

Abstract
Carbonic anhydrase 2 regulates acid-base homeostasis, and recent findings have indicated a correlation between cellular control of acid-base status and the innate defense of the kidney. Mice deficient in carbonic anhydrase 2 (Car2−/− mice) have metabolic acidosis, impaired urine acidification, and are deficient in normal intercalated cells. The objective of the present study was to evaluate the biological consequences of carbonic anhydrase 2 deficiency in a murine model of pyelonephritis. Infection susceptibility and transcription of bacterial response components in Car2−/− mice were compared with wild-type littermate controls. Car2−/− mice had increased kidney bacterial burdens along with decreased renal bacterial clearance after inoculation compared with wild-type mice. Standardization of the urine pH and serum HCO3− levels did not substantially alter kidney infection susceptibility between wild-type and Car2−/− mice; thus, factors other than acid-base status are responsible. Car2−/− mice had significantly increased neutrophil-gelatinase-associated lipocalin mRNA and protein and expression at baseline and a marked decreased ability to upregulate key bacterial response genes during pyelonephritis. Our findings provide in vivo evidence that supports a role for carbonic anhydrase 2 and intercalated cells in promoting renal bacterial clearance. Decreased carbonic anhydrase expression results in increased antimicrobial peptide production by cells other than renal intercalated cells, which is not sufficient to prevent infection after a bacterial challenge.
Keywords: acid-base, intercalated cells, urinary tract infection, inflammation
acid-base regulation is increasingly linked to immune function. Systemic acidosis may impair immune function, antimicrobial peptides have impaired function at extreme urine pH ranges, and intercalated cells (ICs) that regulate kidney acid-base transport are increasingly recognized as innate immune effectors.
The carbonic anhydrase gene family encodes distinct isozymes that differ in tissue-specific expression and cellular localization (40). Carbonic anhydrases are involved in H+ secretion, HCO3− reabsorption, bone resorption, and gastric acidity (40). Carbonic anhydrase 2 (gene: Car2; protein: CA-II) is found in many tissues, including the kidney, bone, brain, eye, stomach, intestine, liver, pancreas, red blood cells, salivary glands, and uterus (30). CA-II is highly expressed in renal ICs (4, 12). Lower CA-II levels of expression are present in collecting duct principal cells (PCs), the loop of Henle, and proximal tubules (3, 5). Mice that lack CA-II have metabolic acidosis, a high urine pH, and are severely depleted in kidney ICs compared with wild-type mice (4).
During an ascending pyelonephritis, pathogens initially encounter the collecting tubule. The collecting tubule contains ICs and PCs. PCs mediate Na+ and water transport, and ICs regulate acid-base transport (20, 38). Escalating evidence shows that IC function as innate immunity effectors in addition to their traditional role in the maintenance of acid-base homeostasis. We (42) have previously characterized ribonuclease 7, a potent antimicrobial peptide with a broad range of activity against uropathogens in the human kidney. Renal ribonuclease 7 expression is limited to ICs, constitutively expressed at baseline, upregulated in response to infection, and secreted into the urine (42, 43). ICs have been demonstrated to express other innate immune proteins, including neutrophil gelatinase-associated lipocalin, also known as lipocalin 2 (gene: Lcn2; protein: Ngal), IL-18 (gene: Il18), and human α-defensin 5 (15, 28, 41). Additionally, uropathogenic Escherichia coli (UPEC) preferentially adhere to the luminal surface of medullary ICs (10). ICs are ideally positioned to defend the kidney from ascending urinary tract infections (UTIs) and activate systemic inflammatory responses (42, 43).
Based on their phenotype of metabolic acidosis, impaired urine acidification, and IC depletion, we hypothesized that CA-II has a critical role in the innate defense of the kidney. The objective of the present study was to evaluate the functional consequences of CA-II depletion on the innate immune response during experimental murine pyelonephritis.
METHODS
Mice.
The Institutional Animal Care and Use Committee of the Nationwide Children's Hospital approved murine research protocol AR12–00035. All animal experiments adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Car2-deficient (Car2−/−) mouse embryos and wild-type (Car2+/+) C57BL/6J mice were purchased from Jackson Laboratory (Bar Harbor, ME). The initial coding sequence of the Car2 gene was replaced with a LacZ marker under the direction of the Car2 promoter (25). Heterozygous (Car2+/−) mice were rederived on a C57BL/6 genetic background at the Transgenic Core at the Research Institute of Nationwide Children's Hospital. Homozygous Car2−/− mice were obtained by crossing Car2+/− mice. Car2+/+ littermates were used as controls. For mouse growth experiments, Car2+/− control mice were also used. The mouse genotype was determined by PCR using tail DNA using the following primer sequences: Car2 wild-type forward primer, 5′-ATGAAGGCTGAACTTAAATC-3′; Car2 mutant forward primer, 5′-TCCACATACACTTCATTCTC-3′, and common reverse primer, 5′-AGGCCTGAGTATTCATTATC-3′.
Infection.
UPEC strain CFT073 was used to infect mice. CFT073 was grown statically in Luria broth (LB) medium overnight at 37°C. Bacteria were pelleted and resuspended in PBS. Female mice aged 7–10 wk were infected by inoculating 1 × 108 CFT073 in 50 μl PBS transurethrally. A second inoculum was performed 3 h later, as previously described (45). Twenty-four hours after the first infection, kidneys and bladders were harvested. Kidneys were homogenized in a bullet blender (Next Advance, Averill Park, NY). The bacterial burden was determined by plating serial dilutions of kidney or bladder homogenates on the LB plate. If UTI confirmation was needed for a PCR experiment, kidneys were bisected longitudinally. Infection was confirmed as half of the kidney, and the other longitudinal half of each kidney was processed for immunostaining or mRNA extraction. Kidney or bladder clearance of bacteria was defined as <50 colony-forming units (CFU)/tissue (44).
Base supplementation.
NaHCO3 (100 mM/l) was added to the drinking water of mice assigned base supplementation from the time of weaning (∼4 wk) until euthanasia (7–10 wk) based on prior reports (32, 37).
LacZ staining.
After euthanasia, mice were perfused with PBS and periodate-lysine-paraformaldehyde (PLP) fixative (2% paraformaldehyde, 75 mM lysine, and 75 mM NaIO4). Kidneys were bisected, fixed in cold PLP fixative for 2 h, and then soaked in 30% sucrose overnight. Kidneys and bladders were embedded in optimal cutting temperature embedding media after treatment. Frozen sections were dried at room temperature for 10 min and then fixed in PLP fixative for 10 min. Kidney slides were incubated with the LacZ substrate X-galactosidase at 4°C overnight. Slides were counterstained with nuclear fast red.
Immunostaining.
After deparaffinization, rehydration, antigen retrieval, and blocking, specimens were incubated with primary antibody overnight at 4°C and incubated for 30 min at room temperature with horseradish peroxidase for immunohistochemistry or fluorophore-conjugated secondary antibody for immunofluorescence. Slides for immunohistochemistry were developed with 3,3′-diaminobenzidine and counterstained with hematoxylin. Slides for immunofluorescence were mounted in Vectashield Mounting Media with 4′,6-diamidino-2-phenylindole (Vector Laboratories, Burlinghame, CA). Imaging was performed with a Keyence BZ-9000 “all in one” microscope and camera (Keyence, Osaka, Japan). Cells were quantified with the automated “dynamic cell count” and “cell count” features of BZ-II Analyzer software (Keyence). Images were adjusted with the BZ-II Analyzer and/or a GNU image manipulation program (GNU Free Software Foundation, Boston, Massachusetts). The antibodies and fluorochromes used are shown in Table 1.
Table 1.
