

Abstract
The objective of the present study was to determine whether mitochondrial function in the cerebral vasculature is maintained after transient middle cerebral artery (MCA) occlusion (tMCAO) in rats. Sprague-Dawley rats were exposed to 90 min of tMCAO followed by 4 or 48 h of reperfusion. MCAs from ischemic (ipsilateral) and nonischemic (contralateral) sides were compared with control MCAs from sham-operated rats. We determined 1) vasoreactivity to diazoxide (DZ; a mitochondrial ATP-activated K+ channel opener), ACh, bradykinin (BK), serotonin, and sodium nitroprusside; 2) levels of mitochondrial and nonmitochondrial proteins and mitochondrial DNA; and 3) vascular levels of tetramethylrhodamine ethyl ester (an indicator of mitochondrial membrane potential). All dilator responses, including those with DZ, were intact 4 h post-tMCAO. Dilator responses to ACh, BK, and sodium nitroprusside were reduced in ipsilateral MCAs at 48 h compared with contralateral MCAs, but DZ responses were comparable with control MCAs. Surprisingly, contralateral responses to ACh, BK, and serotonin were reduced compared with control MCAs at 48 h. Ipsilateral vasodilation to DZ at 48 h was eliminated by endothelial denudation and endothelial nitric oxide synthase (eNOS) inhibition but was only reduced in control MCAs. Mitochondrial proteins, phosphorylated eNOS, mitochondrial DNA, and mitochondrial membrane potential were higher in ipsilateral compared with contralateral MCAs. In conclusion, contrary to conventional wisdom, mitochondria remain functional for at least 48 h after severe ischemic stress in MCAs, and DZ-induced dilation is preserved due to maintained mitochondrial mass, probably in the endothelium, and eNOS signaling. Our findings support the concept that functioning vascular mitochondria are an unexpected target for novel stroke therapies.
Keywords: diazoxide, endothelium, transient middle cerebral artery occlusion, mitochondrial depolarization, vasodilation, experimental strokes
ischemic stroke is a leading cause of human morbidity and mortality (34). Currently, thrombolytic or surgical therapies are the only accepted treatments for occlusive stroke, but clot removal is not prescribed for many patients because the time window for administration is severely limited (14). Until recently, thrombolytic or surgical interventions were only medically endorsed if initiated within 3 h after the occurrence of a stroke (18, 19). New studies, especially those from Europe, have indicated that the therapeutic window of clot resolution can be extended to 4.5 h or more after the onset of stroke, but increasing cerebral vascular dysfunction and neurological damage may reduce beneficial results when initiated at a later time (1, 11, 31, 39). Therefore, new therapies for these later time periods are urgently needed for vulnerable stroke patients. Recently, limited information from several sources has indicated that, contrary to currently held beliefs, significant mitochondrial function remains intact after ischemic stress, and thus mitochondria may provide a novel, but yet unexploited, therapeutic target (28, 38). However, no direct in vivo assessment of vascular mitochondrial function has yet been conducted in the cerebral vasculature after ischemic stress.
Previous studies have shown that selective activation of mitochondrial ATP-sensitive K+ (mitoKATP) channels located on the inner mitochondrial membrane by agents such as diazoxide (DZ) reduces brain infarct volume, preserves the responsiveness of large cerebral arteries, and protects the blood-brain barrier (BBB) when given before transient global ischemia or transient middle cerebral artery (MCA) occlusion (tMCAO) (6, 12, 13, 26). In addition, activation of mitoKATP channels protects the cultured endothelium, neurons, and astroglia against cell death after transient O2-glucose deprivation (OGD) (4, 17, 20, 24, 32). Finally, recent studies have also indicated that pharmacological activation of mitochondria after tMCAO or OGD is as effective as pretreatment, thereby suggesting that significant mitochondrial functionality is present even after severe anoxic/ischemic stress (5). One limitation of in vivo studies is that the location of cellular protection cannot be known with certainty since the neurovascular unit represents many cell types, including neurons, astroglia, microglia, and vascular cells. Nonetheless, several recent studies have demonstrated that the cerebral vasculature, especially the endothelium, plays a critical role in determining neurological outcome after stroke by maintaining appropriate blood flow and the BBB (10, 15). However, no studies have directly examined mitochondrial function in the cerebral vasculature after tMCAO or other experimental models of stroke. Under normal conditions, we and others have shown that mitochondria-dependent vasodilation of large cerebral arteries has both endothelial and vascular smooth muscle (VSM) components that contribute to the overall integrated vascular response (23, 42). Thus, increased nitric oxide (NO) production by the endothelium augments the intrinsic relaxation of the VSM of cerebral arteries after the application of DZ.
We hypothesized that mitochondrial function is maintained after severe ischemic stress in cerebral arteries after tMCAO and represents a previously unappreciated therapeutic target that potentially can protect the cerebral vasculature and brain. Specifically, we investigated 1) the effects of tMCAO on the responses of isolated MCAs to mitochondrial activation as well as to other general, nonmitochondrial-based vasoactive stimuli; 2) the relative contribution of the endothelium and VSM to the integrated cerebral vascular response to mitochondrial activation after tMCAO; and 3) the effects of tMCAO on the expression of mitochondrial and nonmitochondrial proteins and mitochondrial biogenesis in MCAs. We chose to use DZ because of its selectivity to mitochondria, widespread use and tolerance, proven effectiveness for the treatment of nonneurological diseases in people, and ability to cross the BBB (4, 6, 16, 37). We studied cerebral arteries 48 h after tMCAO, at a time when the brain infarct is fully developed, but also examined the arterial responses at 4 h, when post-tMCAO hemodynamics had stabilized.
MATERIALS AND METHODS
Animals.
Male Sprague-Dawley rats (8–10 wk old, n = 124) were obtained from Charles River Laboratories (Wilmington, MA). Animals were housed and cared for according to guidelines of and protocols were approved by the Institutional Animal Care and Use Committee of Tulane University in compliance with National Institutes of Health (NIH) guidelines. Animal care was provided by the Department of Comparative Medicine. Rats were given soft food and water ad libitum.
tMCAO.
