

Abstract
Remodeling in chronic obstructive pulmonary disease (COPD) has at least two dimensions: small airway wall thickening and destruction of alveolar walls. Recently we showed comparable alterations of the extracellular matrix (ECM) compounds collagen, hyaluoran, and elastin in alveolar and small airway walls of COPD patients. The aim of this study was to characterize and assess similarities in alveolar and small airway wall matrix remodeling in chronic COPD models. From this comparative characterization of matrix remodeling we derived and elaborated underlying mechanisms to the matrix changes reported in COPD. Lung tissue sections of chronic models for COPD, either induced by exposure to cigarette smoke, chronic intratracheal lipopolysaccharide instillation, or local tumor necrosis factor (TNF) expression [surfactant protein C (SPC)-TNFα mice], were stained for elastin, collagen, and hyaluronan. Furthermore TNF-α matrix metalloproteinase (MMP)-2, -9, and -12 mRNA expression was analyzed using qPCR and localized using immunohistochemistry. Both collagen and hyaluronan were increased in alveolar and small airway walls of all three models. Interestingly, elastin contents were differentially affected, with a decrease in both alveolar and airway walls in SPC-TNFα mice. Furthermore TNF-α and MMP-2 and -9 mRNA and protein levels were found to be increased in alveolar walls and around airway walls only in SPC-TNFα mice. We show that only SPC-TNFα mice show changes in elastin remodeling that are comparable to what has been observed in COPD patients. This reveals that the SPC-TNFα model is a suitable model to study processes underlying matrix remodeling and in particular elastin breakdown as seen in COPD. Furthermore we indicate a possible role for MMP-2 and MMP-9 in the breakdown of elastin in airways and alveoli of SPC-TNFα mice.
Keywords: chronic obstructive pulmonary disease, elastin, mice model, tumor necrosis factor-α
chronic obstructive pulmonary disease (COPD) is characterized by airflow limitation that is progressive and not fully reversible. The airflow limitation is often ascribed to remodeling, which consists of airway wall thickening by fibrosis and/or emphysema (34). Image analysis is the most commonly used technique to show remodeling. Studies into extracellular matrix (ECM) composition associated with small airway remodeling in COPD and mechanisms involved are, however, few (25, 36). Analysis of matrix composition of remaining alveolar walls in emphysema is even more limited, since research is mainly focused on the process of breakdown per se and the involvement of proteases, particularly matrix metalloproteinases (MMPs).
Small airways and alveoli are historically considered to be two separate compartments that are divergently affected in COPD. In contrast, recent literature indicates similarities in remodeling of alveolar and small airway walls. For instance, elastic fibers are not only found to be decreased in the alveolar but also in small (3) and in large (1) airway walls in COPD. Moreover, signs of destruction of the small airway walls in COPD are obtained by the appearance of fragmentation of the reticular basement membrane (38). In addition, the number of small airways is decreased in patients with COPD (31), indicating emphysema-like destruction of the small airways. On the other hand, some studies suggest that remaining alveolar septae show signs of fibrosis by accumulation of collagen (26), proteoglycans (18, 32), and the glycosaminoglycan hyaluronan (14). We recently showed that matrix of small airway and alveolar walls of COPD patients show very similar features of remodeling by the analysis of elastin, collagen, and hyaluronan (15). Despite these indications of marked similarities in matrix remodeling between small airway and alveolar walls, studies analyzing possible common underlying mechanisms are limited.
Several experimental mouse models of COPD that use several etiological factors are employed to study disease pathology in general and remodeling specifically. Inhalation of cigarette smoke offers use of the primary disease-causing agent to model several key features of the disease, including emphysema and the fibrotic remodeling of the smaller airways (2). The pathophysiological changes are believed to result from the progressive, low-grade inflammation that is elicited by chronic cigarette smoke exposure. Hyaluronan is increased in both alveolar and airway walls of mice exposed to cigarette smoke for 6 mo, and deposition of both collagen and fibronectin is furthermore found in the airway walls (5). In addition, after 6 mo of cigarette smoke exposure, increased levels of fibrilin and collagen III in the parenchyma are shown (27).
Chronic administration of the proinflammatory bacterial compound lipopolysaccharide (LPS), present as part of microbiota in healthy lungs and at enhanced levels during exacerbation of disease, has also been shown to lead to alterations that are characteristic of COPD-like airway remodeling and emphysema (41). Mice chronically exposed to LPS show enlargement of airspaces, which was associated with enhanced whole lung mRNA levels of collagen type I and III and increased levels of collagen IV (7). Transgenic mice that constitutively overexpress tumor necrosis factor (TNF)-α under the surfactant protein C (SP-C) promoter, thus in alveolar type II epithelial cells, have been generated because of the implication of TNF-α in several lung pathological conditions (33). Although initially characterized as a model of fibrosis, since these mice develop chronic neutrophilic inflammation and show increased collagen deposition, subsequent work demonstrates that the phenotype of these transgenic mice included alveolar destruction with increased expiratory static compliance, functional residual capacity, and total lung capacity (17).
To study the mechanisms that underlie remodeling in COPD, as recently described by our group, changes in the matrix compounds collagen, hyaluronan, and elastin are analyzed in both small airways and alveolar walls in the above-mentioned mouse models of COPD. From this comparative characterization of matrix remodeling we derived and elaborated underlying mechanisms to the matrix changes reported in COPD.
METHODS
Smoke model.
Male C57BL/6 mice, 6–8 wk old, were purchased from The Jackson Laboratory (Bar Harbor, ME). The local Ethics Committee for animal experimentation of the faculty of Medicine and Health Sciences (Ghent University, Belgium) approved all in vivo manipulations.