Antibodies used in the present study
| Antibody (Species) | Company | Catalog Number | Dilution |
|---|---|---|---|
| Primary antibodies | |||
| Aquaporin 2 (rabbit, anti-human, ATTO 550 fluorescent label) | Alomone Labs (Jerusulem, Israel) | AQP-002-AO | 1:200 |
| CA-II (rabbit, anti-human) | Epitomics (Burlingame, CA) | S2774 | 1:1,000 |
| Lcn2/Ngal (goat, anti-mouse) | R&D Systems (Minneapolis, MN) | AF1857 | 1:50 |
| Ly6G (rabbit, anti-mouse) | Biolegend (San Diego, CA) | 127607 | 1:50 |
| V-ATPase (goat, anti-human) | Santa Cruz Biotechnology (Santa Cruz, CA) | 21209 | 1:50 |
| Secondary antibodies | |||
| Alexa fluor 488 (donkey, anti-goat) | Jackson ImmunoResearch (West Grove, PA) | 705-546-147 | 1:250 |
| Cy3 (donkey, anti-rat) | Jackson ImmunoResearch | 712-296-150 | 1:1,000 |
| Horseradish peroxidase (goat, anti-rabbit) | Jackson ImmunoResearch | 111-055-003 | 1:500 |
CA-II, carbonic anhydrase 2 (gene: Car2); Lcn2/Ngal, lipocalin 2/neutrophil gelatinase-associated lipocalin.
Blood and urine analysis.
Blood was collected through the submandibular vein and injected into iSTAT EC8+ for HCO3−, K+, and pH measurements (Abbot Point of Care, Princeton, NJ). Urine was collected by bladder massage. Urine was transferred into 1.5-ml tubes containing corn oil. Urine pH was measured with a micro-pH electrode (Mettler Toledo, Columbus, OH). The Comparative Pathology and Mouse Phenotyping Shared Resource at The Ohio State University (Columbus, OH) performed complete blood counts and differentials.
ELISA.
Murine urine Ngal levels were quantified by ELISA (catalog no. AF1857, R&D Systems) and run in duplicate. Urine creatinine levels were measured by a colorimetric assay to account for differences in urine concentration (catalog no. 500701, Cayman Chemical).
Quantitative real-time PCR.
For cDNA generation, RNA was purified from kidneys using the RNeasy kit (Qiagen, Valencia, CA) and quantified with spectrometry. RNA quality/purity was also determined by the ratio of 260 to 280 nm. A value of 2.0 ± 0.1 was considered to be pure. cDNA was generated using the RT First Strand kit (Qiagen) per the manufacturer's protocol.
For gene profiling, cDNA was amplified using the 7500 Real-Time PCR System (Applied Biosystems, Carlsbad, CA). The RT2 profiler antibacterial response (catalog no. PAM148z, Qiagen) was performed according to the manufacturer's instructions. Analysis and data quality control were performed using RT2 profiler PCR Array Data Analysis software (version 3.5, Qiagen). Only results that passed quality checks in PCR array reproducibility, RT efficiency, and genomic DNA contamination were included. Gene expression was normalized using a panel of five housekeeping genes [β-actin (Actb), β2-microglobulin (B2m), Gapdh, glucuronidase-β (Gusb), and heat shock protein 90 kDa α (cytosolic) class B member 1 (Hsp90ab1)].
Statistical analysis.
Statistical analysis was performed, and graphs were made with GraphPad Prism software (GraphPad Software, La Jolla, CA). For the purpose of graphs, negative kidney culture results were assigned a value of 0.1 so that all data points were represented on log-scale scatter plots; the zero value was used for statistical analysis. Groups were analyzed for differences with a two-tailed Student's t-test. Welch's correction was used for bacterial burden analysis, as comparable SDs were not assumed. Proportions and percentages were compared using the VassarStats 2 × 2 contingency table, which assigned the χ2-value, or, if the expected cell frequency was less than five, the Fisher exact probability test was applied (26). Two-way ANOVA was used to measure the effects of two or more variables in weight time course data. If significant differences were identified with ANOVA, post hoc analysis with the Tukey test was used to determine which groups within the sample were different. For the mouse antibacterial response arrays, fold changes of expression between groups and treatments were calculated using SA Biosciences RT2 profiler PCR Array Data Analysis software (11). Statistical significance was assigned for P values of <0.05, and results are expressed as means ± SD.
RESULTS
The phenotype of Car2−/− mice.
To confirm CA-II deletion and phenotype as previously reported by Breton et al. (4), absent CA-II expression was demonstrated with LacZ staining and CA-II antibody immunohistochemistry (Fig. 1). At 8 wk of age, Car2−/− mice were significantly smaller than Car2+/+ mice (14.49 ± 1.66 vs. 20.95 ± 3.68 g, respectively, P < 0.0001). Enumeration of V-ATPase-positive cells demonstrated threefold IC depletion in Car2−/− mice compared with Car2+/+ mice in the cortex and medulla (Fig. 2). Laboratory evaluation (Table 2) demonstrated similar complete blood counts and leukocyte differentials but higher urine pH along with lower serum pH, HCO3−, and K+ in Car2−/− versus Car2+/+ mice. Car2+/− mice had similar weights (20.49 ± 2.97 g, n = 35), serum pH (7.28 ± 0.05, n = 17), and urine pH (5.86 ± 0.25, n = 5) compared with Car2+/+ mice (P = 0.651, 0.447, and 0.57, respectively).
Fig. 1.

Absent carbonic anhydrase 2 (gene: Car2; protein: CA-II) expression. A: absent LacZ staining is present in the cortex (C), outer medulla (OM), and inner medulla (IM) of Car2+/+ mice. LacZ staining (blue) is present in the cortex, outer medulla, and inner medulla of Car2+/− mice but is more pronounced in Car2−/− mice, consistent with expression driven by the LacZ promoter. Magnification: ×4. B: CA-II immunostaining demonstrating similar cell-specific CA-II reactivity (brown) in isolated cells of the collecting ducts (arrows) and lighter, more diffuse proximal tubule staining (arrowheads) in Car2+/+ and Car2+/− mice. No CA-II staining was seen in the cortex, outer medulla, or inner medulla of Car2−/− mice. Magnification: ×40. C: compared with robust CA-II staining (red) in the kidney (right), negligible CA-II staining (red) was present in the Car2+/+ bladder (left) urothelium, with some faint, nonspecific, scattered staining (arrow) on the luminal (*) surface and a rare cell in the smooth muscle layer (arrowhead). No staining was seen in the Car2−/− bladder (right). Bladder magnification: ×40; kidney magnification: ×4. 4′,6-diamidino-2-phenylindole (DAPI; blue) was used to stain nuclei in the bladder images.
Fig. 2.

Quantification of intercalated cells (ICs). ICs were identified by immunofluorescence for V-ATPase immune reactivity (green) and colabeled with DAPI (blue) to identify nuclei and aquaporin 2 (red) to identify collecting duct principal cells. Isolated V-ATPase-positive cells (arrows) were identified in the collecting ducts of the cortex (A) and medulla (D) of Car2+/+ mice. ICs (arrows) in the cortex and medulla were present in tubules positive for aquaporin 2, consistent with collecting ducts, and also in isolated tubules negative for aquaporin 2 in the cortex, consistent with connecting segments. In contrast, Car2−/− mice had infrequent cells that stained for V-ATPase in the cortex (B) and medulla (E). C: ICs comprised 1.27 ± 0.40% and 0.32 ± 0.13% of cortical cells in Car2+/+ and Car2−/− kidneys, respectively (P = 0.004). F: medullary ICs decreased from 2.21 ± 0.58% of total cells in Car2+/+ kidneys to 0.83 ± 0.23% in Car2−/− kidneys (P = 0.004). The automated cell count of ICs was performed on four ×20 images from 2 mice. The presented images are at ×40 magnification, with 20-μm scale bars.
Table 2.