Rats were randomly assigned to two groups: a control (sham-operated) group or an operated group, in which the cerebral vasculature was exposed to tMCAO (36). Rats were subjected to a modified Longa intraluminal filament method to induce ischemia by the occlusion of one of the MCAs. We performed a 90-min period of ischemia under ketamine-xylazine anesthesia consisting of 100 mg/ml ketamine HCl (KetaVed, St. Joseph, MO) and 20 mg/ml AnaSed xylazine (Santa Cruz Biotechnology, Dallas, TX) followed by 48 h of reperfusion without anesthesia. During surgery, supplemental O2 was given, ointment was used to maintain eye lubrication, and a rectal temperature probe and heating pad were used to monitor and maintain body temperature at normal levels. Using an operating microscope for visualization, a midline incision was made in the neck. The right common carotid artery, external carotid artery, and internal carotid artery were isolated from the surrounding tissue from the bifurcation to the base of the skull. A 5-0 silk suture was used to ligate the external carotid artery, and the lingual, maxillary, and occipital artery branches were dissected and coagulated. Afterward, a 5-0 silk suture was placed around the common carotid artery to prevent bleeding. To occlude the MCA, a rubber-coated silicone monofilament (Doccol, Sharon, MA), 3 cm in length and 0.35 or 0.37 mm in diameter, was inserted through the small incision on the common carotid artery into the internal carotid artery and into the circle of Willis, effectively occluding the right MCA. A vascular clamp was used to fix the filament in the vessel to prevent bleeding, and the incision was covered with sterile 0.9% saline-soaked gauze. After 90 min of ischemia, the intraluminal filament was removed, the incision was closed using a wound clip (Fine Science Tools, Foster City, CA), and rats were allowed to recover for 4 or 48 h with free access to soft food and water. Buprenorphine (0.05 mg/kg) was available for postoperative analgesia if needed. The tissue dehydrogenase marker 2,3,5-triphenyltetrazolium chloride (TTC; Sigma-Aldrich, St. Louis, MO) was used to visualize and document the volume of the infarcted brain after the isolation of the MCAs. The reaction between TTC salt and dehydrogenase enzyme resulted in red staining of viable tissue, whereas the infarcted tissue resulted in a lack of staining due to the absence of enzyme activity with which TTC could react. After isolation of the MCAs, 2.0-mm-thick coronal sections were acquired using a rat brain slicer matrix (Zivic, Pittsburgh, PA). Brain slices were placed in a beaker containing a prewarmed (37°C) solution of 2% TTC dissolved in 0.9% saline, which was placed in a 37°C water bath for 20 min. A scan was taken of the slices, and the infarct size was determined using ImageJ Software (NIH). Infarct volumes were established by averaging the infarcted area of the slices and were expressed as a percentage of each hemispheric volume (%infarction). In the control group, rats underwent a sham operation with the same length of anesthesia as the operated group with the midline neck incision performed but without filament insertion (8).
Isolated pressurized artery technique.
We have previously used this method (23). Under 5% isoflurane (VetOne, Boise, ID)-induced anesthesia, rats were decapitated, and brains were removed and transferred to ice-cold 4°C oxygenated (20% O2-5% CO2-75% N2) Ca2+-containing Krebs (Ca2+-Krebs) solution [composed of (in mmol/l) 110 NaCl, 5.0 KCl, 2.5 CaCl2, 1.0 MgSO4, 1.0 KH2PO4, 5.0 glucose, and 24.0 NaHCO3] at 7.4 pH. Microsurgery instruments (Fine Science Tools) and a Nikon SMZ1000 operating microscope were used for the isolation of MCAs in 4°C Ca2+-Krebs solution. An isolated section of the MCA (<1 mm in length) was transferred into a self-heated 37°C organ chamber (Living Systems Instrumentation, St. Albans, VT) containing two glass micropipettes filled with Ca2+-Krebs solution and cannulated on both ends. One of the micropipettes was closed, and the other was connected with silicone tubing to a pressure servo control system (Living Systems Instrumentation), and the temperature was maintained at 37°C. Changes in arteriolar diameter were continuously measured and recorded by a video microscope (Nikon Eclipse TS100, Sony CCD camera, VDA-10 Living Systems Instrumentation Video Dimension Analyzer, LabView 2.0, and HP computer). Spontaneous myogenic tone developed in response to 70 mmHg of intraluminal pressure during a 1-h equilibration period.
Endothelium-dependent responses of MCAs were tested using 10 μmol/l ACh (Sigma-Aldrich) and 10 μmol/l bradykinin (BK; Calbiochem, San Diego, CA), and smooth muscle-dependent functions were characterized using 10 μmol/l serotonin (5-HT; Sigma-Aldrich) and 10 μmol/l sodium nitroprusside (SNP; Sigma-Aldrich). After the addition of each agent, the Ca2+-Krebs solution in the organ chamber was allowed to equilibrate for 15 min.
Vascular responses to 10, 50, and 100 μmol/l DZ (Sigma-Aldrich) were determined on intact and endothelium-denuded MCAs. Endothelial denudation was induced by injecting 1 ml of air through the artery lumen. To investigate the different signal transduction pathways, DZ was applied in the absence and presence of the nonselective NO synthase (NOS) inhibitor Nω-nitro-l-arginine methyl ester hydrochloride (l-NAME) at 100 μmol/l for 30 min (Sigma-Aldrich) and the specific mitoKATP channel inhibitor 5-hydroxydecanoate at 1 mmol/l.
Western blot analysis.
We performed Western blot analysis on isolated MCAs (23, 40). Proteins were extracted by homogenizing MCAs using a Radnoti glass tissue grinder in 4°C Nonidet P-40 lysis buffer (Invitrogen, Frederick, MD) containing a proteinase and phosphatase inhibitor (both at 5 μl/ml, Sigma-Aldrich). The homogenate was centrifuged at 1,000 g for 10 min, and the supernatant was used for gel electrophoresis and Bradford protein assay to determine the protein concentration of the sample. Proteins were separated by 4–20% SDS-PAGE gradient gels and transferred onto polyvinylidene difluoride membranes (Bio-Rad, Hercules, CA). Membranes were blocked with 5% BSA (Sigma-Aldrich) in Tris-buffered saline and 1% Tween 20 (TBST; Sigma-Aldrich) for 1 h at room temperature and then washed three times with TBST. Membranes were incubated overnight at 4°C with the primary antibodies in 1% BSA-TBST blocking solution for the following mitochondrial proteins: ATP synthase, complex V subunit-α at 1:500 dilution (no. 459240, Invitrogen), total dynamin-related protein-1 (DRP-1) at 1:1,000 dilution (no. 611112, BD Transduction, San Jose, CA) and its Ser616-phosphorylated form (pDRP-1) at 1:1,000 dilution (no. 3455, Cell Signaling Technology, Danvers, MA), and voltage-dependent anion channel (VDAC) at 1:1,000 dilution (no. 4866, Cell Signaling Technology). The same incubation conditions were applied for the following nonmitochondrial proteins: total endothelial NOS (eNOS) at 1:500 dilution (no. 610297, BD Transduction Laboratories) and its Ser1177-phosphorylated form (peNOS) at 1:500 dilution (no. 9571, Cell Signaling Technology) and the loading control β-actin at 1:5,000 dilution (no. A5441, Sigma-Aldrich). Membranes were washed three times and incubated with their respective horseradish peroxidase-conjugated secondary anti-goat-rabbit IgG at 1:5,000 dilution (Cell Signaling Technology) or mouse IgG at 1:5,000 (Santa Cruz Biotechnology) in 1% BSA-TBST for 2 h at room temperature. Chemiluminescence (LumiGLO, Gaithersburg, MD) and autoradiography were used to visualize the final reaction. The optical density of each band was quantified and normalized to β-actin immunoband intensity using ImageJ software (%intensity).