Mice (n = 7/group) were exposed whole body to cigarette smoke as described previously (10). Briefly, mice were exposed to the tobacco smoke of five cigarettes (Reference Cigarette 3R4F without filter; University of Kentucky, Lexington, KY) four times a day with 30-min smoke-free intervals, 5 days/wk for 24 wk. An optimal smoke-to-air ratio of 1:6 was obtained. The control mice were exposed to air. After the last exposure (24 h), mice were killed by an intraperitoneal injection of pentobarbital (CEVA-Sanofi, Paris, France).
LPS model.
Male C57BL/6 mice were obtained from Charles River Breeding Laboratories (Maastricht, The Netherlands). Animals were housed individually in standard laboratory cages and allowed food and water ad libitum throughout the experiments. The study protocol was approved by the Institutional Animal Care Committee of Maastricht University, The Netherlands.
Chronic inflammation was induced in 12-wk-old C57BL/6 mice by 24 times intratracheal LPS (10 μg) one time every 96 h according to the previously reported protocol (41). Mice were killed 1 wk after the last instillation. Sham mice received LPS-free sterile 0.9% NaCl instead of LPS.
SPC-TNFα.
SPC-TNFα mice exhibit chronic pulmonary inflammation resulting from overexpression of a TNF-α transgene in SP-C-producing cells (33). Animals were housed four per cage in a room maintained at a constant temperature (20–22°C) in a light-dark 12:12-h schedule according to animal protocols and National Institutes of Health guidelines. Mice were maintained on ad libitum diet (Dyets). For experiments, 12 mo male transgenic mice (n = 7) were compared with age-matched transgene negative littermates (n = 7) (wild type). The animal protocol was approved by the Animal Care and Use Committee of the National Institute on Aging.
Fixation of lungs and staining.
The left lung was fixated by infusion of 4% paraformaldehyde through a tracheal cannula under a constant pressure of 20 cmH2O above the highest point of the lung according to American Thoracic Society/European Respiratory Society guidelines for quantitative assessment of lung structure (19, 35). After excision, the lung was immersed in fresh fixative for 24 h. The lung lobes were embedded in paraffin and cut into 4-mm transverse sections that were randomly selected, and two to four sections were stained for histological analysis.
For elastin staining, slides were incubated for 20 min in Weigert's resorcin-fuchsin (Chroma, Muenster, Germany) at 60–70°C.
Collagen was stained by incubation for 90 min in 0.1% picro Sirius red, known to stain collagen I as well as II and III in saturated aqueous picric acid, pH = 1.5 (Klinipath, Duiven, the Netherlands).
For histolocalization of hyaluronan, 2 μg/ml biotin-labeled hyaluronan-binding protein was used (Calbiochem, Darmstadt, Germany) for 1 h. VECTASTAIN ABComplex/AP system (Vector, Burlingame, CA) was used for enzymatic reactivity and visualized with a Vector Blue alkaline phosphatase substrate kit (Vector). Sections were counterstained with Nuclear Fast Red (Vector).
TNF-α, MMP-2, and MMP-9 were detected using polyclonal antibodies (Abs) against mouse TNF-α, MMP-2, or MMP-9 (R&D, Minneapolis, MN). After application of biotin-conjugated swine anti-rabbit IgG Ab (DakoCytomation, Glostrup, Denmark) and alkaline phosphatase-labeled avidin-biotin complex (Vector), enzymatic reactivity was visualized using the Vector Blue Substrate Kit (Vector). Sections were counterstained with Nuclear Fast Red (Vector) and mounted. Pictures were taken at ×400 magnification using an Eclipse E800 light microscope (Nikon, Melville, NY).
Quantification of matrix.
Sections were scanned using a dot-slide light microscopy slide scanner at ×100 magnification (Olympus, Hamburg, Germany) and analyzed entirely using Leica QWin Pro version 3.5.1 software (Leica Microsystems, Cambridge, UK) by a blinded observer.
Alveolar staining was determined as percentage of stained area to total alveolar tissue area present on the slide. Suitable small airways, defined as smaller than 2 mm diameter and cut in cross sections by a ratio of maximal to minimal internal diameter <2.0, were selected. Staining was calculated as percentage of stained area to total airway wall area. Airway wall thickness was measured from the lumen to the outer margin of the adventitia and calculated as mean wall width by the software.
Quantification of airspace enlargement.
Enlargement of alveolar spaces was determined by quantifying the mean linear intercept (Lm) as described previously (11) using image analysis software (Image J 1.33). Only sections without cutting artifacts, compression, or hilar structures (airway or blood vessel with a diameter larger than 50 μm) were used in the analysis. The Lm was measured by placing a 100 × 100 μm grid over each field. The total length of each line of the grid divided by the number of alveolar intercepts gives the average distance between alveolated surfaces, or the Lm.
qPCR.
Total RNA was isolated from mouse lung tissue using the RNeasy kit (Qiagen), and an equal amount was reverse transcribed into cDNA using a Transcriptor First Strand cDNA Synthesis Kit (Roche). PCR reactions were performed on an ABI 7900HT apparatus (Applied Biosystems, Foster City, CA) using the SYBR green dye (Applied Biosystems). Relative mRNA expression of genes was calculated using the standard curve method. Primer sequences can be found in Table 1.
Table 1.