Laboratory values
| Lab value | Car2+/+ Mice | Car2−/− Mice | P Value |
|---|---|---|---|
| Urine laboratory tests | |||
| pH | 5.73 ± 0.11 | 6.38 ± 0.34 | 0.013* |
| Serum chemistries | |||
| pH | 7.30 ± 0.07 | 7.12 ± 0.03 | <0.001* |
| HCO3−, mmol/l | 19.93 ± 2.61 | 17.24 ± 1.43 | 0.007* |
| K+, mmol/l | 6.32 ± 1.01 | 4.90 ± 1.47 | 0.017* |
| Complete blood counts and differentials | |||
| White blood cells, K/μl | 9.13 ± 0.84 | 8.69 ± 1.06 | 0.754 |
| Neutrophils, K/μl | 1.08 ± 0.10 | 0.96 ± 0.07 | 0.356 |
| Lymphocytes, K/μl | 7.43 ± 0.74 | 7.21 ± 0.94 | 0.861 |
| Monocytes, K/μl | 0.61 ± 0.08 | 0.49 ± 0.07 | 0.292 |
| Eosinophils, K/μl | 0.01 ± 0.00 | 0.02 ± 0.01 | 0.347 |
| Hematocrit, % | 44.93 ± 1.12 | 44.17 ± 1.90 | 0.579 |
Values are means ± SD. For urine laboratory tests, n = 3 Car2+/+ mice and 7 Car2−/− mice except for serum chemistries, where n = 11 mice/genotype; for complete blood counts and differentials, n = 3 mice/genotype.
Statistically significant difference.
Bacterial burden and renal bacteria clearance post-UPEC inoculation.
Infections were variable based on kidney laterality with mean CFU differences between left and right kidneys of 3,892 (range: 0–109,580) in Car2+/+ kidneys and 60,261 (range: 0 to 2.4 × 106) in Car2−/− kidneys (n = 16 mice/genotype). Thus, individual rather than pooled kidneys were evaluated, as previously done by Tittel et al. (45). Figure 3 shows 24- and 48-h kidney and bladder bacterial burdens along with kidney bacterial clearance. Twenty-four hours after inoculation, Car2−/− mice had a mean bacterial burden that was 5-fold higher in the bladder and 15-fold higher in the kidneys compared with Car2+/+ mice. Furthermore, Car2−/− mice were three times less likely to clear bacteria from the kidney at 24 h compared with control mice, and they were eight times less likely to clear at 48 h. Bacteria were largely retained in the bladder of both genotypes. Histological confirmation of infection in UPEC-inoculated mice was confirmed by the presence of neutrophils (Fig. 4).
Fig. 3.
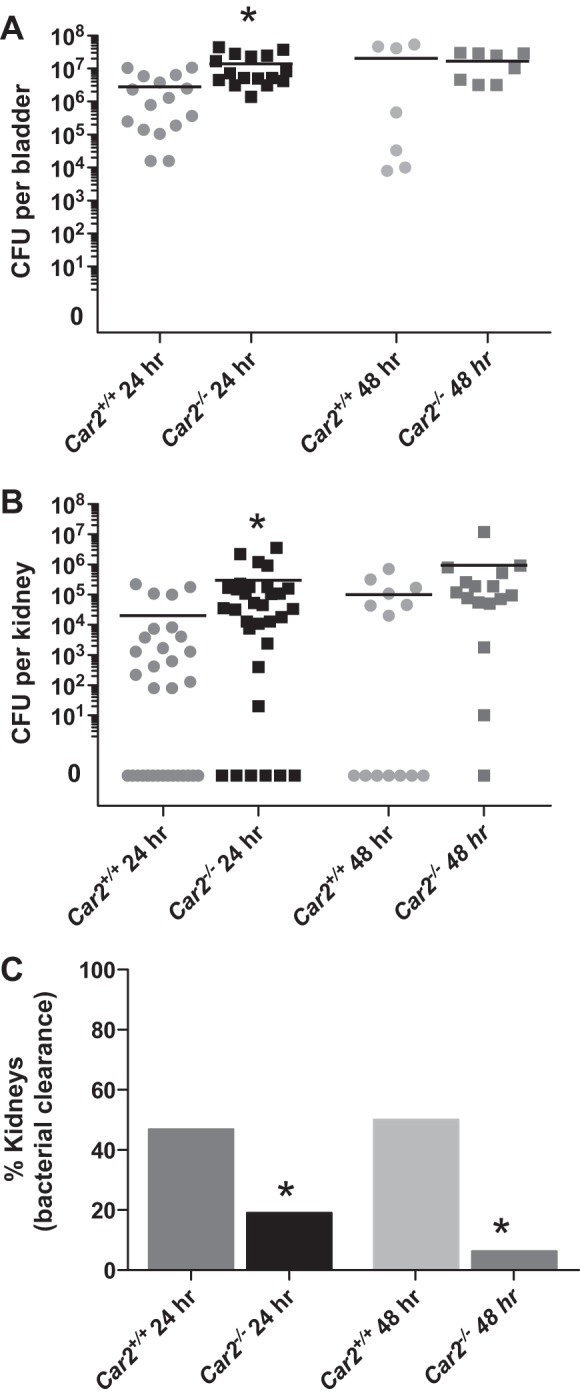
Kidney and bladder bacterial burden. A: mean bladder colony-forming unit (CFU) counts at 24 h afte uropathogenic Escherichia coli (UPEC) inoculation were significantly higher at 1.38 × 107 ± 1.35 × 107 in Car2−/− mice versus 2.83 × 106 ± 3.66 × 106 in Car2+/+ mice (P = 0.006). However, at 48 h after UPEC inoculation, bladder CFUs were similar between Car2+/+ and Car2−/− mice at 2.04 × 107 ± 2.55 × 107 versus 1.68 × 107 ± 1.25 × 107 (P = 0.745). B: mean kidney 24 h postinoculation CFUs were higher in Car2−/− mice compared with Car2+/+ mice at 301,357 ± 749,330 and 19,987 ± 53,965, respectively (P = 0.04). Mean CFUs were also higher at 48 h in Car2−/− mice compared with Car2+/+ mice at 959,488 ± 2.96 × 106 versus 101,357 ± 197,686, respectively. These 48-h kidney CFU changes did not reach statistical significance secondary to a smaller n value and larger SD (P = 0.26). Bacterial clearance from the kidney was evaluated at 24 and 48 h. Kidney clearance of bacteria was achieved in 47% of Car2+/+ kidneys versus 19% of Car2−/− kidneys at 24 h and 50% of Car2+/+ kidneys versus 6% of Car2−/− kidneys at 48 h. Both 24- and 48-h kidney bacterial clearance differences between genotypes were significant, with P values of 0.032 and 0.012, respectively. n = 32 kidneys/genotype at 24 h and 14 kidneys for Car2+/+ genotype along with 16 kidneys for the Car2−/− genotype.
Fig. 4.