Mitochondrial DNA quantification.
DNA was harvested by homogenizing isolated MCAs in 200 μl Nuclei Lysis solution using a DNA extraction kit (Promega, Madison, WI). Levels of mitochondrial (mt)DNA were measured using the mitochondrial cytochrome b gene (MT-CYB, Rn03296746_s1), which was normalized to the nuclear heat shock protein 70 gene (Hspa 1a, Rn00583013_s1, Applied Biosystems) (40). Triplicates of the samples were run using the following protocol: 1 cycle at 95°C for 15.05 min and 45 cycles at 95°C for 15 s and 60°C for 1 min. Gene expression levels were quantified using the ΔΔCT method (where CT is threshold cycle), and mtDNA data were normalized to nuclear DNA and expressed as means ± SE.
Mitochondrial membrane potential.
We investigated mitochondrial membrane potential (ΔΨm) using tetramethylrhodamine ethyl ester (TMRE) dye with excitation at 532 nm and emission at >550 nm (Invitrogen), which accumulates in active, negatively charged mitochondria via the strong electrochemical gradient, resulting in red fluorescence (22). Arteries were loaded with 100 nmol/l TMRE for 30 min followed by a 15-min wash period and then treated with the vehicle control (DMSO), 100 μmol/l DZ, or 500 nmol/l FCCP (Sigma-Aldrich). FCCP is an ionophore uncoupler of oxidative phosphorylation, which eliminates ΔΨm and therefore diminishes mitochondrial TMRE staining. Images were acquired before and after treatments using a Zeiss LSM 7 Live confocal microscope. ImageJ software was used to analyze the recorded fluorescence Z-stack images, and the intensity of fluorescence was determined and expressed as relative fluorescence units (RFUs). The change in TMRE fluorescence was expressed as the percent decrease in control RFUs after DZ/DMSO/FCCP treatments (%RFU).
Data analysis and statistics.
All data are expressed as means ± SE and were analyzed using one-way repeated-measures ANOVA and a Tukey post hoc test; n indicates the number of arteries. P values of <0.05 were considered as statistically significant.
RESULTS
Effectiveness of tMCAO.
All animals used for the stroke experiments had well-defined areas of infarcted tissue on the ischemic side, ipsilateral to the tMCAO without obvious parenchymal damage on the nonischemic, contralateral side after 48 h. Specifically, tMCAO produced an infarction that covered 60.4 ± 2.7% (n = 33, P < 0.05; data not shown) of the ipsilateral hemisphere and involved both cortical and subcortical areas. Nonischemia (control) animals that received a sham operation showed no evidence of neurological impairment or loss of TTC staining.
MCAs 4 h after tMCAO.
Internal diameters for pressurized ipsilateral MCAs were 272 ± 9 µm and 252 ± 11 μm for contralateral MCAs (n = 10 MCAs/group) compared with 231 ± 6 μm for control arteries (n = 22). Both ipsilateral and contralateral arteries generated similar myogenic tone [ipsilateral arteries: 38 ± 6% and contralateral arteries: 44 ± 4%, P = not significant (NS); Fig. 1D].
Fig. 1.

Vascular reactivity at 4 and 48 h after transient middle cerebral artery (MCA) occlusion (tMCAO). Control MCAs showed prominent dilation to ACh (A), bradykinin (BK; B) and sodium nitroprusside (SNP; C) as well as constriction to 70-mmHg intraluminal pressure (D) and serotonin (5-HT; E). tMCAO did not significantly affect these responses at 4 h in either ipsilateral (IPSI) or contralateral (CONTRA) MCAs (n = 10 MCAs/group). However, dilator responses to ACh, BK, and SNP as well as constrictor responses were significantly decreased in ipsilateral arteries at 48 h compared with either control or contralateral arteries. Contralateral MCAs also showed decreased dilation to ACh and BK as well as 5-HT compared with control arteries. Data are expressed as mean ± SE; n = 22 MCAs/group. *P < 0.05, 48-h ipsilateral MCAs vs. 48-h contralateral MCAs; †P < 0.05, 48-h ipsilateral MCAs vs. control MCAs; ‡P < 0.05, 48-h contralateral MCAs vs. control MCAs.
There were no significant differences among responses in ipsilateral and contralateral arteries to 10 μM ACh (ipsilateral arteries: 15 ± 5 μm and contralateral arteries: 13 ± 4 μm, P = NS), BK (ipsilateral arteries: 51 ± 10 μm and contralateral arteries: 45 ± 18 μm, P = NS), SNP (ipsilateral arteries: 100 ± 27 μm and contralateral arteries: 76 ± 20 μm, P = NS), and 5-HT (ipsilateral arteries: −88 ± 10 μm and contralateral arteries: −92 ± 17 μm, P = NS; Fig. 1, A–C and E). Although dilator responses to ipsilateral and contralateral MCAs in response to ACh and BK tended to be lower than 48-h control responses, these were not significant.
Dilator responses of MCAs to DZ with an intact endothelium were similar after tMCAO in both groups (ipsilateral MCA: 10 ± 5, 23 ± 4, and 54 ± 6 μm and contralateral MCAs: 2 ± 3, 18 ± 5, and 43 ± 6 μm for 10, 50, and 100 μmol/l DZ, respectively, P = NS; data not shown).
Denudation of the endothelium decreased DZ-induced dilation on the stroke side in response to 50 and 100 μmol/l DZ (ipsilateral arteries: 4 ± 3, 1 ± 1, and 4 ± 2 μm and contralateral arteries: 1 ± 0, 8 ± 1, and 21 ± 5 μm for 10, 50, and 100 μmol/l DZ, respectively, P < 0.05). In the presence of l-NAME, DZ-induced responses of ipsilateral arteries were decreased to 10 and 50 μmol/l DZ and to 100 μmol/l DZ in contralateral arteries (ipsilateral arteries: −3 ± 3, 2 ± 2, and 46 ± 10 μm and contralateral arteries: 1 ± 4, 11 ± 4, and 25 ± 9 μm for 10, 50, and 100 μmol/l DZ, respectively, P < 0.05; data not shown).
