Furthermore, it has been shown that TG2 has cross-linking-independent phenotypic expressions including GTP-dependent functions (15). Therefore, we have tested two mutually exclusive TG2 functions using mutant constructs, the catalytically inactive form of TG2 (C277V) and GTP-binding-deficient form of TG2 (R580L). We observed that the cross-linking mutant but not the GTP-binding mutant resulted in inhibition of bPASMC proliferation, suggesting that cross-linking activity is required for proliferative phenotype in these cells (Fig. 3). TG2 is known to be activated by Ca2+ via modulating a conformational change in its catalytically active site (46). Numerous studies have reported that calcium homeostasis through depletion of stored Ca2+ and extracellular Ca2+ sources are critical regulators in hypoxia-induced pulmonary vascular remodeling (5, 12, 18, 34). CaSR and TRPV4 participate in Ca2+-induced functional responses in PH (22, 38, 39, 47). Consistently, our studies with the inhibitors of CaSR and TRPV4 channel support a role of Ca2+ in the hypoxia-induced serotonylation of fibronectin, a product of TG2 activity. In this study, we did not measure intracellular Ca2+ concentrations. However, Wang et al. (35) showed that expression of canonical TRP isoforms TRPC1 and TRPC6 was increased in hypoxia exposed murine PASMCs resulting in elevated intracellular Ca2+ concentrations. Therefore, given the complexity of Ca2+ kinetics in PASMCs, further studies are needed to evaluate other pathways of calcium regulation in this process.
Recently, our laboratory showed that TG2 activity was markedly elevated in lungs of a hypoxia/Sugen mouse model of pulmonary hypertension (9). In addition, Baandrup et al. (1) reported that TG2 was significantly upregulated and associated with right ventricular hypertrophy in chronic hypoxic rats. Our present studies show that in PASMCs TG2 is directly responsive to hypoxia by enhancement of both its expression and activity. Consistent with the findings of increased TG2 expression, it has been previously reported in studies of tumor biology that TG2 is one of the genes targeted by the von Hippel-Lindau/hypoxia pathway (37). Furthermore, it has been reported that TG2 and fibronectin are transcriptional targets of HIF-1α that enhances the survival of hypoxic tumor cells (10, 29). In the latter studies, TG2 was shown to induce PI3K signaling activity and when treated with either a TG2 inhibitor or transglutamidation defective TG2 mutant the tumor cells became sensitive to apoptosis (4). Under normoxic conditions, Kumar et al. (17) observed that TG2 induction resulted in NF-κB-mediated HIF-1α transcription and activation via a transglutaminase-independent pathway in breast cancer cells. Furthermore, actions of TG2 have been shown to be associated with altered glucose metabolism and mitochondrial oxygen consumption (16). Our studies show that HIF-1 activation is likely upstream to hypoxia-induced TG2 activity, since ERW1041E did not block HIF-1α accumulation under hypoxic conditions (Fig. 6E). These findings suggest that the relationship of HIF-1 and TG2 is complex and further studies are needed to address the question of whether TG2 induction is involved in HIF-1 activation in normoxic PASMCs.
Schultz et al. (28) have shown that hypoxia and HIF-1α promote growth factor-induced proliferation of hPASMCs. Relevant to these cellular findings, Yu et al. (42) observed that HIF-1α partially deficient mice (HIF-1α +/−) exposed to chronic hypoxia showed significant effects on physiological responses including delayed pulmonary vascular remodeling, pulmonary hypertension, and right ventricular hypertrophy compared with wild-type littermates (HIF-1α +/+). Bonnet et al. (3) showed that HIF-1 is activated as a consequence of downregulation of HIF-1α destabilizing pathways in the 40-wk-old fawn hooded rat and PAH patients. They also observed that intracellular Ca2+ levels are elevated in PASMCs derived from 40-wk-old fawn hooded rats compared with 20-wk-old rats. Recently, Ball et al. (2) reported that mice with a tamoxifen-inducible smooth muscle-specific Cre recombinase expressed deletion of HIF-1α show attenuated pulmonary vascular remodeling and PH in a chronically hypoxic rodent model. Our data indicate that both expression and activity of HIF-1α and TG2 are elevated in hypoxic bPASMCs and suggest that TG2 is downstream to HIF-1α.