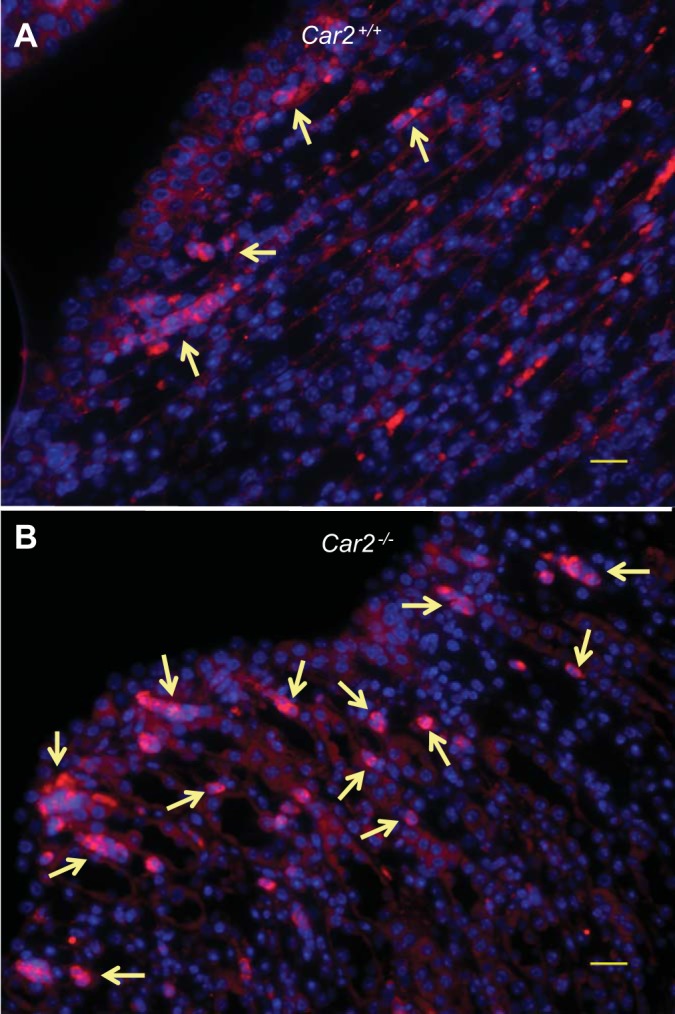
Neutrophil aggregation. A and B: representative pictures of neutrophil aggregation in Car2+/+ mice (A) and Car2−/− mice (B) 24 h after transurethral inoculation with UPEC (magnification: ×40; scale bars = 20 μm). Neutrophils are labeled with Ly6g antibody (red), and nuclei are counterstained with DAPI (blue). Neutrophil aggregation (arrows) was seen in both genotypes, mostly around the periphery of the renal papilla, but neutrophils appeared more numerous and diffuse in Car2−/− kidneys. Images were obtained from 2 kidneys/genotype with 2 sections analyzed per kidney.
Normalization of serum HCO3−.
Because Car2−/− had a lower serum HCO3− and pH than Car2+/+ mice (Table 2), mouse growth, serum laboratory, and 24-h bacterial burden experiments were repeated with NaHCO3 supplemented to Car2−/− mice in their drinking water and compared with a control group of Car2+/+ mice with no base supplementation. The highest tolerable NaHCO3-supplemented water concentration was 100 mmol/l. Serum HCO3− normalized from 17 mmol/l in Car2−/− mice to 20 mmol/l in base-supplemented Car2−/− mice (Table 1 and Fig. 5A). Because mean weights did not differ between Car2+/+ and Car2+/− mice, these genotypes were pooled for time course growth data. The growth of Car2−/− mice with and without base supplementation was compared with that of pooled Car2+/− and Car2+/+ mice from 3 to 12 wk. Car2−/− mice demonstrated improved growth with base supplementation, whereas growth was not affected in control mice (Fig. 5C).
Fig. 5.

Serum HCO3− normalization. A: mean serum HCO3− in base-supplemented Car2−/− mice was 19.98 ± 2.68, consistent with 19.93 ± 2.61 in Car2+/+ mice (P = 0.955). B: serum pH in Car2−/− mice partially corrected from the nonbase-supplemented mean of 7.12 ± 0.03 (Table 2). It was 7.21 ± 0.05 and 7.30 ± 0.07 in base-supplemented Car2−/− and Car2+/+ mice, respectively (P = 0.003). C: Car2−/− mice were smaller at all time points (P < 0.0001). Base-supplemented Car2−/− mice did exhibit significant increased growth at 11 wk (P = 0.047). The growth of Car2−/− mice with (n = 10) and without (n = 9) base supplementation was plotted against the pooled control group of 20 mice with (10 wild-type mice and 10 Car2+/− mice) and without (5 Car2+/+ mice and 15 Car2+/− mice) base supplementation. D: the mean bladder CFU was higher in base-supplemented Car2−/− mice at 1.08 × 107 ± 8.56 × 106 versus 4.53 × 106 ± 4.42 × 106 in Car2+/+ mice (P = 0.0382). E: the mean kidney CFU of 213,329 ± 508,832 in base-supplemented Car2−/− mice was higher than Car2+/+ mice (12,965 ± 36,283), but just missed significance with a P value of 0.056. F: the proportion of kidneys clear of bacteria was lower in base-supplemented Car2−/− mice (2 of 24 mice, 8.3%) than Car2+/+ mice (8 of 16 mice, 50%, P = 0.007).
To analyze the effect of correction of the acid-base disturbances, we performed experimental UTI on base-supplemented Car2−/− mice and nontreated control mice. The mean 24-h CFU was 2.4 times higher in base-supplemented Car2−/− bladders. Compared with Car2+/+ mice, the mean CFU was 16.5 times higher in base-supplemented Car2−/− kidneys (Fig. 5D). Despite missing statistical significance, presumably because of variability in the controls, it was consistent with nonbase-supplemented Car2−/− kidneys (Fig. 3B). Base-supplemented Car2−/− kidneys were six times less likely to be clear of bacteria than Car2+/+ kidneys (Fig. 5E).
Standardization of urine pH.
Because urine pH may affect bacterial growth more than host serum acid-base status, we standardized urine pH between Car2−/− and control mice. To standardize urine pH secondary to the relatively high baseline urine pH in Car2−/− mice (Table 1), control Car2+/+ mice had NaHCO3 added to their drinking water, and the 24-h bacterial burden experiment was repeated (Fig. 6). Car2−/− mice had a mean bacterial burden that was 10.4 times higher in the bladder and 15.3 times higher in the kidney than base-supplemented Car2+/+ mice.
Fig. 6.

Urine pH standardization. A: urine pH was similar between base-supplemented Car2+/+ and Car2−/− mice at 6.16 ± 0.611 and 6.36 ± 0.34, respectively (P = 0.402). B and C: 24-h bladder (B) and kidney (C) bacterial burdens were higher in Car2−/− mice than in base-supplemented Car2+/+ mice. The bladder bacterial burden was 2.39 × 106 ± 4.12 × 106 in base-supplemented Car2+/+ mice and 2.49 × 107 ± 1.47 × 107 in Car2−/− mice (P = 0.001), and the kidney bacterial burden was 24,945 ± 99,862 in base-supplemented Car2+/+ mice and 366,858 ± 665,773 in Car2−/− mice (P = 0.013). D: kidney clearance of bacteria occurred in 2 of 25 (8.0%) of Car2−/− kidneys and 7 of 28 (25%) of base-supplemented Car2+/+ kidneys (P = 0.148).
Mouse bacterial response PCR arrays.
Kidneys from control and UPEC-inoculated Car2+/+ and Car2−/− kidneys were evaluated for differences in 84 key genes involved in the murine antimicrobial response. All genes evaluated in the RT2 PCR array are shown in the Supplemental Material (Supplemental File S1).1 Five kidneys were evaluated for each genotype and infection status. Samples were required to pass the RT2 profiler PCR Array Data Analysis quality control, and two kidneys that did not pass quality control were subsequently excluded. The mean infection burden was similar at 8.0 × 105 (range: 7.6 × 103–2.8 × 106) and 1.3 × 106 (range: 2.6 × 104–2.8 × 106) per kidney in Car2+/+ and Car2−/− UPEC-inoculated mice, respectively (P = 0.60). Heat maps of fold changes in mRNA bacterial response gene expression after UPEC inoculation demonstrated a diminished response in Car2−/− compared with Car2+/+ mice (Fig. 7). Genes that displayed at least a twofold change and that were statistically significantly different (P < 0.05) between infection status and genotype are shown in Table 3. Uninfected Car2−/− mice had increased Lcn2 and cathelicidin expression. The antibacterial response was diminished in infected Car2−/− mice; specifically, Car2−/− mice had a relative inability to upregulate Cd14, Tnf, and Mediterranean fever (Mefv) expression along with relatively decreased interferon regulatory factor 5 (Irf5), NLR family CARD domain-containing protein 4 (Nlrc4), and C-reactive protein (Crp) expression during kidney infection. Although Lcn2 expression was increased in baseline, Car2+/+ mice but not Car2−/− mice could significantly upregulate Lcn2 expression after infection.
