

Abstract
Recent studies suggest that VEGF contributes to hypoxic remodeling of arterial smooth muscle, although hypoxia produces only transient increases in VEGF that return to normoxic levels despite sustained changes in arterial structure and function. To explore how VEGF might contribute to long-term hypoxic vascular remodeling, this study explores the hypothesis that chronic hypoxia produces sustained increases in smooth muscle VEGF receptor density that mediate long-term vascular effects of hypoxia. Carotid arteries from adult sheep maintained at sea level or altitude (3,820 m) for 110 days were harvested and denuded of endothelium. VEGF levels were similar in chronically hypoxic and normoxic arteries, as determined by immunoblotting. In contrast, VEGF receptor levels were significantly increased by 107% (VEGF-R1) and 156% (VEGF-R2) in hypoxic compared with normoxic arteries. In arteries that were organ cultured 24 h with 3 nM VEGF, VEGF replicated effects of hypoxia on abundances of smooth muscle α actin (SMαA), myosin light chain kinase (MLCK), and MLC20 and the effects of hypoxia on colocalization of MLC20 with SMαA, as measured via confocal microscopy. VEGF did not replicate the effects of chronic hypoxia on colocalization of MLCK with SMαA or MLCK with MLC20, suggesting that VEGF's role in hypoxic remodeling is highly protein specific, particularly for contractile protein organization. VEGF effects in organ culture were inhibited by VEGF receptor blockers vatalinib (240 nM) and dasatinib (6.3 nM). These findings support the hypothesis that long-term upregulation of VEGF receptors help mediate sustained effects of hypoxia on the abundance and colocalization of contractile proteins in arterial smooth muscle.
Keywords: chronic hypoxia, confocal colocalization, myosin light chain kinase, regulatory myosin light chain, smooth muscle α-actin
blood vessels regularly undergo structural changes in response to changing patterns of endogenous and exogenous stimuli (4, 40, 47, 48). These continuous changes in blood vessel cellular and extracellular structure are collectively known as remodeling and enable functional adaptation and specialization (24, 42). Vascular remodeling serves both normal physiological adaptations, as with uterine artery remodeling during pregnancy (4), and also with pathological remodeling, as occurs during atherosclerosis (44) and tumorigenesis (7, 9, 34). Whereas vascular remodeling is clearly a prominent and dynamic process in all vascular beds, the factors involved with its initiation and maintenance remain uncertain.
One of the most widely studied initiators of vascular remodeling is hypoxia, which can promote both physiological (13) and pathological (18) changes in vessel structure and function. Reduced oxygen availability can act through multiple pathways to alter the phenotypic characteristics and contractile function of vascular smooth muscle and thereby optimize tissue perfusion and oxygenation (17, 23, 33, 54). Whereas a plethora of evidence supports endothelial (32) and adventitial (14) expansion as major components of vascular remodeling, more recent findings suggest that hypoxic remodeling also involves the medial layer of the artery wall (23, 25). In addition, vascular remodeling appears to involve not only myocytes in the medial layer but also smooth muscle progenitor cells that originate in the bone marrow, migrate to and reside in the adventitial layer of the vessel wall, and then differentiate into myofibroblasts in the medial layer (31). Another key feature of medial smooth muscle cells is that they retain remarkable plasticity and can dedifferentiate from contractile cells into more synthetic, proliferative, and migratory phenotypes in response to many types of environmental stimuli (51). How hypoxia influences smooth muscle plasticity and differentiation remains poorly understood, particularly in nonpulmonary arteries.
Efforts to better understand the mechanisms of hypoxic vascular remodeling have strongly implicated the transcription factor hypoxia inducible factor (HIF) as a primary initiator of multiple gene transcription events that coordinate responses to hypoxia and culminate in increased production of erythropoietin, glycolytic enzymes, and angiogenic factors (5, 38, 43). The most important angiogenic factor induced by HIF is vascular endothelial growth factor (VEGF), which directly stimulates angiogenesis and neovascularization through activation of broadly distributed specific tyrosine kinase receptors (21). Interestingly, hypoxia-induced increases in VEGF typically attain peak values within 48 h and then return back to basal levels within 3 wk of hypoxic exposure (27). Despite the transient nature of the responses of VEGF to hypoxia, the remodeling effects of hypoxia can persist for many weeks and months (20, 28, 35). This pattern of vascular responses to hypoxia raises multiple questions about the role of VEGF in long-term hypoxic vascular remodeling. One possible explanation is that hypoxia promotes short-term increases in VEGF, but long-term increases in VEGF receptors, as suggested to occur in fetal ovine carotid arteries from ewes acclimatized at altitude for 110 days (1). Consistent with this observation, other studies have also shown that hypoxia can increase the density of VEGF R1 and R2 receptors (3, 19, 50).
In light of the published evidence, the current study explores the hypothesis that chronic hypoxia produces a sustained increase in the density of vascular VEGF receptors, which in turn helps mediate the long-term effects of hypoxia on vascular remodeling. This hypothesis builds on our previous work demonstrating that VEGF can contribute to hypoxic vascular remodeling of fetal arteries through alteration of contractile protein abundances and colocalization (1, 8, 23). To test this hypothesis, the experimental approach employed a well-established model of chronic hypoxia in which adult sheep were rendered hypoxic by housing them at an altitude of 3,820 m for 110 days; normoxic controls were maintained at sea level (29). Assessments of hypoxic changes in composition and interactions among contractile proteins focused on 1) SMαA, which is the most abundant contractile protein in smooth muscle cells (52); 2) regulatory myosin light chain (MLC20), which is a key regulator of contraction; and 3) myosin light chain kinase (MLCK), which is the dedicated kinase that phosphorylates and activates MLC20 (16, 46). To assess the direct effects of VEGF on arterial wall remodeling, the experimental approach included organ-culture with VEGF, as previously described in detail (1, 8). Together, these experiments provided a unique perspective of the role of VEGF and VEGF receptors in long-term hypoxic vascular remodeling of large nonpulmonary arteries.
MATERIALS AND METHODS
Protocols, techniques, and procedures used in all animal experiments were approved by the Animal Research Committee of Loma Linda University and also complied with policies and codes of practice recommended by the National Institutes of Health in the NIH Guide for Care and Use of Laboratory Animals.
Tissue harvest and preparation.
Common carotid arteries harvested from adult nonpregnant sheep were used in all experiments. These sheep were killed while fully anesthetized with 100 mg/kg sodium pentobarbital administered intravenously. Control animals (sheep maintained at Loma Linda, CA, altitude 355 m) yielded normoxic arteries, whereas sheep subjected to our chronic hypoxia model provided hypoxic arteries. This model involves acclimatization of nongravid sheep at altitude of 3,820 m above sea level, thereby yielding arterial oxygen tension of 64 ± 2 Torr in the animals. Correspondingly, normoxic arterial oxygen tensions equal 102 ± 2 Torr. Harvested arteries were kept in sterile HEPES buffer solution composed of (in mM) 122.1 NaCl, 25 HEPES, 5.2 KCl, 2.4 MgSO4, 11.1 dextrose, 1.6 CaCl2, and 50 μM EDTA. Loose parenchymal and adventitial tissue as well as blood was removed from the arteries, after which endothelial denudation was achieved via luminal mechanical abrasion. All experimental protocols made use of 3-mm-long arterial segments. Precise arterial length and medial thicknesses were determined using an Olympus U-PMTVC Optical microscope mounted with a Scion Visicapture Twain 1394 Camera for image capture. ImagePro software (v6.0, Media Cybernetics) was used to measure variables from the images captured by the camera.
Contractility studies.