Of the proteins examined, we detected a significant difference (P < 0.05) only for the pDRP-1 levels between ipsilateral and contralateral MCAs, which were more than twofold higher in ipsilateral MCAs (data not shown). We did not determine mtDNA levels or TMRE staining at 4 h.
MCAs 48 h after tMCAO.
The diameters of ipsilateral MCAs were larger when initially visualized after cannulation compared with contralateral and control MCAs (250 ± 4 vs. 233 ± 6 and 231 ± 6 μm, respectively, n = 22 MCAs/group). After pressurization to 70 mmHg and a 60-min equilibration period, ipsilateral MCAs developed less myogenic tone than contralateral and control MCAs (29 ± 3% vs. 35 ± 2% and 39 ± 3%, respectively, P < 0.05, n = 22 MCAs/group; Fig. 1D).
In ipsilateral MCAs, responses to ACh, BK, 5-HT, and SNP (3 ± 2, 27 ± 4, −35 ± 5, and 37 ± 3 μm, respectively, n = 22 MCAs, P < 0.05) were significantly decreased compared with contralateral or control responses (Fig. 1, A–C and E). In addition, contralateral MCA responses (ACh: 11 ± 2 μm, BK: 41 ± 6 μm, 5-HT: −62 ± 6 μm, and SNP: 76 ± 7 μm, n = 22 MCAs, P < 0.05) were significantly decreased compared with control MCAs (ACh: 21 ± 3 μm, BK: 82 ± 7 μm, 5-HT: −85 ± 10 μm, and SNP: 80 ± 8 μm, n = 22 MCAs) for all of the drugs administered except SNP.
Despite the overall reduction of responsiveness of ipsilateral MCAs to other drugs, DZ elicited a significantly larger vasodilation in ipsilateral MCAs to all doses compared with contralateral MCAs, and ipsilateral MCA responses to 50 and 100 μmol/l DZ were similar to those of control MCAs (ipsilateral MCAs: 8 ± 2, 23 ± 3, and 39 ± 5 μm, contralateral MCAs: −5 ± 2, 6 ± 2, and 20 ± 3 μm, and control MCAs: −2 ± 4, 21 ± 6, and 43 ± 7 μm for 10, 50, and 100 μmol/l DZ, respectively, n = 22 MCAs/group, P < 0.05; Fig. 2A). Reduced vasodilation was observed in contralateral MCAs at 50 and 100 μmol/l DZ compared with control arteries. DZ-induced responses to 10, 50, and 100 μmol/l were decreased in the presence of 1 mmol/l 5-hydroxydecanoate in ipsilateral MCAs (4 ± 6, 4 ± 11, and −6 ± 13 μm, respectively, n = 3, P < 0.05), demonstrating the involvement of mitoKATP channels (data not shown).
Fig. 2.

Mitochondrial activation at 48 h after tMCAO of MCAs with an intact endothelium, after endothelium denudation, and in the presence of the nitric oxide synthase (NOS) inhibitor Nω-nitro-l-arginine methyl ester (l-NAME). A: diazoxide (DZ) caused dose-dependent dilation in control arteries. DZ-induced dilation was significantly enhanced in 48-h ipsilateral arteries compared with 48-h contralateral arteries. The 48-h contralateral responses were significantly decreased to 50 and 100 μM DZ compared with control responses. *P < 0.05, 48-h ipsilateral arteries vs. 48-h contralateral arteries; †P < 0.05, 48-h ipsilateral arteries vs. control arteries; ‡P < 0.05, 48-h contralateral arteries vs. control arteries (n = 22 arteries/group). B: denudation of the endothelium eliminated 48-h ipsilateral MCA responses to all DZ concentrations. The 48-h contralateral group also showed significantly decreased responses to 50 and 100 μM DZ. Denudation of the endothelium significantly decreased only 100 μM DZ-induced dilation in the control group. Data are expressed as means ± SE; n = 11. *P < 0.05, intact endothelium vs. denuded endothelium. C: administration of l-NAME decreased DZ-induced dilation in all groups. Data are expressed as means ± SE; n = 11. *P < 0.05, intact endothelium vs. in the presence of l-NAME.
Responses to Ach and BK were eliminated after denudation of the endothelium (data not shown). Endothelium-denuded MCAs had diminished vasodilation in response to all doses of DZ in ipsilateral arteries and to 50 and 100 μmol/l DZ in contralateral MCAs. However, denudation did not fully eliminate dilation in the control group (ipsilateral MCAs: −4 ± 3, 2 ± 3, and 0 ± 3 μm, contralateral MCAs: −3 ± 1, −2 ± 2, and 8 ± 3 μm, and control MCAs: 6 ± 5, 11 ± 4, and 16 ± 4 μm for 10, 50, and 100 μmol/l DZ, respectively, n = 11 MCAs/group, P < 0.05; Fig. 2B).
Cotreatment of l-NAME with DZ to endothelium-intact arteries resulted in significantly reduced vasodilation in ipsilateral and control MCAs to 50 and 100 μmol/l DZ, although there was no significant decrease in the responses of contralateral MCAs to DZ (ipsilateral MCAs: 5 ± 3, 7 ± 3, and 20 ± 6 μm, contralateral MCAs: −3 ± 2, 1 ± 3, and 13 ± 5 μm, and control MCAs: −2 ± 4, 2 ± 2, and 19 ± 4 μm for 10, 50, and 100 μmol/l DZ, respectively, n = 11 MCAs/group, P < 0.05; Fig. 2C).
Protein expression.
All mitochondrial protein levels examined were significantly (P < 0.05) increased in ipsilateral MCAs, including VDAC (77 ± 20%), complex V subunit-α (227 ± 22%), DRP-1 (309 ± 24%), and pDRP-1 (131 ± 40%) compared with contralateral MCAs (VDAC: 33 ± 5%, complex V: 119 ± 19%, DRP-1: 101 ± 11%, and pDRP-1: 44 ± 13%) and control MCAs (VDAC: 55 ± 3%, complex V: 96 ± 12%, DRP-1: 103 ± 10%, and pDRP-1: 34 ± 10%; Fig. 3, A–D). Levels of total eNOS (ipsilateral MCAs: 106 ± 22%, contralateral MCAs: 97 ± 19%, and control MCAs: 82 ± 15%) were unchanged (Fig. 4E); however, peNOS expression levels were significantly increased in ipsilateral MCAs (73 ± 22%, P < 0.05) compared with contralateral MCAs (21 ± 6%) and control MCAs (27 ± 4%; Fig. 3F). In each group, we used 33 MCAs to perform the Western blot experiments.
Fig. 3.