Fig. 7.

Bacterial response heat map. A heat map representation of the Car2−/− and Car2+/+ mouse kidney bacterial response is shown. Increased and decreased fold changes in kidney mRNA expression are represented by red and green squares, respectively. A: direct comparison of infected Car2+/+ and infected Car2−/− mice revealed a marked increase in genes with a relative decrease in bacterial response gene mRNA expression in Car2−/− mice. B: gene layout of the heat maps.
Table 3.
RT2 bacterial response array results
| Gene Name | Gene Symbol | Functional Gene Grouping | Fold Change | P Value |
|---|---|---|---|---|
| Expression in Car2−/−mice relative to Car2+/+ mice | ||||
| Lipocalin 2 | Lcn2 | Antimicrobial peptide | +4.05 | 0.03 |
| Cathelicidin antimicrobial peptide | Camp | Antimicrobial peptide | +2.03 | 0.04 |
| Expression in infected Car2−/− mice relative to infected Car2+/+ mice | ||||
| C-reactive protein, pentraxin-related | Crp | Bacterial pattern recognition receptors and the inflammatory response | −3.9 | 0.045 |
| Expression in infected Car2+/+mice relative to Car2+/+ mice | ||||
| Lipocalin 2 | Lcn2 | Antimicrobial peptide | +4.5 | 0.04 |
| CD14 antigen | Cd14 | Inflammatory response | +4.5 | 0.04 |
| Tumor necrosis factor | Tnf | Inflammatory response | +4.4 | 0.04 |
| Mediterranean fever | Mefv | Inflammatory response | +20.9 | 0.03 |
| Expression in infected Car2−/−mice relative to Car2−/− mice | ||||
| Interferon regulatory factor 5 | Irf5 | Cytokines and chemokines | −3.5 | 0.04 |
| NLR family, CARD domain containing 4 | Nlrc4 | Inflammasomes and the inflammatory Response | −3.5 | 0.03 |
| C-reactive protein, pentraxin-related | Crp | Bacterial pattern recognition receptors and the inflammatory response | −3.5 | 0.01 |
Ngal immunoreactivity and ELISA.
Ngal (protein product of Lcn2) immunostaining in Car2+/+ and Car2−/− mice with and without infection is shown in Fig. 7. Isolated Car2+/+ and Car2−/− proximal tubular cells were positive for Ngal at baseline and during infection. Scattered immunoreactivity for Ngal was present in focal areas of Car2−/− but not Car2+/+ medullas at baseline. Medullary Ngal staining increased in both genotypes with infection but remained focal. Staining was often present in cells with apical aquaporin 2 staining, consistent with PCs (Fig. 8, I and J). In the absence of infection, urine Ngal/creatinine levels were higher in Car2−/− mice than in Car2+/+ mice at 20.81 ± 5.032 pg/mg (n = 6) versus 10.28 ± 1.393 pg/mg (n = 10), respectively (P = 0.026).
Fig. 8.
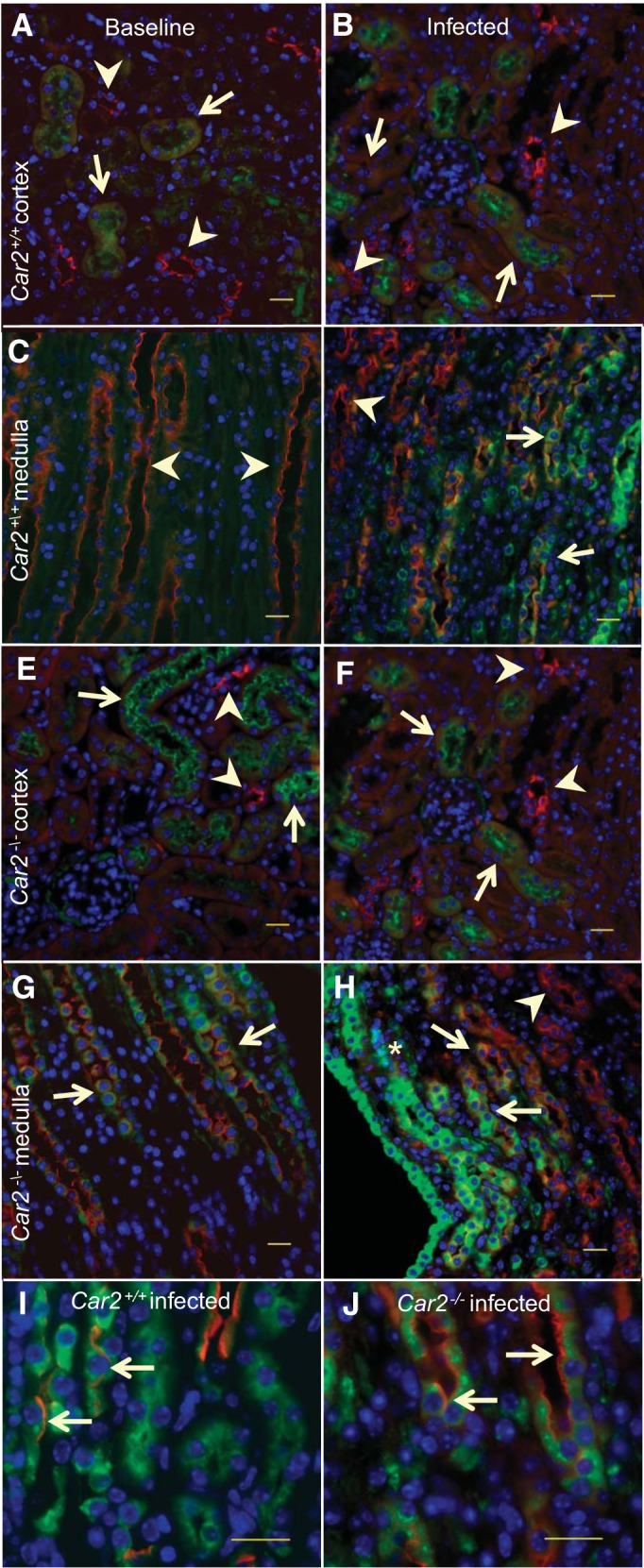
Neutrophil gelatinase-associated lipocalin (Ngal) immunoreactivity. Kidney Ngal (green) immunolabeling staining at baseline and during infection labeled for collecting ducts with aquaporin 2 (red). In the Car2+/+ cortex at baseline (A) and during infection (B), occasional proximal tubules demonstrated low immunoreactivity (arrows), but collecting ducts were negative (arrowheads). At baseline (C), no Car2+/+ medullary staining for Ngal was found in collecting ducts (arrowhead), but during infection (D), some collecting ducts stained for Ngal (arrows), whereas others did not (arrowheads). In Car2−/− mice at baseline (E) and during infection (F), cortical Ngal immunoreactivity was present in occasional proximal tubules (arrows) but not collecting ducts (arrowheads). At baseline (G), some occasional Car2−/− medullary collecting ducts immunolabeled with Ngal, with the intensity of the fluorescence increasing during infection (H). Areas of marked Ngal staining were scattered, usually in the area of neutrophil aggregations (*). Aquaporin-2-positive cells were also positive for Ngal (arrows) in Car2+/+ (I) and Car2−/− (J) infected medullas, indicating that principal cells can express Ngal in response to infection. Magnification: ×40 in A–D and ×100 in I and J. Three sections from 2 mice per genotype and infection status were reviewed. All scale bars = 20 μm.
DISCUSSION
We have demonstrated a significant increase in kidney bacterial burden and diminished antibacterial response in Car2−/− mice compared with Car2+/+ mice. We further confirmed the previous findings that Car2−/− mice have at least a threefold decrease in the percentage of ICs. CA-II helps generates the needed H+ for H+ V-ATPase; thus, even ICs that are present should conceptually have impaired function (9). Of note, aveolar macrophage antibacterial activity is impaired with V-ATPase inhibition, providing an example of overlap between acid-base and immune regulation (2).