Primer sequences
| Gene | Forward Primer 5′ to 3′ | Reverse Primer 5′ to 3′ |
|---|---|---|
| TNFα | CAGCGCTGAGGTCAATCTGCC | TGCCCGGACTCCGCAA |
| MMP2 | CGATGTCGCCCCTAAAACAG | CTTGAGGGTATCTTTCAGCACAAA |
| MMP9 | TCTTCCCCAAAGACCTGAAAAC | GCCCGGGTGTAACCATAGC |
| MMP12 | TGAGGCAGAAACGTGGACTAAA | ATTGACTTTGGATTATTGGAATGCT |
| Rpl13A | CACTCTGGAGGAGAAACGGAAGG | GCAGGCATGAGGCAAACAGTC |
TNF, tumor necrosis factor; MMP, matrix metalloproteinase.
Statistics.
First data were tested for normal distribution using the Shapiro-Wilk test. Normally distributed data were tested using ANOVA (SPSS20), and data were expressed as means and SE.
Data that were not normally distributed were analyzed nonparametrically. Between-group comparisons were analyzed using the Kruskal-Wallis test, followed where appropriate by the Mann-Whitney U-test (SPSS20). Data were expressed as medians and interquartile range.
A P value <0.05 was considered statistically significant.
RESULTS
All three mouse models showed features of COPD.
Pulmonary emphysema is characterized by the destruction of alveolar walls because of damage to the lung parenchyma, leading to enlargement of alveolar spaces. Therefore, we quantified emphysematous lesions by measuring Lm. Exposure to cigarette smoke, LPS, and overexpression of TNF-α induced pulmonary emphysema, as evidenced by a significant increase in the Lm as shown in Fig. 1. Furthermore, small airway wall width tended to be increased in all three models (air 23.1 ± 4.6, cigarette smoke 26.6 ± 8.7 P = 0.13; NaCl 20.6 ± 3.7, LPS 23.5 ± 5.6 P = 0.12; wild type 17.8 ± 5.0, SPC-TNFα 20.7 ± 6.6 P = 0.15) although these increases were not statistically significant.
Fig. 1.
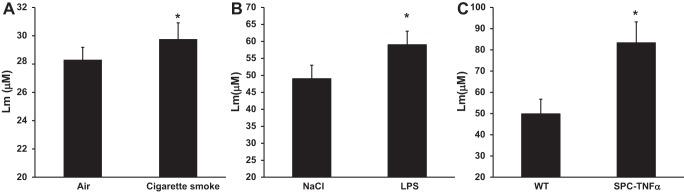
Airspace enlargement in mouse models of chronic obstructive pulmonary disease (COPD). Effect of cigarette smoke (A), lipopolysaccharide (LPS, B) and tumor necrosis factor (TNF)-α (C) overexpression on airspace enlargement as measured by the mean linear intercept (Lm). Results are expressed as means + SE; *P < 0.05 compared with respective control group as tested by ANOVA.
Airway and alveolar wall collagen content was significantly increased in the three mouse models of COPD.
To examine whether deposition of collagens (type I, II, and/or III) occurred in alveolar and small airways of the various mouse models, collagens were stained using picro Sirius red and analyzed quantitatively. Deposition of collagens was observed in the pleura, perivascular, and peribronchiolar in the basal membrane and connective tissue, and in the alveolar walls. Because comparing matrix remodeling in alveolar and airway walls was of interest in this study, collagen staining was quantified in both compartments. Interestingly, in all three models both alveolar and airway wall collagen content was increased significantly compared with control conditions (Fig. 2, A-D).
Fig. 2.

Increased collagen content in both alveolar and small airway walls of COPD mouse models. Relative area fraction of quantitatively measured collagen content in alveolar walls and small airway walls of cigarette smoke-exposed (A), LPS-treated (B), and surfactant protein C (SP-C)-TNFα (C) mice. D: representative images of each experimental group. Arrows indicate collagen deposition (red). Results are expressed as medians + interquartile range (IQR); *P < 0.05 compared with respective control group as tested using Mann-Whitney U-test.
Hyaluronan was increased significantly in alveolar and airway walls in mouse models of COPD.
Previously, we have shown increased hyaluronan in alveolar and airway walls of mice exposed to cigarette smoke. Therefore, in addition to collagen, alterations in hyaluronan amounts were investigated in alveolar and small airway walls of the various mouse models. Hyaluronan was stained using a biotinylated hyaluronan-binding protein and analyzed quantitatively. Histochemical localization of hyaluronan was seen in the alveolar walls; more prominent staining was observed in airway walls and blood vessels, which was highly comparable to the localization of collagen. Enhanced hyaluronan levels were shown in both alveolar and small airway walls in the different models compared with control mice, where the most pronounced effect was seen in alveolar walls of the SPC-TNFα model (Fig. 3, A-D).
Fig. 3.

Increased hyaluronan content in alveolar and small airway walls in mouse models of COPD. Relative area fraction of quantitatively measured hyaluronan content in alveolar walls and small airway walls of cigarette smoke-exposed (A), LPS-treated (B), and SPC-TNFα (C) mice. D: representative images of each experimental group. Arrows indicate hyaluronan deposition (blue). Results are expressed as medians + IQR; *P < 0.05 compared with respective control group as tested using Mann-Whitney U-test.
Divergent effects on elastin content in airway and alveolar walls in cigarette smoke-, LPS-, and TNF-α-induced COPD models.
