

Abstract
Angiotensin II (ANG II) plays a role in muscle wasting and remodeling; however, little evidence shows its direct effects on specific muscle functions. We presently investigated the acute in vitro effects of ANG II on resting ionic conductance and calcium homeostasis of mouse extensor digitorum longus (EDL) muscle fibers, based on previous findings that in vivo inhibition of ANG II counteracts the impairment of macroscopic ClC-1 chloride channel conductance (gCl) in the mdx mouse model of muscular dystrophy. By means of intracellular microelectrode recordings we found that ANG II reduced gCl in the nanomolar range and in a concentration-dependent manner (EC50 = 0.06 μM) meanwhile increasing potassium conductance (gK). Both effects were inhibited by the ANG II receptors type 1 (AT1)-receptor antagonist losartan and the protein kinase C inhibitor chelerythrine; no antagonism was observed with the AT2 antagonist PD123,319. The scavenger of reactive oxygen species (ROS) N-acetyl cysteine and the NADPH-oxidase (NOX) inhibitor apocynin also antagonized ANG II effects on resting ionic conductances; the ANG II-dependent gK increase was blocked by iberiotoxin, an inhibitor of calcium-activated potassium channels. ANG II also lowered the threshold for myofiber and muscle contraction. Both ANG II and the AT1 agonist L162,313 increased the intracellular calcium transients, measured by fura-2, with a two-step pattern. These latter effects were not observed in the presence of losartan and of the phospholipase C inhibitor U73122 and the in absence of extracellular calcium, disclosing a Gq-mediated calcium entry mechanism. The data show for the first time that the AT1-mediated ANG II pathway, also involving NOX and ROS, directly modulates ion channels and calcium homeostasis in adult myofibers.
Keywords: angiotensin II, chloride channel conductance, AT1 receptor, protein kinase C, NADPH oxidase
in fast-twitch muscle fibers, the macroscopic chloride conductance (gCl), due to the expression/activity of ClC-1 channel, accounts for ∼80% of the total resting ionic conductance and ensures the electrical stability of myofibers (4, 9, 67, 68). In fact, the large gCl prevents membrane overexcitability due to potassium accumulation in transverse tubules during firing by reducing the length constant of electrotonic signal propagation (4, 9, 54). In spite of the important progress made in the last years in measuring ClC-1 chloride currents (25, 28, 48, 60), macroscopic recordings of muscle gCl still provide crucial information about the function, pharmacology, and biochemical modulation of these channels in native skeletal muscle (8, 19, 20, 22).
ClC-1 channel plays a key role in skeletal muscle function and plasticity. In fact, loss-of-function mutations of this channel, and the consequent reduction of gCl, account for the pathological hyperexcitability and delayed muscle relaxation in myotonic syndromes (8, 47, 68). Concurrently, changes of gCl take part in acute and chronic mechanotransduction signaling; in fact the modulation of sarcolemmal excitability, via fine tuning of contractile response, can have an impact on myofiber phenotype and metabolism via muscular gene reprogramming. In particular, both the expression of ClC-1 channel and gCl are phenotype dependent, being greater in fast-twitch vs. slow-twitch myofibers (56). In addition, changes in expression and/or function of the ClC-1 channel play a key role in the myofiber phenotype transition observed in various conditions. For instance, an early increase of gCl is observed in rat soleus muscle after 3 days of hindlimb unloading (HU), as a model of muscle disuse, preceding the HU induced slow-to-fast phenotype transition (56, 57). Mechanotransduction is aberrant in muscle disorders due to defects in cytoskeletal scaffolding, i.e., in dystrophic conditions due to the absence of the structural protein dystrophin. Interestingly, this progressive myopathy is characterized by an impairment of gCl occurring in strict correlation with mechanical challenge; i.e., gCl is reduced in fast-twitch hindlimb extensor digitorum longus (EDL) muscle of dystrophin-deficient mdx mouse by protocols of in vivo exercise otherwise ineffective in wild-type muscles (11, 21). Previous studies showed that the dynamic changes of gCl observed in the various conditions are in part the result of its modulation via specific biochemical pathways. Indeed, a decrease of gCl can result in the consequence of phosphorylation signaling brought about by calcium-dependent protein kinase C (PKC), and a relationship between gCl and intracellular calcium concentration ([Ca2+]i) exists (20, 22). In particular, pathophysiological conditions of skeletal muscle with a reduced value of gCl, such as aging, drug-induced myopathies, and, again, muscular dystrophy, are characterized by elevated [Ca2+]i (11, 21, 22, 31, 44), while the increase of gCl, occurring in slow-twitch muscle during HU, parallels a decrease of both [Ca2+]i and PKC signaling (30, 56, 57). The pathway for activation of the calcium-dependent PKC occurs via a G protein (20); few endogenous ligands can activate the receptor-mediated PLC/PKC signaling pathways able to modulate gCl, further supporting the key role of the latter for muscle physiology (55, 58, 72). For instance, ATP-mediated activation of P2Y1 purinergic receptor acutely modulates gCl, likely for adapting muscle performance during exercise and fatigue (23, 72). As anticipated, gCl is selectively reduced in dystrophic mdx muscle upon mechanical stress and this alteration can be due to the action of proinflammatory mediators. In fact gCl reduction in mdx muscles is contrasted by anti-inflammatory agents, while tumor necrosis factor-α (TNF-α), a key modulator of dystrophic muscle necrosis, partially decreases gCl via PKC activation (11, 18, 21, 58). We recently observed that a treatment with enalapril, an inhibitor of the angiotensin-converting enzyme (ACE), while reducing the presence of markers of oxidative stress and inflammation in mdx mouse muscles, also leads to a dose-dependent restoration of gCl (14). This result lead us to hypothesize a possible role of angiotensin II (ANG II) signaling in ClC-1 channel modulation. ANG II is known for its actions in cardiovascular system and its involvement in heart disease; however, it has been claimed that ANG II exerts prooxidant, proinflammatory, and profibrotic action in several tissues, among which is skeletal muscle (13, 69, 73). Increasing evidence supports a key role of enhanced activation of systemic and local renin-angiotensin system (RAS) and ANG II in aberrant remodeling and wasting conditions of skeletal muscle, including muscular dystrophy (13, 41, 43, 70). Other than in microvasculature, the presence of ANG II receptors type 1 (AT1) and 2 (AT2) in myofibers and muscle cell lines has been described, although controversy is still unresolved about the role of these tissue receptors in mediating the ANG II actions in mature skeletal muscle (41, 43, 45, 69, 78). Importantly, ANG II via AT1 receptor activates canonical Gq protein PLC/PKC signaling, which also leads to activation of NADPH-oxidase (NOX) in most of the tissues where AT1 receptors are expressed. This pathway accounts for production of reactive oxygen species (ROS) and activation of redox-sensitive cellular process, including the regulation of ionic homeostasis, as in renal podocytes (1, 41, 43, 63). Activation of NOX in skeletal muscle by systemic ANG II has also been observed, and overexpression of NOX is responsible of oxidative stress occurring in dystrophic muscle (41, 43, 50, 73, 74). Based on these findings we tested the working hypothesis that ANG II is a novel endogenous ligand involved in inflammation and ROS-mediated modulation of skeletal muscle chloride channel conductance. To this aim we assessed the acute effects and signaling pathways of ANG II on resting gCl of mouse EDL muscle fibers by means of electrophysiological recordings and the use of specific pharmacological tools. Considering the novelty of the experimental study and the possible cross talk of the ANG II signaling pathway with other myofiber effectors, we also evaluated in parallel the effect of ANG II and other tools on resting conductance to potassium ions (gK), excitation-contraction coupling, and calcium homeostasis, integrating the electrophysiological recordings with cytofluorimetric assay and contraction recordings. The results showed for the first time a direct role of ANG II, via the AT1 receptor, in chloride channel modulation and in calcium entry in adult myofiber, which also involves the NOX and ROS pathway.
MATERIALS AND METHODS
All experiments were conducted in accordance with the Italian Guidelines for the Care and Use of Laboratory Animals (D.L. 116/92), which conform with the European Community Directive published in 1986 (86/609/EEC), and received approval from the local Institutional Animal Care and Use Committees.
In vitro/ex vivo experiments.
Four- to six-month-old male wild-type (WT) mice (C57/BL10ScSn; Charles River, Jackson Laboratories) were used for all the experiments. The animals were anesthetized with 1.2 g/kg urethane ip injection. EDL muscle of one hindlimb was removed and rapidly placed in the recording chambers filled with normal physiological solutions for electrophysiological recordings. The contralateral was used for fura-2 microfluorescence studies or for isometric contraction recordings or snap frozen in liquid nitrogen and stored at −80°C until use for real-time quantitative (q)PCR analysis, based on experimental needs. EDL muscles from age-matched male mdx mice (Charles River, Jackson Laboratories) were used as a positive control for qPCR experiments and were spared samples from other studies.
Electrophysiological recordings by intracellular microelectrodes.