Protein expression 48 h after tMCAO. A–F: representative Western blots (top) and summary data (bottom) of 32-kDa voltage-dependent anion channel (VDAC; A), 46.9-kDa complex V subunit-α (B), 82.8-kDa mitochondrial fission protein dynamin-related protein-1 (DRP-1; C), phosphorylated (p)DRP-1 (D), 140-kDa total endothelial NOS (eNOS; E), and peNOS (F). Histograms showing that all of the mitochondrial proteins and peNOS levels were significantly increased in 48-h ipsilateral MCAs compared with 48-h contralateral and control MCAs. Data are expressed as means ± SE; n = 33 MCAs/group. *P < 0.05, 48-h ipsilateral MCAs vs. 48-h contralateral MCAs; †P < 0.05, 48-h ipsilateral MCAs vs. control MCAs; ‡P < 0.05, 48-h contralateral MCAs vs. control MCAs.
Fig. 4.
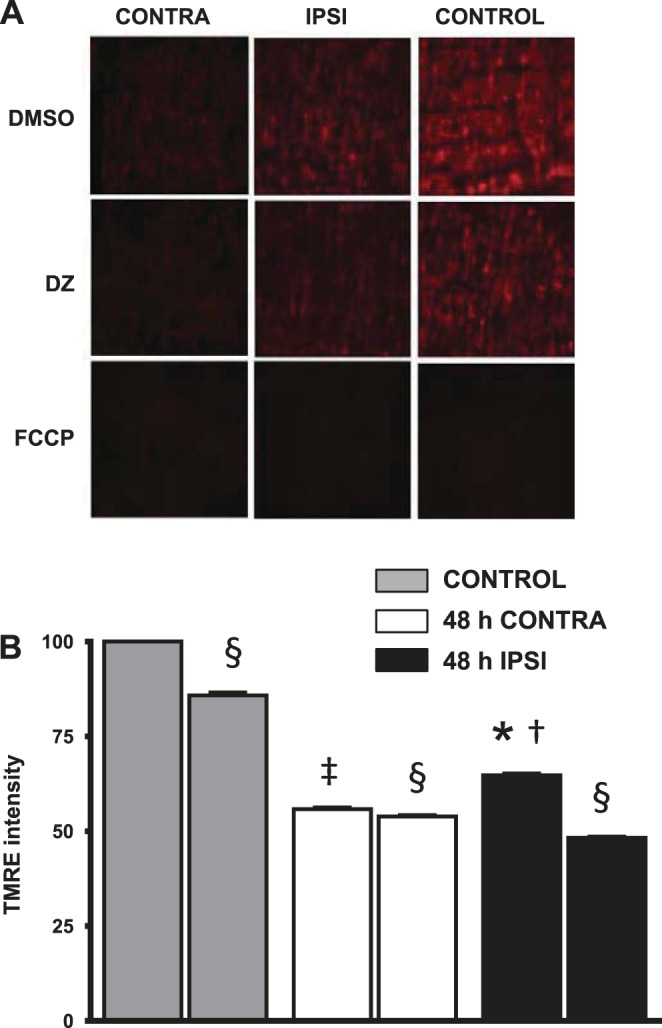
Tetramethylrhodamine ethyl ester (TMRE) staining 48 h after tMCAO. A and B: representative images of TMRE-loaded MCA sections (A) and summary data (B) showing treatments with DMSO (vehicle), DZ, and FCCP. Ischemia-reperfusion resulted in a decrease in TMRE staining compared with control MCAs. However, TMRE staining in ipsilateral MCAs was significantly increased compared with contralateral MCAs. DZ depolarized the mitochondria, producing a significant decrease in TMRE fluorescence, which had a similar magnitude in ipsilateral compared with control MCAs. Although statistically significant, the decrease in TMRE fluorescence in contralateral MCAs was small. Data are expressed as means ± SE; n = 4. *P < 0.05, 48-h ipsilateral MCAs vs. 48-h contralateral MCAs; †P < 0.05, 48-h ipsilateral MCAs vs. control MCAs; ‡P < 0.05, 48-h contralateral MCAs vs. control MCAs; §P < 0.05, DZ treatment vs. baseline.
TMRE staining.
Basal ipsilateral TMRE intensity (65 ± 0.5%, P < 0.05, n = 4) in a standard field of observation after tMCAO was increased compared with contralateral intensity (56 ± 0.4%, n = 4). Furthermore, DZ significantly decreased the TMRE intensity in all groups (ipsilateral group: 48 ± 0.3%, contralateral group: 54 ± 0.4%, and control group: 86 ± 0.8%, P < 0.05), indicating mitochondrial membrane depolarization, but the decrease in TMRE intensity was significantly greater for ipsilateral compared with contralateral arteries (Fig. 4, A and B).
mtDNA.
Ischemia-reperfusion resulted in a significantly increased mtDNA-to-nuclear DNA ratio in ipsilateral MCAs (102 ± 16%, P < 0.05) compared with contralateral MCAs (44 ± 2%, n = 20 MCAs/group) normalized to control MCAs (Fig. 5). However, there was no significant difference between ipsilateral and control groups. The mtDNA-to-nuclear DNA ratio in contralateral MCAs was significantly decreased compared with control MCAs (100%; Fig. 5).
Fig. 5.
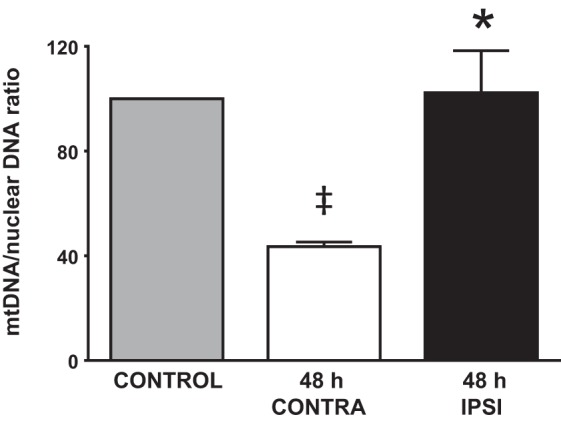
Increased mitochondrial (mt)DNA 48 h after tMCAO. The graph shows significantly larger mtDNA expression in ipsilateral MCAs after tMCAO compared with contralateral MCAs. Levels of mtDNA were similar in ipsilateral and control MCAs. Data are expressed as means ± SE; n = 20 in each group. *P < 0.05, 48-h ipsilateral MCAs vs. 48-h contralateral MCAs; ‡P < 0.05, 48-h contralateral MCAs vs. control MCAs.
