

Abstract
Platelet-derived growth factor (PDGF) and transforming growth factor-β (TGF-β) signaling are required for hepatic stellate cell (HSC) activation under pathological conditions such as liver metastatic tumor growth. These two signaling pathways are functionally divergent; PDGF signaling promotes proliferation and migration of HSCs, and TGF-β induces transdifferentiation of quiescent HSCs into myofibroblasts. Although PDGF signaling is implicated in TGF-β-mediated epithelial mesenchymal transition of tumor cells, the role of PDGF receptors in TGF-β activation of HSCs has not been investigated. Here we report that PDGF receptor-α (PDGFR-α) is required for TGF-β signaling of cultured human HSCs although HSCs express both PDGF-α and -β receptors. PDGFR-α knockdown inhibits TGF-β-induced phosphorylation and nuclear accumulation of SMAD2 with no influence on AKT or ERK phosphorylation associated with noncanonical TGF-β signaling. PDGFR-α knockdown suppresses TGF-β receptor I (TβRI) but increases TβRII gene transcription. At the protein level, PDGFR-α is recruited to TβRI/TβRII complexes by TGF-β stimulation. PDGFR-α knockdown blocks TGF-β-mediated internalization of TβRII and induces accumulation of TβRII at the plasma membrane, thereby inhibiting TGF-β phosphorylation of SMAD2. Functionally, knockdown of PDGFR-α reduces paracrine effects of HSCs on colorectal cancer cell proliferation and migration in vitro. In mice and patients, colorectal cancer cell invasion of the liver induces upregulation of PDGFR-α of HSCs. In summary, our finding that PDGFR-α knockdown inhibits SMAD-dependent TGF-β signaling by repressing TβRI transcriptionally and blocking endocytosis of TGF-β receptors highlights a convergence of PDGF and TGF-β signaling for HSC activation and PDGFR-α as a therapeutic target for liver metastasis and other settings of HSC activation.
Keywords: tumor microenvironment, myofibroblasts, colorectal liver metastasis, receptor endocytosis and trafficking, gene transcription
development of liver metastasis is dependent on paracrine interactions between tumor cells and the hepatic microenvironment. In response to tumor invasion of the liver, hepatic stellate cells (HSCs), which are resident liver-specific pericytes, are activated into tumor-associated myofibroblasts that in turn promote tumor implantation and metastatic growth in the liver (14, 30). HSC activation is a complex process requiring coordinated activation of multiple intracellular signaling events, including transforming growth factor-β (TGF-β)- and platelet-derived growth factor (PDGF)-mediated signaling, which present as therapeutic targets for liver metastases and other diseases associated with HSC activation (9, 10, 14, 22).
TGF-β is a key cytokine that induces activation of HSCs into myofibroblasts (9, 19, 22). The prototypic cell surface receptors for TGF-β are TGF-β receptor I (TβRI) and II (TβRII). Ligand binding induces TβRII to associate with and phosphorylate TβRI, thereby activating the kinase domain of TβRI. The activated TβRI then phosphorylates SMAD family proteins allowing them to execute their nuclear translocation and modification of gene transcription within the nucleus (23, 24, 32). Endocytosis of TGF-β receptors is required for SMAD phosphorylation (7, 34). In addition to this canonical SMAD-dependent signaling pathway, TGF-β also activates noncanonical TGF-β pathways by phosphorylating AKT and ERK (25, 31).
PDGF ligands and their receptor-mediated signaling mediate HSC mitogenesis and motility. PDGF ligands consist of PDGF-A, B, C, and D, which interact with cell surface receptors termed as PDGF receptor-α (PDGFR-α) and -β (PDGFR-β). PDGF-A binds to PDGFR-α only, PDGF-B binds to both receptor isoforms, PDGF-C appears to bind to PDGFR-α, and PDGF-D binds to PDGFR-β predominantly (2, 17). PDGFs are secreted as homo- or heterodimers, and their ligation with receptors induces dimeric complexes, such as PDGFR-αα and PDGFR-ββ homodimers, and PDGFR-αβ heterodimers, which are implicated in diverse physiological and pathophysiological processes (2, 18). Downstream signaling pathways activated by PDGFs include Ras-MAPK, phosphatidylinositol 3-kinase (PI3K), and phospholipase C (PLC)-γ pathways (26).
Coordinated activation of both TGF-β and PDGF signaling that leads to expression of genes responsive for phenotypic changes of HSCs is requisite for the HSC activation process (22). Because both TGF-β and PDGF receptors transmit extracellular stimuli into gene transcription of HSCs, it is likely that these two signaling pathways may interconnect in HSCs. Although upregulation of PDGFR-α or PDGFR-β by TGF-β stimulation was reported in fibroblasts or tumor cell lines (11, 16, 35), it is less investigated whether PDGF receptors regulate the TGF-β-mediated HSC activation process. Here we report that both PDGFR-α and PDGFR-β are expressed in human HSCs and that only PDGFR-α is required for SMAD-dependent TGF-β signaling of HSCs. PDGFR-α knockdown inhibits SMAD-dependent TGF-β signaling via repressing TβRI gene transcription and blocking TGF-β-mediated internalization of TGF-β receptors posttranscriptionally. Additionally, knockdown of PDGFR-α in HSCs impairs the effects of HSCs on promoting the proliferation and migration of tumor cells in vitro. Furthermore, colorectal cancer cell invasion of the liver induces PDGFR-α expression in HSCs, supporting PDGFR-α as a therapeutic target for liver diseases associated with HSC activation.
MATERIALS AND METHODS
Cell lines, antibodies, and reagents.
Human primary HSCs were from ScienCell Research laboratories (Carlsbad, CA). They were cultured in DMEM supplemented with 10% FBS and antibiotics (penicillin and streptomycin) and used up to 10 passages. LX2 cells were a gift from Dr. S. Friedman (33). HT29 human colon cancer cells were from ATCC (Manassas, VA) and cultured in the same DMEM medium. All cells used were tested to be free of mycoplasma infection. Antibodies used are as follows: anti-PDGFR-α (3164; Cell Signaling, Danvers, MA), anti-PDGFR-β (28E1) (3169; Cell Signaling), anti-P-SMAD2 (S465/467) (3101; Cell Signaling), anti-P-AKT (S473) (D9E) XP (4060; Cell Signaling), phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) XP (4370; Cell Signaling), anti-TβRII (K105) (3713; Cell Signaling), anti-DYKDDDK Tag (FLAG) (2368; Cell Signaling), anti- early endosomal antigen-1 (EEA-1) (610456; BD Transduction Laboratory, San Jose, CA), anti-α-smooth muscle actin (SMA) (A5228; Sigma-Aldrich, St. Louis. MO), anti-α-SMA (ab5694; Abcam, Cambridge, MA), anti-SMAD2/3 (3102; Cell Signaling), anti- hemagglutinin (HA) (12CA5) (Roche Diagnostics, Indianapolis, Indiana), anti-HA (c29F4) (3724; Cell Signaling), anti-GAPDH (G8140; US Biological, Salem, MA). Recombinant human TGF-β1 was purchased from R & D Systems (100-B) (Minneapolis, MN), and a dose of 2.5 ng/ml was used to treat HSCs.