A primary component of human renal CA-II deficiency and/or IC dysfunction is distal renal tubular acidosis (dRTA). dRTA results when urinary acidification in the collecting tubule is impaired, resulting in systemic metabolic acidosis and total body K+ depletion (33). Furthermore, urine pH remains inappropriately high due to the inability to secrete protons distally. Car2−/− mice have metabolic acidosis and hypokalemia along with relatively alkaline urine, consistent with dRTA. The association of CA-II deficiency and UTI risk is supported by reports of increased UTIs in humans with dRTA. First, Halperin et al. (17) reported that “most” of 10 unrelated dRTA patients had a history of UTI. Next, children and adults with familial hypomagnesaemia-hypercalciuria, a rare condition in which dRTA is a component, had UTIs in 3 of 4 subjects (75%) and 10 of 18 subjects (55%) in case series (34, 35). Finally, in a case series of patients with medullary sponge, a condition associated with dRTA, 14 of 21 female patients (67%) and 4 of 26 of male patients (15%) presented with bacteriuria (7, 19). The increased UTI susceptibility in Car2−/− mice we have identified appears consistent with our initial hypothesis that proper IC number and function is a crucial component of the innate defense of the urinary tract. Potential mechanisms for the increased UTI risk in Car2−/− mice include an increased infection risk from metabolic acidosis, changes in bacterial growth secondary to urine pH, increased infection susceptibility due to CA-II deficiency in other organ systems, IC depletion, or abnormal renal expression of CA-II across the kidneys and urinary tract. In the present study, we attempted to address all possible mechanisms to establish the significance of CA-II and ICs in the innate defense of the kidney.
Whether altered acid-base homeostasis contributes to infection risk is controversial. Immune function is supported by increased human neutrophil activation and macrophage phagocytosis during acidosis (29, 46). However, decreased neutrophil chemotaxis and increased infection-related hemodialysis mortality with acidosis support increased infection susceptibly (39, 47). In the present study, we demonstrated impaired renal bacterial clearance and an approximately >15-fold increased bacterial burden in Car2−/− compared with Car2+/+ kidneys with and without base supplementation. Additionally base-supplemented Car2−/− bladders had increased bacterial burdens, despite negligible CA-II bladder expression. Thus, factors other than base supplementation or local expression of CA-II are likely responsible for the infection risk. The increase in serum HCO3− was biologically relevant as base supplementation did result in improved growth in Car2−/− mice; however, this observation must be tempered as base supplementation only partially normalized the serum pH.
Alkaline urine pH from defective distal acidification is a characteristic of dRTA. Historically, acidification of urine with agents such as ascorbic acid has been proposed as a UTI treatment but has not been conclusively demonstrated to be effective (16). Furthermore, strains of uropathogens may have variable pH sensitivity (22). Our group has demonstrated that certain urinary antimicrobial peptides can have a wide variety of activity at different urinary pHs. Specifically, activity is reduced at extremes of pH and optimum around physiologic 7.4 (48). The host must balance altering growth conditions for bacteria while not detrimentally affecting its innate defenses. Car2−/− and base-supplemented Car2+/+ mice had similar mean pH readings, yet Car2−/− mice had significantly higher kidney and bladder bacterial burdens. Either factors other than urine pH are responsible for the UTI susceptibility in Car2−/− mice or the microenvironment around the collecting duct is altered by “local” pH adjustments not reflected by the overall urine pH.
Infection susceptibility due to CA-II deficiency in other organ systems is a potential mechanism for UTI risk in Car2−/− mice. The phenotype of Car2−/− mice has previously been reported to include impaired bone formation along with gastric cysts and pit cell hyperplasia (24, 27). The likelihood is low that these extrarenal manifestations contribute to UTI risk. Neutrophils contain CA-I and CA-II at a 3:1 concentration; however, the similar serum neutrophil number in Car2+/+ and Car2−/− mice does not suggest an inherent neutrophil deficiency in these cells (6).
Although systemic metabolic acidosis, impaired urine acidification, and extrarenal abnormalities associated with CA-II deficiency might contribute to infection risk, our findings suggest that they are not the primary etiology of UTI risk in Car2−/− mice. Therefore, we propose that loss of innate immune effectors produced by ICs contributes to UTI risk in Car2−/− mice. We (42, 43) have previously demonstrated that human ICs secrete RNase 7 into the urine and that inactivation of urinary RNase 7 results in a marked increase in bacterial growth. Ideally, we would evaluate RNase7 mRNA and protein expression as a target for decreased innate immune effectors with IC deficiency. However, no known mouse ortholog of RNase 7 has been identified (36). Car2−/− mice have increased mRNA expression of antibacterial peptides (Lcn2) at baseline. Lcn2/Ngal has previously been demonstrated to be expressed postrenal injury by renal ICs, the loop of Henle, and, in some reports, the proximal tubule (18, 28). Regardless of the etiology of the baseline increased Lcn2 expression in Car2−/− mice, it is not sufficient to prevent increased infection susceptibility and is associated with a decreased ability to upregulate Lcn2 expression in response to infection.
A marked inability to upregulate the bacterial response after UPEC inoculation was identified. In addition to their antimicrobial roles, cathelicidin and Ngal have previously been demonstrated to have anti-inflammatory properties. Cathelicidin blocks dendritic cell Toll-like receptor 4 activation (13). During pneumococcal pneumonia, Ngal attenuates the inflammatory response and bacterial clearance (49). Historically, CRP production was presumed restricted to the liver, but renal cortical epithelial cells have been identified as a second site for production (21). CRP mediates phagocytosis; therefore, the decreased Crp expression in Car2−/− mice is consistent with increased bacterial burden postinfection. Bacterial lipopolysaccharides induce inflammation by Cd14, Tnf, and Mefv (14, 23, 31). Inflammasomes, including those assembled by Nlcr4, are cytoplasmic complexes that recognize bacterial infections and activate cytokine production (50). The ability of Car2+/+ but not Car2−/− mice to significantly upregulate Cd14, Tnf, Nlcr4, and Mefv after UPEC inoculation further implicates a decreased inflammatory response with CA-II deficiency.
Renal ICs have several similarities to innate immune cells from other organs. Examples of epithelial cells with important innate immunity roles include type II pneumocytes of the lung and Paneth cells of the small intestine. ICs share common structural features with these aforementioned epithelial cells, including optimal strategic locations to prevent infection dissemination, apical microvilli, prominent endoplasmic reticulum and/or mitochrondria, and the production of innate immune proteins (1, 8).
While our study offers an exciting new correlation between CA-II deficiency and pyelonephritis risk, we do acknowledge some limitations. First, although serum HCO3− in Car2−/− mice normalized with base supplementation, the serum pH only partially corrected. Our antibacterial response PCR array evaluated whole kidney mRNA for differences between Car2−/− and wild-type mice. Because ICs, the primary site for renal CA-II expression, only account for a small percentage of total kidney cells, significant differences may be diluted out by the overall contribution of other cells. Inflammatory infiltrates during renal function have focal differences in occurrence and intensity; thus, we often had relatively large SDs, which resulted in a marked change in fold expression between genotypes and infection status but did not reach statistical significance. It is possible that key regulatory genes with statistically nonsignificant and/or lower fold changes have considerable biological relevance. The antibacterial response arrays only evaluate for differences between 84 key genes. While these genes are important in general in response to infection, IC-specific pathways and genes may not have been captured on the arrays. Because CA-II deficiency is global, extrarenal contributions to the observed phenotype cannot be completely ruled out. An important future research direction will be the generation of conditional knockout animals in which CA-II deficiency is limited to ICs. Such model systems can be used to conclusively demonstrate whether ICs are kidney epithelial innate immune cells, similar to Paneth cells of the small intestine. Additionally, whether altered CA-II expression impairs circulating neutrophil, macrophage, or dendritic cell function warrants extensive evaluation.