Cobalt chloride has been found to mimic actions of hypoxia by rapid stabilization, accumulation of HIF-1α, and subsequent dimerization with HIF-1β in the nucleus, resulting in transactivation of several target genes (13, 32). Responses of cellular TG2 activity to cobalt chloride paralleled those of hypoxia in the systems we studied, making this compound a useful substitute for hypoxia. Our studies with PX-478, an inhibitor of constitutive and hypoxia-induced HIF-1α (14), showed that TG2 transcription is dependent on HIF-1 activity in bPASMCs. In addition, we observed that fibronectin transcription is also dependent on HIF-1 activity in bPASMCs. Consistently, previous studies observed that TG2 and fibronectin expression are coregulated in multiple cell-types (45).
Consistent with our findings showing a smooth muscle cellular response to hypoxia, recent studies by other investigators have also reported that hPASMCs proliferate in response to hypoxia (6, 26, 43). However, these studies did not include evaluations of TG2. For our studies we specifically selected PASMCs from patients with PAH because of their relatively robust growth compared with control cells. This rapid growth of PASMCs from patients with PAH compared with normal PASMCs is noteworthy and reasonable to further investigate. Chen et al. (7) reported that thioredoxin-1 activity is upregulated in hypoxia-induced hPASMC growth and in lungs of hypoxia-exposed mice. In addition, they also observed that thioredoxin-1 inhibition abrogated HIF activation, Akt phosphorylation, and consequent PASMC proliferation. Since thioredoxin is a known activator of TG2 (11), it might be possible that thioredoxin and HIF-1α effects are mediated through TG2 in hypoxia-induced PASMC responses; however, further studies are needed to explore this possibility. Recently, Zemskov et al. (45) observed that TG2 expression is upregulated in response to platelet-derived growth factor (PDGF) treatment and is involved in PDGF receptor activation and function in human aortic smooth muscle cells. Future studies are needed to address this phenomenon in PASMCs. Finally, we do not presently know the relationship of TG2 to proteins that are directly involved in smooth muscle cell cycling and proliferation. It is recognized that TG2 may exert both pro- and antiapoptotic effects in cells depending on the cell type and cross-linking or GTP-binding properties of TG2 (8). An analysis of the direct relationship of TG2 to hypoxia-induced smooth muscle cell proliferation will require a better understanding of the mechanism by which hypoxia itself initiates cellular proliferation.
Taken together with the established roles of HIF-1α and intracellular calcium homeostasis in hypoxia-induced pulmonary hypertension, it is reasonable to conclude that TG2 plays an important role in pulmonary vascular remodeling associated with PASMC proliferation. In conclusion, the results of our studies suggest a novel mechanism as illustrated simplistically in Fig. 8 to highlight this importance of TG2.
Fig. 8.
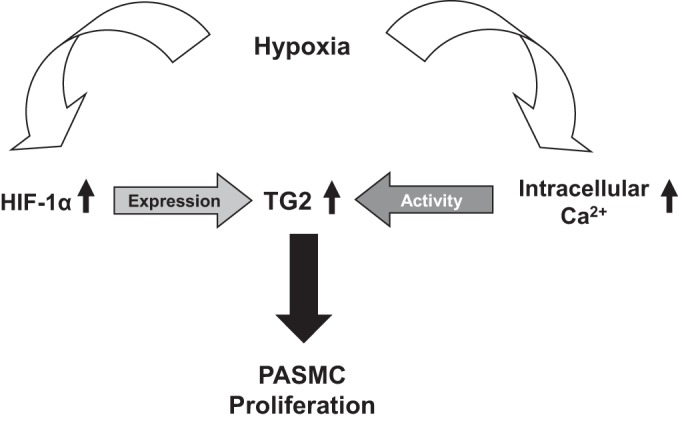
Scheme of hypoxia-induced TG2 expression and activity in PASMCs.
GRANTS
This study was supported by NIH Research Grants RO1HL107713 (B. L. Fanburg), R01DK063158 (C. Khosla), and HL60917, HL115008, and HL103453 (S. A. A. Comhair).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
K.C.P., D.T., R.R.W., C.K., S.A.C., and B.L.F. conception and design of research; K.C.P., R.R.W., A.J.H., and T.L. performed experiments; K.C.P., A.J.H., and T.L. analyzed data; K.C.P., D.T., R.R.W., A.J.H., T.L., and B.L.F. interpreted results of experiments; K.C.P. prepared figures; K.C.P. and B.L.F. drafted manuscript; K.C.P., D.T., A.J.H., C.K., and B.L.F. edited and revised manuscript; K.C.P., D.T., R.R.W., A.J.H., T.L., C.K., S.A.C., and B.L.F. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Cornelius Klöck and Brad Palanski (Stanford University, Stanford, CA) for providing us reagents (ERW1041E and 5-BP). In addition, we thank Dr. Richard Cerione (Cornell University, NY) for the generous gift of TG2 mutants (C277V; R580L).