Retroviral and lentiviral transduction of HSCs.
Retroviruses encoding TβRII-HA or TβRI-FLAG were generated and harvested by transfecting 293T cells with plasmids as we previously described (19). shRNA lentiviral constructs that target against PDGFR-α or PDGFR-β were purchased from Sigma Mission library. The sequence for PDGFR-α shRNA is as follows: NM_006206.3–4080s1c1 (CCGGCTACTACTGTTATCAGTAATGCTCGAGCATTACTGATAACAGTAGTAGTTTTTTG); sequence for PDGFR-α shRNA-1: NM_006206.3–4363s1c1 (CCGGCCCTCTGAAATAATGGGATTACTCGAGTAATCCCATTATTTCAGAGGGTTTTTTG); sequence for PDGFR-β shRNA: NM_002609.3–3724s1c1 (CCGGGCTGGAACAGTTGCCGGATTCCTCGAGGAATCCGGCAACTGTTCCAGCTTTTTTG). Lentiviral packaging and harvesting were done according to standard protocols recommended by the manufacturer. Transduction of HSCs with viral particles was carried out by incubating cells with viral supernatant (25%) containing polybrene (8 μg/ml) overnight at 37°C. Seventy-two hours later cells were harvested for further experiments.
Immunofluorescence, confocal microscopy, and image quantification.
To stain HSCs or frozen liver sections, samples were first fixed with 3% paraformaldehyde followed by permeabilization with 0.2% Triton X-100. Cells or sections were incubated with primary antibodies for 2 h at room temperature or overnight at 4°C, and appropriate Alexa Fluor-conjugated second antibodies (Invitrogen, Thermo Fisher Scientific, Waltham, MA) were used for secondary detection. Cell nuclei were counterstained with Hoechst dye or DAPI (1:2,000). Fluorescence confocal microscopy was performed using a LSM 510 Laser Scanning Microscope (Carl Zeiss, Jena, Germany) using a ×63 or ×40 lens (15, 19).
Double immunofluorescence (IF) for TβRII-HA and EEA-1 and quantitation of TβRII/EEA-1 colocalization were performed as we described previously (19). In brief, serum-starved HSCs were pretreated with cycloheximide (40 μg/ml) for 1 h and incubated with TGF-β1 (2.5 ng/ml) at 4°C for 30 min to ensure TGF-β1/receptor binding. Cells were then washed with PBS and incubated at 37°C for various times for double IF and confocal microscopy. Confocal images were first split into a single color format for counting vesicles positive for TβRII-HA (green) (group A). Vesicles positive for both TβRII-HA and EEA-1 (yellow) were detected and counted in a merged picture (group B). Percentage of TβRII-HA/EEA-1 colocalization = B/A × 100%. More than six representative cells in each group were selected for quantification and statistical analysis.
Immunoprecipitation and Western blot.
Buffer containing 1% Nonidet P-40 and 5% glycerol was used as lysis buffer for immunoprecipitation (IP), and RIPA lysis and extraction buffer containing 1% Nonidet P-40, 1% sodium deoxycholate, and 0.1% SDS were used for Western blot (WB). For IP, 200–500 μg of total protein, 1–2 μg of a primary antibody, and 30 μl slurry of protein G Sepharose 4 Fast Flow (GE Healthcare Life Sciences, Pittsburgh, PA) were used for each precipitation. After Sepharose beads were washed four times with lysis buffer, precipitated proteins were eluted with Laemmli sample buffer (Bio-Rad, Hercules, CA) and loaded onto PAGE gels for WB. WB was carried out as we previously described (5). Densitometric analysis was performed using Image J software (NIH).
Biotinylation to quantitate plasma membrane TβRII and its degradation.
To quantitate plasma membrane TβRII, cell surface proteins were first labeled with cell nonpermeable biotin (EZ-Link Sulfo-NHS-Biotin, 21217; Thermo Fisher Scientific; 1 mg/ml) at 4°C for 15–30 min. After cells were washed three times with PBS containing 0.1 M glycine, they were harvested for streptavidin-agarose (S1638; Sigma-Aldrich) pull down followed by WB for TβRII. Densitometric analysis of WB blots was done using the Image J software. Quantitation of degradation of plasma membrane TβRII in response to TGF-β stimulation was performed as described (19). In brief, HSCs expressing TβRII-HA were serum starved and labeled with biotin. After they were incubated with TGF-β1 (2.5 ng/ml) at 4°C for 30 min for TGF-β1/receptor binding, they were washed with PBS to remove free TGF-β1 and moved into 37°C incubator for various times of incubation. Cells were then harvested for streptavidin-agarose pull down followed by TβRII WB to quantitate biotinylated TβRII that remained internalized but was spared from degradation.
Quantitative RT-PCR.
RNeasy kit (Qiagen, Venlo, Netherlands) was used for RNA isolation from HSCs, and reverse transcription was done using Oligo (dT) 12–18 Primer and SuperScript III Reverse Transcriptase (Invitrogen, Thermo Fisher Scientific). Real-time PCR was performed using a SYBR Green PCR Master Mix (Applied Biosystems, Thermo Fisher Scientific) in a 7500 Real-Time PCR System (Applied Biosystems). GAPDH was used as an internal control. Data were expressed as fold changes of mRNA of interest to mRNA of GAPDH. Primers used in this study were purchased from Qiagen (QT00083412, QT00014350, and QT00079247).
Boyden chamber assay and cell proliferation assay.
To assess paracrine effects of HSCs on tumor migration and proliferation, HSCs with or without PDGFR-α knockdown were serum starved, and conditioned media were collected. To assess the role of the conditioned media on HT29 cell migration, 15,000 tumor cells were seeded into each upper well of a Boyden chamber (NeuroProbe, Gaithersburg, MD), and basal medium or conditioned medium was added into each lower chamber as a chemoattractant. Four hours later, cells that had migrated to the lower surface of the filter were stained with DAPI for fluorescence microscopy. Further analysis and quantification were done as previously described (5). For cell proliferation assay, a CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay kit (Promega, Madison, WI) was used. In brief, 5,000 HT29 cells that were suspended in 100 μl of conditioned medium were seeded into each well of a 96-well plate, and cell numbers of HT29 were determined at different time points (24, 48, 72 h) according to a manufacturer-recommended protocol.
Experimental liver metastasis model and liver biopsies of patients.
All animal protocols were reviewed and approved by IACUC of Mayo Clinic. IF on clinical samples was approved by IRB of Mayo Clinic after written informed consents of patients were obtained. Portal vein implantation of HT29 human colorectal cancer cells into SCID mouse liver (01S11; Frederick National Laboratory) was done as described (5, 19). In brief, 1 × 106 or 2 × 106 HT29 human colorectal cancer cells suspended in 100 μl PBS were injected into portal vein of a mouse that was under whole body anesthesia. Mice were killed at day 6 after tumor implantation, and their livers were snap frozen in optimal cutting temperature embedding compound for cryo-sectioning and IF analysis. Liver biopsies of patients with colorectal cancer were from a Mayo Clinic tissue collection, and the clinical features of the patients were described in a recent publication (19).