In conclusion, after transurethral UPEC inoculation, Car2−/− mice have increased kidney and bladder bacterial burdens, decreased kidney bacterial clearance, and a marked decreased inflammatory response. The findings of this study expand on the past descriptions of acid-base regulation and renal innate immunity to determine the functional immune consequences of CA-II deficiency. Car2−/− mice had decreased IC endowment, which indicates that Car2 is important to not just acid-base homeostasis but also to proper IC development and/or function. In light of the previous data, we propose that CA-II deficiency, potentially due to IC depletion, increases pyelonephritis risk.
GRANTS
A. L. Schwaderer and D. S. Hains are supported by National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant 1-RC4-DK-090937-01. G. J. Schwartz is supported by NIDDK Grant DK-050603.
DISCLOSURES
G. J. Schwartz has consulted for Novartis. Otherwise, the authors have no conflicting financial interests.
AUTHOR CONTRIBUTIONS
Author contributions: D.S.H., X.C., and A.L.S. conception and design of research; D.S.H., X.C., V.S., E.B.-B., W.F., R.E., B.B., and A.L.S. performed experiments; D.S.H., X.C., V.S., E.B.-B., W.F., R.E., B.B., G.J.S., and A.L.S. analyzed data; D.S.H., X.C., V.S., E.B.-B., W.F., B.B., G.J.S., and A.L.S. interpreted results of experiments; D.S.H., X.C., and A.L.S. prepared figures; D.S.H. and A.L.S. drafted manuscript; D.S.H., X.C., V.S., E.B.-B., R.E., B.B., G.J.S., and A.L.S. edited and revised manuscript; D.S.H., X.C., V.S., E.B.-B., W.F., R.E., B.B., G.J.S., and A.L.S. approved final version of manuscript.
ACKNOWLEDGMENTS
Some of the data reported in the present study have been submitted in abstract form to American Society of Nephrology 2014 Renal Week (Abstract 364).
Footnotes
Supplemental Material for this article is available at the American Journal of Physiology-Renal Physiology website.
REFERENCES
- 1.Bevins CL, Salzman NH. Paneth cells, antimicrobial peptides and maintenance of intestinal homeostasis. Nat Rev Microbiol 9: 356–368, 2011 [DOI] [PubMed] [Google Scholar]
- 2.Bidani A, Reisner BS, Haque AK, Wen J, Helmer RE, Tuazon DM, Heming TA. Bactericidal activity of alveolar macrophages is suppressed by V-ATPase inhibition. Lung 178: 91–104, 2000 [DOI] [PubMed] [Google Scholar]
- 3.Breton S. The cellular physiology of carbonic anhydrases. J Pancreas 2: 159–164, 2001 [PubMed] [Google Scholar]
- 4.Breton S, Alper SL, Gluck SL, Sly WS, Barker JE, Brown D. Depletion of intercalated cells from collecting ducts of carbonic anhydrase II-deficient (CAR2 null) mice. Am J Physiol Renal Fluid Electrolyte Physiol 269: F761–F774, 1995 [DOI] [PubMed] [Google Scholar]
- 5.Brown D, Kumpulainen T, Roth J, Orci L. Immunohistochemical localization of carbonic anhydrase in postnatal and adult rat kidney. Am J Physiol Renal Fluid Electrolyte Physiol 245: F110–F118, 1983 [DOI] [PubMed] [Google Scholar]
- 6.Campbell AR, Andress DL, Swenson ER. Identification and characterization of human neutrophil carbonic anhydrase. J Leukoc Biol 55: 343–348, 1994 [DOI] [PubMed] [Google Scholar]
- 7.Carboni I, Andreucci E, Caruso MR, Ciccone R, Zuffardi O, Genuardi M, Pela I, Giglio S. Medullary sponge kidney associated with primary distal renal tubular acidosis and mutations of the H+-ATPase genes. Nephrol Dial Transplant 24: 2734–2738, 2009 [DOI] [PubMed] [Google Scholar]
- 8.Castranova V, Rabovsky J, Tucker JH, Miles PR. The alveolar type II epithelial cell: a multifunctional pneumocyte. Toxicol Appl Pharmacol 93: 472–483, 1988 [DOI] [PubMed] [Google Scholar]
- 9.Chambrey R, Kurth I, Peti-Peterdi J, Houillier P, Purkerson JM, Leviel F, Hentschke M, Zdebik AA, Schwartz GJ, Hubner CA, Eladari D. Renal intercalated cells are rather energized by a proton than a sodium pump. Proc Natl Acad Sci USA 110: 7928–7933, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chassin C, Goujon JM, Darche S, du Merle L, Bens M, Cluzeaud F, Werts C, Ogier-Denis E, Le Bouguenec C, Buzoni-Gatel D, Vandewalle A. Renal collecting duct epithelial cells react to pyelonephritis-associated Escherichia coli by activating distinct TLR4-dependent and -independent inflammatory pathways. J Immunol 177: 4773–4784, 2006 [DOI] [PubMed] [Google Scholar]
- 11.Cheeseman MT, Tyrer HE, Williams D, Hough TA, Pathak P, Romero MR, Hilton H, Bali S, Parker A, Vizor L, Purnell T, Vowell K, Wells S, Bhutta MF, Potter PK, Brown SD. HIF-VEGF pathways are critical for chronic otitis media in Junbo and Jeff mouse mutants. PLOS Genet 7: e1002336, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Da Silva N, Pisitkun T, Belleannee C, Miller LR, Nelson R, Knepper MA, Brown D, Breton S. Proteomic analysis of V-ATPase-rich cells harvested from the kidney and epididymis by fluorescence-activated cell sorting. Am J Physiol Cell Physiol 298: C1326–C1342, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Nardo A, Braff MH, Taylor KR, Na C, Granstein RD, McInturff JE, Krutzik S, Modlin RL, Gallo RL. Cathelicidin antimicrobial peptides block dendritic cell TLR4 activation and allergic contact sensitization. J Immunol 178: 1829–1834, 2007 [DOI] [PubMed] [Google Scholar]
- 14.Diaz A, Hu C, Kastner DL, Schaner P, Reginato AM, Richards N, Gumucio DL. Lipopolysaccharide-induced expression of multiple alternatively spliced MEFV transcripts in human synovial fibroblasts: a prominent splice isoform lacks the C-terminal domain that is highly mutated in familial Mediterranean fever. Arthritis Rheum 50: 3679–3689, 2004 [DOI] [PubMed] [Google Scholar]
- 15.Gauer S, Sichler O, Obermuller N, Holzmann Y, Kiss E, Sobkowiak E, Pfeilschifter J, Geiger H, Muhl H, Hauser IA. IL-18 is expressed in the intercalated cell of human kidney. Kidney Int 72: 1081–1087, 2007 [DOI] [PubMed] [Google Scholar]
- 16.Geerlings SE, Brouwer EC, Gaastra W, Verhoef J, Hoepelman AI. Effect of glucose and pH on uropathogenic and non-uropathogenic Escherichia coli: studies with urine from diabetic and non-diabetic individuals. J Med Microbiol 48: 535–539, 1999 [DOI] [PubMed] [Google Scholar]
- 17.Halperin ML, Goldstein MB, Haig A, Johnson MD, Stinebaugh BJ. Studies on the pathogenesis of type I (distal) renal tubular acidosis as revealed by the urinary Pco2 tensions. J Clin Invest 53: 669–677, 1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han M, Li Y, Liu M, Li Y, Cong B. Renal neutrophil gelatinase associated lipocalin expression in lipopolysaccharide-induced acute kidney injury in the rat. BMC Nephrol 13: 25, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Harrison AR, Rose GA. Medullary sponge kidney. Urol Res 7: 197–207, 1979 [DOI] [PubMed] [Google Scholar]
- 20.Hasler U, Mordasini D, Bens M, Bianchi M, Cluzeaud F, Rousselot M, Vandewalle A, Feraille E, Martin PY. Long term regulation of aquaporin-2 expression in vasopressin-responsive renal collecting duct principal cells. J Biol Chem 277: 10379–10386, 2002 [DOI] [PubMed] [Google Scholar]