REFERENCES
- 1.Baandrup JD, Markvardsen LH, Peters CD, Schou UK, Jensen JL, Magnusson NE, Orntoft TF, Kruhoffer M, Simonsen U. Pressure load: the main factor for altered gene expression in right ventricular hypertrophy in chronic hypoxic rats. PLoS One 6: e15859, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ball MK, Waypa GB, Mungai PT, Nielsen JM, Czech L, Dudley VJ, Beussink L, Dettman RW, Berkelhamer SK, Steinhorn RH, Shah SJ, Schumacker PT. Regulation of hypoxia-induced pulmonary hypertension by vascular smooth muscle hypoxia-inducible factor-1alpha. Am J Respir Crit Care Med 189: 314–324, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, Haromy A, Harry G, Moudgil R, McMurtry MS, Weir EK, Archer SL. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 113: 2630–2641, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Boroughs LK, Antonyak MA, Cerione RA. A novel mechanism by which tissue transglutaminase activates signaling events that promote cell survival. J Biol Chem 289: 10115–10125, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Broughton BR, Jernigan NL, Norton CE, Walker BR, Resta TC. Chronic hypoxia augments depolarization-induced Ca2+ sensitization in pulmonary vascular smooth muscle through superoxide-dependent stimulation of RhoA. Am J Physiol Lung Cell Mol Physiol 298: L232–L242, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen B, Calvert AE, Cui H, Nelin LD. Hypoxia promotes human pulmonary artery smooth muscle cell proliferation through induction of arginase. Am J Physiol Lung Cell Mol Physiol 297: L1151–L1159, 2009 [DOI] [PubMed] [Google Scholar]
- 7.Chen B, Nelin VE, Locy ML, Jin Y, Tipple TE. Thioredoxin-1 mediates hypoxia-induced pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol 305: L389–L395, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Datta S, Antonyak MA, Cerione RA. GTP-binding-defective forms of tissue transglutaminase trigger cell death. Biochemistry 46: 14819–14829, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.DiRaimondo TR, Klock C, Warburton R, Herrera Z, Penumatsa K, Toksoz D, Hill N, Khosla C, Fanburg B. Elevated transglutaminase 2 activity is associated with hypoxia-induced experimental pulmonary hypertension in mice. ACS Chem Biol 9: 266–275, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jang GY, Jeon JH, Cho SY, Shin DM, Kim CW, Jeong EM, Bae HC, Kim TW, Lee SH, Choi Y, Lee DS, Park SC, Kim IG. Transglutaminase 2 suppresses apoptosis by modulating caspase 3 and NF-kappaB activity in hypoxic tumor cells. Oncogene 29: 356–367, 2010 [DOI] [PubMed] [Google Scholar]
- 11.Jin X, Stamnaes J, Klock C, DiRaimondo TR, Sollid LM, Khosla C. Activation of extracellular transglutaminase 2 by thioredoxin. J Biol Chem 286: 37866–37873, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karaki H, Ozaki H, Hori M, Mitsui-Saito M, Amano K, Harada K, Miyamoto S, Nakazawa H, Won KJ, Sato K. Calcium movements, distribution, and functions in smooth muscle. Pharmacol Rev 49: 157–230, 1997 [PubMed] [Google Scholar]
- 13.Kim KS, Rajagopal V, Gonsalves C, Johnson C, Kalra VK. A novel role of hypoxia-inducible factor in cobalt chloride- and hypoxia-mediated expression of IL-8 chemokine in human endothelial cells. J Immunol 177: 7211–7224, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Koh MY, Spivak-Kroizman T, Venturini S, Welsh S, Williams RR, Kirkpatrick DL, Powis G. Molecular mechanisms for the activity of PX-478, an antitumor inhibitor of the hypoxia-inducible factor-1alpha. Mol Cancer Ther 7: 90–100, 2008 [DOI] [PubMed] [Google Scholar]
- 15.Kumar A, Xu J, Sung B, Kumar S, Yu D, Aggarwal BB, Mehta K. Evidence that GTP-binding domain but not catalytic domain of transglutaminase 2 is essential for epithelial-to-mesenchymal transition in mammary epithelial cells. Breast Cancer Res 14: R4, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar S, Donti TR, Agnihotri N, Mehta K. Transglutaminase 2 reprogramming of glucose metabolism in mammary epithelial cells via activation of inflammatory signaling pathways. Int J Cancer 134: 2798–2807, 2014 [DOI] [PubMed] [Google Scholar]