For contractility measurements, artery segments measuring 3-mm in length were mounted on paired tungsten wires between a low-compliance isometric force transducer and a rod attached to a micrometer used for precise variation of resting tension. Each segment was equilibrated at 38.5°C (normal ovine core temperature) for 1 h in calcium replete Na bicarbonate Krebs solution (pH 7.4) composed of (in mM) 122 NaCl, 25.6 NaHCO3, 5.17 KCl, 2.56 dextrose, 2.49 MgSO4, 1.60 CaCl2, 0.114 ascorbic acid, and 0.027 EGTA. Artery segments were kept viable by continuous bubbling of the incubation buffer with 95% oxygen with 5% carbon dioxide. Working diameters (D) required to attain strain ratios of 1.5, 1.8, 2.1, 2.3, 2.7, 3.0, and 3.3 were calculated relative to unstressed artery diameter, Do (measured at a passive tension of 0.03 g). Contractile responses at each stretch ratio to isotonic potassium Krebs composed of (in mM) 122 KCl, 11.1 dextrose, 5.16 NaCl, 2.50 MgSO4, 2.15 NaHCO3, 1.60 CaCl2, 0.114 ascorbic acid, and 0.027 EDTA were recorded after equilibration in Na Krebs. After each maximum contraction, arteries were washed and left to stabilize in Na Krebs before the next stretch ratio was achieved. Once contractions in response to the highest stretch ratio were recorded, the active component of the artery was eliminated by freezing in liquid nitrogen and left to equilibrate in 3 mM EGTA solution. Passive arterial stresses were subsequently measured at each strain ratio starting with the highest stretch ratio to the lowest. Spontaneous myogenic tone was defined as the difference in tone generated before and after freezing the artery segment, whereas arterial stiffness was calculated from the passive stresses using the Young's modulus model as previously described (1, 8).
Fluorescent immunohistochemistry.
Freshly harvested as well as organ-cultured arteries were fixed overnight in EM-grade 4% paraformaldehyde (Electron Microscopy Sciences, #15713S) and then embedded in paraffin before sectioning at 5 μm. Immunohistochemical processing of the tissues commenced with initial deparaffinization in Histoclear solution (National Diagnostic, Atlanta, GA, #HS-200) and rehydration in graded concentrations of alcohol in descending order. The tissue samples were subsequently microwaved in a 6.03 pH citrate buffer to recover antigenicity. Permeabilization of the sections in 0.1% Triton X-100 (Sigma Aldrich, St. Louis, MO, #T-8787) was done before incubation in 1% bovine serum albumin (Santa Cruz Biotechnology, Santa Cruz, CA, #SC-2323) to ensure blockade of nonspecific interactions.
Confocal microscopy.
Using the 5 μm arterial sections generated as previously described in Fluorescent immunohistochemistry, double staining of the sections was done using primary antibodies reactive with three different pairs of contractile proteins (SMαA-MLCK, SMαA-MLC20, and MLCK-MLC20). After an overnight primary incubation, the sections were washed in phosphate buffer solution (PBS) and left to equilibrate with species-matched secondary antibodies labeled with Dylight 488 (SMαA) and 633 (MLCK) for SMαA-MLCK colocalization, 488 (SMαA) and 633 (MLC20) for SMαA-MLC20 colocalization, and 488(MLC20) and 633 (MLCK) for MLCK-MLC20 colocalization. Secondary incubation was done for 2 h at room temperature in a dark room to minimize photo bleaching. Imaging was done with an Olympus FV1000 at a lateral resolution of 200 nm, optical section width of 700 nm, and a numerical aperture of 18. Colocalization indexes were completed using a nonparametric quadrant analysis as previously described (11). Sources and titer ratios of the primary antibodies include: Sigma-Aldrich for monoclonal anti-MLC20 (#M4401) at 1:300, monoclonal anti-SMαA (A5228) at 1:200, Abcam (Cambridge, MA) polyclonal anti- SMαA (Ab 5694) at 1:100, and Santa Cruz Biotechnology for polyclonal MYLK (SC-25428) at 1:50. Whereas all software-based image analysis was performed on raw images, the brightness of some images shown in the figures was increased to improve visual clarity and resolution; such changes are indicated in the corresponding figures.
Westerns.
Carotid artery segments of known weights were homogenized using a glass-on-glass method in a high urea (8 M) extraction buffer containing (in mM) 500 NaCl, 23 glycine, 20 Tris, 10 EGTA, and 10% glycerol at pH 8.6. Protease inhibitor cocktail (Sigma-Aldrich, #M1745) at 5 μl/ml of buffer was also added to the extraction buffer. After the tissues were ground in the extraction buffer, the homogenate samples were centrifuged at 5,000 g for 20 min. Protein concentrations were determined using the Bio-Rad Bradford assay. Post centrifugation, homogenates along with progressively increasing concentrations of standards used to calibrate target protein abundance were separated with an SDS-PAGE set-up. Separated proteins were electrophoretically transferred from the gel matrix onto nitrocellulose membranes at 200 mA current for 1.5 h in Towbin's buffer containing 192 mM glycine, 25 mM Tris, and 10% to 20% methanol. The transfer process was conducted with ice packs on both sides of the transfer tank to mitigate the heat generated via electrophoresis. After transfer onto nitrocellulose, the membranes were blocked for 1 h with Tris buffered saline containing 5% nonfat dry milk (M-TBS) while applying gentle shaking. Afterward, membranes were washed in a detergent (0.1% Tween-20) containing MTBS and then incubated with primary antibodies for 3 h using 1:3,000 for SMαA, 1:10,000 for MLCK, 1:200 for MLC20 [all three from Sigma Aldrich as stated in Confocal microscopy and 1:750 for VEGF-A165 (from Abcam, #AB119)]. After primary incubation, secondary antibody conjugated to Dylight 800 (Pierce Chemical, Rockford, IL, #46422) was applied to the membranes for 90 min before imaging was completed via a LI-COR Bioscience Odyssey system.
Tissue homogenization for quantifying VEGF receptors was initiated via a glass pestle and mortar using an extraction buffer containing (in mM) 500 NaCl, 50 Tris, and 5 EDTA at pH 7.4. Six different protease inhibitors including (in μM) 500 AEBSF, 400 pepstatin-A, 20 bestatin, 10 E-64, 7.5 leupeptin, and 7 aprotinin (all purchased from Sigma-Aldrich) were also added to the buffer. A 1:50 tissue extraction buffer ratio was used. Next, centrifugation of the homogenate was done at 100,000 g for 1 h at 4°C, after which the pellet was resuspended into the buffer at a 1:10 ratio with the addition of (in mM) 150 NaCl, 50 Tris, 10 DTT, 1% Triton X-100, 0.5% Na deoxycholate, 0.2% SDS, and 10% glycerol with same concentrations of protease inhibitors listed above. After this, the homogenate was ultrasonicated at 20% amplitude to shear DNA for 6 times at 5 s each, centrifuged again at 10,000 g for 15 min. Collected supernatants were assayed for total protein concentrations via Bradford's protein assay. Separation of the proteins was completed using a 5% SDS-PAGE with the addition of β-mercaptoethanol (BME- 35 mM) in the upper part of the tank with the buffer. As stated above, standard pooled reference samples were included in the separation gel lanes. Proteins from the electrophoresis gel were transferred onto a nitrocellulose membrane using Towbin's buffer at 350 mA for 1.5 h. Towbin's buffer used here is same as the one described previously but with the addition of 35 mM of BME, 0.01% SDS, and 20% methanol only. With the proteins now successfully transferred on membranes, blocking was done with MTBS for 60 min at room temperature while applying gentle shaking. Membranes were subsequently washed in 0.1% Tween containing MTBS. Incubation with primary antibodies was done for 3 h using dilutions for VEGF R2 receptor (SC-504) and for VEGF R1 receptor (SC-316) both purchased from Santa Cruz using titers of 1:200. After primary incubation, membranes were washed for 5 min × 6 before secondary incubation commenced for 90 min. The same secondary antibody as the one described in the previous paragraph was used. Next, membranes were washed in TBS only for 5 min × 6 before the membranes were imaged with a LI-COR Bioscience's Odyssey system.