DISCUSSION
There are several new findings from this study. First, contrary to generally held expectations, we found that mitochondria-dependent cerebral arterial dilation to DZ was intact and comparable with control values on the ipsilateral side after tMCAO at both 4 and 48 h. Thus, despite a prolonged period of ischemia, overall mitochondrial responses in cerebral arteries were preserved, whereas other nonmitochondrial dilator responses, especially at 48 h, were severely impaired. Second, the vascular endothelium appeared to play a dominant role in the preserved mitochondria-derived vasodilation on the ipsilateral side at 48 h. Third, preserved MCA dilation to DZ at 48 h on the ipsilateral side was associated with strong evidence of maintained or increased mitochondrial mass as well as preserved mitochondrial depolarization to DZ, as shown by TMRE measurements. An unexpected finding was that cerebral vascular responsiveness in MCAs on the side opposite the ischemia was substantially impaired at 48 h, indicating that the cerebral vascular dysfunction extends beyond the ischemic zone. We also found that contralateral MCAs showed decreased responsiveness to all of the agents examined at 48 h compared with control arteries. Thus, even in cerebral arteries distant from previously occluded arteries, vascular dysfunction was present, thereby identifying the serious limitation of the practice of only comparing ipsilateral and contralateral MCAs, as in some stroke studies. These results clearly illustrate the diversity of responses to ischemic stress that occur in different segments of the cerebral vasculature and provide very strong support for the concept that mitochondria, probably specific to the endothelium, represent a novel therapeutic target for the treatment of stroke patients, especially when clot resolution is delayed beyond the conventional 3 h.
Relatively large cerebral arteries, including MCAs, play an important role in maintaining cerebral blood flow and contributing to changes in myogenic activity to diverse stimuli under normal and pathological conditions (3, 30). We found that under basal conditions, ipsilateral MCAs had a significantly increased vascular diameter compared with contralateral and control MCAs and a significantly impaired myogenic response 48 h after tMCAO to 70-mmHg intravascular pressure. At this juncture, severe vascular dysfunction in the form of reduced MCA responsiveness occurred to several dilator or constrictor agents on the ipsilateral side. Our results are generally, but not totally, in agreement with those of previous investigators who showed variable effects on cerebral vascular responsiveness to various stimuli at various times after tMCAO (7–9, 27, 29). The variability of reported results is probably due to the use of different techniques, species, and protocols. For example, we show that despite the same duration of tMCAO, dilator and constrictor responses of ipsilateral MCAs to all examined stimuli were largely intact at 4 h but severely impaired at 48 h.
We appear to be among the first to show prominent impaired vasodilator and vasoconstrictor responses of contralateral MCAs that were never occluded compared with control MCAs from normal rats. Our finding suggests that transient ischemic stress produces endothelial and VSM dysfunction even in cerebral arteries that are somewhat distant from occluded arteries. During our review of the literature, we found only limited reports of cerebral vascular dysfunction on the contralateral side after tMCAO since responses in other studies were not normally compared with sham-operated, control arteries. The reasons for the relative vascular dysfunction on the contralateral side are unclear at this time and deserve systematic investigation since similar effects may be present in nonoccluded arteries in people suffering from a stroke and thus may affect the extent of functional vascular and neuronal recovery.
We elicited endothelium-dependent dilator responses using ACh or BK, and the relaxation responses of MCAs to these agents were greatly reduced 48 h after tMCAO on the ipsilateral side, as were endothelium-independent dilator and constrictor responses to SNP and 5-HT, respectively. Therefore, we expected ipsilateral MCA responses to DZ to be similarly impaired since dilation to this stimulus has previously been found to be due to contributions from both the endothelium and VSM (23). In addition, in an earlier report (22), we found that dilation to DZ was impaired in another disease condition: insulin resistance. Consistent with our previous studies in normal animals, DZ was a potent vasodilator in control MCAs and endothelium denudation and l-NAME treatment attenuated but did not block dilation of cerebral arteries to DZ (12, 23). DZ-induced dilation in ipsilateral MCAs was maintained at similar levels as control arteries at not only 4 h but also 48 h after tMCAO, but not due to dual contributions from VSM and the endothelium to DZ, as seen in control arteries and as previously reported (16). Thus, the removal of endothelium virtually eliminated MCA dilation to DZ at all doses examined. Similarly, inhibition of endothelium-dependent dilator stimuli by the administration of a NOS inhibitor caused a significant decrease in the responses of MCAs to DZ. Taken together, the data imply that the endothelium is the primary determinant of retained or enhanced dilation to DZ after IR but also indicate that its role is complex. For example, l-NAME administration might inhibit both coupled and uncoupled eNOS, such that both dilator (NO) and constrictor (superoxide anion) influences from the endothelium may be altered. In ipsilateral MCAs at 4 h after tMCAO, there was a tendency for enhanced dilation to DZ with l-NAME treatment, possibly implicating a role of uncoupled eNOS during this time period. Nonetheless, an increase in peNOS in ipsilateral MCAs provides further support for an augmented role of the endothelium in mediating DZ-induced dilation after tMCAO and may also demonstrate the basis for an amplification of mitochondria-initiated signaling. The endothelium is the most accessible component of the cerebral circulation for the systemic administration of drugs and other agents, such as stem cells, and thus is a potential target for new stroke therapies (15, 41).
Supporting our vascular reactivity experiments with DZ, we found evidence of maintained or increased mitochondrial mass in ipsilateral arteries after tMCAO. First, the expression level of highly conserved VDAC, one of the major proteins in mitochondria-mediated apoptosis, was increased in ipsilateral MCAs compared with contralateral and control MCAs. VDAC is located in the outer mitochondrial membrane, which provides transport pathways for respiratory chain substrates required for oxidative phosphorylation (21, 25, 33, 44). Our results are supported by results from other studies (33, 44). Furthermore, we found that protein levels of complex V subunit-α, located on the inner mitochondrial membrane, were significantly increased after ischemia-reperfusion in ipsilateral MCAs compared with control and contralateral MCAs. Complex V produces ATP from ADP via phosphorylation in the presence of the respiratory chain's electron transport-generated proton gradient across the inner mitochondrial membrane (35). These findings concerning VDAC and complex V subunit-α are in agreement with our previous findings on rat primary cortical neurons after OGD (40). However, there were no differences between protein levels in the contralateral and control groups. One possible cause of the increased ATP synthase expression may be a required amplified function to restore oxidative phosphorylation, which is a time-dependent mechanism. The increased levels of VDAC and complex V support the concept that total mitochondrial mass is increased after transient ischemia due to mitochondrial biogenesis. Corresponding with evidence of increased mitochondrial mass were elevated levels of fission proteins DRP-1 and pDRP-1 in ipsilateral MCAs (2, 40). Our second level of evidence of maintained or increased mitochondrial mass in ipsilateral arteries after tMCAO is that mtDNA levels were higher in ipsilateral compared with contralateral MCAs. Similarly, Yin et al. (43) showed increased mtDNA and numbers of mitochondria in the brain 24 h after hypoxic-ischemic brain injury. We have shown similar findings in cultured neurons after OGD (40). Third, we observed a greater intensity of the mitochondria-specific TMRE fluorescence in ipsilateral compared with contralateral MCAs. Evaluation of TMRE responses to DZ indicated a greater decrease in the ipsilateral ΔΨm compared with contralateral arteries, thereby providing information independent of vascular responsiveness that the mitochondria retained normal functionality. However, DZ caused a similar decrease in ipsilateral and control ΔΨm, which reflects our vascular data where DZ-induced vasodilation was similar in ipsilateral and control MCAs. The greater amount of TMRE staining in control MCAs than in ipsilateral MCAs may indicate that although mitochondrial proteins and perhaps mitochondrial numbers are augmented after tMCAO, all mitochondria, especially in VSM, might not be fully functioning. Nonetheless, increased biosynthesis of mitochondria, as demonstrated by increased mitochondrial proteins and mtDNA as well as the remodeling protein pDRP-1, together with enhanced levels of activated eNOS, appears to be sufficient for the preservation of MCA responses to DZ 48 h after tMCAO.