- 17.Kumar S, Mehta K. Tissue transglutaminase constitutively activates HIF-1alpha promoter and nuclear factor-kappaB via a non-canonical pathway. PLoS One 7: e49321, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lin MJ, Leung GP, Zhang WM, Yang XR, Yip KP, Tse CM, Sham JS. Chronic hypoxia-induced upregulation of store-operated and receptor-operated Ca2+ channels in pulmonary arterial smooth muscle cells: a novel mechanism of hypoxic pulmonary hypertension. Circ Res 95: 496–505, 2004 [DOI] [PubMed] [Google Scholar]
- 19.Liu Y, Wei L, Laskin DL, Fanburg BL. Role of protein transamidation in serotonin-induced proliferation and migration of pulmonary artery smooth muscle cells. Am J Respir Cell Mol Biol 44: 548–555, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lorand L, Graham RM. Transglutaminases: crosslinking enzymes with pleiotropic functions. Nat Rev Mol Cell Biol 4: 140–156, 2003 [DOI] [PubMed] [Google Scholar]
- 21.McDaniel SS, Platoshyn O, Wang J, Yu Y, Sweeney M, Krick S, Rubin LJ, Yuan JX. Capacitative Ca2+ entry in agonist-induced pulmonary vasoconstriction. Am J Physiol Lung Cell Mol Physiol 280: L870–L880, 2001 [DOI] [PubMed] [Google Scholar]
- 22.Miao HZ, Li B, Xu FX, Jin L, Wang GZ, Lin Y, Deng ZH, Xiao W, Li GW. [Role of MEK1/ERK1, 2 pathways in the calcium-sensing receptor mediation of hypoxia-induced proliferation of rat pulmonary artery smooth muscle cells]. Zhonghua Yi Xue Za Zhi 93: 606–609, 2013 [PubMed] [Google Scholar]
- 23.Penumatsa KC, Fanburg BL. Transglutaminase 2-mediated serotonylation in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 306: L309–L315, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Preston IR, Hill NS, Warburton RR, Fanburg BL. Role of 12-lipoxygenase in hypoxia-induced rat pulmonary artery smooth muscle cell proliferation. Am J Physiol Lung Cell Mol Physiol 290: L367–L374, 2006 [DOI] [PubMed] [Google Scholar]
- 25.Preston IR, Sagliani KD, Warburton RR, Hill NS, Fanburg BL, Jaffe IZ. Mineralocorticoid receptor antagonism attenuates experimental pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 304: L678–L688, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raghavan A, Zhou G, Zhou Q, Ibe JC, Ramchandran R, Yang Q, Racherla H, Raychaudhuri P, Raj JU. Hypoxia-induced pulmonary arterial smooth muscle cell proliferation is controlled by forkhead box M1. Am J Respir Cell Mol Biol 46: 431–436, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Salvaterra CG, Goldman WF. Acute hypoxia increases cytosolic calcium in cultured pulmonary arterial myocytes. Am J Physiol Lung Cell Mol Physiol 264: L323–L328, 1993 [DOI] [PubMed] [Google Scholar]
- 28.Schultz K, Fanburg BL, Beasley D. Hypoxia and hypoxia-inducible factor-1α promote growth factor-induced proliferation of human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol 290: H2528–H2534, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3: 721–732, 2003 [DOI] [PubMed] [Google Scholar]
- 30.Shimoda LA, Semenza GL. HIF and the lung: role of hypoxia-inducible factors in pulmonary development and disease. Am J Respir Crit Care Med 183: 152–156, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shimoda LA, Sham JS, Sylvester JT. Altered pulmonary vasoreactivity in the chronically hypoxic lung. Physiol Res 49: 549–560, 2000 [PubMed] [Google Scholar]
- 32.Srinivasan S, Dunn JF. Stabilization of hypoxia-inducible factor-1alpha in buffer containing cobalt chloride for Western blot analysis. Anal Biochem 416: 120–122, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Stiebellehner L, Frid MG, Reeves JT, Low RB, Gnanasekharan M, Stenmark KR. Bovine distal pulmonary arterial media is composed of a uniform population of well-differentiated smooth muscle cells with low proliferative capabilities. Am J Physiol Lung Cell Mol Physiol 285: L819–L828, 2003 [DOI] [PubMed] [Google Scholar]