Organ culture.
To determine the effects of VEGF and its receptor antagonists compared with those of hypoxia, arterial segments from both normoxic and hypoxic sheep were maintained in media (Dulbecco's modified Eagle media) containing 70 μg/ml of Gentamycin (Gibco, Carlsbad, CA, #15750–060), 4 mM glutamine (Sigma Aldrich, St. Louis, MO #G7513), 3.7 g/l of Na2HCO3, 2% antibiotic-antimycotic solution (Gibco, #15240–096), 1% nonessential amino acid solution (Sigma Aldrich, #M7145), 0.5% amino acid solution (Sigma Aldrich, #M5550) in a 12-well untreated plate. Incubation was done in a humidified incubator with 5% CO2 at 37°C. Control arteries were defined as those kept in media for an additional 24 h after the initial 24 h, whereas those incubated in media containing 3 ng/ml of VEGF, 240 nM of vatalinib, and 6.3 nM of dasatinib served to determine the effect of VEGF and VEGF tyrosine kinase receptors. Treatment groups were defined as those that were kept in media for an initial 24 h and then an additional 24 h in media containing the various drugs.
Data analysis and statistics.
Each animal killed contributed to segments used in fresh, control, VEGF, and VEGF receptor antagonist treatment groups. Normal distribution of data sets was tested using the D'Agostino-Pearson analysis, whereas contractile stress measurements, wall thicknesses, and stiffness were compared using analysis of variance (ANOVA). Within ANOVA, homogeneity was verified using a Bartlett's-Cochran test, and statistical power was at least 0.8. In calculating contractile stresses, a robust computation involving force generated by graded strains (tension in grams × acceleration due to gravity) per unit area (length × working wall thickness, μm) was used.
For Western blotting, unknown concentrations of protein of interest were measured against a standard curve generated from a pooled reference of ovine adult carotid tissues. Importantly, exactly the same standards were used on all gels for each individual protein examined. The standard protein masses loaded per lane ranged from 0.16 to 5.00 μg for MLCK, 0.16 to 5.00 μg for MLC20, 0.01 to 0.50 μg for SMαA, 0.63 to 15.00 μg for VEGF protein, 4 to 338 μg for VEGF-R2, and 1.2 to 97.7 μg for VEGF-R1. For these proteins, the unknown masses loaded per lane ranged from 1.7 to 30.0 μg for MLCK, 1.7 to 2.0 μg for MLC20, 0.17 to 0.20 μg for SMαA, 3 to 19 μg for VEGF, 40 to 85 μg for VEGF-R2, and 15 to 60 μg for VEGF-R1. For all gels, the mass of protein loaded was optimized to fall within the dynamic range defined by the standards loaded on the same gel. The standard curve mass values were used to estimate the coefficients of the logistic equation (mass determined optical density), and then the inverse form of this equation (optical density determined mass) was used to directly calculate the equivalent mass for each band measured in each gel. This equivalent mass for each unknown was then normalized to the mass of protein loaded. In this way, we determined a value by direct calculation that quantified the abundance of each band relative to the mass loaded, and it was this final value that was compared among the different experimental groups and conditions. In this way, differences in standard curve range and mass loaded were fully accounted for in all calculations of abundance. These approaches controlled for membrane-to-membrane variations in antibody avidity, blocking efficiency, and intensity of illumination during quantitation. This approach also enabled calculation of unknowns as relative abundances in units of micrograms unknown per micrograms standard. One limitation of this approach was the absence of loading controls, which would imply that pipetting errors could go unnoticed. These errors, however, would still contribute to overall measurement errors. Relative abundances from normoxic and hypoxic groups were compared using two-way ANOVA. To facilitate comparisons between the effects of chronic hypoxia and responses to VEGF, the relative abundances for all contractile proteins, as measured by immunoblot, were normalized relative to values measured in serum-starved normoxic arteries in the absence of VEGF.
RESULTS
A total of 10 normoxic and 11 hypoxic sheep were used in this study. The normoxic sheep contributed 108 arterial segments and hypoxic ones contributed 91 segments. In all cases, n denotes number of sheep used in each protocol and P < 0.05 represents comparative results of statistical significance. Data here were presented as mean ± SE or mean ± SD as indicated.
Chronic hypoxia alters arterial structure and contractility.
Chronic hypoxia significantly increased medial thickness by 24% (Fig. 1, left) and increased arterial stiffness by 23% (Fig. 1, middle). Conversely, chronic hypoxia significantly decreased the stretch ratio required to attain peak myogenic tone by 20% (Fig. 1, right) and decreased peak myogenic tone by 43% (Fig. 1, right).
Fig. 1.
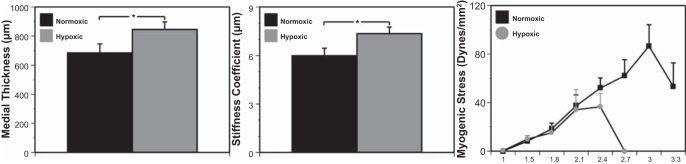
Arterial structure and contractility are altered by chronic hypoxia. Arteries from sheep subjected to hypoxic acclimatization exhibited increased medial thickness (left) and stiffness (middle) compared with the normoxic control group. In contrast, stress strain experiments showed a decreased maximum stretch ratio as well as peak myogenic tone in arteries from hypoxic sheep compared with normoxic control sheep (right). Results are presented as means ± SE for n = 8 for normoxic arteries and n = 11 for hypoxic arteries. *P < 0.05 denotes comparative significant differences.
Chronic hypoxia alters smooth muscle contractile protein expression.
Chronic hypoxia exerted unique protein-specific changes in abundances of contractile proteins. Quantification via immunoblotting showed that hypoxia increased SMαA by 54%, decreased MLCK by 47%, and increased MLC20 by 69% (Fig. 2).
Fig. 2.

Chronic hypoxia differentially alters smooth muscle contractile protein expression. Quantification of smooth muscle contractile proteins via Western immunoblotting assays revealed a significantly higher level of smooth muscle alpha actin (SMαA; left) and 20 kDa regulatory myosin light chain (MLC20, right) in hypoxic arteries compared with normoxic control arteries. In sharp contrast, myosin light chain kinase (MLCK) abundance decreased (middle) with chronic hypoxia compared with normoxic values. These results suggest a highly protein-specific alteration of hypoxia in these ovine carotid arteries. Immunohistochemistry images, enhanced for brightness and contrast at a value of 60, show similar trends as Western blotting for all three contractile proteins (bottom). Results here are presented as means ± SE for n = 7 for all normoxic groups and n = 6 for all hypoxic groups. Comparative significant differences: *P < 0.05.
Effects of chronic hypoxia on contractile protein colocalization.
Chronic hypoxia did not change MLCK-SMαA colocalization (Fig. 3, left), increased MLC20-SMαA colocalization by 704% (Fig. 3, middle), and increased MLCK-MLC20 colocalization by 335% (Fig. 3, right).
Fig. 3.

Effects of chronic hypoxia on contractile protein colocalization. Interactions between smooth muscle contractile proteins assayed through confocal colocalization microscopy revealed a 704% increase in MLC20-SMαA colocalization and a 335% increase in MLCK-MLC20 colocalization in hypoxic compared with normoxic arteries. For MLCK-SMαA colocalization, however, coefficients were similar in both normoxic and hypoxic arteries. Immunohistochemistry images show similar patterns of change (bottom). Results are presented as means ± SE for n ≥ 5 for all experimental groups. Comparative significant differences: *P < 0.05.