In wild-type and control-treated animals, elastin was notable in alveolar walls, surrounding the epithelial layer of airway walls and around blood vessels. In contrast with collagen and hyaluronan, here we observed differences in elastin staining between the three models. Cigarette smoke exposure did not affect elastin content in either the alveolar or airway walls. In LPS-treated mice, the elastin content of alveolar was unaffected, whereas it was significantly increased in airway walls. In contrast, in SPC-TNFα mice, elastin content of both airway walls and remaining alveolar walls was significantly decreased compared with wild-type mice (Fig. 4).
Fig. 4.

Divergent effects on elastin content in airway and alveolar walls in COPD mouse models. Relative area fraction of quantitatively measured elastin content in alveolar and small airway walls of cigarette smoke-exposed (A), LPS-treated (B), and SPC-TNFα (C) mice. D: representative images of each experimental group. Arrows indicate elastin content (dark purple). Results are expressed as medians + IQR; *P < 0.05 compared with respective control group as tested using Mann-Whitney U-test.
TNF-α expression was not enhanced in the chronic cigarette smoke and chronic LPS models.
TNF-α, a proinflammatory cytokine, is known to suppress tropoelastin mRNA in lung fibroblasts but also promotes breakdown through enhanced release of elastolytic enzymes. To examine if the presence of TNF-α is related to the uniquely decreased elastin content of SPC-TNFα mice, next the expression of TNF-α in all three models was assessed. mRNA levels of TNF-α showed a tendency to increase; however, this increase was not significant in cigarette smoke- and LPS-exposed mice compared with their respective controls. In contrast, a 32-fold significant increase was found in the SPC-TNFα mice in agreement with the model (Table 2). Furthermore, TNF-α was localized in lung tissue using immunohistochemistry. As shown in Fig. 5, in the SPC-TNFα mice, TNF-α staining was found in alveolar walls and around airway walls. In cigarette smoke- and LPS-exposed mice, on the other hand, no significant TNF-α presence could be detected (data not shown).
Table 2.
Relative TNF-α mRNA expression
| Model | Relative mRNA Expression vs. Control | P Value |
|---|---|---|
| Cigarette smoke | 2.1 ± 0.3 | 0.160 |
| LPS | 3.2 ± 1.5 | 0.091 |
| SPC-TNFα | 32.0 ± 7.4 | 0.004 |
Values are means ± SE.
LPS, lipopolysaccharide; SPC, surfactant protein C.
Fig. 5.
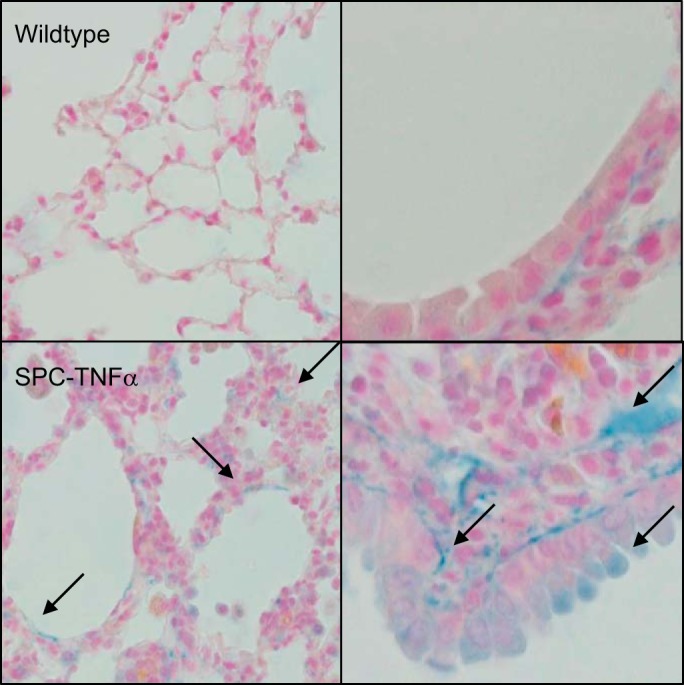
Increased TNF-α staining in alveolar and airway walls of SPC-TNFα mice. Photomicrographs at ×400 magnification of TNF-α staining (in blue) and counterstaining in red in lung tissue of one representative mouse of each condition. Arrows indicate TNF-α staining (blue).
Expression of MMP-2 and MMP-9 was uniquely upregulated in SPC-TNFα mice.
Because MMP-2, -9, and -12 are known to be involved in the breakdown of elastin and to be induced by TNF-α, expression of these proteases was investigated in the mouse models. In mice exposed to cigarette smoke only, MMP-12 mRNA was found to be increased (Fig. 6A), whereas, in the LPS model, none of the MMPs investigated was significantly changed (Fig. 6B). Interestingly, in SPC-TNFα mice where reduction of elastin in small airway and remaining alveolar walls was found, both MMP-2 and MMP-9 mRNA levels were increased significantly (Fig. 6C). In addition, to study protein levels of MMP-2 and MMP-9, immunohistochemistry was performed. MMP-2 and MMP-9 staining was present in macrophages, alveolar walls, and around bronchial walls in SPC-TNFα mice but was absent in the other models (Fig. 7).
Fig. 6.
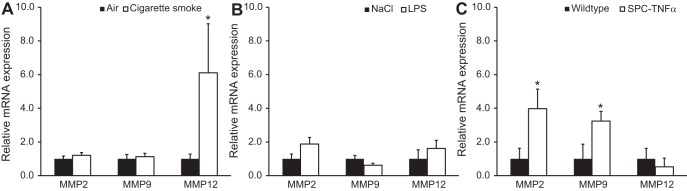
Matrix metalloproteinase (MMP)-2, -9, and -12 mRNA expression in mouse models of COPD. Relative mRNA expression of MMP-2, -9, and -12 in the smoke-exposed (A), LPS-treated (B), and SPC-TNFα (C) mice as measured by qPCR. Data were corrected for the housekeeping gene Rpl13A and expressed as means + SE; *P < 0.05 compared with respective control group as tested by ANOVA.