Statistical analysis.
All data are shown as means ± SD. Two-tailed Student's t-test or ANOVA followed by post hoc test was used to evaluate difference among groups using GraphPad Prism 5 software (GraphPad Software, La Jolla, CA). P < 0.05 was considered statistically different.
RESULTS
Knockdown of PDGFR-α but not PDGFR-β inhibits TGF-β-mediated phosphorylation of SMAD2 and nuclear accumulation of SMAD2 in HSCs.
To explore a possible role of PDGFR-α and PDGFR-β in TGF-β1 signaling of HSCs, we transduced HSCs with lentiviruses encoding nontargeting shRNA (NT shRNA, control) or a shRNA-targeting against, either PDGFR-α (PDGFR-α shRNA) or PDGFR-β (PDGFR-β shRNA). Cells were serum starved and stimulated with TGF-β1 for different times, 0, 5, and 30 min and 24 h. Although TGF-β1 stimulation induces phosphorylation of both SMAD2 and 3, we used SMAD2 phosphorylation as a readout of TGF-β1 signaling owing to the more reliable antibody reagents available for detection of SMAD2. In control HSCs, SMAD2 phosphorylation was readily detected at 5 min after TGF-β1 stimulation (Fig. 1A), and it reached a higher level at 30 min and still remained at a detectable level even at 24 h after TGF-β1 stimulation. In PDGFR-α knockdown HSCs, TGF-β1-induced SMAD2 phosphorylation was drastically reduced at these time points (Fig. 1A, third row). By contrast, TGF-β1-induced SMAD2 phosphorylation was unaffected in PDGFR-β knockdown HSCs compared with control HSCs (Fig. 1A, third row). Unexpectedly, PDGFR-α protein was prominently downregulated at 24 h after TGF-β1 stimulation in PDGFR-β knockdown HSCs (Fig. 1A, first row) although this was not pursued as a focus of the present study. A different PDGFR-α shRNA that targeted against a different region of human PDGFR-α transcript confirmed the inhibitory effect of PDGFR-α knockdown on SMAD2 phosphorylation (Supplemental Fig. S1; supplemental material for this article is available online at the Journal of American Physiology Gastrointestinal and Liver Physiology website). Additionally, we tested the role of PDGFR-α knockdown in LX2 cells, a well-characterized immortalized human HSC line. PDGFR-α knockdown also inhibited TGF-β-mediated SMAD2 phosphorylation in LX2 cells (Fig. 1B). Thus knockdown of PDGFR-α inhibits TGF-β phosphorylation of SMAD2 in HSCs.
Fig. 1.

Knockdown of platelet-derived growth factor receptor (PDGFR)-α but not PDGFR-β in hepatic stellate cells (HSCs) inhibits SMAD-dependent transforming growth factor (TGF)-β signaling. A: HSCs that were transduced with lentiviruses encoding non-targeting shRNA (NT shRNA, control), PDGFR-α shRNA, or PDGFR-β shRNA were serum starved and stimulated with TGF-β1 (2.5 ng/ml) for indicated times. Cells were harvested for Western blot (WB). Compared with control HSCs, TGF-β-induced P-SMAD2 was drastically reduced by PDGFR-α knockdown but not by PDGFR-β knockdown. The blot was reprobed for GAPDH for a loading control. Data are representative of multiple experiments with consistent results. B: LX2 cells that were treated as described in A were collected for WB for P-SMAD2; GAPDH WB was used as a loading control. PDGFR-α knockdown inhibited TGF-β-induced P-SMAD2 in LX2 cells. Data represent multiple replicates. C: control or PDGFR-α knockdown HSCs that were serum starved and treated with TGF-β1 (2.5 ng/ml) for 60 min were harvested for immunofluorescence (IF) for SMAD2. Nuclear SMAD2 IF intensity was quantitated by Image J software (NIH). TGF-β1 induced nuclear accumulation of SMAD2 in control HSCs, and this effect of TGF-β1 was significantly inhibited by PDGFR-α knockdown. Cell nuclei were counterstained by Hoechst dye. Bar = 50 μm. *P < 0.05 by ANOVA. D: HSCs that were treated as described in A were harvested for WB for phosphorylation of AKT and ERK. PDGFR-α knockdown did not significantly affect TGF-β1-induced AKT or ERK phosphorylation. Data represent 3 repeats with similar results.
Because phosphorylated SMAD2 subsequently translocates into nucleus, where SMAD complexes regulate gene transcription, we performed IF to investigate whether PDGFR-α knockdown influenced TGF-β1-induced accumulation of SMAD2 in the nucleus. HSCs that were serum starved and stimulated with TGF-β1 for 60 min were harvested for SMAD2 IF. As shown in Fig. 1C, left, TGF-β1 stimulation induced nuclear localization of SMAD2 in control HSCs. This effect of TGF-β1 was inhibited in PDGFR-α knockdown HSCs (Fig. 1C, right). IF quantification of TGF-β1-treated cells by Image J software revealed that the average nuclear SMAD IF intensity was reduced by more than 70% by PDGF-α knockdown (P < 0.05, by ANOVA, n > 42 cells per group) (Fig. 1C, bottom). Thus PDGFR-α is required for TGF-β1-mediated phosphorylation and nuclear accumulation of SMAD2.
Knockdown of PDGFR-α does not influence noncanonical TGF-β signaling of HSCs.
TGF-β1 also activates non-SMAD-dependent pathways, including PI3K and Ras-MARK signaling pathways (25, 31). Therefore, we performed WB to test whether PDGFR-α knockdown influenced TGF-β1-mediated phosphorylation of AKT and ERK in HSCs. As shown in Fig. 1D, TGF-β1 induced phosphorylation of AKT at 5 and 30 min and phosphorylation of ERK at 5 min after TGF-β1 stimulation in control HSCs. These effects of TGF-β1, however, were not significantly changed in PDGFR-α knockdown HSCs (Fig. 1D), suggesting that PDGFR-α is only required for TGF-β-mediated SMAD-dependent signaling of HSCs.
Knockdown of PDGFR-α differentially regulates TβRII and TβRI protein levels of HSCs.
To understand how PDGFR-α regulates TGF-β-induced SMAD2 phosphorylation, we first compared total SMAD2 and 3 protein levels in control and PDGFR-α knockdown HSCs using WB. As shown in Fig. 2A, PDGFR-α knockdown did not reduce protein levels of total SMAD2 and 3. The total SMAD2 levels in PDGFR-α knockdown HSCs were even slightly higher than those of control HSCs. Next we compared total TβRII protein levels by WB using an antibody recognizing endogenous TβRII of HSCs (19). To this end, control and PDGFR-α knockdown HSCs were harvested at 72 h after shRNA lentiviral transduction. Surprisingly, we found that endogenous TβRII protein levels were significantly increased by PDGFR-α knockdown (Fig. 2B, P < 0.05 by t-test, n = 7). Because the level of endogenous TβRI was too low to detect by commercial antibodies, we transduced HSCs with lentiviruses encoding TβRI-FLAG fusion proteins and quantitated TβRI-FLAG fusion proteins using anti-FLAG antibody. As detected by WB, PDGFR-α knockdown, however, significantly reduced total TβRI-FLAG protein levels of HSCs (Fig. 2C, P < 0.05 by t-test, n = 4 repeats). Furthermore, we performed similar experiments in LX2 cells and confirmed that PDGFR-α knockdown differentially regulated TGF-β receptor I and II in LX2 cells (Fig. 2D).