Effects of chronic hypoxia on VEGF and VEGF receptor expression.
Chronic hypoxia did not alter VEGF abundance (Fig. 4), but increased VEGF-R2 receptors by 156% and VEGF-R1 receptors by 107% (Fig. 5). Standard errors were 0.4 and 0.77 for (normoxic and hypoxic − total VEGF-R2, respectively) and 0.07 and 0.16 (normoxic and hypoxic 250 kDa VEGF-R1).
Fig. 4.
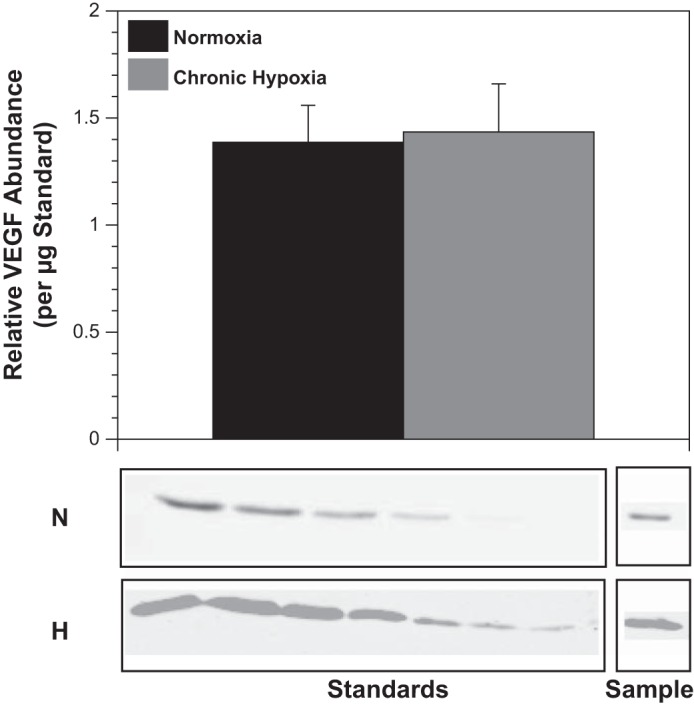
Chronic hypoxia does not significantly increase VEGF expression. Western blot quantification of endogenous VEGF levels in arteries harvested from sheep acclimatized to chronic hypoxia for 110 days yielded values similar to those obtained in arteries harvested from normoxic control sheep. This result suggests that initial increases in VEGF levels (well documented) were not sustained chronically. Results are presented as means ± SE for n = 7 and 6 for normoxic and hypoxic arteries, respectively.
Fig. 5.

Chronic hypoxia upregulates VEGF receptors. Whereas Western blot quantification revealed that VEGF levels were not altered by chronic hypoxia, abundances of VEGF tyrosine kinase receptors were significantly increased by chronic hypoxia. VEGF R1 levels were increased by 107% and VEGF R2 levels were increased by 156% in hypoxic arteries. Receptor levels presented here were measured relative to amounts in a standard pool of normoxic carotid arteries. In light of the fact that both fetal and adult samples were run on the same gel with the same standards, the lanes with the adult samples are shown here adjacent to the previously published standard and fetal bands (1) for all 4 groups. Bar graphs for KDR represent averages of all glycosylated bands, but for Flt-1 only the 250-kDa band was used for quantification because the 230-kDa band was absent in the hypoxic arteries. Results are presented as mean ± SD for n = 6 for all experimental groups. Comparative significant differences: *P < 0.05 via ANOVA.
VEGF-induced changes in contractile protein interactions are maintained through VEGF receptor upregulation.
With MLC20-SMαA colocalization as a reporter assay, exogenous VEGF (3 ng/ml) increased MLC20-SMαA colocalization by 377%, and this effect was completely blocked in cocultures with the VEGF receptor antagonists' vatalinib (240 nM) and dasatinib (6.3 nM) (Fig. 6).
Fig. 6.

Chronic hypoxia alters contractile protein colocalization through upregulation of VEGF receptors. VEGF in culture produced significant increases (377%) in colocalization between MLC20 and SMαA. Cocultures with VEGF receptor antagonists vatalinib (240 nM) and dasatinib (6.3 nM) completely blocked the potent effect of VEGF on these contractile proteins. These results suggest that VEGF-induced changes in interactions between the contractile proteins are mediated through the receptors. Results are presented as means ± SE for n = 5 in all experimental groups. Comparative significant differences: *P < 0.05 via ANOVA.
Effect of VEGF in culture on contractile protein abundances.
Low dose (3 ng/ml) VEGF had no significant effect on SMαA, MLCK, or MLC20 abundances in cultured normoxic arteries. In hypoxic arteries, VEGF increased SMαA by 51% and MLC20 by 161% but decreased MLCK by 91%, thus replicating the effects of hypoxia on all three contractile proteins (Fig. 7).
Fig. 7.

VEGF replicates hypoxic effects on SMαA, MLCK, and MLC20 expression. Organ culture of endothelium-denuded common carotid arteries with low-dose VEGF (3 ng/ml) increased SMαA by 52% (left) and MLC20 by 161% (right) but decreased MLCK by 91% (middle). VEGF thus replicates effects of chronic hypoxia for all three key contractile proteins examined, suggesting that VEGF could mediate hypoxic changes in contractile protein expression in ovine carotid smooth muscle cells. Data are presented as means ± SE for n = 7 and 6 for normoxic and hypoxic arteries, respectively. Comparative significant differences: *P < 0.05 via ANOVA.
Effects of VEGF on contractile protein colocalization.
VEGF increased MLCK-SMαA colocalization by 108% and increased MLC20-SMαA colocalization by 377% but had no effect on MLCK-MLC20 colocalization. VEGF produced effects similar to hypoxia only for MLC20-SMαA colocalization (Fig. 8).
Fig. 8.

VEGF replicates hypoxic effects on MLC20-SMαA colocalization. Organ culture of normoxic arteries with 3 ng/ml of VEGF increased MLCK-SMαA and MLC20-SMαA colocalization. For MLCK-MLC20 colocalization, VEGF had no significant effect on their colocalization coefficient (top). Similarly, immunohistochemistry images, enhanced for brightness at a value of 60, show corresponding changes (bottom). These results show that VEGF replicates hypoxic effects on MLC20-SMαA colocalization, suggesting that VEGF could mediate hypoxic alteration of contractile protein interaction in a highly protein-specific manner. Data here are presented as means ± SE for n ≥ 5 for all experimental groups. Comparative significant differences: *P < 0.05 via ANOVA.
DISCUSSION
This study offers four main original findings. First, long-term hypoxia (110 days at 3,820 m) increased medial wall thicknesses and stiffness but decreased myogenic tone. Second, long-term hypoxia had no effect on tissue VEGF levels but increased abundances of both the R1 and R2 subtypes of VEGF receptors. Third, VEGF in culture replicated effects of long-term hypoxia on abundances of SMαA, MLCK, and MLC20. Fourth, VEGF in culture replicated effects of long-term hypoxia on contractile protein colocalization only for MLC20 with SMαA, suggesting that VEGF mediates hypoxic vascular remodeling more in terms of abundance than organization of contractile proteins and that factors other than VEGF also contribute to hypoxic vascular remodeling. Overall, this study demonstrates that hypoxic vascular remodeling involves a long-term upregulation of VEGF receptors that can explain some of the effects of chronic hypoxia on contractile protein abundance, organization, and function.
Long-term hypoxia alters arterial structure and contractility.