Fig. 7.
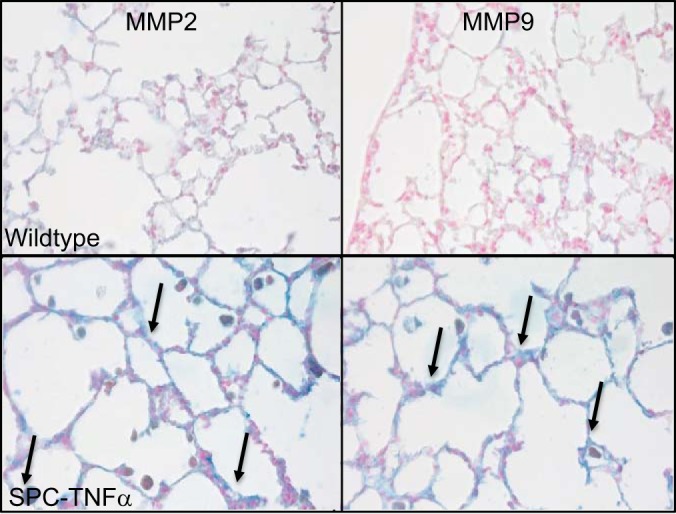
Increased MMP-2 and MMP-9 staining in alveolar and airway walls of SPC-TNFα mice. Photomicrographs at ×400 magnification of MMP-2 or MMP-9 staining (in blue) and counterstaining in red in lung tissue of one representative mouse of each condition. Arrows indicate MMP-2 or MMP-9 staining (blue).
DISCUSSION
In this study, three different mouse models all showing features of COPD were used to study ECM remodeling. We demonstrated that the changes in three matrix components observed in small airway and alveolar walls are largely comparable within each mouse model of COPD but that alterations in elastin are different between cigarette smoke-exposed, LPS-instilled, and SPC-TNFα mice. In particular, increased collagen and hyaluronan contents were found in both small airway and remaining alveolar walls in all models. The elastin contents were, however, differentially affected, with an absence of change in both alveolar and small airway walls in the smoke-exposed model, an increase in elastin in the airways of LPS-instilled mice, and a decreased elastin content of both alveolar and airway walls in SPC-TNFα mice. Furthermore TNF-α and MMP-2 and -9 mRNA and protein levels were found to be increased in alveolar walls and around airway walls in SPC-TNFα mice only.
Descriptions of the pathology of COPD often contrast the loss of elastin and destruction of the alveolar walls in the lung parenchyma to the fibrosis seen in the small airway walls. Several studies have, however, also reported increased levels of collagen in alveolar walls in animal models of emphysema and in lung tissue of patients with emphysema (26, 28). In the present study, all three mouse models of COPD were characterized by a significantly increased collagen content of both alveolar and small airway walls. In this study, we could not distinguish between the different types of collagen, however, increased levels of collagen III but not collagen I were shown before in the alveolar tissue of smoke-exposed mice (27), suggesting that the increase in collagen in our study is because of increased collagen III. In the chronic LPS model, procollagen I and III mRNA levels were increased in whole lung lysates (7). Furthermore, in mice genetically overexpressing TNF-α, total collagen content was increased along airways and in thickened septae (29). Importantly, collagen deposition can be regarded not only as evidence of fibrosis, but abnormal collagen remodeling could also explain lung functional changes and emphysema development and progression through mechanical forces. In elastase-treated mice, for instance, the increased collagen content of alveoli was related to breaking of parenchymal fibers during stretching at significantly lower tension, probably because of an altered collagen I-to-collagen III ratio (21).
Within the lung, collagen and elastin fibers are embedded in a hydrated gel of which glycosaminoglycans are the major constituents (20). Hyaluronan is the major glycosaminoglycan in lung tissue and has diverse functions in lung homeostasis and pulmonary disease. Furthermore, interactions between proteoglycans and MMPs are mediated by glycosaminoglycans, which alter MMP activity (30). We previously showed that hyaluronan was increased in sputum of COPD patients and in both small airway and alveolar walls of COPD patients (14, 15). Furthermore, in cigarette smoke-exposed mice, there was an increase in hyaluronan deposition in the alveolar and small airway wall after 4 wk of exposure that was not progressive in the 6 mo exposure model of emphysema (6). We hereby extend these findings to two other mouse models for COPD, showing an elevated hyaluronan content in alveolar and airway walls in all three models. Future studies into the size of the hyaluronan fragments deposited are needed, since different sizes exert different functions. High molecular weight fragments are important in hydration and provision of a matrix, whereas low molecular weight fragments can induce inflammatory responses (22).
On the other hand, in COPD patients, it was shown that elastic fibers are reduced not only in the alveoli but also in the small airways (3, 15) and, in addition, emphysema-like disappearance of small airways (31), suggesting that similar destructive processes occur in the airways and in the lung parenchyma. While inflammatory processes, for instance, increases in CD8+ T lymphocytes and macrophages are present in both alveolar and airway walls of COPD patients, it would not be surprising that similar changes occur in both alveolr and airway walls regarding extracellular matrix remodeling. Parallel changes in airways and alveoli were present with respect to enhanced collagen and hyaluronan deposition within the models in this study. In the current study, also, elastin content was investigated in both airway and alveolar walls in three mouse models for COPD. No changes in elastin were found in mice chronically exposed to cigarette smoke. In the chronic intratracheal LPS model, no changes were found in alveolar walls, but elastin content was increased in airway walls, which can possibly be explained by increased fiber assembly. Elastin content was significant lower in SPC-TNFα mice, both in airway walls and remaining alveolar walls. The loss of elastin in lungs of SPC-TNFα mice is in line with increased static expiratory compliance in these mice at an age of 6 mo (17).