EDL muscle was transferred into a 20-ml muscle bath containing 95% O2-5% CO2-gassed physiological solutions (see compositions below), gently fixed by tendons at the resting length, and maintained at 30°C. Muscle chamber was accessible for two sliding micromanipulators (Zeiss) that allowed fine positioning and impalements, under a stereomicroscope (Nikon SMZ800), of glass capillary micropipettes into single muscle fibers for microelectrodes electrophysiological recordings in current-clamp configuration. Electrodes were Ag/AgCl wires or pellets in contact with salt-conducting solutions filling the micropipettes. These were made from borosilicate glass capillary (OD: 1.0 mm; Hilgenberg) using an horizontal puller (DMZ Universal Puller Zeitz). The micropipette tip was ∼0.5–1 μm. Micropipettes were filled with 2 M K-citrate for the current-injecting microelectrode and with 3 M KCl for the microelectrode recording resting membrane potential and electrotonic responses. Electrode resistance was ∼15–20 MΩ. An Ag/AgCl bath electrode allowed estimation of electric signals with respect to ground. The current pulse generation, acquisition of the voltage records, and calculation of the fiber constants were done under computer control via a specifically home-designed software and a National Instrument motherboard and D/A and A/D interface (National Instrument BNC-2090). Then, computer-generated constant square-wave current pulses of fixed amplitude were delivered at low frequency (<1 Hz) to the current-injecting microelectrode after D/A conversion and successive integration and amplification by an homemade current/voltage clamp box. The membrane responses recorded by the second microelectrode were amplified by a 705-WPI amplifier (WPI, Sarasota, FL) and acquired after A/D conversion for real-time calculation of membrane cable parameters. These latter were obtained by measuring the attenuation of the electrotonic potential in response to standard hyperpolarizing current pulse at two distances between the recording and the current-injecting electrode, according to the cable equation (7, 9, 19). In particular the maximal electrotonic potential (V1) was obtained with minimal distance between the two microelectrodes (distance 1 = 0.05 mm) while an attenuated electrotonic potential (V2) was recorded at a second distance (distance 2) ranging between 0.7 and 1.4 mm. The distance between microelectrodes was read for each fiber impalement via a microscope ocular micrometer. Particular care was taken at verifying that resting membrane potential of myofiber was constant between the two measurements; no holding current injection was needed or used. Current pulses and resting and electrotonic membrane potential were continuously monitored via an oscilloscope-configured computer screen. The basic cable model parameters, measured according to the theory of an infinite linear cable, were the space constant (λ), the fiber input resistance (Rin), and the time constant (τ). The constant λ was experimentally determined as the logarithmic decay of the electrotonic potential with the distance from the current electrode; Rin was determined as the steady-state electrotonic potential divided by the current intensity; and τ was determined to be the 84% rise time of the electrotonic potential, which is theoretically equal to the time constant for charging both surface and tubular membrane components (7, 9, 42, 52). This approach can be used without modification for the calculation of cable parameters of muscle fibers in different experimental situations provided that the length of the fibers allows the appreciation of the logarithmic decay of the electrotonic potential on which the method of steady-state spatial decay is based (7, 9, 19). However, final values strictly depend on experimental conditions and methods of calculations (54); therefore, caution is required to maintain experimental conditions constant throughout. The values recorded in the present study were in line with previous recordings from our and others laboratories (5, 19, 52). The mean length of EDL muscle fiber for the present experiments was calculated as the muscle resting length at the tendon insertions divided per 0.44 (6) and was ∼5 mm, which is about 10-fold higher than myofiber length constant in normal physiological solution (λ = 0.56 ± 0.008 mm; n = 54 myofibers/10 preparations). With the assumption of a constant value of EDL fibers myoplasmic resistivity (Ri) of 140 Ω·cm2, it was possible to calculate the membrane cable parameters, and in particular membrane resistance (Rm = 0.2 × Rin × λ × π × dcalc), fiber diameter {dcalc= [4 × Ri × λ/π × (20 × Rin)1/2]}, and total membrane capacitance [Cm = (τ/0.2 × Rin × λ)] (7, 19, 52). The total membrane conductance (gm) was calculated as 1/Rm in normal physiological solution, whereas 1/Rm calculated in a chloride-free solution was the potassium conductance (gK). gCl was calculated as the mean gm minus the mean gK (8, 19, 21).
The mechanical threshold (MT) was determined in EDL myofibers in normal physiological solution using the same electrophysiological setup but configured for two-microelectrode “point” voltage-clamp recordings, in line with previous descriptions (21, 27, 36). In brief, the two microelectrodes were inserted within 0.05 mm of each other into the central region of a randomly selected superficial fiber that was continuously viewed using the stereomicroscope (×100 magnification). After myofiber impalement and in the voltage-clamp mode, the holding potential was set at −90 mV. This value was chosen because it was close to the physiological resting membrane potential but negative enough to ensure resting conditions of voltage-dependent processes during intervals between depolarizing command pulses. The latter of variable duration (from 5 to 500 ms) were given at a rate of ∼0.3 Hz. Tetrodotoxin (3 μM) was continuously present during recordings to prevent action potential generation (18, 21, 36). At each duration, the command voltage was increased using an analog and/or digital control until contraction, as movement of fiber edge, was observed opposite the current electrode, according to what has been described in detail elsewhere (27), and then backed down until the contraction just disappeared. A digital sample-and-hold millivoltmeter stored the value of the threshold membrane potential at this point. We estimated the uncertainty of any single measurement to be 1–2 mV. The distance between the electrodes, far below the myofiber length constant, allows isopotential conditions at the site of determinations and then rules out possible bias on the threshold measurement due to changes in length constant produced by any compound. The longer the depolarizing pulse, the more negative is the voltage for contraction until a constant value is reached, due to the equilibrium between the speed for calcium release and that for calcium reuptake. The threshold membrane potential V (mV) for each fibers was averaged at each pulse duration t (milliseconds), and then mean values were plotted against duration, giving a “strength-duration” relationship. A fit estimate of the rheobase voltage (R) and of the rate constant (1/τ) to reach the rheobase was obtained by nonlinear least-squares algorithm using the following equation: V = [H-R exp (t/τ)]/[1-exp (t/τ)], where H is the holding potential (mV), R is the rheobase (mV), and τ is the time constant. In the fitting algorithm, each point was weighed by the reciprocal of the variance of that mean V and the best-fit estimates of the parameters R and τ were made (18, 21). We used this procedure to be able to incorporate all of our determination points and their associated errors into our estimate of R under each condition.
Fura-2 calcium imaging.
EDL muscles were pinned in a dissecting dish containing 95% O2-5% CO2-gassed normal physiological solution at room temperature (∼22°C) and small bundles of 10–15 fibers, arranged in a single layer, were dissected lengthwise, tendon to tendon, with the use of microscissors, as described elsewhere (11, 30). Calcium measurements were performed using the membrane-permeant Ca2+ indicator fura-2 acetoxymethyl ester (fura-2 AM; Molecular Probes-Invitrogen). Loading of muscle fibers was performed for 1 h at 25°C in normal physiological solution containing 5 μM fura-AM mixed to 0.05% (vol/vol) Pluronic F-127 (Molecular Probes). After loading, muscle fibers were washed with normal physiological solutions, continuously gassed with 95% O2 and 5% CO2 and maintained at 30°, and mounted in a modified RC-27NE experimental chamber (Warner Instruments, Hamden, CT) on the stage of an inverted Eclipse TE300 microscope (Nikon) with a ×40 Plan-Fluor objective (Nikon). Fluorescence measurements were made using a QuantiCell 900 integrated imaging system (Visitech International, Sunderland, UK) as previously described (11, 30). During experiments, pairs of background subtracted images of fura-2 fluorescence (510 nm) after excitation at 340 and 380 nm were acquired and ratiometric images (340/380 nm) were calculated for each muscle fiber of the preparation using QC2000 software. For each fiber, a constant window of ∼1.5–2 mm2 in the central region, was used for the assessments. Subsequently, fluorescence ratio values were converted to the resting cytosolic calcium [Ca2+]i (nM), according to Fraysse et al. (30) and Grynkiewicz et al. (34), using a calibration procedure previously described.
Isometric contraction.
EDL muscle was securely tight at tendon insertion and placed in a muscle chamber containing the normal physiological solution continuously gassed with 95% O2-5% CO2 (pH 7.2–7.4) and maintained at 27°C. The muscle was connected by one tendon at the dual-mode muscle lever-system transducer (300C dual mode transducer; Aurora Scientific). Electrical stimulation field was obtained by two axial platinum wires connected to a stimulator (LE 12406; 2Biological Instruments). After an equilibration period (30 min), the preparation was stretched to its optimal length (L0; measured with an external caliper), i.e., the length producing the maximal twitch to a 0.2-ms pulse of the maximal effective voltage (40–50 V). The stimulation protocol consisted in a force-frequency curve constructed as follows: 350-ms trains of 0.2-ms maximal pulses from 10 to 140 Hz for determination of maximal tetanic tension and frequency for half-maximal activation (Hz50). Data were collected with DMCv 4.1.6 and analyzed using DMAv 3.1.2 Lab-view software (Aurora Scientific). At the end of experiments, samples were removed, cleaned from tendons, dried, and weighted. Absolute values of tension were normalized by cross-sectional area as previously described (6, 11, 14).
Solutions and drugs.
The normal physiological solution had the following composition (mM): 148 NaCl, 4.5 KCl, 2.0/2.5 CaCl2, 1.0 MgCl2, 0.44 NaH2PO4, 12.0 NaHCO3, and 5.55 glucose. The calcium-free solution has the same composition as the normal one except that CaCl2 (2.5 mM) was omitted and 10 mM EGTA were added. The chloride-free solution was made by equimolar substitution of nonpermeable methylsulfate salts for NaCl and KCl and nitrate salts for CaCl2 and MgCl2. The solutions were continuously gassed with 95% O2-5% CO2 (pH 7.2–7.4) and maintained at 30°C for all the duration of the experiment (20).
The compounds tested were ANG II, chelerythrine, losartan, PD123,319, N-acetyl cysteine (NAC), apocynin, L162,313, U-0126, SB-203580, iberiotoxin (Sigma-Aldrich), and U73122 (Calbiochem).
Stock solutions were prepared in distilled water or DMSO, based on solubility. The final concentrations were obtained with adequate dilutions of microliter amounts of concentrated stock solutions, with normal or chloride-free physiological solution, according to the experimental need. Final concentrations of DMSO never exceeded 0.1% according to previous studies (20). ANG II was incubated at different concentrations and for 15–20 min before the electrophysiological recordings were performed. All other compounds were tested at fixed concentrations, based on previous results and data in the literature. Antagonists were preincubated for 30 min before microelectrode recordings in the absence and in the presence of ANG II.
Isolation of total RNA, reverse transcription, and real-time PCR.