Fig. 2.

Knockdown of PDGFR-α differentially regulates TGF-β receptor I and II (TβRI and II). A: at 72 h after shRNA lentiviral transduction, control or PDGFR-α knockdown HSCs were serum starved, treated with TGF-β1 for indicated times, and harvested for WB for SMAD2 and SMAD3. PDGFR-α knockdown reduced TGF-β1-induced P-SMAD2, slightly increased total SMAD2, and did not significantly influence total SMAD3 protein levels. Data represent 3 repeats. B: HSCs that were treated as described in A were harvested for endogenous TβRII by WB. GAPDH WB was used as a loading control. Densitometry of WB is shown on the right. PDGFR-α knockdown significantly increased total endogenous TβRII protein levels. *P < 0.05 by t-test, n = 7 repeats. C: HSCs expressing TβRI-FLAG fusion proteins by retroviral transduction were subjected to shRNA lentiviral transduction. At 72 h after shRNA lentiviral transduction, HSCs were collected for WB using anti-FLAG. GAPDH WB was used as a loading control. PDGFR-α knockdown significantly reduced total TβRI-FLAG fusion protein levels in HSCs. *P < 0.05 by t-test, n = 4 repeats. D: same experiments were performed with LX2 cells. PDGFR-α knockdown significantly increased endogenous TβRII and reduced TβRI-FLAG levels in LX2 cells. *P < 0.05 by t-test, n = 3 repeats.
TGF-β stimulation induces trimeric protein complexes containing TβRII, TβRI, and PDGFR-α.
Although the suppressed TβRI protein levels by PDGFR-α knockdown may contribute in part to the inhibited SMAD2 phosphorylation phenotype of PDGFR-α knockdown cells, TβRII upregulation by PDGFR-α knockdown, however, was very intriguing, which led us to test whether the upregulated TβRII was biologically active or not. To this end, we performed the experiments below to explore a possible role of PDGFR-α for TβRII endocytosis, trafficking, and degradation, which are critical events for TGF-β signaling.
We first knocked down TβRII by TβRII shRNA to validate the role of TβRII for TGF-β-mediated SMAD2 phosphorylation in HSCs. As revealed by WB, knockdown of TβRII drastically inhibited TGF-β-mediated SMAD2 phosphorylation, confirming a critical role of TβRII for TGF-β signaling of HSCs (Fig. 3A). We next studied subcellular localization of PDGFR-α and TβRII in HSCs by IF and confocal microscopy. Because none of the commercial anti-TβRII antibodies were suitable for IF, we transduced HSCs with retroviruses encoding TβRII-HA and detected TβRII-HA fusion proteins by HA IF (19). As revealed by double IF, PDGFR-α and TβRII-HA colocalized at the plasma membrane (arrowheads) and in endocytic vesicles (arrows) (Fig. 3B). Additionally, we found that endogenous TβRII was coprecipitated with PDGFR-α from HSC lysates when we performed IP using anti-PDGFR-α (Fig. 3B, bottom). Furthermore, we performed IP with HSCs treated with or without TGF-β for 10 min and found that TGF-β stimulation increased interactions between PDGFR-α and TβRII (Fig. 3C). Thus PDGFR-α interacts with endogenous TβRII in HSCs with TGF-β increasing their interactions.
Fig. 3.

TGF-β1 induces TβRII/TβRI/PDGFR-α trimeric protein complexes. A: HSCs transduced with either NT shRNA or TβRII shRNA lentiviruses were serum starved and stimulated by TGF-β1 for 30 min. Cells were harvested for WB. TβRII knockdown inhibited TGF-β1-mediated SMAD2 phosphorylation in HSCs. B, top: HSCs expressing TβRII-HA fusion proteins by retroviral transduction were subjected to double IF for PDGFR-α (green) and TβRII-HA (red). These 2 proteins colocalized at the plasma membrane (arrowheads) and in intracellular vesicles (arrows). Cell nuclei were counterstained by Hoechst dye. Bar = 20 μm. Bottom: HSC lysates were subjected to immunoprecipitation (IP) using anti-PDGFR-α, coprecipitated endogenous TβRII was detected by WB for TβRII. Endogenous PDGFR-α and TβRII interacted in HSCs. C: control or PDGFR-α knockdown HSCs that were serum starved and stimulated by TGF-β1 for 10 min were harvested for IP using anti-PDGFR-α. TGF-β1 stimulation for 10 min increased PDGFR-α/TβRII interactions. Data are representative of multiple repeats. D: HSCs expressing TβRI-FLAG fusion proteins were serum starved and treated with TGF-β1 for 10 min. Cells were then harvested for IP using anti-FLAG. TGF-β1 stimulation induced formation of TβRII/TβRI complexes. E: HSCs treated as described in D were harvested for IP using anti-FLAG, and coprecipitated endogenous PDGFR-α and TβRII were detected by WB. TGF-β1 stimulation induced formation of TβRII/TβRI/PDGFR-α trimeric protein complexes. Data are representative of multiple repeats.
Next we transduced HSCs with TβRI-FLAG retroviruses and studied protein interactions between TβRII, TβRI, and PDGFR-α in TGF-β-stimulated HSCs using IP. Consistent with the accepted model, TGF-β stimulation for 10 min induced formation of TβRII/TβRI protein complexes in HSCs (Fig. 3D). Furthermore, as revealed by IP using anti-FLAG, TGF-β stimulation for 10 min induced TβRI to associate with PDGFR-α and TβRII (Fig. 3E), suggesting that TGF-β induces TβRII/TβRI/PDGFR-α trimeric protein complexes in HSCs. Thus our data support a model whereby PDGFR-α may be recruited to TGF-β receptors by TGF-β to influence internalization and trafficking of TGF-β receptors.
PDGFR-α knockdown inhibits TGF-β-mediated TβRII internalization and induces accumulation of TβRII at the plasma membrane.
TGF-β stimulation induces interaction of TβRII and TβRI at the plasma membrane. Subsequently, the receptor complexes are internalized and sorted into early endosomes, where the receptors interact with and phosphorylate SMADs (23, 32). Because of technical reasons, TβRII but not TβRI has been studied as a receptor that undergoes ligand-dependent and -independent internalization, trafficking, and recycling (7, 20). On the basis of this, we performed experiments to study the role of PDGFR-α in TGF-β-mediated internalization and trafficking of TβRII in HSCs. To this end, HSCs were first transduced with retroviruses encoding TβRII-HA and then transduced with either NT or PDGFR-α shRNA lentiviruses. After cells were serum starved and treated with TGF-β1 for various times, they were collected for double IF for HA (for TβRII-HA fusion proteins) and EEA-1 (marker for the early endosomes). Double IF and quantitative data revealed that, in control cells, the rate of colocalization of TβRII and EEA-1 was highest at 5 min after TGFβ1 stimulation, and it gradually decreased at 15 and 30 min thereafter (Fig. 4A). In PDGFR-α knockdown cells, TβRII/EEA-1 colocalization was significantly reduced at both 5 and 15 min after TGF-β1 stimulation compared with control HSCs (Fig. 4A, P < 0.05 by ANOVA, n = 6). A lack of a time-dependent increase of TβRII/EEA-1 colocalization in PDGFR-α knockdown cells indicated that PDGFR-α knockdown indeed blocked TGF-β-mediated internalization of TβRII.