TNF-α is known to be induced in sputum of patients with stable COPD (24). The current study demonstrated that, next to the unique decreased elastin content seen in the remaining walls in SPC-TNFα mice, TNF-α expression was not increased in the chronic cigarette smoke and LPS models. Furthermore, no induction of MMP-2 and MMP-9 mRNA and protein levels was observed in these models. The increased expression of MMP-12, which is mainly produced by macrophages, found in cigarette smoke-exposed mice is in line with previously published data (4, 23). Furthermore, studies show that MMP-12 knockout mice are protected against inflammation in an acute cigarette smoke exposure model (23) and a role for MMP-12 in induction of emphysema by overexpression (37). However, the increased MMP-12 levels observed in the chronic cigarette smoke exposure model here were not associated with alterations in the elastin content of airway walls and remaining alveolar wall, whereas MMP-12 expression was not enhanced in SPC-TNFα mice which did display loss of elastin. Because the current study focused on identifying mechanisms underlying elastin breakdown as seen in COPD (3), MMP-12 was not investigated further. However, because MMP-12 has been found to be upregulated in sputum of COPD patients (13), it is likely to play a role in COPD pathophysiology, which should be elucidated further. Furthermore, future studies are needed to study the role of neutrophil elastase, which can cause breakdown of elastin as well. In the current study, lungs were, however, lavaged, removing neutrophis and thus neutrophil elastase from the tissue.
TNF-α is known to suppress tropoelastin levels in lung fibroblast by itself and furthermore to promote elastin breakdown through enhanced release of elastolytic enzymes (39). TNF-α upregulates MMPs, including MMP-2 and MMP-9, which are known to have elastinolytic activity, and overexpression of these MMPs causes emphysematous changes in mice (16). SPC-TNFα mice displayed increased levels of MMP-2 and MMP-9 mRNA expression and increased MMP-2 and MMP-9 protein in both alveolar and airway walls. These results are in line with a study that found increased MMP-2 and MMP-9 activity in bronchoalveolar lavage (BAL) fluid of SPC-TNFα mice (17, 40). A role for TNF-α in tissue remodeling is furthermore evident from a study that demonstrated that TNF-α receptor knock-out mice were protected from cigarette smoke-induced elastin breakdown as measured by attenuated desmosine levels in BAL (8). In addition, an MMP-9/12 inhibitor could prevent increases in levels of desmosine in lavage of smoke-exposed guinea pigs (9). Interestingly, COPD patients, which received the MMP-9/12 inhibitor AZD1236 for 6 wk, had significantly reduced plasma desmosine levels compared with placebo-treated patients (12). Therefore, the induction of the TNF-α-MMP-2/9 axis in the airway walls and remaining alveolar walls that was found in SPC-TNFα mice can possibly explain the breakdown of elastin in these compartments.
In the current study, we show that only SPC-TNFα mice show changes in elastin remodeling in both airway and alveolar walls, which is comparable to what we observed in COPD patients (15). These findings reveal that the SPC-TNFα model is suitable to study processes underlying matrix remodeling and, in particular, elastin breakdown as seen in COPD. However, despite the fact that Fujita et al. reported that these mice had no lung abnormalities 7 days after birth, our findings should be confirmed using an inducible TNF-α transgene to exclude interference in lung development by TNF-α (17). Furthermore, we indicate a role for MMP-2 and MMP-9 in the breakdown of elastin in airways and alveoli of SPC-TNFα mice. Further investigation into MMP expression in relation to elastin breakdown at earlier time points is needed to elucidate this mechanism.
GRANTS
For the SPC-TNFα study, funding was provided by the Intramural Research Program of the National Institute on Aging.
DISCLOSURES
No conflicts of interest, financial or otherwise are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: I.M.E., M.A.D., E.F.W., and N.L.R. conception and design of research; I.M.E., E.M.M., K.R.B., and J.H.J.V. performed experiments; I.M.E. analyzed data; I.M.E., M.A.D., and N.L.R. interpreted results of experiments; I.M.E. prepared figures; I.M.E. drafted manuscript; I.M.E., M.A.D., E.M.M., E.F.W., and N.L.R. edited and revised manuscript; I.M.E., M.A.D., R.d.C., K.R.B., J.H.J.V., E.F.W., and N.L.R. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Fabienne Wijshof and Mike Eggen for technical support in doing the experiments. Furthermore, we are grateful for the help of Carine Peutz and Jack Cleutjens with validating the stainings and designing of the matrix scoring program. We thank Dawn Boyer, Dawn Phillips, Justine Lucas, and Dawn Nines for the outstanding husbandry work.