For each EDL muscle sample, total RNA was isolated by RNeasy Fibrous Tissue Mini Kit (Qiagen catalog no. 74704) and quantified by using a spectrophotometer (ND-1000 NanoDrop; Thermo Scientific). To perform reverse transcription, for each sample, 400 ng of total RNA were added to 1 μl dNTP mix 10 mM each (Roche catalog no. 11277049001) and 1 μl random hexamers 50 μM (Life Technologies catalog no. n808-0127) and incubated at 65°C for 5 min. Afterward, 4 μl 5× First Standard Buffer (Life Technologies catalog no. Y02321), 2 μl 0.1 M DTT (Life Technologies catalog no. Y00147), and 1 μl Recombinant RNasin Ribonuclease Inhibitor 40 U/μl (Promega catalog no. N2511) were added and incubated at 42°C for 2 min. To each solution, 1 μl Super Script II Reverse Trascriptase 200 U/μl (Life Technologies catalog no. 18064–014) was added and then incubated at 25°C for 10 min, at 42°C for 50 min and at 70°C for 15 min. Real-time PCR was performed in triplicate using the Applied Biosystems Real-time PCR 7500 Fast System, MicroAmp Fast Optical 96-Well Reaction Plate 0.1 ml (Life Technologies catalog no. 4346906), and MicroAmp Optical Adhesive Film (Life Technologies catalog no. 4311971). Each reaction was carried on as single plex reaction. The setup of reactions consisted 8 ng of cDNA, 0.5 μl of TaqMan Gene Expression Assays, (Life Technologies), 5 μl of TaqMan Universal PCR master mix No AmpErase UNG (2×) (Life Technologies catalog no. 4324018), and Nuclease-Free Water not DEPC-Treated (Life Technologies catalog no. AM9930) for a final volume of 10 μl. Under the following RT-TaqMan-PCR conditions: step 1: 95°C for 20 s; step 2: 95°C for 3 s; and step 3: 60°C for 30 s; steps 2 and 3 were repeated 40 times. The results were compared with relative standard curve obtained by five points of 1:4 serial dilutions. The mRNA expression of the genes was normalized to the best housekeeping genes glyceraldehyde-3-phosphate dehydrogenase (GAPDH) selected from beta-actin (Actb) and beta-2 microglobulin (b2m) and GAPDH. TaqMan Hydrolysis primer and probe gene expression assays were ordered with the following assay IDs: angiotensin I-converting enzyme (peptidyl-dipeptidase A) 1 (Ace) ID: Mm00802048_m1; angiotensin II receptor, type 1a and 1b (Atg1a/b) ID: Mm00558224_s1; angiotensinogen (Agt) ID: Mm00599662_m1; tubulin, alpha 1B (Tuba1b) ID: Mm01964369_g1; nicotinamide adenine dinucleotide phosphate oxidase 2 (Cybb) ID: Mm01287743_m1; 2bm ID:Mm00437762_m1; Gapdh ID: Mm99999915_g1; and Actb ID: Mm00607939_s1.
Statistical analysis.
All data are expressed as means ± SE. The SE estimate for gCl was obtained as previously described (19, 21). Statistical analysis for direct comparison between two groups of data means was performed by unpaired Student's t-test. The MT values are expressed as both means ± SE for the absolute values of voltage threshold at each pulse duration. Fitted rheobase (R) and rate constant (1/τ) parameters ± SE were determined from the variance-covariance matrix in the nonlinear least square fitting algorithm (18, 21). For these fitted parameters, the statistical significance between groups was estimated by using the above tests using these SEs and a number of degrees of freedom equal to the total number of threshold values determining the curves minus the number of means minus two for the free parameters (18, 21). Differences between groups were evaluated for statistical significance (P value <0.05) by Student's t-test.
RESULTS
Effect of in vitro application of ANG II on cable properties and component ionic conductance of EDL muscle fibers.
EDL muscle fibers were exposed to different concentrations of ANG II (range from 0.01 to 3 μM). ANG II increased membrane resistance (Rm) in a concentration-dependent manner up to 0.1 μM, while at higher concentrations a plateau effect was observed (Table 1). For instance Rm was 522 ± 35 Ω·cm2 (n = 3/19) and 531 ± 19 Ω·cm2 (n = 3/22) at 0.1 and 1 μM, respectively. Figure 1A shows the time-dependent change of Rm for the population of fibers incubated or not with 0.3 μM ANG II. As can be seen, after 20 min of incubation, all fibers exposed to ANG II had higher values of Rm and these latter remained constant over time (up to 90 min) (Fig. 1A). The effect of ANG II was fully reversible. For instance Rm was 629 ± 68 Ω·cm2 (N/n = 2/11) after the application of 0.3 μM of ANG II and returned to 377 ± 31 Ω·cm2 (n = 2/12) after washout, a value fully overlapping the basal value in normal physiological solution (351 ± 8 Ω·cm2; N/n = 2/12). Table 1 also shows the values of membrane capacitance (Cm) that were in line with those previously observed by us in EDL myofibers of mice the same age (19). These values are slightly higher with respect to those found in other studies. This could be related to the different experimental conditions as well as to differences in myofiber diameter and in the ratio between sarcolemma and transverse tubule membrane compartments, due to specie-specific or age-related differences (15, 24, 64). Importantly, no significant effects of ANG II were observed on either Cm or fiber diameter (Table 1). The Rm increase by ANG II in EDL myofibers was due to a significant, concentration-dependent reduction of total membrane ionic conductance (Table 2). In fast-twitch skeletal muscle fibers, such as EDL, the total resting gm is due to the resting conductance to chloride ions (gCl) for ∼80%, while the rest is due to potassium conductance (gK), the contribution to conductance of other ionic species being very small, if any. The evaluation of the ANG II effects in chloride-free solution actually showed a decrease in Rm, due to a concentration-dependent increase in gK, appreciable from 0.01 μM onward (Table 2). Therefore, the decrease in gm was entirely due to a graded concentration-related inhibition of gCl by ANG II (Fig. 1C and Table 2). In particular, ANG II lead to a 40% decrease of gCl at 0.3 μM, with a maximal block of 50% at the higher concentrations, i.e., 3 μM, while at the same ANG II concentration gK was increased about twofold. The concentration for half-maximal block (IC50) of gCl calculated from the fit of data points in the concentration range reaching the plateau was 0.06 μM (Fig. 1C), while the half-effective concentration (EC50) for increasing gK was 0.26 μM (Fig. 1D). The addition of 100 μM of S(-)-2-(p-chlorophenoxy) propionic acid, a direct blocker of skeletal muscle chloride channels (20), reduced the residual conductance observed in normal physiological solution in the presence of the maximal concentration of ANG II down to 652 ± 22 μS/cm2 (n = 15), a value overlapping the gK values in the presence of ANG II described in Table 2. This supports that the residual conductance was still due to chloride ions.
Table 1.
Effect of in vitro application of ANG II on passive cable parameters of EDL muscle fibers of WT mice
| Experimental Conditions | N/n | dcalc, μm | Cm, μF/cm2 | Rm, Ω·cm2 |
|---|---|---|---|---|
| Normal physiological solution | 15/81 | 48.3 ± 1.2 | 9.2 ± 1 | 356 ± 4 |
| + 0.01 μM ANG II | 3/23 | 52.7 ± 2.8 | 6.1 ± 0.8 | 391 ± 12* |
| + 0.03 μM ANG II | 3/27 | 47.4 ± 2.1 | 7.3 ± 1.0 | 420 ± 19* |
| + 0.1 μM ANG II | 3/19 | 45.1 ± 3.0 | 9.8 ± 1.8 | 522 ± 35* |
| + 0.3 μM ANG II | 6/42 | 50.0 ± 2.2 | 9.6 ± 1.1 | 529 ± 20* |
| + 1 μM ANG II | 3/22 | 53.3 ± 2.4 | 7.7 ± 1.3 | 531 ± 19* |
| + 3 μM ANG II | 3/19 | 46.0 ± 2 | 7.6 ± 1.5 | 582 ± 30* |
In each experimental condition, the values are means ± SE from n number of fibers sampled from N number of preparations tested. The following parameters are shown: dcalc, calculated fiber diameter; Cm, membrane capacitance; and Rm, membrane resistance. EDL, extensor digitorum longus; WT, wild type.
Significant difference with respect to the control value by Student's t-test (0.005 < P < 0.05).
Fig. 1.

Effect of in vitro application of ANG II on membrane resistance, resting membrane potential, and resting conductance to chloride and potassium ions of extensor digitorum longus (EDL) muscle fibers of wild-type (WT) mice. A: membrane resistance (Rm) plotted as a function of time (in min). The chart shown the Rm values in the absence (●) and in the presence of ANG II (0.3 μM; ○). B: resting membrane potential in fiber sampled for the Rm values in the absence and in the presence of ANG II (0.3 μM) both in normal physiological solution and in chloride-free solution. Values are means ± SE from 59–81 fibers from 6–15 preparations. C and D: percent block of chloride conductance (gCl) and percent increase of potassium conductance (gK), respectively, as a function of increasing concentrations of ANG II. Each point is the means ± SE from 12–56 fibers from 3–4 muscle preparations. All values are statistically different with respect to the value in the absence of ANG II (0.001<P < 0.01 by Student's t-test).
Table 2.
Effect of in vitro application of ANG II on resting ionic conductance of EDL muscle fibers of WT mice
| Experimental Conditions | N/n | gm, μS/cm2 | N′/n′ | gK, μS/cm2 | gCl, μS/cm2 |
|---|---|---|---|---|---|
| Normal physiological solution | 15/81 | 2,841 ± 29 | 9/56 | 343 ± 13 | 2,498 ± 24 |
| + 0.01 μM ANG II | 3/23 | 2,607 ± 74* | 3/12 | 458 ± 8* | 2,149 ± 61* |
| + 0.03 μM ANG II | 3/27 | 2,483 ± 89* | 3/18 | 427 ± 30* | 2,056 ± 80* |
| + 0.1 μM ANG II | 3/19 | 2,033 ± 103* | 3/20 | 530 ± 35* | 1,503 ± 76* |
| + 0.3 μM ANG II | 6/42 | 1,976 ± 59* | 4/41 | 517 ± 17* | 1,459 ± 44* |
| + 1 μM ANG II | 3/22 | 1,935 ± 72* | 3/22 | 602 ± 39* | 1,333 ± 58* |
| + 3 μM ANG II | 3/19 | 1,812 ± 100* | 3/29 | 671 ± 26* | 1,141 ± 66* |
Values are means ± SE from n and n′ number of fibers sampled from N and N′ number of preparations tested in normal and chloride free physiological solutions, respectively. The following parameters are shown: gm, total membrane conductance; gK, potassium conductance; and gCl, chloride conductance.
Significant difference with respect to the control value by Student's t-test (0.005 < P < 0.05).
Resting membrane potential (RMP) values were in line with those described by others, with slight differences due to the different experimental conditions (26, 27, 35). However, RMP was not modified by ANG II in either normal and chloride-free physiological solutions (Fig. 1B), and no basal holding current was used.