Fig. 4.

PDGFR-α knockdown inhibits TGF-β-mediated internalization of TβRII and induces accumulation of TβRII at the plasma membrane. A: HSCs expressing TβRII-HA by retroviral transduction were transduced with either NT shRNA or PDGFR-α shRNA lentiviruses. After cells were serum starved and pretreated with cycloheximide (CHX) (40 μg/ml) for 1 h, they were incubated with TGF-β1 at 4°C for 30 min for ligand/receptor binding. After the washing off of free TGF-β1, cells were incubated at 37°C for indicated times and harvested for double IF for HA (green) and early endosomal antigen-1 (EEA-1) (red). Colocalization of these 2 proteins (arrows) was quantitated, and quantitative data are shown on the bottom. TGF-β1 stimulation for 5 or 15 min increased TβRII/EEA-1 colocalization in control HSCs, and this effect of TGF-β1 was significantly inhibited by PDGFR-α knockdown. Bar = 20 μm. *P < 0.05 by ANOVA, n = 6 cells each group. Data represent 3 independent experiments with consistent results. B: plasma membrane TβRII of HSCs was detected by biotinylation of cell surface proteins followed by streptavidin-agarose pull down and WB for TβRII. PDGFR-α knockdown significantly increased cell surface TβRII of HSCs. *P < 0.05 by t-test, n = 5 repeats.
On the basis of the above finding, we next analyze plasma membrane TβRII using biotinylation of cell surface protein approach to test a hypothesis that PDGFR-α knockdown may induce accumulation of TβRII at the plasma membrane of HSCs. As revealed by the biotinylation assays, plasma membrane TβRII levels of PDGFR-α knockdown HSCs were indeed significantly higher than those of control HSCs (Fig. 4B, P < 0.05 by t-test, n = 5). Thus knockdown of PDGFR-α in HSCs blocks TGF-β-mediated internalization of TβRII, which subsequently results in the accumulation of TβRII at the plasma membrane.
PDGFR-α knockdown inhibits TGF-β downregulation of TβRII in HSCs.
Endocytic TβRII follows two trafficking routes after internalization; a fraction of TβRII is sorted to late endosome/lysosomes or proteasomes for degradation, and another is transported back to the plasma membrane for recycling (19, 20, 23, 28). Consistent with this concept, TGF-β stimulation induces time-dependent downregulation of TβRII in HSCs (19). On the basis of these, we next tested a hypothesis that PDGFR-α knockdown may inhibit TGF-β1 downregulation of TβRII in HSCs because PDGFR-α knockdown blocked TβRII internalization. Two experiments were performed to test this hypothesis: 1) HSCs that were pretreated with cycloheximide to inhibit synthesis of TβRII were serum starved and stimulated by TGF-β1 for different times, and the total TβRII levels of cell lysates were quantitated by WB; 2) HSCs that were subjected to biotinylation of cell surface proteins were serum starved and stimulated with TGF-β, and streptavidin agarose pull down followed by TβRII WB was used to analyze degradation of cell surface TβRII. As shown in Fig. 5A, TGF-β1 stimulation for 60 and 120 min induced obvious reduction of total TβRII protein levels in control HSCs, and this effect of TGF-β1 was significantly inhibited in PDGFR-α knockdown HSCs (Fig. 5A, P < 0.05 by ANOVA, n = 3 repeats). Consistently, TGF-β1 stimulation for 60 and 120 min downregulated biotinylated TβRII in control HSCs, and this effect of TGF-β1 was significantly inhibited in PDGFR-α knockdown HSCs (Fig. 5B, P < 0.05 by ANOVA, n = 3). These data suggest that PDGFR-α knockdown indeed inhibits TGF-β1 downregulation of TβRII, which may contribute in part to the increase of TβRII protein levels of PDGFR-α knockdown HSCs.
Fig. 5.

PDGFR-α knockdown inhibits TGF-β1 downregulation of TβRII. A: HSCs expressing TβRII-HA were transduced with NT shRNA or PDGFR-α shRNA lentiviruses. Cells that were serum starved, pretreated with CHX (40 μg/ml) for 1 h, and incubated with TGFβ-1 for indicated times were harvested for TβRII WB. WB data were analyzed by the Image J software for TβRII degradation. PDGFR-α knockdown significantly inhibited TGF-β1 downregulation of total TβRII in HSCs. *P < 0.05 by ANOVA, n = 3 repeats. B: serum-starved HSCs with their cell surface proteins prelabeled with biotin were incubated with TGF-β1 for indicated times. Cells were harvested for streptavidin-agarose pull down and TβRII WB to determine the degradation of plasma membrane TβRII. Densitometric analysis is shown on the bottom. PDGFR-α knockdown significantly inhibited TGF-β1 downregulation of cell surface TβRII. *P < 0.05 by ANOVA, n = 3 repeats.
PDGFR-α knockdown differentially regulates gene transcription of TGF-β receptors.
We also performed real-time quantitative RT-PCR to test whether PDGFR-α knockdown regulated gene transcription of TGF-β receptors. TβRI, TβRII, and GAPDH primers that were specifically designed for SYBR Green-based quantitative RT-PCR were obtained from Qiagen. We found that PDGFR-α knockdown differentially regulated mRNA transcripts of native TGF-β receptors in HSCs; it increased gene transcript of TβRII and reduced gene transcript of TβRI (Fig. 6, A and B, P < 0.05 by t-test, n > 3 repeats). Interestingly, knockdown of PDGFR-β, which had no influence on TGF-β signaling (Fig. 1A), also significantly inhibited gene transcription of TβRI (Fig. 6B), suggesting that reduction of TβRI transcript was not a sole reason for impaired TGF-β signaling of PDGFR-α knockdown HSCs. PDGFR-α knockdown may inhibit TGF-β signaling in HSCs by at least two mechanisms: 1) suppressing TβRI gene transcription, and 2) blocking TGF-β-mediated internalization and trafficking of TGF-β receptors.
Fig. 6.
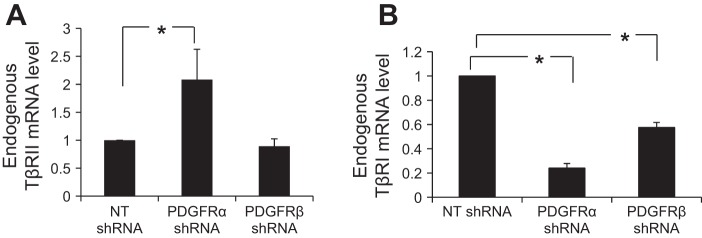
PDGFR-α knockdown differentially regulates gene transcription of TβRI and TβRII. A: real-time RT-PCR was performed with RNA of control or PDGFR-α knockdown HSCs. Knockdown of PDGFR-α but not PDGFR-β significantly increased transcription of endogenous TβRII compared with control HSCs. *P < 0.05 by ANOVA, n = 3 repeats. B: knockdown of either PDGFR-α or PDGFR-β significantly reduced endogenous TβRI mRNA levels compared with control HSCs. *P < 0.05 by ANOVA, n = 4 repeats.