Chronic hypoxia typically promotes increased wall thickness in most arteries (29, 42) due largely to expansion of the adventitial layer (14). However, resident cells in the medial layer can also undergo hypertrophy and hyperplasia upon exposure to hypoxia (42). Medial hyperplasia can also result from hypoxia induced dedifferentiation of contractile smooth muscle cells into synthetic cells that can proliferate, migrate, and secrete extracellular matrix proteins (6). Adventitial fibroblasts and smooth muscle progenitors can also invade the media and differentiate into myofibroblasts (31), adding further to hypoxic increases in medial thickness. The current findings (Fig. 1) emphasize that chronic hypoxia can increase medial thickness, with simultaneous increases in passive arterial stiffness suggesting either an increase in the collagen-to-elastin ratio or an increase in collagen cross linking (17, 56). Chronic hypoxia also depressed myogenic reactivity and, in contrast to the response of fetal arteries (1), significantly left-shifted the stress-strain relation (Fig. 1). Because stiffness was increased, these results together suggest that hypoxia decreased either the efficiency of mechanotransduction, the strength of its coupling to the myogenic response, or overall contractile capacity (2, 15, 30). In light of previous results suggesting that hypoxia can promote the transformation of smooth muscle phenotype from a contractile to a more synthetic phenotype (6) and thereby decrease contractility (23), our next series of experiments examined the effects of hypoxia on the abundance and organization of three key contractile proteins.
Chronic hypoxia differentially modulates contractile protein abundances.
SMαA is the most abundant contractile protein in vascular smooth muscle and is the main component of the thin filament cytoskeleton essential for force production via interactions with activated myosin cross bridges (26, 60). In contrast to our findings in fetal arteries (1), chronic hypoxia significantly increased SMαA abundance (Fig. 2). This suggests the presence of a hypoxia responsive element (HRE) in the SMαA promoter, but no published evidence supports this hypothesis. Alternatively, the translation efficiency for SMαA could be increased by hypoxia. Although miRNAs can significantly modulate actin abundance and assembly (62), evidence that hypoxia can stimulate the actions of these miRNAs is lacking. Given that SMαA is expressed early in differentiation of smooth muscle (41), hypoxic increases in SMαA might also reflect increased smooth muscle proliferation (10, 56). Aside from the hypoxic mechanisms that increase SMαA abundance, the direction of this change cannot explain the parallel hypoxic inhibition of myogenic contractility (Fig. 1).
Regulatory myosin light chain (20 kDa myosin light chain, MLC20) is another critically important smooth muscle contractile protein; phosphorylation of its serine-19 is prerequisite for contraction (59). As for SMαA, hypoxia increased MLC20 abundance, suggesting hypoxic enhancement of the transcription or translation of MLC20 mRNA. Evidence supporting hypoxic modulation of transcription or translation of MLC20 mRNA, however, has not been reported. Alternatively, hypoxic increases in smooth muscle proliferation (10, 56) could explain increased MLC20. As for SMαA, increased MLC20 is inconsistent with the observed hypoxic depression of myogenic contractility (Fig. 1), unless these increases are occurring more in noncontractile (synthetic) than in fully differentiated contractile smooth muscle (49).
MLCK is the dedicated kinase that phosphorylates serine-19 of MLC20 to initiate smooth muscle contraction (22). In contrast to SMαA and MLC20, chronic hypoxia decreased MLCK by 47% (Fig. 2), suggesting hypoxic inhibition of transcription or translation of MLCK mRNA; as for SMαA and MLC20, the published literature provides no support for such effects. Hypoxia might promote MLCK degradation and turnover, as suggested by organ culture studies (8), but not by in vivo studies of long-term hypoxia. As for SMαA and MLC20, hypoxia-induced shifts in smooth muscle phenotype may help explain marked hypoxic decreases in MLCK, particularly if MLCK is expressed late in contractile differentiation (53). Although the hypoxic mechanisms that mediate protein-specific effects on SMαA, MLC20, and MLCK remain unclear, these results suggest that hypoxic attenuation of myogenic contractility can be explained, in part, by decreased MLCK abundance.
Chronic hypoxia differentially modulates contractile protein organization.
Smooth muscle contraction involves rapid and efficient interactions among multiple contractile proteins whose distribution and organization, as well as their abundances, powerfully influence contractile force (59). Under some conditions, phosphorylated MLC20 uncouples from force generation, although all essential contractile proteins are present at appropriate abundances (36, 37). Such observations support the hypothesis that contractile protein organization is an important determinant of contractility that is physiologically regulated, particularly during differentiation and functional maturation (63). To explore the hypothesis that chronic hypoxia modulates contractility through changes in protein organization, our approach focused on colocalization of several pairs of contractile proteins. Consistent with hypoxic increases in SMαA and MLC20, hypoxia increased MLC20-SMαA colocalization (Fig. 3). In contrast, chronic hypoxia had no effect on MLCK-SMαA colocalization, although hypoxia dramatically reduced MLCK abundance (Fig. 2) and in the fetus increased MLCK-SMαA colocalization (1). More importantly, hypoxia increased MLCK-MLC20 colocalization, suggesting that MLCK lost during hypoxic adaptation came primarily from a fraction not colocalized with either SMαA or MLC20. This interpretation is consistent with the hypothesis that MLCK serves important nonkinase and possibly structural functions within smooth muscle (39, 61) that can be killed during hypoxic adaptation. The data also suggest that contractility is diminished but preserved in hypoxic arteries through increased colocalization of the remaining MLCK with MLC20. Taken together, these results emphasize that the organization of contractile proteins may be more important than their abundances for generating contractile force.
Effects of long-term hypoxia on VEGF and VEGF receptor levels.
To explore mechanisms mediating hypoxic changes in vascular structure and function, we examined involvement of VEGF, a well-documented mediator of hypoxic angiogenesis (21, 57) that can influence the abundances and organization of contractile proteins in normoxic (8) and hypoxic (23) arteries. Consistent with reports that chronic hypoxia transiently increases VEGF (1, 12), tissue levels were unchanged after 110 days of chronic hypoxia (Fig. 4) but were several-fold greater than in fetal arteries (1). Conversely, chronic hypoxia elevated abundances of the VEGF-R1 and -R2 receptors (Fig. 5), but the R1 receptor increase was markedly smaller than the 8-fold increase observed in fetal arteries (1). These findings support the hypothesis that hypoxic remodeling of adult arteries is mediated in the short term (<21 days) through increases in VEGF levels and in the long term (>28 days) through increases in VEGF receptor levels, as suggested in fetal arteries (1).
To confirm that VEGF receptors can mediate modulation of contractile protein organization, separate organ culture experiments quantified effects of VEGF on contractile protein colocalization in the presence and absence of VEGF receptor antagonists. Given that hypoxia increased MLC20-SMαA colocalization (Fig. 3), we used this pair to monitor the effects of a physiological concentration of VEGF (3 ng/ml), as previously reported (1, 23). Whereas VEGF increased MLC20-SMαA colocalization 3.5-fold, inclusion of the VEGF receptor antagonists vatalinib (240 nM) and dasatinib (6.3 nM) at effective concentrations (23) completely inhibited the effects of VEGF on MLC20-SMαA colocalization (Fig. 6). Consistent with previous studies (1, 55, 58), these results support the hypothesis that contributions of VEGF to vascular remodeling during hypoxic acclimatization can occur via upregulated VEGF-R1 and -R2 levels. To further examine how VEGF could contribute to hypoxic vascular remodeling, our experimental approach next focused on effects of organ culture with VEGF on contractile protein abundance and organization.
VEGF differentially modulates contractile protein abundances and colocalization.