Evaluation of receptor and signaling pathways involved in the modulation of gCl and gK by ANG II.
To gain insight into the mechanism underlying the decrease of gCl and the increase of gK by ANG II, a series of experiments were carried out with various pharmacological tools in both normal and chloride-free solutions to estimate specific actions on the component ionic conductance. For these experiments the standard concentration of ANG II used as reference was 0.3 μM, able to produce a repeatable and significant 40% reduction of gCl and a 50% increase of gK (Fig. 2, A and B).
Fig. 2.

Pharmacological evaluation of the signaling mechanism of ANG II on gCl and gK. A–D: effect of ANG II (0.3 μM), alone and after the incubation with various agents, on component ionic conductance of EDL muscle fibers, as follows. A: effect of the selective antagonists of ANG II receptors type 1 (AT1; losartan, 0.3 μM), AT2 receptors (PD123,319, 0.3 μM), and the PKC inhibitor (chelerythrine, 1 μM) on chloride conductance (gCl). Values are means ± SE from the number of fibers indicated in the parentheses from 2–10 preparations. The effects of losartan, PD123,319, and chelerythrine alone are also reported. *Significant difference with respect to the control value by Student's t-test (0.005 < P < 0.001). B: effect of the selective antagonists of AT1 (losartan, 0.3 μM), AT2 receptors (PD 123,319, 0.3 μM), and the PKC inhibitor (chelerythrine, 1 μM) on potassium conductance (gK). Values are means ± SE from the number of fibers indicated in the parentheses from 2–8 preparations. The effect of losartan, PD123,319, and chelerythrine alone are also reported. *Significant difference with respect to the control value by Student's t-test (P < 0.001). C: effect of the antioxidant N-acetyl cysteine (NAC, 5 mM), NOX inhibitor (apocynin, 10 μM), ERK1/2 inhibitor (U-0126, 5 μM), and p38MAPK inhibitor (SB-203580, 5 μM) on the total membrane conductance (gm). Values are means ± SE from the number of fibers indicated in the parentheses from 2–7 preparations. The effect of NAC, apocynin, U-0126 and SB-203580 alone are also reported. *Significant difference with respect to the control value by Student's t-test (P < 0.001). D: effect of the antioxidant NAC (5 mM) and iberiotoxin (IbTX, 0.4 μM), an inhibitor of calcium-activated potassium channels, on potassium conductance (gK). Values are means ± SE from the number of fibers indicated in the parentheses from 2 preparations. *Significant difference with respect to the control value by Student's t-test (P < 0.001).
As can be seen in Fig. 2A, the effect of 0.3 μM ANG II was completely inhibited in the presence of an equimolar concentration of the AT1 blocker losartan. Conversely, no antagonism was observed with the AT2 blocker PD123,319 (0.3 μM). None of the antagonists produced per se any change of gCl (Fig. 2A). To verify if ANG II modulates gCl by activation of a PKC-dependent signaling pathway (20), we evaluated the effect of ANG II in the presence of chelerythrine (1 μM), a PKC inhibitor. The latter completely antagonized ANG II effects, while it did produce a slight but significant effect on gCl per se (Fig. 2A). Similar antagonistic effects by losartan and chelerythrine of ANG II action were observed on gK (Fig. 2B).
The above experiments suggest that both gCl and GK conductance are modulated by the same signaling pathway. Based on this and considering that the significant reduction of gm by ANG II is totally due to reduction of gCl, which shunts the increase in gK, part of the following experiments were conducted in normal physiological solution to simplify the experimental protocols and avoid unnecessary overuse of animals. In fact, the use of pharmacological tools with unclear reversible effects in normal physiological solutions could have biased the measurements of gK in chloride-free medium. In particular, we evaluated the effect of 0.3 μM ANG II in the presence of apocynin (10 μM), an inhibitor of NOX. Interestingly, apocynin fully contrasted the effect of ANG II on gm in EDL fibers (Fig. 2C). Similarly, the incubation of NAC (5 mM), a known ROS scavenger, fully contrasted the reduction of gm by 0.3 μM ANG II (Fig. 2C). Again, neither apocynin nor NAC per se exerted any effect on gm (Fig. 2C).
No antagonism of ANG II effects on gm was found by the preincubation of U-0126 (5 μM), an inhibitor of ER kinase 1/2 (ERK), or of SB-203580 (5 μM), an inhibitor of p38MAP kinase, therefore ruling out the activation of parallel signaling of the AT1 receptor (Fig. 2C) (50).
At the light of the above results, few experiments were performed in chloride-free physiological solution for additional investigation of ANG II on gK. Importantly, the ROS scavenger NAC inhibited ANG II action on gK, again corroborating the involvement of the same pathway acting on gCl (Fig. 2D). In consideration of the above results and in the attempt to characterize the channels involved in the ANG II increase in gK, we tested the effect of iberiotoxin, a specific blocker of calcium-activated potassium channels (BK). Interestingly, the application of 0.4 μM iberiotoxin fully counteracted the ANG II increase of gK (Fig. 2D), while it was without any effect in the absence of ANG II (data not shown).
To have a biochemical correlate of the above signaling we measured by qRT-PCR the expression of the AT1a/b receptor, angiotensinogen (Agt), and angiotensin I converting enzyme (ACE), plus the expression of NOX2 (gp91phox) and its coregulator tubulin α-1b in EDL muscle. For a better quantification, the same genes were measured in parallel in dystrophic mdx EDL muscle, based on our recent confirmation of a higher expression of NOX-2 and tubulin α-1b in gastrocnemius muscle of this genotype (12, 59, 74). The AT1a/b receptor, angiotensinogen, and ACE were expressed in EDL muscle and, surprisingly (70), at comparable level in WT and mdx mice (Fig. 3); as expected NOX2 (gp91phox) and tubulin α-1b were markedly overexpressed in mdx vs. WT animals.
Fig. 3.
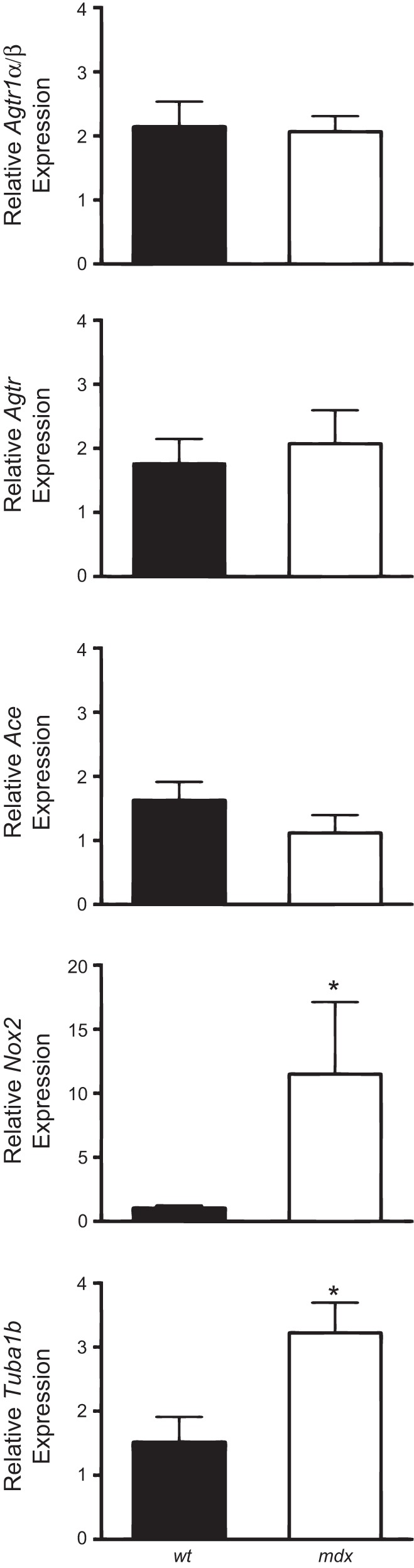
Expression of genes involved in ANG II signaling pathway. Histograms show quantification of transcript levels with quantitative PCR for angiotensin II receptor type 1a and type 1b (Agtr1a/b), angiotensinogen (Agt), angiotensin-converting enzyme (Ace), gp91-phox (Nox2), and tubulin α 1b (Tuba1b) normalized by the GAPDH gene, in EDL muscle of WT and mdx mice. In each graph, the bars are the means ± SE from the 4–6 animals. *Significant differences between groups by Student's t-test (P < 0.05).
Effect of ANG II on excitation contraction-coupling and intracellular calcium transients.
We next assessed whether ANG II signaling in EDL myofibers also involves a modulation of calcium homeostasis. We first evaluated the effect of ANG II on the voltage threshold for mechanical activation (MT), an integrated electrophysiological index of excitation contraction-coupling and of calcium homeostasis (21, 30). In particular, the application of depolarizing steps of increasing durations shifts the voltage for fiber contraction toward a more negative potential until a constant rheobase voltage is reached. At the rheobase voltage, a steady-state is reached between the calcium released from and stored back in the sarcoplasmic reticulum (SR), also in relation to resting calcium levels. In the presence of 0.3 μM ANG II, a marked shift of strength-duration curve describing the MT toward a more negative potential was observed in EDL myofibers. This is indicative of more calcium being available for contraction, either resulting from greater release, slower reuptake, or higher basal cytosolic levels. The shift of the threshold potential values was significant and clearly appreciable at all durations of the depolarizing step (Fig. 4A). The calculated rheobase value, in the presence of ANG II, was −72.5 ± 1.3 mV, significantly more negative than the value recorded in the absence of ANG II (−68 ± 1.7 mV; P < 0.001, by Student's t-test; Table 3). Interestingly, the effect of ANG II on MT was again inhibited by a prior incubation with losartan 0.3 μM but not with 0.3 μM PD123,319 (Fig. 4, B and C). The preincubation of chelerythrine similarly inhibited ANG II actions (Table 3). In light of the findings of previous paragraph, we evaluated the possible involvement of ROS in ANG II-induced changes of MT, due to the ability of ROS to contribute to a greater calcium release by SR for oxidation of ryanodine receptor (RyR; Ref. 37). As shown in Fig. 4D, NAC (5 mM) shifted the rheobase voltage toward more positive values with respect to those recorded in normal physiological solutions (Fig. 4D and Table 3). However, the preincubation of NAC was not able to prevent the effect of ANG II on the rheobase voltage of EDL myofibers (Table 3). No effect by any drug has been observed on the time constant values of MT (data not shown).