To our knowledge, this is the first study to report preserved mitochondria-derived vascular responses in the ischemic side and diminished responses in the contralateral side in MCAs after 90 min of ischemia and 4 and 48 h of reperfusion. Our results support the concept that the endothelium exerts a dominant compensatory role in maintaining mitochondrial function after prolonged, transient cerebral ischemia. Although the underlying mechanisms are not totally clear, these results may contribute to a greater understanding of the ongoing changes after ischemic stress as well as aid in the development of potential therapies to help people suffering from strokes. Thus, targeting mitochondria in the immediate postischemic period may not only improve cerebral blood flow but may also protect the neurovascular unit from further damage and cell death and thereby decrease stroke-related morbidity and mortality. In addition, our finding that both previously occluded as well as nonoccluded arteries were affected differently by tMCAO indicates that distinct therapeutic approaches are needed to preserve cerebral vascular function after strokes.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL-077731, HL-030260, HL-065380, and HL-093554.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: I.R. and D.W.B. conception and design of research; I.R., P.V.G.K., and S.D. performed experiments; I.R. analyzed data; I.R. and D.W.B. interpreted results of experiments; I.R. prepared figures; I.R. drafted manuscript; I.R., P.V.G.K., S.D., and D.W.B. edited and revised manuscript; I.R., P.V.G.K., S.D., and D.W.B. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Nancy Busija for editorial assistance. The authors also thank Dr. Zakaria Abd Elmageed for help with the PCR experiments and Dan Liu and Korey A. Walter for technical help.
REFERENCES
- 1.Ahmed N, Wahlgren N, Grond M, Hennerici M, Lees KR, Mikulik R, Parsons M, Roine RO, Toni D, Ringleb P, SITS Investigators. Implementation and outcome of thrombolysis with alteplase 3–4.5 h after an acute stroke: an updated analysis from SITS-ISTR. Lancet Neurol 9: 866–874, 2010. [DOI] [PubMed] [Google Scholar]
- 2.Barsoum MJ, Yuan H, Gerencser AA, Liot G, Kushnareva Y, Graber S, Kovacs I, Lee WD, Waggoner J, Cui J, White AD, Bossy B, Martinou JC, Youle RJ, Lipton SA, Ellisman MH, Perkins GA, Bossy-Wetzel E. Nitric oxide-induced mitochondrial fission is regulated by dynamin-related GTPases in neurons. EMBO J 25: 3900–3911, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Busija DW. The regulation of cerebral blood flow. In: Cerebral Autoregulation, edited by Phillis JW. New York: CRC, 1993, p. 46. [Google Scholar]
- 4.Busija DW, Gaspar T, Domoki F, Katakam PV, Bari F. Mitochondrial-mediated suppression of ROS production upon exposure of neurons to lethal stress: mitochondrial targeted preconditioning. Adv Drug Deliv Rev 60: 1471–1477, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Busija DW, Katakam PV. Mitochondrial mechanisms in cerebral vascular control: shared signaling pathways with preconditioning. J Vasc Res 51: 175–189, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Busija DW, Lacza Z, Rajapakse N, Shimizu K, Kis B, Bari F, Domoki F, Horiguchi T. Targeting mitochondrial ATP-sensitive potassium channels–a novel approach to neuroprotection. Brain Res Brain Res Rev 46: 282–294, 2004. [DOI] [PubMed] [Google Scholar]
- 7.Cipolla MJ, Bullinger LV. Reactivity of brain parenchymal arterioles after ischemia and reperfusion. Microcirculation 15: 495–501, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cipolla MJ, Curry AB. Middle cerebral artery function after stroke: the threshold duration of reperfusion for myogenic activity. Stroke 33: 2094–2099, 2002. [DOI] [PubMed] [Google Scholar]
- 9.Cipolla MJ, Lessov N, Clark WM, Haley EC., Jr Postischemic attenuation of cerebral artery reactivity is increased in the presence of tissue plasminogen activator. Stroke 31: 940–945, 2000. [DOI] [PubMed] [Google Scholar]
- 10.del Zoppo GJ. Virchow's triad: the vascular basis of cerebral injury. Rev Neurol Dis 5, Suppl 1: S12–S21, 2008. [PMC free article] [PubMed] [Google Scholar]
- 11.Del Zoppo GJ, Saver JL, Jauch EC, Adams HP, Jr; American Heart Association Stroke Council. Expansion of the time window for treatment of acute ischemic stroke with intravenous tissue plasminogen activator: a science advisory from the American Heart Association/American Stroke Association. Stroke 40: 2945–2948, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Domoki F, Kis B, Nagy K, Farkas E, Busija DW, Bari F. Diazoxide preserves hypercapnia-induced arteriolar vasodilation after global cerebral ischemia in piglets. Am J Physiol Heart Circ Physiol 289: H368–H373, 2005. [DOI] [PubMed] [Google Scholar]
- 13.Domoki F, Perciaccante JV, Veltkamp R, Bari F, Busija DW. Mitochondrial potassium channel opener diazoxide preserves neuronal-vascular function after cerebral ischemia in newborn pigs. Stroke 30: 2713–2719, 1999. [DOI] [PubMed] [Google Scholar]
- 14.Elijovich L, Chong JY. Current and future use of intravenous thrombolysis for acute ischemic stroke. Curr Atheroscler Rep 12: 316–321, 2010. [DOI] [PubMed] [Google Scholar]
- 15.Fagan SC, Hess DC, Hohnadel EJ, Pollock DM, Ergul A. Targets for vascular protection after acute ischemic stroke. Stroke 35: 2220–2225, 2004. [DOI] [PubMed] [Google Scholar]