Organ culture with VEGF increased SMαA and MLC20 abundances in a manner similar to the effects of chronic hypoxia (Fig. 7), suggesting that VEGF could contribute to hypoxic vascular remodeling through stimulation of smooth muscle proliferation, resulting in increased expression of SMαA and MLC20 (10, 56). Aside from similar effects of VEGF and chronic hypoxia on SMαA and MLC20, these effects are inconsistent with the decreased myogenic contractility (26) observed in chronically hypoxic arteries (Fig. 1); some additional influence must be involved. In contrast to SMαA and MLC20, MLCK abundance was markedly decreased by organ culture with VEGF, as with chronic hypoxia (Fig. 7), suggesting that hypoxic increases in VEGF may drive decreases in MLCK and thereby explain reduced myogenic contractility observed in hypoxic arteries.
Smooth muscle contractility is determined not only by contractile protein abundances but also by their organization (60). Indeed, our recent findings demonstrate that abundance and organization of ovine contractile proteins are independently regulated (1, 23, 45). To explore the hypothesis that VEGF contributes to hypoxic changes in contractile protein organization, we quantified colocalization between SMαA, MLCK, and MLC20 before and after organ culture with VEGF. As with chronic hypoxia, organ culture with VEGF increased MLC20-SMαA colocalization (Fig. 8). However, hypoxia had no effect on MLCK-SMαA colocalization (Fig. 3), but VEGF increased this colocalization (Fig. 8). Similarly, VEGF had no effect on MLCK-MLC20 colocalization although chronic hypoxia increased this interaction and so too did VEGF in fetal arteries (1). Given these results, VEGF could explain effects of chronic hypoxia on MLC20-SMαA colocalization, but not on MLCK colocalization with either SMαA or MLC20. These results reinforce the views that the respective effects of VEGF and chronic hypoxia are distinct and highly protein specific and that contractile protein abundance and colocalization are independently regulated. These results also demonstrate that VEGF can explain some but not all of the effects of chronic hypoxia on contractile protein abundance and organization and thereby support the hypotheses that factors other than VEGF participate in hypoxic vascular remodeling.
Overview
The present study reinforces the view that hypoxic increases in VEGF are transient (Fig. 4), but that hypoxic remodeling persists even after VEGF returns to normoxic levels (Fig. 1). These sustained remodeling effects of chronic hypoxia appear to be mediated, in part, by increased densities of the VEGF-R1 and R2 receptors in arterial smooth muscle (Fig. 5). In turn, activation of VEGF receptors can alter abundances of SMαA, MLC20, and MLCK as well as MLC20-SMαA colocalization (Figs. 6–8) in a pattern strikingly similar to that produced by chronic hypoxia (Figs. 2 and 3). In further support of a role for VEGF in hypoxic remodeling, both chronic hypoxia (Fig. 1) and a low physiological concentration of VEGF (8) can attenuate myogenic contractility in ovine arteries. Together, these findings suggest that hypoxic remodeling is mediated in the short term by increased VEGF but in the long term by increased VEGF receptor densities and that this pathway contributes to hypoxic changes in the abundances of SMαA, MLC20 and MLCK as well as MLC20-SMαA colocalization (Fig. 9). In contrast, MLCK-SMαA and MLCK-MLC20 colocalizations are affected differently by chronic hypoxia and VEGF (Fig. 9), suggesting that VEGF may contribute more to hypoxic changes in contractile protein abundances than their organization. Because effects of VEGF and chronic hypoxia on contractile protein organization are closely aligned in fetal ovine arteries (1), the present findings further demonstrate that the role of VEGF in hypoxic remodeling is highly age dependent for contractile protein organization. Overall, the age-dependent effects of both chronic hypoxia and VEGF were most prominent for SMαA abundance and the colocalization of MLCK with MLC20, which may help explain why myogenic stress relations were so different in fetal and adult arteries. Whereas the mechanisms coupling chronic hypoxia to distinct changes in contractile protein organization remain uncertain, the present results strongly suggest that hypoxic changes in the abundances of SMαA, MLC20, and MLCK and organization of SMαA with MLC20 involve sustained increases in VEGF receptors in arterial smooth muscle. Whereas this study demonstrates direct effects of hypoxia and VEGF on smooth muscle remodeling, additional influences of the vascular endothelium on these processes remain uncertain but promising topics for further investigation. Indeed, the combined results strongly suggest that in vivo experiments designed to test the effects of VEGF receptor antagonists on hypoxic vascular remodeling are well justified.
Fig. 9.
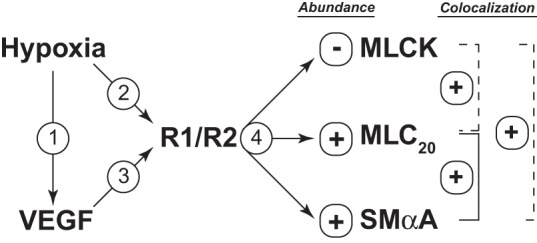
Experimental design and overview. To test the hypothesis that long-term upregulation of VEGF receptors mediates sustained arterial remodeling in ovine adult sheep, we first measured VEGF levels (arrow 1), which returned to normoxic levels after 110 days of hypoxia. In contrast, hypoxia produced sustained increases in the abundances of both VEGF-R1 and VEGF-R2 in endothelium-denuded carotid artery smooth muscle (arrow 2). Stimulation of smooth muscle VEGF receptor in organ culture (arrow 3) increased the abundance of SMαA and MLC20 (arrow 4) and increased their colocalization. VEGF also decreased the abundance of MLCK. These effects of VEGF replicated the effects of chronic hypoxia, as indicated by the solid lines. In contrast to VEGF, however, hypoxia was without effect on colocalization of MLCK with SMαA but increased colocalization of MLCK with MLC20, as indicated by the dashed lines. Together, the findings support the hypothesis that hypoxic upregulation of VEGF receptors contribute to sustained hypoxic arterial remodeling in adult sheep carotid arteries, particularly in relation to contractile protein abundances.
GRANTS
The work reported in this manuscript was supported by National Heart, Lung, and Blood Institute Grants HL54120 and HL64867, National Institute of Child Health and Human Development Grant HD31266, National Institute of Neurological Disorders and Stroke Grant NS076945, and the Loma Linda University School of Medicine.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: O.A. and W.J.P. conception and design of research; O.A., V.B., and J.M.W. performed experiments; O.A., J.M.W., and W.J.P. analyzed data; O.A., M.C.H., J.M.W., and W.J.P. interpreted results of experiments; O.A. and W.J.P. prepared figures; O.A. and W.J.P. drafted manuscript; O.A., M.C.H., and W.J.P. edited and revised manuscript; O.A., M.C.H., and W.J.P. approved final version of manuscript.