Fig. 4.

Effect of ANG II on the mechanical threshold of EDL muscle fibers of WT mice. Each panel shows the voltages for fiber contraction (mechanical threshold) at each pulse duration in different experimental conditions. For some data points the standard error bar is not visible being smaller than symbol size. The fit of the experimental data led to the calculation of rheobase voltage values reported in Table 3. A: effect of ANG II (0.3 μM) (open triangles). Values are means ± SE from 10-22 fibers from 2-3 preparations. B: effect of the selective antagonists for AT1 (losartan, 0.3 μM) alone (open triangles) and in the presence of ANG II (0.3 μM) (gray square). Values are means ± SE from 8-13 fibers from 2-3 preparations. C: effect of the selective antagonist of the AT2 receptor (PD 123,319, 0.3 μM) alone (open triangles) and in the presence of ANG II (0.3 μM) (gray square). Values are means ± SE from 8-12 fibers from 2 preparations. D: effect of the reactive oxygen species scavenger, N-acetyl cysteine (NAC; 5 mM) alone (open triangles) and in the presence of ANG II (0.3 μM) (gray square). Values are means ± SE from 10-21 fibers from 3 preparations. In each panel, the curve in the absence of any compound is also shown (filled circles).
Table 3.
Effect of ANG II on fitted rheobase voltages of EDL muscle fibers of WT mice
| EDL Muscle | Fitted Rheobase Voltage, mV |
|---|---|
| Normal physiological solution | −68.0 ± 1.7 |
| + 0.03 μM ANG II | −72.5 ± 1.3* |
| Normal physiological solution | −68.0 ± 1.2 |
| + 0.3 μM losartan | −68.0 ± 1.2 |
| + 0.3 μM losartan + 0.3 μM ANG II | −69.4 ± 1.3 |
| Normal physiological solution | −68.6 ± 0.9 |
| + 0.3 μM PD123,319 | −68.5 ± 1.9 |
| + 0.3 μM PD123,319 + 0.3 μM ANG II | −71.9 ± 1.3* |
| Normal physiological solution | −67.3 ± 1.9 |
| + 5 mM NAC | −64.7 ± 1.3 |
| + 5 mM NAC + ANG II 0.3 μM ANG II | −70.4 ± 1.3 |
| Normal physiological solution | −66.5 ± 2.0 |
| + 1 μM chelerythrine | −66.9 ± 1.6 |
| + 1 μM chelerythrine + 0.3 μM ANG II | −67.1 ± 1.7 |
For each experimental condition are shown the rheobase voltages (±SE) obtained from the fit of the strength-duration curves in Fig. 4 to the equation described in materials and methods. The threshold membrane potential values used for the fitting process are means ± SE from 6–22 fibers from 2–3 preparations. NAC, N-acetyl cysteine.
Significant difference with respect to the control value by Student's t-test (0.005 < P < 0.05).
The functional outcome of the observed effect on MT was evaluated by assessing the acute effect of ANG II on isometric contraction. A 30-min incubation with either 0.3 or 1 μM ANG II did not exert changes in maximal tetanic force (data not shown). Interestingly, the force-frequency relationship, a calcium-dependent parameter, was significantly shifted toward lower frequencies, in line with the lower threshold for contraction found with MT recordings (Fig. 5). In particular the frequency for half-maximal activation (Hz50) was 66.8 ± 2.8 and 53.6 ± 2.8 before and after the application of 0.3 μM ANG II (n = 3; P < 0.02 by Student's t-test), respectively. No additional shift was observed at the higher ANG II concentration.
Fig. 5.
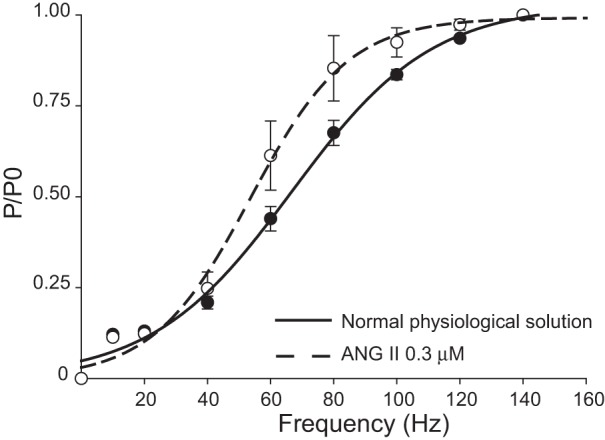
Effect of ANG II on force-frequency (P/P0) relationship of EDL muscle of WT mice. Effect of ANG II (0.3 μM, dashed line) with respect to normal physiological solution (continuous line) on force-frequency relationship of isolated EDL muscles stimulated at increasing frequencies (10–140 Hz). Values for each frequency stimulation are the means ± SE from 3 preparations.
To verify whether the ANG II-induced MT lowering was mirrored by changes in resting [Ca2+]i, cytofluorimetric evaluations were performed with the fluorescent calcium probe fura-2. In agreement with previous studies (11, 30, 49), resting [Ca2+]i of fast-twitch EDL myofibers was in the concentration range of 0.03–0.04 μM. Acute application of ANG II, at the concentration of 1 μM, triggered a sustained rise of [Ca2+]i that ranged between 1.8 and 2.5-fold with respect to the resting value. This effect occurred in 75% of the sampled fiber preparations. Intracellular microelectrode recordings were performed to measure resting membrane potential values in randomly selected bundles used for fura-2 experiments. The values were −65.7 ± 3.1 mV (n fibers = 16) in normal physiological solution and −65.2 ± 3.1 mV (n fibers = 18) in fura-2-containing solution, therefore ruling out the possible occurrence of depolarization due to preparation or fura-2. Interestingly, as shown in Fig. 6A, the [Ca2+]i increase induced by ANG II exhibited a peculiar time course, characterized by a two-step slow rising phase (lasting 20 min), which finally led to the large increase of [Ca2+]i observed. When the drug was removed, [Ca2+]i in part rapidly decreased (Fig. 6A) and subsequently slowly returned close to the basal resting [Ca2+]i (in ∼20 min; data not shown). At a lower concentration (0.5 μM), ANG II was less effective, producing an increase of [Ca2+]i of ∼40% (Fig. 6B). Due to the complexity of the observed signal, we evaluated the involvement of the AT1 receptor by the use of the nonpeptidic AT1 agonist L162,313 and of losartan. L162,313 caused a change of [Ca2+]i resembling that induced by ANG II (Fig. 6A). Particularly, at 0.03 μM and 0.1 μM, L162,313 triggered an increase in [Ca2+]i of ∼21 and 60% above the resting level, respectively. Again, this effect was observed in ∼70% of the incubated fibers (Fig. 6B). As shown in Fig. 6, the presence of 0.3 μM losartan, which alone had no effect on resting [Ca2+]i, completely antagonized the 0.1-μM L162,313-induced [Ca2+]i increase. Furthermore, preincubation with 10 μM U73122, a PLC inhibitor that by itself had no effect on resting [Ca2+]i, was able to antagonize the effect of 0.1 μM L162,313 on [Ca2+]i (Fig. 6C). All these results suggested the involvement of the canonical AT1 receptor signaling pathway also in the regulation of calcium homeostasis. To better investigate the source for the calcium increase, a set of experiments were performed in a calcium-free solution. Interestingly, the effects of L162,313 were fully abolished, suggesting a key role of calcium entry. Since it has been recently demonstrated that in podocytes generation of ROS is an essential part for the PLC-dependent cascade initiated by ANG II acting on AT1 receptor leading to increased calcium influx in these cells (1), we also evaluated the effect of L162,313 in the presence of ROS scavenger NAC. In accordance with the demonstrated dependence of resting calcium on the RyR1 redox state (37, 71), preincubation of NAC caused per se a significant decrease of resting [Ca2+]i. Interestingly, the effect of ANG II was completely antagonized by NAC.
Fig. 6.

Effect of ANG II and L162,313 on calcium homeostasis of mouse skeletal muscle fibers. A: representative traces of the calcium increase induced by the addition of either 1 μM ANG II (on left) or L162,313 (on right). B: bars show the mean value of intracellular Ca2+ concentration ([Ca2+]i) from the number of fibers indicated in the parentheses, at least from 3 animals, recorded in the absence and presence of either ANG II (0.5 and 1 μM) or L162,313 (0.03 and 0.1 μM). The data are expressed as the means ± SE. C: summary of the 0.1 μM L162,313-induced response in presence of external calcium (2.5 mM Ca2+), in the absence of external calcium (Ø Ca2+) and in the presence of losartan (0.3 μM), U73122 (10 μM), and NAC (5 mM). Each bar is the means ± SE from the number of fibers indicated in the parentheses, from at least 3 animals. Losartan, U73122, and NAC were incubated for 10 min before the addition of L162,313; their own effects are also reported. *Significant difference with respect to control value (by Student's t-test, P < 0.05).
DISCUSSION
The present results provide the first evidence that, in mouse skeletal muscle fibers, ANG II reduces, via AT1 receptor, the activity of the channels accounting for resting gCl. The inhibition of gCl is paralleled by an increase in gK that slightly counterbalanced the effect of ANG II on total membrane conductance.
Our study focused on the possible role of ANG II in modulating the function of muscle gCl due to its involvement in several pathophysiological condition of skeletal muscle and based on increasing evidence that ANG II can play a key role in pathophysiological states of skeletal muscle. RAS and ANG II might be involved in skeletal muscle wasting and regeneration impairment, via ROS production, activation of ubiquitin-mediated protein degradation, and interference with metabolism and insulin-like growth factor I signaling (41, 43, 50, 63, 73). Accordingly, a role of RAS in heart and skeletal muscle damage in muscular dystrophy has been proposed (13, 14, 70). Many of the above actions have been observed upon in vivo manipulation of the RAS system or in cell lines, and little few evidence is available about direct actions of ANG II on adult myofibers (43, 45, 69, 78). The ability of an in vivo enalapril treatment to contrast gCl impairment in dystrophic mdx animals was the starting point of the present study, based on the hypothesis that gCl may be a direct target of ANG II actions in adult myofibers (14).