REFERENCES
- 1.Annoni R, Lancas T, Tanigawa RY, de Medeiros Matsushita M, de Morais Fernezlian S, Bruno A, da Silva LF, Roughley PJ, Battaglia S, Dolhnikoff M, Hiemstra PS, Sterk PJ, Rabe KF, Mauad T. Extracellular matrix composition in chronic obstructive pulmonary disease. Eur Respir J 40: 1362–1373, 2012 [DOI] [PubMed] [Google Scholar]
- 2.Bartalesi B, Cavarra E, Fineschi S, Lucattelli M, Lunghi B, Martorana PA, Lungarella G. Different lung responses to cigarette smoke in two strains of mice sensitive to oxidants. Eur Respir J 25: 15–22, 2005 [DOI] [PubMed] [Google Scholar]
- 3.Black PN, Ching PS, Beaumont B, Ranasinghe S, Taylor G, Merrilees MJ. Changes in elastic fibres in the small airways and alveoli in COPD. Eur Respir J 31: 998–1004, 2008 [DOI] [PubMed] [Google Scholar]
- 4.Bracke K, Cataldo D, Maes T, Gueders M, Noel A, Foidart JM, Brusselle G, Pauwels RA. Matrix metalloproteinase-12 and cathepsin D expression in pulmonary macrophages and dendritic cells of cigarette smoke-exposed mice. Int Arch Allergy Immunol 138: 169–179, 2005 [DOI] [PubMed] [Google Scholar]
- 5.Bracke KR, Dentener MA, Papakonstantinou E, Vernooy JH, Demoor T, Pauwels NS, Cleutjens J, van Suylen RJ, Joos GF, Brusselle GG, Wouters EF. Enhanced deposition of low-molecular-weight hyaluronan in lungs of cigarette smoke-exposed mice. Am J Respir Cell Mol Biol 42: 753–761, 2010 [DOI] [PubMed] [Google Scholar]
- 6.Bracke KR, Dentener MA, Papakonstantinou E, Vernooy JH, Demoor T, Pauwels NS, Cleutjens J, van Suylen RJ, Joos GF, Brusselle GG, Wouters EF. Enhanced deposition of low-molecular-weight hyaluronan in lungs of cigarette smoke-exposed mice. Am J Respir Cell Mol Biol 42: 753–761, 2010 [DOI] [PubMed] [Google Scholar]
- 7.Brass DM, Hollingsworth JW, Cinque M, Li Z, Potts E, Toloza E, Foster WM, Schwartz DA. Chronic LPS inhalation causes emphysema-like changes in mouse lung that are associated with apoptosis. Am J Respir Cell Mol Biol 39: 584–590, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Churg A, Dai J, Tai H, Xie C, Wright JL. Tumor necrosis factor-alpha is central to acute cigarette smoke-induced inflammation and connective tissue breakdown. Am J Respir Crit Care Med 166: 849–854, 2002 [DOI] [PubMed] [Google Scholar]
- 9.Churg A, Wang R, Wang X, Onnervik PO, Thim K, Wright JL. Effect of an MMP-9/MMP-12 inhibitor on smoke-induced emphysema and airway remodelling in guinea pigs. Thorax 62: 706–713, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D'Hulst AI, Vermaelen KY, Brusselle GG, Joos GF, Pauwels RA. Time course of cigarette smoke-induced pulmonary inflammation in mice. Eur Respir J 26: 204–213, 2005 [DOI] [PubMed] [Google Scholar]
- 11.D'Hulst AI, Vermaelen KY, Brusselle GG, Joos GF, Pauwels RA. Time course of cigarette smoke-induced pulmonary inflammation in mice. Eur Respir J 26: 204–213, 2005 [DOI] [PubMed] [Google Scholar]
- 12.Dahl R, Titlestad I, Lindqvist A, Wielders P, Wray H, Wang M, Samuelsson V, Mo J, Holt A. Effects of an oral MMP-9 and -12 inhibitor, AZD1236, on biomarkers in moderate/severe COPD: a randomised controlled trial. Pulm Pharmacol Ther 25: 169–177, 2012 [DOI] [PubMed] [Google Scholar]
- 13.Demedts IK, Morel-Montero A, Lebecque S, Pacheco Y, Cataldo D, Joos GF, Pauwels RA, Brusselle GG. Elevated MMP-12 protein levels in induced sputum from patients with COPD. Thorax 61: 196–201, 2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dentener MA, Vernooy JH, Hendriks S, Wouters EF. Enhanced levels of hyaluronan in lungs of patients with COPD: relationship with lung function and local inflammation. Thorax 60: 114–119, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eurlings IM, Dentener MA, Cleutjens JP, Peutz CJ, Rohde GG, Wouters EF, Reynaert NL. Similar matrix alterations in alveolar and small airway walls of COPD patients (Abstract). BMC Pulm Med 14: 90, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Foronjy R, Nkyimbeng T, Wallace A, Thankachen J, Okada Y, Lemaitre V, D'Armiento J. Transgenic expression of matrix metalloproteinase-9 causes adult-onset emphysema in mice associated with the loss of alveolar elastin. Am J Physiol Lung Cell Mol Physiol 294: L1149–L1157, 2008 [DOI] [PubMed] [Google Scholar]
- 17.Fujita M, Shannon JM, Irvin CG, Fagan KA, Cool C, Augustin A, Mason RJ. Overexpression of tumor necrosis factor-alpha produces an increase in lung volumes and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 280: L39–L49, 2001 [DOI] [PubMed] [Google Scholar]
- 18.Hallgren O, Nihlberg K, Dahlback M, Bjermer L, Eriksson LT, Erjefalt JS, Lofdahl CG, Westergren-Thorsson G. Altered fibroblast proteoglycan production in COPD (Abstract). Respir Res 11: 55, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hsia CC, Hyde DM, Ochs M, Weibel ER. An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am J Respir Crit Care Med 181: 394–418, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hukins DW. Connective Tissue Matrix. London, UK: Macmillan Education, 1984 [Google Scholar]