REFERENCES
- 1.Adeoye OO, Butler SM, Hubbell MC, Semotiuk A, Williams JM, Pearce WJ. Contribution of increased VEGF receptors to hypoxic changes in fetal ovine carotid artery contractile proteins. Am J Physiol Cell Physiol 304: C656–C665, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ainslie KM, Garanich JS, Dull RO, Tarbell JM. Vascular smooth muscle cell glycocalyx influences shear stress-mediated contractile response. J Appl Physiol 98: 242–249, 2005 [DOI] [PubMed] [Google Scholar]
- 3.Ara J, Shukla P, Frank M. Enhanced expression of the Flt-1 and Flk-1 receptor tyrosine kinases in a newborn piglet model of ischemic tolerance. J Neurochem 124: 735–746, 2013 [DOI] [PubMed] [Google Scholar]
- 4.Ashton SV, Whitley GS, Dash PR, Wareing M, Crocker IP, Baker PN, Cartwright JE. Uterine spiral artery remodeling involves endothelial apoptosis induced by extravillous trophoblasts through Fas/FasL interactions. Arterioscler Thromb Vasc Biol 25: 102–108, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bao B, Ahmad A, Kong D, Ali S, Azmi AS, Li Y, Banerjee S, Padhye S, Sarkar FH. Hypoxia induced aggressiveness of prostate cancer cells is linked with deregulated expression of VEGF, IL-6 and miRNAs that are attenuated by CDF. PLos One 7: e43726, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beamish JA, He P, Kottke-Marchant K, Marchant RE. Molecular regulation of contractile smooth muscle cell phenotype: implications for vascular tissue engineering. Tissue Eng 16: 467–491, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Briet M, Burns KD. Chronic kidney disease and vascular remodelling: molecular mechanisms and clinical implications. Clin Sci (Lond) 123: 399–416, 2012 [DOI] [PubMed] [Google Scholar]
- 8.Butler SM, Abrassart JM, Hubbell MC, Adeoye O, Semotiuk A, Williams JM, Mata-Greenwood E, Khorram O, Pearce WJ. Contributions of VEGF to age-dependent transmural gradients in contractile protein expression in ovine carotid arteries. Am J Physiol Cell Physiol 301: C653–C666, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carmeliet P. Angiogenesis in health and disease. Nat Med 9: 653–660, 2003 [DOI] [PubMed] [Google Scholar]
- 10.Chanakira A, Dutta R, Charboneau R, Barke R, Santilli SM, Roy S. Hypoxia differentially regulates arterial and venous smooth muscle cell proliferation via PDGFR-beta and VEGFR-2 expression. Am J Physiol Heart Circ Physiol 302: H1173–H1184, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Charles SM, Zhang L, Cipolla MJ, Buchholz JN, Pearce WJ. The roles of cytosolic Ca2+ concentration and myofilament Ca2+ sensitization in age-dependent cerebrovascular myogenic tone. Am J Physiol Heart Circ Physiol 299: H1034–H1044, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chavez JC, Agani F, Pichiule P, LaManna JC. Expression of hypoxia-inducible factor-1alpha in the brain of rats during chronic hypoxia. J Appl Physiol 89: 1937–1942, 2000 [DOI] [PubMed] [Google Scholar]
- 13.Clyman RI, Seidner SR, Kajino H, Roman C, Koch CJ, Ferrara N, Waleh N, Mauray F, Chen YQ, Perkett EA, Quinn T. VEGF regulates remodeling during permanent anatomic closure of the ductus arteriosus. Am J Physiol Regul Integr Comp Physiol 282: R199–R206, 2002 [DOI] [PubMed] [Google Scholar]
- 14.Davie NJ, Crossno JTJ, Frid MG, Hofmeister SE, Reeves JT, Hyde DM, Carpenter TC, Brunetti JA, McNiece IK, Stenmark KR. Hypoxia-induced pulmonary artery adventitial remodeling and neovascularization: contribution of progenitor cells. Am J Physiol Lung Cell Mol Physiol 286: L668–L678, 2004 [DOI] [PubMed] [Google Scholar]
- 15.Davis MJ, Wu X, Nurkiewicz TR, Kawasaki J, Davis GE, Hill MA, Meininger GA. Integrins and mechanotransduction of the vascular myogenic response. Am J Physiol Heart Circ Physiol 280: H1427–H1433, 2001 [DOI] [PubMed] [Google Scholar]
- 16.De Lanerolle P, Paul RJ. Myosin phosphorylation/dephosphorylation and regulation of airway smooth muscle contractility. Am J Physiol Lung Cell Mol Physiol 261: L1–L14, 1991 [DOI] [PubMed] [Google Scholar]
- 17.Dodson RB, Rozance PJ, Fleenor BS, Petrash CC, Shoemaker LG, Hunter KS, Ferguson VL. Increased arterial stiffness and extracellular matrix reorganization in intrauterine growth-restricted fetal sheep. Pediatr Res 73: 147–154, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Galis ZS, Khatri JJ. Matrix metalloproteinases in vascular remodeling and atherogenesis: the good, the bad, and the ugly. Circ Res 90: 251–262, 2002 [PubMed] [Google Scholar]
- 19.Gerber HP, Condorelli F, Park J, Ferrara N. Differential transcriptional regulation of the two vascular endothelial growth factor receptor genes. Flt-1, but not Flk-1/KDR, is up-regulated by hypoxia. J Biol Chem 272: 23659–23667, 1997 [DOI] [PubMed] [Google Scholar]
- 20.Hampl V, Bibova J, Ostadalova I, Povysilova V, Herget J. Gender differences in the long-term effects of perinatal hypoxia on pulmonary circulation in rats. Am J Physiol Lung Cell Mol Physiol 285: L386–L392, 2003 [DOI] [PubMed] [Google Scholar]
- 21.Hoeben A, Landuyt B, Highley MS, Wildiers H, Van Oosterom AT, De Bruijn EA. Vascular endothelial growth factor and angiogenesis. Pharmacol Rev 56: 549–580, 2004 [DOI] [PubMed] [Google Scholar]
- 22.Hong F, Haldeman BD, Jackson D, Carter M, Baker JE, Cremo CR. Biochemistry of smooth muscle myosin light chain kinase. Arch Biochem Biophys 510: 135–146, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hubbell MC, Semotiuk AJ, Thorpe RB, Adeoye OO, Butler SM, Williams JM, Khorram O, Pearce WJ. Chronic hypoxia and VEGF differentially modulate abundance and organization of myosin heavy chain isoforms in fetal and adult ovine arteries. Am J Physiol Cell Physiol 303: C1090–C1103, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Intengan HD, Schiffrin EL. Vascular remodeling in hypertension: roles of apoptosis, inflammation, and fibrosis. Hypertension 38: 581–587, 2001 [DOI] [PubMed] [Google Scholar]
- 25.Ishida A, Murray J, Saito Y, Kanthou C, Benzakour O, Shibuya M, Wijelath ES. Expression of vascular endothelial growth factor receptors in smooth muscle cells. J Cell Physiol 188: 359–368, 2001 [DOI] [PubMed] [Google Scholar]
- 26.Kim HR, Appel S, Vetterkind S, Gangopadhyay SS, Morgan KG. Smooth muscle signalling pathways in health and disease. J Cell Mol Med 12: 2165–2180, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuo N, Benhayon D, Przybylski R, Martin R, LaManna J. Prolonged hypoxia increases vascular endothelial growth factor mRNA and protein in adult mouse brain. J Appl Physiol 86: 260–264, 1999 [DOI] [PubMed] [Google Scholar]
- 28.Lewis AM, Mathieu-Costello O, McMillan PJ, Gilbert RD. Effects of long-term, high-altitude hypoxia on the capillarity of the ovine fetal heart. Am J Physiol Heart Circ Physiol 277: H756–H762, 1999 [DOI] [PubMed] [Google Scholar]
- 29.Longo LD, Hull AD, Long DM, Pearce WJ. Cerebrovascular adaptations to high-altitude hypoxemia in fetal and adult sheep. Am J Physiol Regul Integr Comp Physiol 264: R65–R72, 1993 [DOI] [PubMed] [Google Scholar]
- 30.Martinez-Lemus LA, Wu X, Wilson E, Hill MA, Davis GE, Davis MJ, Meininger GA. Integrins as unique receptors for vascular control. J Vasc Res 40: 211–233, 2003 [DOI] [PubMed] [Google Scholar]