By the use of various pharmacological tools we found that ANG II modulates gCl with a complex signaling, involving activation of PKC, as well as of NOX and ROS. The biochemical modulation of mammalian muscle gCl by PKC-mediated phosphorylation events is supported by several observations (5, 20, 22, 57). Studies from our laboratory showed that direct activation of PKC with phorbol esters or inhibition of phosphatases with okadaic acid, potently reduce gCl, suggesting that a balance between phosphorylation-dephosphorylation pathways is required to maintain the basal physiological high values of gCl (20,57). Change in the expression of PKC α, ε, and θ parallels the phenotype-dependent change in gCl observed in disuse conditions (57). Importantly, PKC activators/inhibitors have been shown to modulate ClC-1 currents in voltage-clamp experiments in native and heterologous expression system (39, 62). According to these observations, site-directed mutagenesis studies highlighted that Thr891-Ser892-Thr893 at the COOH-terminal region of ClC-1 are involved in the PKC phosphorylation, which, in turn, affects channel gating; a phosphorylation mechanism controlling the trafficking of the channels from an intracellular pool to the membrane surface has also been proposed (39, 53, 62). We have also previously shown that the PKC-mediated modulation of gCl occurs via a Gq protein, suggesting the involvement of an endogenous mediator (22). This first result has then been corroborated by the identification of endogenous ligands of G-coupled receptors, such as P2Y1 and ghrelin receptors, able to modulate the ClC-1 channel (55, 72). The present results show, for the first time, that ANG II is an important key modulator of ClC-1 channels, via activation of the AT1 receptor, corroborating that the modulation of gCl by endocrine/autocrine signals plays a crucial role for adapting muscle function to physiological requests or as part of signaling in excitation-transcription coupling (54, 56).
The AT1 receptor signals via canonical Gq/PLC pathway. As a classical paradigm, such a transduction signal produces diacylglycerol (DAG) and inositol-1,4,5-trisphosphate (IP3). While recent evidence demonstrated that IP3 plays a marginal, if any, role in releasing Ca2+ from muscle SR stores (3), DAG can directly activate PKC and also reinforce the signaling by stimulating receptor-mediated calcium entry via activation of transient receptor potential (TRP) channels, such as TRPC5 and 6 (1, 38). In support of this view we found that ANG II caused, via the AT1 receptor, a “two-step shape” increase of calcium transient, which was completely abolished by the removal of external calcium. Then our results are consistent with an AT1-mediated activation of a phospholipid and calcium-dependent PKC, that finally shifts the equilibrium toward an increased phosphorylation state of the ClC-1 channel. The observed plateau effect on gCl by ANG II is also in line with a physiological and reversible activation of a G protein-coupled phosphorylation pathway.
In addition, the present results add clear evidence that the production of ROS via NOX works as fundamental part of the pathway leading to the observed reduction of gCl by ANG II in myofibers as in other experimental conditions (1, 41, 43, 73). This is emphasized by the observation that effects of ANG II are completely antagonized by both the classical antioxidant NAC and by the NOX inhibitor apocynin, as well as by the PKC inhibitor, although the occurrence of a parallel ROS-mediated signaling cannot be fully ruled out.
ANG II may regulate NOX in skeletal muscle via PKC activation; this step is required for p47phox phosphorylation and in turn for the correct assembly and activation of NOX (65, 75). ROS can trigger and/or amplify ANG II signaling pathway. In fact, a direct modulation of gCl by ROS can occur by involving oxidation of critical cysteine residues of ClC-1 channel, as described on heterologously expressed systems (2, 79). PKC isoforms are also redox sensitive, and oxidation of accessory subunits has been shown to lead to a cofactor independent activation of PKC in PC12 cells (33). Therefore, a possible reinforcing loop generated by ROS on PKC activation is feasible and may also account for the ability of both NAC and chelerythrine to antagonize the ANG II-related increase of gK. This latter effect is in fact due to the involvement of large conductance big potassium channels (BK), which are calcium-activated and also modulated by PKC (40). The reduction of gCl, or more generally the concerted modulation of resting ionic conductance, via ANG II-induced activation of PKC and ROS signaling, may contribute to muscle fatigue and have an aggravating role in excitability disorders (23, 29, 43, 72). This observation, along with the advancements in measuring ClC-1 currents by voltage-clamp recordings, paves the way for future studies of the ANG II effects on channel biophysics (25, 28, 48, 60).
The results also disclosed a wider involvement of ANG II-mediated AT1 signaling on membrane electrical properties and excitation-contraction coupling, which added novel findings to the main objective of the study on chloride channel conductance. Other than the modulation of gK, ANG II, via the AT1 receptor, shifted MT toward more negative potentials; in parallel a shift of the frequency-force curve of isometric contraction toward lower stimulation frequencies was observed. The mechanism underlying the ANG II effect on MT is puzzling and likely independent of the effect on gCl. In fact the myofibers of myotonic animals have, contrary to simple expectation, a shift of MT toward more positive potentials, and similar modifications can be observed with drugs that inhibit gCl (10, 36). In particular we found that NAC does not antagonize ANG II and can per se shift MT toward a more positive potential. A basal oxidation of RyR1 receptor may account for the NAC effect, since this mechanism induces a sustained calcium release; this mechanism can also account for the slight differences in MT found between species or in different studies (17, 27, 37, 46, 71). Accordingly, NOX is localized in the transverse tubule and its involvement in calcium release has been described (37). This hypothesis is supported by our observation that NAC per se also reduced basal calcium in fura-2 experiments. However, NAC antagonized the ANG II effects on calcium homeostasis but not on MT, therefore ruling out a role of ROS on RyR, DHPR, or contractile proteins in the observed shift of MT by ANG II (49). Therefore, it is likely to suggest other mechanisms for the ANG II effects on excitation-contraction coupling, such as the PKC-mediated phosphorylation of key elements, as supported by the chelerythrine antagonism (76). These mechanisms need to be further investigated also in relation to possible ANG II impact on muscle pathophysiology. Part of the differential effects observed can be related to the dual step in calcium increase that may in turn act on calcium-sensitive targets, i.e., BK channels and PKC, at different concentrations and time scales. The absence of both peaks in calcium-free medium and the antagonism by losartan, the PLC inhibitor, and NAC allows the hypothesis that the calcium entry takes place via two autoreinforcing mechanisms, i.e., a first step related to DAG followed by a second phase, mediated by ROS (1, 38). However, and in line with the minor, if any, role of external calcium for muscle contraction, no changes were observed in EDL muscle force during contraction.
Focused experiments are required to identify the channel involved in calcium entry under ANG II stimulation and to better clarify the mechanism involved. Importantly, an enhanced calcium entry via TRP-like mechanosensitive channels does occur in skeletal muscle fibers, especially in dystrophic conditions that are characterized by enhanced ROS production (59, 61, 66). Also, an ANG II-independent activation of AT1 receptor by mechanical stress has been described and does contribute to heart failure (80), supporting the potential alternative involvement of the system in myopathic state related to altered mechanotransduction.
A possible scheme of the ANG II signaling in myofiber is shown in Fig. 7. Importantly, the effective concentrations of ANG II in our study were similar to those found by others and slightly higher with respect to those calculated in binding studies (1, 16, 32). Therefore, the effects observed are in line with pathophysiological concentration of ANG II in vivo, especially for autocrine/paracrine actions. In fact tissue levels are in the nanomolar range and then up to a 10-fold higher than those detectable in plasma, considering also the extremely short half-life of ANG II (51).
Fig. 7.

Proposed model of the ANG II signaling on chloride channels of skeletal muscle fiber. The binding of ANG II to AT1-R leads to Gq protein-coupled activation (Gq) of phospholipase C (PLC), triggering a classical transduction mechanism, resulting in increase in cytosolic Ca2+ and activation of a Ca2+-diacylglycerol (DAG)-dependent PKC, which in turn phosphorylates ClC-1. These events can also result in activation of NOX with production of ROS. Putative target sites for ROS are shown on ClC-1 channel and ryanodine receptor (RyR1). However, PKC is another potential target of ROS, with consequent enzyme activation and initiation of a reinforcing loop. Furthermore, DAG and ROS can both act on a cationic channels, likely belonging to the transient receptor potential (TRP) family, which likely accounts for the enhanced Ca2+ entry by ANG II. The increase of cytosolic Ca2+ may then coactivate PKC, contribute to excitation-contraction coupling and activate of Ca2+-activated K+ (BK) channel. Arrows indicate a general stimulating action (dashed lines for additional potential signaling). ClC-1 channels are indicated both in transverse (t)-tubule membrane and at surface membrane as their localization is still unresolved, while NOX is indicated in t-tubules based on Hidalgo et al. (37). The presence of AT1 receptor at sarcolemma is hypothesized in relation to ANG II molecular dimension and limited access of t-tubules by external modulators. The position of TRP-like and BK channels at the surface membrane is arbitrarily related to graphical reasons and is therefore purely speculative.
Our study provides key evidence about acute ANG II signaling in adult myofibers and supports the view that gCl and ClC-1 channel are sensitive indexes to monitor oxidative stress as well as therapeutic efficacy of antioxidant drugs in myopathic states. The long-term impact of modulation of gCl, calcium homeostasis, and excitation-contraction coupling by ANG II in muscle dysfunction, in oxidative-stress-induced muscle wasting, and in metabolism-remodeling uncoupling deserves to be further investigated (41, 43, 69, 77).
GRANTS
This work was supported by a grant from the Duchenne Parent Project Netherland (DPP/NL) and MIUR-PRIN Project No. 20108YB5W3_004.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: A.C. and A.D.L. conception and design of research; A.C., A.L., E.C., M.C., A.G., A.S., P.M., R.F.C., and G.M.C. performed experiments; A.C., A.L., E.C., A.M.M., R.F.C., and G.M.C. analyzed data; A.C., A.L., and A.D.L. interpreted results of experiments; A.C., R.F.C., and G.M.C. prepared figures; A.C. and A.D.L. drafted manuscript; A.L. and A.D.L. edited and revised manuscript; S.P. and A.D.L. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Diana Conte Camerino for critical comments on the manuscript.