PDGFR-α knockdown impairs paracrine effects of HSCs on proliferation and migration of colorectal cancer cells.
Cancers of the gastrointestinal tract, such as colorectal tumors, frequently metastasize to the liver. To test the role of PDGFR-α of activated HSCs in colorectal tumor growth, we collected conditioned media from both control HSCs and PDGFR-α knockdown HSCs. These conditioned media were used as stimulants in cell proliferation assays and chemoattractants in Boyden chamber assays. As shown in Fig. 7, A and B, conditioned media from both control and PDGFR-α knockdown HSCs stimulated proliferation and migration of HT29 cells in vitro. However, the conditioned medium of PDGFR-α knockdown HSCs was less effective to promote the proliferation and migration of HT29 cells compared with that of control HSCs (Fig. 7, A and B). Thus, through a paracrine mechanism, PDGFR-α of HSCs promotes tumor cell proliferation and migration in vitro, which may be important for implantation and metastatic tumor growth in the liver.
Fig. 7.

PDGFR-α knockdown impairs paracrine effects of HSCs on tumor cell proliferation and migration in vitro. A: conditioned media collected from control and PDGFR-α knockdown HSCs were used as stimulants for HT29 proliferation assay. The conditioned medium of PDGFR-α knockdown HSCs was less effective on HT29 proliferation in vitro compared with that of control HSCs. *P < 0.05 by ANOVA; n = 3 repeats with similar results. B: conditioned media collected from control and PDGFR-α knockdown HSCs were used as chemoattractants for HT29 in Boyden chamber assays. The conditioned medium of PDGFR-α knockdown HSCs exhibited a reduced effect on promoting HT29 migration compared with that of control HSCs. *P < 0.05 by ANOVA; n = 3 repeats. Bar = 100 μm.
Colorectal cancer cells induce upregulation of PDGFR-α in HSCs in mice and patients.
For in vivo relevance of PDGFR-α of HSCs in HSC activation and liver metastatic growth, we implanted HT29 human colorectal cancer cells into the liver of SCID mice by portal vein injection. Livers of SCID mice containing HT29 micrometastases were isolated at day 6 after implantation for double IF for PDGFR-α and α-SMA. The HT29 micrometastases were identified by positive IF staining for STEM121 (Fig. 8A, bottom). STEM121 is an extensively used antibody for detection of human-derived cells in mice, owing to its specific reaction with a human cytoplasmic antigen. Colonization of HT29 cells in the mouse liver induced activation of HSCs as demonstrated by α-SMA IF (red) signals surrounding the HT29 micrometastases (Fig. 8A, arrows). A fraction of these activated HSCs also express high levels of PDGFR-α (green) (Fig. 8A, arrows). By contrast, quiescent HSCs in unaffected liver tissues were negative for both PDGFR-α and α-SMA (Fig. 8A, arrowheads). Furthermore, quantitative data revealed that PDGFR-α IF densities of activated HSCs were three time higher than those of quiescent HSCs (P < 0.05 by t-test, n = 6) (Fig. 8B).
Fig. 8.

Colorectal cancer cells induce upregulation of PDGFR-α and α-smooth muscle actin (SMA) in HSCs in mice and patients. A: HT29 human colorectal cancer cells were implanted into the livers of SCID mice by portal vein injection. Liver sections containing HT29 micrometastases were subjected to IF for α-SMA, PDGFR-α, STEM121 (human specific antigen), and hematoxylin and eosin (H.E.) staining. Tumor cells induced adjacent HSCs to coexpress α-SMA (red) and PDGFR-α (green) (arrows). Quiescent HSCs were negative for both proteins (arrowheads). Cell nuclei were counterstained by Hoechst dye (blue). T, tumor cells; bar = 20 μm. B: PDGFR-α IF signals were quantitated by the Image J software. PDGFR-α IF intensity of activated HSCs was 3 times higher than that of quiescent HSCs. *P < 0.05 by t-test; n = 6. qHSCs, quiescent HSCs; aHSCs, activated HSCs. C: liver biopsies of patients with colorectal cancer were stained for α-SMA and PDGFR-α. Colorectal cancer cells induced upregulation of α-SMA and PDGFR-α in adjacent HSCs (yellow, arrows). T, tumor cells; bar = 20 μm. D: myofibroblasts of established colorectal liver metastases expressed abundant PDGFR-α (arrowheads). T, tumor; S, stroma. Bar = 100 μm.
Next, the same double IF technique was performed on liver biopsies of patients with colorectal cancer that were obtained from a Mayo Clinic tissue collection (19). The colorectal liver metastases were positive for carcinoembryonic antigen, a clinical diagnostic marker of colon and rectal cancers (Supplemental Fig. S2). Consistent with the mouse model, high levels of PDGFR-α IF signals (green) were detected in activated HSCs in liver biopsies of patients with colorectal cancer (Fig. 8C, arrows), which overlapped with α-SMA IF signals (red) (Fig. 8C, arrows). Furthermore, in established colorectal liver metastases, tumor-associated myofibroblasts were strongly positive for both PDGFR-α and α-SMA (Fig. 8D, arrowheads). Thus colorectal cancer invasion of the liver induces upregulation of PDGFR-α in adjacent HSCs, which may be a requisite step for myofibroblastic activation of HSCs in the hepatic tumor microenvironment and a therapeutic target for liver metastasis.
DISCUSSION
In this study, we identified that PDGFR-α but not PDGFR-β participated in TGF-β1 signaling of HSCs. PDGFR-α knockdown inhibited TGF-β1-mediated SMAD2 phosphorylation and nuclear accumulation SMAD2 with no influence on noncanonical TGF-β signaling, such as activation of AKT and ERK. Mechanistically, we demonstrated that PDGFR-α knockdown inhibited SMAD2-dependent TGF-β signaling by at least two mechanisms: 1) suppressing TβRI by reducing its gene transcription and 2) blocking the internalization and trafficking of TβRII at the protein level. PDGFR-α is a novel binding partner of TβRII that is recruited to TβRII/TβRI protein complexes by TGF-β stimulation, which in turn promotes internalization and trafficking of TGF-β receptors (Fig. 9). It is yet unknown how PDGFR-α interacts with TGF-β receptors such as TβRII. For example, which region of TβRII mediates TβRII/PDGFR-α binding? Does TβRII/PDGFR-α binding occur at the intracellular or extracellular region of these two receptors? Does TβRII/PDGFR-α binding influence kinase activity of TβRII and TβRI? Elucidation of these mechanisms may provide opportunities for targeting TβRII/PDGFR-α interactions for suppressing myofibroblastic activation of HSCs and the hepatic tumor microenvironment.
Fig. 9.