- 34.Wan J, Yamamura A, Zimnicka AM, Voiriot G, Smith KA, Tang H, Ayon RJ, Choudhury MS, Ko EA, Wang J, Wang C, Makino A, Yuan JX. Chronic hypoxia selectively enhances L- and T-type voltage-dependent Ca2+ channel activity in pulmonary artery by upregulating Cav1.2 and Cav32. Am J Physiol Lung Cell Mol Physiol 305: L154–L164, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang J, Weigand L, Lu W, Sylvester JT, Semenza GL, Shimoda LA. Hypoxia inducible factor 1 mediates hypoxia-induced TRPC expression and elevated intracellular Ca2+ in pulmonary arterial smooth muscle cells. Circ Res 98: 1528–1537, 2006 [DOI] [PubMed] [Google Scholar]
- 36.Wei L, Warburton RR, Preston IR, Roberts KE, Comhair SA, Erzurum SC, Hill NS, Fanburg BL. Serotonylated fibronectin is elevated in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 302: L1273–L1279, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wykoff CC, Pugh CW, Maxwell PH, Harris AL, Ratcliffe PJ. Identification of novel hypoxia dependent and independent target genes of the von Hippel-Lindau (VHL) tumour suppressor by mRNA differential expression profiling. Oncogene 19: 6297–6305, 2000 [DOI] [PubMed] [Google Scholar]
- 38.Xia Y, Fu Z, Hu J, Huang C, Paudel O, Cai S, Liedtke W, Sham JS. TRPV4 channel contributes to serotonin-induced pulmonary vasoconstriction and the enhanced vascular reactivity in chronic hypoxic pulmonary hypertension. Am J Physiol Cell Physiol 305: C704–C715, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamura A, Guo Q, Yamamura H, Zimnicka AM, Pohl NM, Smith KA, Fernandez RA, Zeifman A, Makino A, Dong H, Yuan JX. Enhanced Ca2+-sensing receptor function in idiopathic pulmonary arterial hypertension. Circ Res 111: 469–481, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yamamura A, Yamamura H, Guo Q, Zimnicka AM, Wan J, Ko EA, Smith KA, Pohl NM, Song S, Zeifman A, Makino A, Yuan JX. Dihydropyridine Ca2+ channel blockers increase cytosolic [Ca2+] by activating Ca2+-sensing receptors in pulmonary arterial smooth muscle cells. Circ Res 112: 640–650, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoo JO, Yi SJ, Choi HJ, Kim WJ, Kim YM, Han JA, Ha KS. Regulation of tissue transglutaminase by prolonged increase of intracellular Ca2+, but not by initial peak of transient Ca2+ increase. Biochem Biophys Res Commun 337: 655–662, 2005 [DOI] [PubMed] [Google Scholar]
- 42.Yu AY, Shimoda LA, Iyer NV, Huso DL, Sun X, McWilliams R, Beaty T, Sham JS, Wiener CM, Sylvester JT, Semenza GL. Impaired physiological responses to chronic hypoxia in mice partially deficient for hypoxia-inducible factor 1alpha. J Clin Invest 103: 691–696, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yu XM, Wang L, Li JF, Liu J, Li J, Wang W, Wang J, Wang C. Wnt5a inhibits hypoxia-induced pulmonary arterial smooth muscle cell proliferation by downregulation of β-catenin. Am J Physiol Lung Cell Mol Physiol 304: L103–L111, 2013 [DOI] [PubMed] [Google Scholar]
- 44.Yu Y, Fantozzi I, Remillard CV, Landsberg JW, Kunichika N, Platoshyn O, Tigno DD, Thistlethwaite PA, Rubin LJ, Yuan JX. Enhanced expression of transient receptor potential channels in idiopathic pulmonary arterial hypertension. Proc Natl Acad Sci USA 101: 13861–13866, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zemskov EA, Mikhailenko I, Smith EP, Belkin AM. Tissue transglutaminase promotes PDGF/PDGFR-mediated signaling and responses in vascular smooth muscle cells. J Cell Physiol 227: 2089–2096, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang J, Lesort M, Guttmann RP, Johnson GV. Modulation of the in situ activity of tissue transglutaminase by calcium and GTP. J Biol Chem 273: 2288–2295, 1998 [DOI] [PubMed] [Google Scholar]
- 47.Zhang J, Zhou J, Cai L, Lu Y, Wang T, Zhu L, Hu Q. Extracellular calcium-sensing receptor is critical in hypoxic pulmonary vasoconstriction. Antioxid Redox Signal 17: 471–484, 2012 [DOI] [PubMed] [Google Scholar]