REFERENCES
- 1.Anderson M, Roshanravan H, Khine J, Dryer SE. Angiotensin II activation of TRPC6 channels in rat podocytes requires generation of reactive oxygen species. J Cell Physiol 229: 434–442, 2014 [DOI] [PubMed] [Google Scholar]
- 2.Arthur PG, Grounds MD, Shavlakadze T. Oxidative stress as a therapeutic target during muscle wasting: considering the complex interactions. Curr Opin Clin Nutr Metab Care 11: 408–416, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Blaauw B, Del Piccolo P, Rodriguez L, Hernandez Gonzalez VH, Agatea L, Solagna F, Mammano F, Pozzan T, Schiaffino S. No evidence for inositol 1,4,5-trisphosphate-dependent Ca2+ release in isolated fibers of adult mouse skeletal muscle. J Gen Physiol 140: 235–241, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bretag AH. Muscle chloride channels. Physiol Rev 67: 618–724, 1987 [DOI] [PubMed] [Google Scholar]
- 5.Brinkmeier H, Jockusch H. Activators of protein kinase C induce myotonia by lowering chloride conductance in muscle. Biochem Biophys Res Commun 148: 1383–1389, 1987 [DOI] [PubMed] [Google Scholar]
- 6.Brooks SV, Faulkner JA. Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol 404: 71–82, 1988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bryant SH, Conte-Camerino D. Chloride channel regulation in the skeletal muscle of normal and myotonic goat. Pflügers Arch 417: 605–610, 1991 [DOI] [PubMed] [Google Scholar]
- 8.Bryant SH, Morales-Aguilera A. Chloride conductance in normal and myotonic muscle fibres and the action of monocarboxylic aromatic acids. J Physiol 219: 367–383, 1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bryant SH. Cable properties of external intercostal muscle fibres from myotonic and nonmyotonic goats. J Physiol 204: 539–550,1969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bryant SH. Myotonia in the goat. Ann NY Acad Sci 317: 314–325, 1979 [DOI] [PubMed] [Google Scholar]
- 11.Burdi R, Rolland JF, Fraysse B, Litvinova K, Cozzoli A, Giannuzzi V, Liantonio A, Camerino GM, Sblendorio V, Capogrosso RF, Palmieri B, Andreetta F, Confalonieri P, De Benedictis L, Montagnani M, De Luca A. Multiple pathological events in exercised dystrophic mdx mice are targeted by pentoxifylline: outcome of a large array of in vivo and ex vivo tests. J Appl Physiol 106: 1311–1324, 2009 [DOI] [PubMed] [Google Scholar]
- 12.Camerino GM, Cannone M, Giustino A, Massari AM, Capogrosso RF, Cozzoli A, De Luca A. Gene expression in mdx mouse muscle in relation to age and exercise: aberrant mechanical-metabolic coupling and implications for preclinical studies in Duchenne muscular dystrophy. Hum Mol Genet pii: ddu287, 2014 [DOI] [PubMed] [Google Scholar]
- 13.Cohn RD, van Erp C, Habashi JP, Soleimani AA, Klein EC, Lisi MT, Gamradt M, ap Rhys CM, Holm TM, Loeys BL, Ramirez F, Judge DP, Ward CW, Dietz HC. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat Med 13: 204–210, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cozzoli A, Nico B, Sblendorio VT, Capogrosso RF, Dinardo MM, Longo V, Gagliardi S, Montagnani M, De Luca A. Enalapril treatment discloses an early role of angiotensin II in inflammation- and oxidative stress-related muscle damage in dystrophic mdx mice. Pharmacol Res 64: 482–492, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dangain J, Vrbová G. Effect of chronic electrical stimulation at low frequency on the passive membrane properties of muscle fibers from dystrophic mice. Exp Neurol 79: 630–640, 1983 [DOI] [PubMed] [Google Scholar]
- 16.De León H, Garcia R. Angiotensin II receptor subtypes in rat renal preglomerular vessels. Receptor 2: 253–260, 1992 [PubMed] [Google Scholar]
- 17.De Luca A, Conte Camerino D. Effects of aging on the mechanical threshold of rat skeletal muscle fibers. Pflügers Arch 420: 407–409, 1992 [DOI] [PubMed] [Google Scholar]
- 18.De Luca A, Nico B, Liantonio A, Didonna MP, Fraysse B, Pierno S, Burdi R, Mangieri D, Rolland JF, Camerino C, Zallone A, Confalonieri P, Andreetta F, Arnoldi E, Courdier-Fruh I, Magyar JP, Frigeri A, Pisoni M, Svelto M, Conte Camerino D. A multidisciplinary evaluation of the effectiveness of cyclosporine A in dystrophic mdx mice. Am J Pathol 166: 477–489, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.De Luca A, Pierno S, Camerino DC. Electrical properties of diaphragm and EDL muscles during the life of dystrophic mice. Am J Physiol Cell Physiol 272: C333–C340, 1997 [DOI] [PubMed] [Google Scholar]
- 20.De Luca A, Pierno S, Liantonio A, Camerino C, Camerino DC. Phosphorylation and IGF-1 mediated dephosphorylation pathways control the activity and the pharmacological properties of skeletal muscle chloride channels. Br J Pharmacol 125: 477–482, 1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.De Luca A, Pierno S, Liantonio A, Cetrone M, Camerino C, Fraysse B, Mirabella M, Servidei S, Rüegg UT, Conte Camerino D. Enhanced dystrophic progression in mdx mice by exercise and beneficial effects of taurine and insulin-like growth factor-1. J Pharmacol Exp Ther 304: 453–463, 2003 [DOI] [PubMed] [Google Scholar]
- 22.De Luca A, Tricarico D, Pierno S, Conte Camerino D. Aging and chloride channel regulation in rat fast-twitch muscle fibres. Pflügers Arch 427: 80–85, 1994 [DOI] [PubMed] [Google Scholar]
- 23.de Paoli FV, Broch-Lips M, Pedersen TH, Nielsen OB. Relationship between membrane Cl− conductance and contractile endurance in isolated rat muscles. J Physiol 591: 531–545, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Delbono O, Renganathan M, Messi ML. Regulation of mouse skeletal muscle L-type Ca2+ channel by activation of the insulin-like growth factor-1 receptor. J Neurosci 17: 6918–6928, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.DiFranco M, Herrera A, Vergara JL. Chloride currents from the transverse tubular system in adult mammalian skeletal muscle fibers. J Gen Physiol 137: 21–41, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donaldson PJ, Leader JP. Intracellular ionic activities in the EDL muscle of the mouse. Pflügers Arch 400: 166–170, 1994 [DOI] [PubMed] [Google Scholar]
- 27.Dulhunty AF. Effects of membrane potential on mechanical activation in skeletal muscle. J Gen Physiol 79: 233–251, 1982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dutka TL, Murphy RM, Stephenson DG, Lamb GD. Chloride conductance in the transverse tubular system of rat skeletal muscle fibres: importance in excitation-contraction coupling and fatigue. J Physiol 586: 875–887, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fauler M, Jurkat-Rott K, Lehmann-Horn F. Membrane excitability and excitation-contraction uncoupling in muscle fatigue. Neuromuscul Disord 22, Suppl 3: S162–S167, 2012 [DOI] [PubMed] [Google Scholar]
- 30.Fraysse B, Desaphy JF, Pierno S, De Luca A, Liantonio A, Mitolo CL, Camerino DC. Decrease in resting calcium and calcium entry associated with slow-to-fast transition in unloaded rat soleus muscle. FASEB J 17: 1916–1918, 2003 [DOI] [PubMed] [Google Scholar]
- 31.Fraysse B, Desaphy JF, Rolland JF, Pierno S, Liantonio A, Giannuzzi V, Camerino C, Didonna MP, Cocchi D, De Luca A, Conte Camerino D. Fiber type-related changes in rat skeletal muscle calcium homeostasis during aging and restoration by growth hormone. Neurobiol Dis 21: 372–380, 2006 [DOI] [PubMed] [Google Scholar]
- 32.Goodfriend TL, Elliott ME, Catt KJ. Angiotensin receptors and their antagonists. N Engl J Med 334: 1649–1654, 1996 [DOI] [PubMed] [Google Scholar]
- 33.Gopalakrishna R, Gundimeda U, Schiffman JE, McNeill TH. A direct redox regulation of protein kinase C isoenzymes mediates oxidant-induced neuritogenesis in PC12 cells. J Biol Chem 283: 14430–14444, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem 260: 3440–3450, 2005 [PubMed] [Google Scholar]
- 35.Head SI. Membrane potential, resting calcium and calcium transients in isolated muscle fibres from normal and dystrophic mice. J Physiol 469: 11–19, 1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Heiny JA, Jong D, Bryant SH, Conte-Camerino D, Tortorella V. Enantiomeric effects on excitation-contraction coupling in frog skeletal muscle by a chiral phenoxy carboxylic acid. Biophys J 57: 147–152, 1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hidalgo C, Sanchez G, Barrientos G, Aracena-Parks P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S-glutathionylation. J Biol Chem 281: 26473–26482, 2006 [DOI] [PubMed] [Google Scholar]
- 38.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature 397: 259–263, 1999 [DOI] [PubMed] [Google Scholar]
- 39.Hsiao KM, Huang RY, Tang PH, Lin MJ. Functional study of CLC-1 mutants expressed in Xenopus oocytes reveals that a C-terminal region Thr891-Ser892-Thr893 is responsible for the effects of protein kinase C activator. Cell Physiol Biochem 25: 687–694, 2010 [DOI] [PubMed] [Google Scholar]
- 40.Hypolite JA, Lei Q, Chang S, Zderic SA, Butler S, Wein AJ, Malykhina AP, Chacko S. Spontaneous and evoked contractions are regulated by PKC-mediated signaling in detrusor smooth muscle: involvement of BK channels. Am J Physiol Renal Physiol 304: F451–F462, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inoue N, Kinugawa S, Suga T, Yokota T, Hirabayashi K, Kuroda S, Okita K, Tsutsui H. Angiotensin II-induced reduction in exercise capacity is associated with increased oxidative stress in skeletal muscle. Am J Physiol Heart Circ Physiol 302: H1202–H1210, 2012 [DOI] [PubMed] [Google Scholar]