Working model: PDGFR-α is a novel binding partner of TGF-β receptors that is required for TGF-β signaling of HSCs. PDGFR-α is recruited to TβRI/TβRII complexes by TGF-β stimulation. Through interacting with TβRII, PDGFR-α promotes internalization and trafficking of TGF-β receptors into the early endosomes, where phosphorylation of SMADs occurs and TGF-β signaling is activated. Knockdown of PDGFR-α blocks endocytosis of TGF-β receptors, thereby inhibiting phosphorylation of SMADs. PM, plasma membrane.
In the metastasis mouse model and patients with colorectal cancer, cancer cells induced coactivation of α-SMA and PDGFR-α proteins in adjacent HSCs. In vitro, PDGFR-α knockdown inhibited TGF-β1-mediated internalization of TGF-β1 receptors and TGF-β1 signaling, indicating that PDGFR-α is a requisite for HSCs to undergo TGF-β-mediated myofibroblastic activation in the hepatic tumor microenvironment. Additionally, conditioned medium of PDGFR-α knockdown HSCs exhibited reduced effects on promoting HT29 colorectal cancer cell proliferation and migration in vitro, highlighting PDGFR-α of HSCs as a therapeutic target for liver metastasis. Interestingly, TGF-β ligands have been identified as one of the transcriptional targets of PDGFR-mediated signaling (4, 29). Because PDGF-AA, PDGF-AB, and PDGF-BB are also major mitogens and chemoattractants for activated HSC/myofibroblasts with their effects mediated in part by PDGFR-α on downstream PLC-γ, PI3K, and Ras-MARK signaling (21, 22), agents that target PDGFR-α may exert potent effects on inhibiting transdifferentiation, proliferation, and migration of HSCs.
PDGFR-α but not PDGFR-β participates in SMAD-dependent TGF-β signaling of HSCs, indicating a functional divergence of these two PDGF receptors. Although a functional cooperation of these two subunits of PDGFR-αβ receptor was shown in a pig aortic endothelial cell line (8), PDGFR-α and PDGFR-β in fact played opposite roles in promoting anchorage-independent growth phenotype of NIH3T3 cells in vitro (36). Furthermore, gene knockout mice for each receptor isoform revealed distinct roles of PDGFR-α and PDGFR-β in vivo. PDGF-B/PDGFR-β played a major role in recruitment of mural cells to the blood vessel during vessel development and maturation, and PDGF-A/PDGFR-α had a broad role in organ development, including the development of central nervous system, lung, gonad, gut, and kidneys during embryogenesis (3). The different responses of these two PDGF receptors were likely due to the innate differences in available adaptor molecules that transmit their signals in different cell types (36).
We currently do not know why and how PDGFR-α knockdown differentially regulates gene transcription of TβRI and TβRII. Sp1 transcription factor and p300 transcriptional coactivator are known to regulate TβRI gene transcription (1, 13). It is possible that PDGFR-α knockdown may repress TβRI by downregulating transcription factors such as Sp1 or p300. TGF-β downstream signaling is known to repress TβRII transcription to provide a negative feedback mechanism on TGF-β stimulation (27). Because TGF-β signaling is inhibited by PDGFR-α knockdown, it is likely that this negative feedback of TGF-β on TβRII transcription was abolished, which may result in increased TβRII gene transcription phenotype of PDGFR-α knockdown HSCs.
In summary, in addition to the previously recognized role of PDGFR-α as a receptor for mitogens and chemoattractants of HSCs, we have identified a new role for PDGFR-α in SMAD-dependent TGF-β signaling of HSCs. At the transcriptional level, PDGFR-α differentially regulates gene transcription of TβRI and TβRII. At the posttranscriptional level, PDGFR-α interacts with TβRII, which is required for internalization and trafficking of TβRII and TGF-β signaling of HSCs. Our data further strengthen PDGFR-α as a therapeutic target for liver diseases associated with HSC activation. The approved anticancer drug Imatinib (Gleeve, ST1–571) and Sunitinib (6), as well as Crenolanib besylate, which is currently under investigation (12), are examples of drugs that show effects on suppressing the kinase activity of PDGFR-α. Because activating mutations of PDGFR-α are associated with certain type of cancers, combined use of these drugs with inhibitors targeting TGF-β signaling may provide benefits on targeting both tumor cells and the tumor microenvironment. Furthermore, our study is also highly relevant to other diseases that are associated with myofibroblastic activation, including liver, lung, and renal fibrosis.
GRANTS
This work was supported by a P/F Award and the Clinical and Cell Biology Cores of the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567), NIH grants R01 CA160069 to N. Kang, and R01 DK059615 and AA021171 to V. H. Shah, and a startup fund to N. Kang at the Hormel Institute. K. Tu is funded by China Scholarship Council (CSC).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Author contributions: C.L., J.L., X.X., L.G., K.T., and Q.L. performed experiments; C.L., J.L., X.X., L.G., K.T., Q.L., and N.K. analyzed data; C.L., J.L., and N.K. prepared figures; J.L. and N.K. drafted manuscript; V.H.S. and N.K. edited and revised manuscript; V.H.S. and N.K. approved final version of manuscript; N.K. conception and design of research; N.K. interpreted results of experiments.
ACKNOWLEDGMENTS
We thank Drs. Kah Whye Peng, Yasuhiro Ikeda, and Carmelo Bernabeu for providing reagents.