- 21.Jabs WJ, Logering BA, Gerke P, Kreft B, Wolber EM, Klinger MH, Fricke L, Steinhoff J. The kidney as a second site of human C-reactive protein formation in vivo. Eur J Immunol 33: 152–161, 2003 [DOI] [PubMed] [Google Scholar]
- 22.Kaye D. Antibacterial activity of human urine. J Clin Invest 47: 2374–2390, 1968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kotlyarov A, Neininger A, Schubert C, Eckert R, Birchmeier C, Volk HD, Gaestel M. MAPKAP kinase 2 is essential for LPS-induced TNF-α biosynthesis. Nat Cell Biol 1: 94–97, 1999 [DOI] [PubMed] [Google Scholar]
- 24.Leppilampi M, Parkkila S, Karttunen T, Gut MO, Gros G, Sjoblom M. Carbonic anhydrase isozyme-II-deficient mice lack the duodenal bicarbonate secretory response to prostaglandin E2. Proc Natl Acad Sci USA 102: 15247–15252, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Longo-Guess CM, Gagnon LH, Fritzsch B, Johnson KR. Targeted knockout and lacZ reporter expression of the mouse Tmhs deafness gene and characterization of the hscy-2J mutation. Mamm Genome 18: 646–656, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lowry R. VassarStats: Website for Statistical Computation (online) http://vassarstats.net/ [4 September 2014]. [Google Scholar]
- 27.Margolis DS, Szivek JA, Lai LW, Lien YH. Phenotypic characteristics of bone in carbonic anhydrase II-deficient mice. Calcified Tissue Int 82: 66–76, 2008 [DOI] [PubMed] [Google Scholar]
- 28.Paragas N, Qiu A, Zhang Q, Samstein B, Deng SX, Schmidt-Ott KM, Viltard M, Yu W, Forster CS, Gong G, Liu Y, Kulkarni R, Mori K, Kalandadze A, Ratner AJ, Devarajan P, Landry DW, D'Agati V, Lin CS, Barasch J. The Ngal reporter mouse detects the response of the kidney to injury in real time. Nat Med 17: 216–222, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Park SY, Bae DJ, Kim MJ, Piao ML, Kim IS. Extracellular low pH modulates phosphatidylserine-dependent phagocytosis in macrophages by increasing stabilin-1 expression. J Biol Chem 287: 11261–11271, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Purkerson JM, Schwartz GJ. The role of carbonic anhydrases in renal physiology. Kidney Int 71: 103–115, 2007 [DOI] [PubMed] [Google Scholar]
- 31.Rietschel ET, Schletter J, Weidemann B, El-Samalouti V, Mattern T, Zahringer U, Seydel U, Brade H, Flad HD, Kusumoto S, Gupta D, Dziarski R, Ulmer AJ. Lipopolysaccharide and peptidoglycan: CD14-dependent bacterial inducers of inflammation. Microb Drug Resist 4: 37–44, 1998 [DOI] [PubMed] [Google Scholar]
- 32.Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, Hashim AI, Morse DL, Raghunand N, Gatenby RA, Gillies RJ. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res 69: 2260–2268, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol 13: 2160–2170, 2002 [DOI] [PubMed] [Google Scholar]
- 34.Rodriguez-Soriano J, Vallo A. Pathophysiology of the renal acidification defect present in the syndrome of familial hypomagnesaemia-hypercalciuria. Pediatr Nephrol 8: 431–435, 1994 [DOI] [PubMed] [Google Scholar]
- 35.Rodriguez-Soriano J, Vallo A, Garcia-Fuentes M. Hypomagnesaemia of hereditary renal origin. Pediatr Nephrol 1: 465–472, 1987 [DOI] [PubMed] [Google Scholar]
- 36.Rosenberg HF. RNase A ribonucleases and host defense: an evolving story. J Leukoc Biol 83: 1079–1087, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Royaux IE, Wall SM, Karniski LP, Everett LA, Suzuki K, Knepper MA, Green ED. Pendrin, encoded by the Pendred syndrome gene, resides in the apical region of renal intercalated cells and mediates bicarbonate secretion. Proc Natl Acad Sci USA 98: 4221–4226, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schuster VL, Fejes-Toth G, Naray-Fejes-Toth A, Gluck S. Colocalization of H+-ATPase and band 3 anion exchanger in rabbit collecting duct intercalated cells. Am J Physiol Renal Fluid Electrolyte Physiol 260: F506–F517, 1991 [DOI] [PubMed] [Google Scholar]
- 39.Simchowitz L, Cragoe EJ., Jr Regulation of human neutrophil chemotaxis by intracellular pH. J Biol Chem 261: 6492–6500, 1986 [PubMed] [Google Scholar]
- 40.Sly WS, Hu PY. Human carbonic anhydrases and carbonic anhydrase deficiencies. Annu Rev Biochem 64: 375–401, 1995 [DOI] [PubMed] [Google Scholar]
- 41.Spencer JD, Hains DS, Porter E, Bevins CL, DiRosario J, Becknell B, Wang H, Schwaderer AL. Human α defensin 5 expression in the human kidney and urinary tract. PLOS ONE 7: e31712, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Spencer JD, Schwaderer AL, Dirosario JD, McHugh KM, McGillivary G, Justice SS, Carpenter AR, Baker PB, Harder J, Hains DS. Ribonuclease 7 is a potent antimicrobial peptide within the human urinary tract. Kidney Int 80: 174–180, 2011 [DOI] [PubMed] [Google Scholar]
- 43.Spencer JD, Schwaderer AL, Wang H, Bartz J, Kline J, Eichler T, DeSouza KR, Sims-Lucas S, Baker P, Hains DS. Ribonuclease 7, an antimicrobial peptide upregulated during infection, contributes to microbial defense of the human urinary tract. Kidney Int 83: 615–625, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Svensson M, Irjala H, Alm P, Holmqvist B, Lundstedt AC, Svanborg C. Natural history of renal scarring in susceptible mIL-8Rh−/− mice. Kidney Int 67: 103–110, 2005 [DOI] [PubMed] [Google Scholar]
- 45.Tittel AP, Heuser C, Ohliger C, Knolle PA, Engel DR, Kurts C. Kidney dendritic cells induce innate immunity against bacterial pyelonephritis. J Am Soc Nephrol 22: 1435–1441, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Trevani AS, Andonegui G, Giordano M, Lopez DH, Gamberale R, Minucci F, Geffner JR. Extracellular acidification induces human neutrophil activation. J Immunol 162: 4849–4857, 1999 [PubMed] [Google Scholar]
- 47.Vashistha T, Kalantar-Zadeh K, Molnar MZ, Torlen K, Mehrotra R. Dialysis modality and correction of uremic metabolic acidosis: relationship with all-cause and cause-specific mortality. Clin J Am Soc Nephrol 8: 254–264, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang H, Schwaderer AL, Kline J, Spencer JD, Kline D, Hains DS. Contribution of structural domains to the activity of ribonuclease 7 against uropathogenic bacteria. Antimicrob Agents Chemother 57: 766–774, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Warszawska JM, Gawish R, Sharif O, Sigel S, Doninger B, Lakovits K, Mesteri I, Nairz M, Boon L, Spiel A, Fuhrmann V, Strobl B, Muller M, Schenk P, Weiss G, Knapp S. Lipocalin 2 deactivates macrophages and worsens pneumococcal pneumonia outcomes. J Clin Invest 123: 3363–3372, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhao Y, Yang J, Shi J, Gong YN, Lu Q, Xu H, Liu L, Shao F. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477: 596–600, 2011 [DOI] [PubMed] [Google Scholar]