- 21.Ito S, Ingenito EP, Brewer KK, Black LD, Parameswaran H, Lutchen KR, Suki B. Mechanics, nonlinearity, and failure strength of lung tissue in a mouse model of emphysema: possible role of collagen remodeling. J Appl Physiol 98: 503–511, 2005 [DOI] [PubMed] [Google Scholar]
- 22.Jiang D, Liang J, Noble PW. Hyaluronan as an immune regulator in human diseases. Physiol Rev 91: 221–264, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.John G, Kohse K, Orasche J, Reda A, Schnelle-Kreis J, Zimmermann R, Schmid O, Eickelberg O, Yildirim AO. The composition of cigarette smoke determines inflammatory cell recruitment to the lung in COPD mouse models. Clin Sci 126: 207–221, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumor necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 153: 530–534, 1996 [DOI] [PubMed] [Google Scholar]
- 25.Kranenburg AR, Willems-Widyastuti A, Moori WJ, Sterk PJ, Alagappan VK, de Boer WI, Sharma HS. Enhanced bronchial expression of extracellular matrix proteins in chronic obstructive pulmonary disease. Am J Clin Pathol 126: 725–735, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Lang MR, Fiaux GW, Gillooly M, Stewart JA, Hulmes DJ, Lamb D. Collagen content of alveolar wall tissue in emphysematous and non-emphysematous lungs. Thorax 49: 319–326, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lopes FD, Toledo AC, Olivo CR, Prado CM, Leick EA, Medeiros MC, Santos AB, Garippo A, Martins MA, Mauad T. A comparative study of extracellular matrix remodeling in two murine models of emphysema. Histol Histopathol 28: 269–276, 2013 [DOI] [PubMed] [Google Scholar]
- 28.Lucey EC, Goldstein RH, Stone PJ, Snider GL. Remodeling of alveolar walls after elastase treatment of hamsters. Results of elastin and collagen mRNA in situ hybridization. Am J Respir Crit Care Med 158: 555–564, 1998 [DOI] [PubMed] [Google Scholar]
- 29.Lundblad LK, Thompson-Figueroa J, Leclair T, Sullivan MJ, Poynter ME, Irvin CG, Bates JH. Tumor necrosis factor-alpha overexpression in lung disease: a single cause behind a complex phenotype. Am J Respir Crit Care Med 171: 1363–1370, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Malla N, Berg E, Uhlin-Hansen L, Winberg JO. Interaction of pro-matrix metalloproteinase-9/proteoglycan heteromer with gelatin and collagen. J Biol Chem 283: 13652–13665, 2008 [DOI] [PubMed] [Google Scholar]
- 31.McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott WM, Sanchez PG, Wright AC, Gefter WB, Litzky L, Coxson HO, Pare PD, Sin DD, Pierce RA, Woods JC, McWilliams AM, Mayo JR, Lam SC, Cooper JD, Hogg JC. Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med 365: 1567–1575, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Merrilees MJ, Ching PS, Beaumont B, Hinek A, Wight TN, Black PN. Changes in elastin, elastin binding protein and versican in alveoli in chronic obstructive pulmonary disease (Abstract). Respir Res 9: 41, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miyazaki Y, Araki K, Vesin C, Garcia I, Kapanci Y, Whitsett JA, Piguet PF, Vassalli P. Expression of a tumor necrosis factor-alpha transgene in murine lung causes lymphocytic and fibrosing alveolitis. A mouse model of progressive pulmonary fibrosis. J Clin Invest 96: 250–259, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nakano Y, Muller NL, King GG, Niimi A, Kalloger SE, Mishima M, Pare PD. Quantitative assessment of airway remodeling using high-resolution CT. Chest 122: 271S–275S, 2002 [PubMed] [Google Scholar]
- 35.Ochs M, Muhlfeld C. Quantitative microscopy of the lung: a problem-based approach. Part 1: basic principles of lung stereology. Am J Physiol Lung Cell Mol Physiol 305: L15–L22, 2013 [DOI] [PubMed] [Google Scholar]
- 36.Papakonstantinou E, Karakiulakis G. The ‘sweet’ and ‘bitter’ involvement of glycosaminoglycans in lung diseases: pharmacotherapeutic relevance. Br J Pharmacol 157: 1111–1127, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Qu P, Du H, Wang X, Yan C. Matrix metalloproteinase 12 overexpression in lung epithelial cells plays a key role in emphysema to lung bronchioalveolar adenocarcinoma transition. Cancer Res 69: 7252–7261, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sohal SS, Reid D, Soltani A, Ward C, Weston S, Muller HK, Wood-Baker R, Walters EH. Reticular basement membrane fragmentation and potential epithelial mesenchymal transition is exaggerated in the airways of smokers with chronic obstructive pulmonary disease. Respirology 15: 930–938, 2010 [DOI] [PubMed] [Google Scholar]
- 39.Sproul EP, Argraves WS. A cytokine axis regulates elastin formation and degradation. Matrix Biol 32: 86–94, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomson EM, Williams A, Yauk CL, Vincent R. Overexpression of tumor necrosis factor-alpha in the lungs alters immune response, matrix remodeling, and repair and maintenance pathways. Am J Pathol 180: 1413–1430, 2012 [DOI] [PubMed] [Google Scholar]
- 41.Vernooy JH, Dentener MA, van Suylen RJ, Buurman WA, Wouters EF. Long-term intratracheal lipopolysaccharide exposure in mice results in chronic lung inflammation and persistent pathology. Am J Respir Cell Mol Biol 26: 152–159, 2002 [DOI] [PubMed] [Google Scholar]