REFERENCES
- 1.Ammanamanchi S, Brattain MG. Restoration of transforming growth factor-beta signaling through receptor RI induction by histone deacetylase activity inhibition in breast cancer cells. J Biol Chem 279: 32620–32625, 2004 [DOI] [PubMed] [Google Scholar]
- 2.Andrae J, Gallini R, Betsholtz C. Role of platelet-derived growth factors in physiology and medicine. Genes Dev 22: 1276–1312, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Betsholtz C. Insight into the physiological functions of PDGF through genetic studies in mice. Cytokine Growth Factor Rev 15: 215–228, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Chen YT, Chang FC, Wu CF, Chou YH, Hsu HL, Chiang WC, Shen J, Chen YM, Wu KD, Tsai TJ, Duffield JS, Lin SL. Platelet-derived growth factor receptor signaling activates pericyte-myofibroblast transition in obstructive and post-ischemic kidney fibrosis. Kidney Int 80: 1170–1181, 2011 [DOI] [PubMed] [Google Scholar]
- 5.Decker NK, Abdelmoneim SS, Yaqoob U, Hendrickson H, Hormes J, Bentley M, Pitot H, Urrutia R, Gores GJ, Shah VH. Nitric oxide regulates tumor cell cross-talk with stromal cells in the tumor microenvironment of the liver. Am J Pathol 173: 1002–1012, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Demetri GD. Differential properties of current tyrosine kinase inhibitors in gastrointestinal stromal tumors. Semin Oncol 38, Suppl 1: S10–S19, 2011 [DOI] [PubMed] [Google Scholar]
- 7.Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol 5: 410–421, 2003 [DOI] [PubMed] [Google Scholar]
- 8.Emaduddin M, Ekman S, Ronnstrand L, Heldin CH. Functional co-operation between the subunits in heterodimeric platelet-derived growth factor receptor complexes. Biochem J 341: 523–528, 1999 [PMC free article] [PubMed] [Google Scholar]
- 9.Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology 134: 1655–1669, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gialeli C, Nikitovic D, Kletsas D, Theocharis AD, Tzanakakis GN, Karamanos NK. PDGF/PDGFR signaling and targeting in cancer growth and progression: focus on tumor microenvironment and cancer-associated fibroblasts. Curr Pharm Des 20: 2843–2848, 2014 [DOI] [PubMed] [Google Scholar]
- 11.Gotzmann J, Fischer AN, Zojer M, Mikula M, Proell V, Huber H, Jechlinger M, Waerner T, Weith A, Beug H, Mikulits W. A crucial function of PDGF in TGF-beta-mediated cancer progression of hepatocytes. Oncogene 25: 3170–3185, 2006 [DOI] [PubMed] [Google Scholar]
- 12.Heinrich MC, Griffith D, McKinley A, Patterson J, Presnell A, Ramachandran A, Debiec-Rychter M. Crenolanib inhibits the drug-resistant PDGFRA D842V mutation associated with imatinib-resistant gastrointestinal stromal tumors. Clin Cancer Res 18: 4375–4384, 2012 [DOI] [PubMed] [Google Scholar]
- 13.Ji C, Casinghino S, McCarthy TL, Centrella M. Multiple and essential Sp1 binding sites in the promoter for transforming growth factor-beta type I receptor. J Biol Chem 272: 21260–21267, 1997 [DOI] [PubMed] [Google Scholar]
- 14.Kang N, Gores GJ, Shah VH. Hepatic stellate cells: partners in crime for liver metastases? Hepatology 54: 707–713, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kang N, Yaqoob U, Geng Z, Bloch K, Liu C, Gomez T, Billadeau D, Shah V. Focal adhesion assembly in myofibroblasts fosters a microenvironment that promotes tumor growth. Am J Pathol 177: 1888–1900, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leof EB, Proper JA, Goustin AS, Shipley GD, DiCorleto PE, Moses HL. Induction of c-sis mRNA and activity similar to platelet-derived growth factor by transforming growth factor beta: a proposed model for indirect mitogenesis involving autocrine activity. Proc Natl Acad Sci USA 83: 2453–2457, 1986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li X, Eriksson U. Novel PDGF family members: PDGF-C and PDGF-D. Cytokine Growth Factor Rev 14: 91–98, 2003 [DOI] [PubMed] [Google Scholar]
- 18.Li X, Ponten A, Aase K, Karlsson L, Abramsson A, Uutela M, Backstrom G, Hellstrom M, Bostrom H, Li H, Soriano P, Betsholtz C, Heldin CH, Alitalo K, Ostman A, Eriksson U. PDGF-C is a new protease-activated ligand for the PDGF alpha-receptor. Nat Cell Biol 2: 302–309, 2000 [DOI] [PubMed] [Google Scholar]
- 19.Liu C, Billadeau DD, Abdelhakim H, Leof E, Kaibuchi K, Bernabeu C, Bloom GS, Yang L, Boardman L, Shah VH, Kang N. IQGAP1 suppresses TbetaRII-mediated myofibroblastic activation and metastatic growth in liver. J Clin Invest 123: 1138–1156, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell H, Choudhury A, Pagano RE, Leof EB. Ligand-dependent and -independent transforming growth factor-beta receptor recycling regulated by clathrin-mediated endocytosis and Rab11. Mol Biol Cell 15: 4166–4178, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pinzani M. PDGF and signal transduction in hepatic stellate cells. Front Biosci 7: d1720–d1726, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Pinzani M, Marra F, Carloni V. Signal transduction in hepatic stellate cells. Liver 18: 2–13, 1998 [DOI] [PubMed] [Google Scholar]
- 23.Rahimi RA, Leof EB. TGF-beta signaling: a tale of two responses. J Cell Biochem 102: 593–608, 2007 [DOI] [PubMed] [Google Scholar]
- 24.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113: 685–700, 2003 [DOI] [PubMed] [Google Scholar]
- 25.Suzuki K, Wilkes MC, Garamszegi N, Edens M, Leof EB. Transforming growth factor beta signaling via Ras in mesenchymal cells requires p21-activated kinase 2 for extracellular signal-regulated kinase-dependent transcriptional responses. Cancer Res 67: 3673–3682, 2007 [DOI] [PubMed] [Google Scholar]
- 26.Trojanowska M. Role of PDGF in fibrotic diseases and systemic sclerosis. Rheumatology 47, Suppl 5: v2–v4, 2008 [DOI] [PubMed] [Google Scholar]
- 27.Truty MJ, Lomberk G, Fernandez-Zapico ME, Urrutia R. Silencing of the transforming growth factor-beta (TGFbeta) receptor II by Kruppel-like factor 14 underscores the importance of a negative feedback mechanism in TGFbeta signaling. J Biol Chem 284: 6291–6300, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tu K, Li J, Verma VK, Liu C, Billadeau DD, Lamprecht G, Xiang X, Guo L, Dhanasekaran R, Roberts LR, Shah VH, Kang N. VASP promotes TGF-beta activation of hepatic stellate cells by regulating Rab11 dependent plasma membrane targeting of TGF-beta receptors. Hepatology. 2014. June 10. 10.1002/hep.27251 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tuuminen R, Nykanen AI, Krebs R, Soronen J, Pajusola K, Keranen MA, Koskinen PK, Alitalo K, Lemstrom KB. PDGF-A, -C, and -D but not PDGF-B increase TGF-beta1 and chronic rejection in rat cardiac allografts. Arterioscler Thromb Vasc Biol 29: 691–698, 2009 [DOI] [PubMed] [Google Scholar]
- 30.Vidal-Vanaclocha F. The prometastatic microenvironment of the liver. Cancer Microenviron 1: 113–129, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wilkes MC, Leof EB. Transforming growth factor beta activation of c-Abl is independent of receptor internalization and regulated by phosphatidylinositol 3-kinase and PAK2 in mesenchymal cultures. J Biol Chem 281: 27846–27854, 2006 [DOI] [PubMed] [Google Scholar]
- 32.Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-beta receptor. Nature 370: 341–347, 1994 [DOI] [PubMed] [Google Scholar]
- 33.Xu L, Hui AY, Albanis E, Arthur MJ, O'Byrne SM, Blaner WS, Mukherjee P, Friedman SL, Eng FJ. Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut 54: 142–151, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu P, Liu J, Derynck R. Post-translational regulation of TGF-beta receptor and Smad signaling. FEBS Lett 586: 1871–1884, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamakage A, Kikuchi K, Smith EA, LeRoy EC, Trojanowska M. Selective upregulation of platelet-derived growth factor alpha receptors by transforming growth factor beta in scleroderma fibroblasts. J Exp Med 175: 1227–1234, 1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu J, Deuel TF, Kim HR. Platelet-derived growth factor (PDGF) receptor-alpha activates c-Jun NH2-terminal kinase-1 and antagonizes PDGF receptor-beta -induced phenotypic transformation. J Biol Chem 275: 19076–19082, 2000 [DOI] [PubMed] [Google Scholar]