- 42.Jack BJ, Noble D, Tsien RW. Electric Current Flow in Excitable Cells. Oxford, UK: Oxford Univ Press, 1975, p. XVI [Google Scholar]
- 43.Kackstein K, Teren A, Matsumoto Y, Mangner N, Möbius-Winkler S, Linke A, Schuler G, Punkt K, Adams V. Impact of angiotensin II on skeletal muscle metabolism and function in mice: contribution of IGF-1, Sirtuin-1 and PGC-1α. Acta Histochem 115: 363–370, 2013 [DOI] [PubMed] [Google Scholar]
- 44.Liantonio A, Giannuzzi V, Picollo A, Babini E, Pusch M, Conte Camerino D. Niflumic acid inhibits chloride conductance of rat skeletal muscle by directly inhibiting the CLC-1 channel and by increasing intracellular calcium. Br J Pharmacol 150: 235–247, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Linderman JR, Greene AS. Distribution of angiotensin II receptor expression in the microcirculation of striated muscle. Microcirculation 8: 275–281, 2001 [DOI] [PubMed] [Google Scholar]
- 46.Liu G, Pessah IN. Molecular interaction between ryanodine receptor and glycoprotein triadin involves redox cycling of functionally important hyperreactive sulfhydryls. J Biol Chem 269: 33028–33034, 1994 [PubMed] [Google Scholar]
- 47.Lossin C, George AL., Jr Myotonia congenita. Adv Genet 63: 25–55, 2008 [DOI] [PubMed] [Google Scholar]
- 48.Lueck JD, Rossi AE, Thornton CA, Campbell KP, Dirksen RT. Sarcolemmal-restricted localization of functional CLC-1 channels in mouse skeletal muscle. J Gen Physiol 136: 597–613, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Moopanar TR, Allen DG. Reactive oxygen species reduce myofibrillar Ca2+ sensitivity in fatiguing mouse skeletal muscle at 37 degrees C. J Physiol 564: 189–199, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Morales MG, Vazquez Y, Acuña MJ, Rivera JC, Simon F, Salas JD, Alvarez Ruf J, Brandan E, Cabello-Verrugio C. Angiotensin II-induced pro-fibrotic effects require p38MAPK activity and transforming growth factor beta 1 expression in skeletal muscle cells. Int J Biochem Cell Biol 44: 1993–2002, 2012 [DOI] [PubMed] [Google Scholar]
- 51.Nishiyama A, Seth DM, Navar LG. Renal interstitial fluid angiotensin I and angiotensin II concentrations during local angiotensin-converting enzyme inhibition. J Am Soc Nephrol 13: 2207–2212, 2002 [DOI] [PubMed] [Google Scholar]
- 52.Palade PT, Barchi RL. On the inhibition of muscle membrane chloride conductance by aromatic carboxylic acids. J Gen Physiol 69: 879–896, 1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Papponen H, Kaisto T, Myllylä VV, Myllylä R, Metsikkö K. Regulated sarcolemmal localization of the muscle-specific ClC-1 chloride channel. Exp Neurol 191: 163–173, 2005 [DOI] [PubMed] [Google Scholar]
- 54.Pedersen TH, de Paoli FV, Flatman JA, Nielsen OB. Regulation of ClC-1 and KATP channels in action potential-firing fast-twitch muscle fibers. J Gen Physiol 134: 309–322, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pierno S, De Luca A, Desaphy JF, Fraysse B, Liantonio A, Didonna MP, Lograno M, Cocchi D, Smith RG, Camerino DC. Growth hormone secretagogues modulate the electrical and contractile properties of rat skeletal muscle through a ghrelin-specific receptor. Br J Pharmacol 139: 575–584, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pierno S, Desaphy JF, Liantonio A, De Bellis M, Bianco G, De Luca A, Frigeri A, Nicchia GP, Svelto M, Léoty C, George AL, Jr, Camerino DC. Change of chloride ion channel conductance is an early event of slow-to-fast fibre type transition during unloading-induced muscle disuse. Brain 125: 1510–1521, 2002 [DOI] [PubMed] [Google Scholar]
- 57.Pierno S, Desaphy JF, Liantonio A, De Luca A, Zarrilli A, Mastrofrancesco L, Procino G, Valenti G, Conte Camerino D. Disuse of rat muscle in vivo reduces protein kinase C activity controlling the sarcolemma chloride conductance. J Physiol 584: 983–995, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pierno S, Nico B, Burdi R, Liantonio A, Didonna MP, Cippone V, Fraysse B, Rolland JF, Mangieri D, Andreetta F, Ferro P, Camerino C, Zallone A, Confalonieri P, De Luca A. Role of tumour necrosis factor alpha, but not of cyclo-oxygenase-2-derived eicosanoids, on functional and morphological indices of dystrophic progression in mdx mice: a pharmacological approach. Neuropathol Appl Neurobiol 33: 344–359, 2007 [DOI] [PubMed] [Google Scholar]
- 59.Prosser BL, Khairallah RJ, Ziman AP, Ward CW, Lederer WJ. X-ROS signalling in the heart and skeletal muscle: stretch-dependent local ROS regulates [Ca(2+)](i). J Mol Cell Cardiol 58: 172–181, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pusch M, Steinmeyer K, Jentsch TJ. Low single channel conductance of the major skeletal muscle chloride channel, ClC-1. Biophys J 66: 149–152, 1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rolland JF, De Luca A, Burdi R, Andreetta F, Confalonieri P, Conte Camerino D. Overactivity of exercise-sensitive cation channels and their impaired modulation by IGF-1 in mdx native muscle fibers: beneficial effect of pentoxifylline. Neurobiol Dis 24: 466–474, 2006 [DOI] [PubMed] [Google Scholar]
- 62.Rosenbohm A, Rudel R, Fahlke C. Regulation of the human skeletal muscle chloride channel hClC-1 by protein kinase C. J Physiol 514: 677–685, 1999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Russell ST, Eley H, Tisdale MJ. Mechanism of attenuation of angiotensin-II-induced protein degradation by insulin-like growth factor-I (IGF-I). Cell Signal 19: 1583–1595, 2007 [DOI] [PubMed] [Google Scholar]
- 64.Schneider MF, Chandler WK. Effects of membrane potential on the capacitance of skeletal muscle fibers. J Gen Physiol 67: 125–163, 1976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Seshiah PN, Weber DS, Rocic P, Valppu L, Taniyama Y, Griendling KK. Angiotensin II stimulation of NAD(P)H oxidase activity: upstream mediators. Circ Res 91: 406–413, 2002 [DOI] [PubMed] [Google Scholar]
- 66.Shkryl VM, Martins AS, Ullrich ND, Nowycky MC, Niggli E, Shirokova N. Reciprocal amplification of ROS and Ca(2+) signals in stressed mdx dystrophic skeletal muscle fibers. Pflügers Arch 458: 915–928, 2009 [DOI] [PubMed] [Google Scholar]
- 67.Steinmeyer K, Klocke R, Ortland C, Gronemeier M, Jockusch H, Gründer S, Jentsch TJ. Inactivation of muscle chloride channel by transposon insertion in myotonic mice. Nature 354: 304–308b, 1991 [DOI] [PubMed] [Google Scholar]
- 68.Steinmeyer K, Ortland C, Jentsch TJ. Primary structure and functional expression of a developmentally regulated skeletal muscle chloride channel. Nature 354: 301–304a, 1991 [DOI] [PubMed] [Google Scholar]
- 69.Sukhanov S, Semprun-Prieto L, Yoshida T, Michael Tabony A, Higashi Y, Galvez S, Delafontaine P. Angiotensin II, oxidative stress and skeletal muscle wasting. Am J Med Sci 342: 143–147, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sun G, Haginoya K, Dai H, Chiba Y, Uematsu M, Hino-Fukuyo N, Onuma A, Iinuma K, Tsuchiya S. Intramuscular renin-angiotensin system is activated in human muscular dystrophy. J Neurol Sci 280: 40–48, 2009 [DOI] [PubMed] [Google Scholar]
- 71.Sun J, Xin C, Eu JP, Stamler JS, Meissner G. Cysteine-3635 is responsible for skeletal muscle ryanodine receptor modulation by NO. Proc Natl Acad Sci USA 98: 11158–11162, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Voss AA. Extracellular ATP inhibits chloride channels in mature mammalian skeletal muscle by activating P2Y1 receptors. J Physiol 587: 5739–5752, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wei Y, Sowers JR, Nistala R, Gong H, Uptergrove GM, Clark SE, Morris EM, Szary N, Manrique C, Stump CS. Angiotensin II-induced NADPH oxidase activation impairs insulin signaling in skeletal muscle cells, J. Biol. Chem 281 35137–35146, 2006 [DOI] [PubMed] [Google Scholar]
- 74.Whitehead NP, Yeung EW, Froehner SC, Allen DG. Skeletal muscle NADPH oxidase is increased and triggers stretch-induced damage in the mdx mouse. PLoS One 5: e15354, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Williams HC, Griendling KK. NADPH oxidase inhibitors: new antihypertensive agents? J Cardiovasc Pharmacol 50: 9–16, 2007 [DOI] [PubMed] [Google Scholar]
- 76.Woo JS, Hwang JH, Ko JK, Weisleder N, Kim do H, Ma J, Lee EH. S165F mutation of junctophilin 2 affects Ca2+ signalling in skeletal muscle. Biochem J 427: 125–134, 2010 [DOI] [PubMed] [Google Scholar]
- 77.Yoshida T, Galvez S, Tiwari S, Rezk BM, Semprun-Prieto L, Higashi Y, Sukhanov S, Yablonka-Reuveni Z, Delafontaine P. Angiotensin II inhibits satellite cell proliferation and prevents skeletal muscle regeneration. J Biol Chem 288: 23823–23832, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zhang L, Du J, Hu Z, Han G, Delafontaine P, Garcia G, Mitch WE. IL-6 and serum amyloid A synergy mediates angiotensin II-induced muscle wasting. J Am Soc Nephrol 20: 604–612, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhang XD, Tseng PY, Chen TY. ATP inhibition of CLC-1 is controlled by oxidation and reduction. J Gen Physiol 132: 421–428, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zou Y, Akazawa H, Qin Y, Sano M, Takano H, Minamino T, Makita N, Iwanaga K, Zhu W, Kudoh S, Toko H, Tamura K, Kihara M, Nagai T, Fukamizu A, Umemura S, Iiri T, Fujita T, Komuro I. Mechanical stress activates angiotensin II type 1 receptor without the involvement of angiotensin II. Nat Cell Biol 6: 499–506, 2004 [DOI] [PubMed] [Google Scholar]