- 31.Marx SO, Totary-Jain H, Marks AR. Vascular smooth muscle cell proliferation in restenosis. Circ Cardiovasc Interv 4: 104–111, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mata-Greenwood E, Meyrick B, Soifer SJ, Fineman JR, Black SM. Expression of VEGF and its receptors Flt-1 and Flk-1/KDR is altered in lambs with increased pulmonary blood flow and pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 285: L222–L231, 2003 [DOI] [PubMed] [Google Scholar]
- 33.McQuillan LP, Leung GK, Marsden PA, Kostyk SK, Kourembanas S. Hypoxia inhibits expression of eNOS via transcriptional and posttranscriptional mechanisms. Am J Physiol Heart Circ Physiol 267: H1921–H1927, 1994 [DOI] [PubMed] [Google Scholar]
- 34.Ment LR, Stewart WB, Fronc R, Seashore C, Mahooti S, Scaramuzzino D, Madri JA. Vascular endothelial growth factor mediates reactive angiogenesis in the postnatal developing brain. Brain Res Dev Brain Res 100: 52–61, 1997 [DOI] [PubMed] [Google Scholar]
- 35.Mlynarczyk M, Imamura T, Umezaki H, Kaushal KM, Zhang L, Ducsay CA. Long-term hypoxia changes myometrial responsiveness and oxytocin receptors in the pregnant ewe: differential effects on longitudinal versus circular smooth muscle. Biol Reprod 69: 1500–1505, 2003 [DOI] [PubMed] [Google Scholar]
- 36.Morgado M, Cairrao E, Santos-Silva AJ, Verde I. Cyclic nucleotide-dependent relaxation pathways in vascular smooth muscle. Cell Mol Life Sci 69: 247–266, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morgan KG, Gangopadhyay SS. Invited Review: Cross-bridge regulation by thin filament-associated proteins. J Appl Physiol 91: 953–962, 2001 [DOI] [PubMed] [Google Scholar]
- 38.Mu D, Jiang X, Sheldon RA, Fox CK, Hamrick SE, Vexler ZS, Ferriero DM. Regulation of hypoxia-inducible factor 1alpha and induction of vascular endothelial growth factor in a rat neonatal stroke model. Neurobiol Dis 14: 524–534, 2003 [DOI] [PubMed] [Google Scholar]
- 39.Nakamura A, Xie C, Zhang Y, Gao Y, Wang HH, Ye LH, Kishi H, Okagaki T, Yoshiyama S, Hayakawa K, Ishikawa R, Kohama K. Role of non-kinase activity of myosin light-chain kinase in regulating smooth muscle contraction, a review dedicated to Dr. Setsuro Ebashi. Biochem Biophys Res Commun 369: 135–143, 2008 [DOI] [PubMed] [Google Scholar]
- 40.Osol G, Mandala M. Maternal uterine vascular remodeling during pregnancy. Physiology (Bethesda) 24: 58–71, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Owens GK, Loeb A, Gordon D, Thompson MM. Expression of smooth muscle-specific alpha-isoactin in cultured vascular smooth muscle cells: relationship between growth and cytodifferentiation. J Cell Biol 102: 343–352, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Packer CS, Roepke JE, Oberlies NH, Rhoades RA. Myosin isoform shifts and decreased reactivity in hypoxia-induced hypertensive pulmonary arterial muscle. Am J Physiol Lung Cell Mol Physiol 274: L775–L785, 1998 [DOI] [PubMed] [Google Scholar]
- 43.Pages G, Pouyssegur J. Transcriptional regulation of the Vascular Endothelial Growth Factor gene–a concert of activating factors. Cardiovasc Res 65: 564–573, 2005 [DOI] [PubMed] [Google Scholar]
- 44.Pasterkamp G. Imaging of de novo atherosclerotic arterial remodeling: clinical sense or research sensibility? J Am Coll Cardiol 53: 1716–1717, 2009 [DOI] [PubMed] [Google Scholar]
- 45.Pearce WJ, Khorram O. Maturation and differentiation of the fetal vasculature. Clin Obstet Gynecol 56: 537–548, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pfitzer G. Invited review: regulation of myosin phosphorylation in smooth muscle. J Appl Physiol 91: 497–503, 2001 [DOI] [PubMed] [Google Scholar]
- 47.Pries AR, Reglin B, Secomb TW. Remodeling of blood vessels: responses of diameter and wall thickness to hemodynamic and metabolic stimuli. Hypertension 46: 725–731, 2005 [DOI] [PubMed] [Google Scholar]
- 48.Reglin B, Secomb TW, Pries AR. Structural adaptation of microvessel diameters in response to metabolic stimuli: where are the oxygen sensors? Am J Physiol Heart Circ Physiol 297: H2206–H2219, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rensen SS, Doevendans PA, van Eys GJ. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Neth Heart J 15: 100–108, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sandner P, Wolf K, Bergmaier U, Gess B, Kurtz A. Hypoxia and cobalt stimulate vascular endothelial growth factor receptor gene expression in rats. Pflügers Arch 433: 803–808, 1997 [DOI] [PubMed] [Google Scholar]
- 51.Schwartz SM, Campbell GR, Campbell JH. Replication of smooth muscle cells in vascular disease. Circ Res 58: 427–444, 1986 [DOI] [PubMed] [Google Scholar]
- 52.Skalli O, Pelte MF, Peclet MC, Gabbiani G, Gugliotta P, Bussolati G, Ravazzola M, Orci L. Alpha-smooth muscle actin, a differentiation marker of smooth muscle cells, is present in microfilamentous bundles of pericytes. J Histochem Cytochem 37: 315–321, 1989 [DOI] [PubMed] [Google Scholar]
- 53.Steinbach SK, El-Mounayri O, DaCosta RS, Frontini MJ, Nong Z, Maeda A, Pickering JG, Miller FD, Husain M. Directed differentiation of skin-derived precursors into functional vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 31: 2938–2948, 2011 [DOI] [PubMed] [Google Scholar]
- 54.Stenmark KR, Fagan KA, Frid MG. Hypoxia-induced pulmonary vascular remodeling: cellular and molecular mechanisms. Circ Res 99: 675–691, 2006 [DOI] [PubMed] [Google Scholar]
- 55.Takagi H, King GL, Ferrara N, Aiello LP. Hypoxia regulates vascular endothelial growth factor receptor KDR/Flk gene expression through adenosine A2 receptors in retinal capillary endothelial cells. Invest Ophthalmol Vis Sci 37: 1311–1321, 1996 [PubMed] [Google Scholar]
- 56.Thompson JA, Richardson BS, Gagnon R, Regnault TR. Chronic intrauterine hypoxia interferes with aortic development in the late gestation ovine fetus. J Physiol 589: 3319–3332, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tipoe GL, Lau TY, Nanji AA, Fung ML. Expression and functions of vasoactive substances regulated by hypoxia-inducible factor-1 in chronic hypoxemia. Cardiovasc Hematol Agents Med Chem 4: 199–218, 2006 [DOI] [PubMed] [Google Scholar]
- 58.Tuder RM, Flook BE, Voelkel NF. Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or to chronic hypoxia. Modulation of gene expression by nitric oxide. J Clin Invest 95: 1798–1807, 1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Walsh MP. Vascular smooth muscle myosin light chain diphosphorylation: mechanism, function, and pathological implications. IUBMB Life 63: 987–1000, 2011 [DOI] [PubMed] [Google Scholar]
- 60.Walsh MP, Cole WC. The role of actin filament dynamics in the myogenic response of cerebral resistance arteries. J Cereb Blood Flow Metab 33: 1–12, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang HH, Nakamura A, Matsumoto A, Yoshiyama S, Qin X, Ye LH, Xie C, Zhang Y, Gao Y, Ishikawa R, Kohama K. Nonkinase activity of MLCK in elongated filopodia formation and chemotaxis of vascular smooth muscle cells toward sphingosylphosphorylcholine. Am J Physiol Heart Circ Physiol 296: H1683–H1693, 2009 [DOI] [PubMed] [Google Scholar]
- 62.Xin M, Small EM, Sutherland LB, Qi X, McAnally J, Plato CF, Richardson JA, Bassel-Duby R, Olson EN. MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev 23: 2166–2178, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yablonka-Reuveni Z, Christ B, Benson JM. Transitions in cell organization and in expression of contractile and extracellular matrix proteins during development of chicken aortic smooth muscle: evidence for a complex spatial and temporal differentiation program. Anat Embryol (Berl) 197: 421–437, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]