- 16.Garrett BN, Kaplan NM. Efficacy of slow infusion of diazoxide in the treatment of severe hypertension without organ hypoperfusion. Am Heart J 103: 390–394, 1982. [DOI] [PubMed] [Google Scholar]
- 17.Gaspar T, Kis B, Snipes JA, Lenzser G, Mayanagi K, Bari F, Busija DW. Transient glucose and amino acid deprivation induces delayed preconditioning in cultured rat cortical neurons. J Neurochem 98: 555–565, 2006. [DOI] [PubMed] [Google Scholar]
- 18.Hacke W, Donnan G, Fieschi C, Kaste M, von Kummer R, Broderick JP, Brott T, Frankel M, Grotta JC, Haley EC, Jr, Kwiatkowski T, Levine SR, Lewandowski C, Lu M, Lyden P, Marler JR, Patel S, Tilley BC, Albers G, Bluhmki E, Wilhelm M, Hamilton S; ATLANTIS Trials Investigators; ECASS Trials Investigators; NINDS rt-PA Study Group Investigators. Association of outcome with early stroke treatment: pooled analysis of ATLANTIS, ECASS, and NINDS rt-PA stroke trials. Lancet 363: 768–774, 2004. [DOI] [PubMed] [Google Scholar]
- 19.Hacke W, Kaste M, Bluhmki E, Brozman M, Davalos A, Guidetti D, Larrue V, Lees KR, Medeghri Z, Machnig T, Schneider D, von Kummer R, Wahlgren N, Toni D; ECASS Investigators. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med 359: 1317–1329, 2008. [DOI] [PubMed] [Google Scholar]
- 20.Horiguchi T, Kis B, Rajapakse N, Shimizu K, Busija DW. Opening of mitochondrial ATP-sensitive potassium channels is a trigger of 3-nitropropionic acid-induced tolerance to transient focal cerebral ischemia in rats. Stroke 34: 1015–1020, 2003. [DOI] [PubMed] [Google Scholar]
- 21.Jonas EA. Molecular participants in mitochondrial cell death channel formation during neuronal ischemia. Exp Neurol 218: 203–212, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katakam PV, Domoki F, Snipes JA, Busija AR, Jarajapu YP, Busija DW. Impaired mitochondria-dependent vasodilation in cerebral arteries of Zucker obese rats with insulin resistance. Am J Physiol Regul Integr Comp Physiol 296: R289–R298, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Katakam PV, Wappler EA, Katz PS, Rutkai I, Institoris A, Domoki F, Gaspar T, Grovenburg SM, Snipes JA, Busija DW. Depolarization of mitochondria in endothelial cells promotes cerebral artery vasodilation by activation of nitric oxide synthase. Arterioscler Thromb Vasc Biol 33: 752–759, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kis B, Nagy K, Snipes JA, Rajapakse NC, Horiguchi T, Grover GJ, Busija DW. The mitochondrial KATP channel opener BMS-191095 induces neuronal preconditioning. Neuroreport 15: 345–349, 2004. [DOI] [PubMed] [Google Scholar]
- 25.Lemasters JJ, Holmuhamedov E. Voltage-dependent anion channel (VDAC) as mitochondrial governator–thinking outside the box. Biochim Biophys Acta 1762: 181–190, 2006. [DOI] [PubMed] [Google Scholar]
- 26.Lenzser G, Kis B, Bari F, Busija DW. Diazoxide preconditioning attenuates global cerebral ischemia-induced blood-brain barrier permeability. Brain Res 1051: 72–80, 2005. [DOI] [PubMed] [Google Scholar]
- 27.Marrelli SP, Khorovets A, Johnson TD, Childres WF, Bryan RM., Jr P2 purinoceptor-mediated dilations in the rat middle cerebral artery after ischemia-reperfusion. Am J Physiol Heart Circ Physiol 276: H33–H41, 1999. [DOI] [PubMed] [Google Scholar]
- 28.Mattson MP, Culmsee C, Yu ZF. Apoptotic and antiapoptotic mechanisms in stroke. Cell Tissue Res 301: 173–187, 2000. [DOI] [PubMed] [Google Scholar]
- 29.Mayhan WG, Amundsen SM, Faraci FM, Heistad DD. Responses of cerebral arteries after ischemia and reperfusion in cats. Am J Physiol Heart Circ Physiol 255: H879–H884, 1988. [DOI] [PubMed] [Google Scholar]
- 30.Mayhan WG, Faraci FM, Baumbach GL, Heistad DD. Effects of aging on responses of cerebral arterioles. Am J Physiol Heart Circ Physiol 258: H1138–H1143, 1990. [DOI] [PubMed] [Google Scholar]
- 31.Montano A, Staff I, McCullough LD, Fortunato G. Community implementation of intravenous thrombolysis for acute ischemic stroke in the 3- to 4.5-hour window. Am J Emerg Med 31: 1707–1709, 2013. [DOI] [PubMed] [Google Scholar]
- 32.Nagy K, Kis B, Rajapakse NC, Bari F, Busija DW. Diazoxide preconditioning protects against neuronal cell death by attenuation of oxidative stress upon glutamate stimulation. J Neurosci Res 76: 697–704, 2004. [DOI] [PubMed] [Google Scholar]
- 33.Peixoto PM, Dejean LM, Kinnally KW. The therapeutic potential of mitochondrial channels in cancer, ischemia-reperfusion injury, and neurodegeneration. Mitochondrion 12: 14–23, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics–2012 update: a report from the American Heart Association. Circulation 125: e2–e220, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sack MN. Mitochondrial depolarization and the role of uncoupling proteins in ischemia tolerance. Cardiovasc Res 72: 210–219, 2006. [DOI] [PubMed] [Google Scholar]
- 36.Shimizu K, Lacza Z, Rajapakse N, Horiguchi T, Snipes J, Busija DW. MitoKATP opener, diazoxide, reduces neuronal damage after middle cerebral artery occlusion in the rat. Am J Physiol Heart Circ Physiol 283: H1005–H1011, 2002. [DOI] [PubMed] [Google Scholar]
- 37.Szewczyk A. Intracellular targets for antidiabetic sulfonylureas and potassium channel openers. Biochem Pharmacol 54: 961–965, 1997. [DOI] [PubMed] [Google Scholar]
- 38.Szewczyk A, Wojtczak L. Mitochondria as a pharmacological target. Pharmacol Rev 54: 101–127, 2002. [DOI] [PubMed] [Google Scholar]
- 39.Wahlgren N, Ahmed N, Davalos A, Hacke W, Millan M, Muir K, Roine RO, Toni D, Lees KR; SITS investigators. Thrombolysis with alteplase 3–4.5 h after acute ischaemic stroke (SITS-ISTR): an observational study. Lancet 372: 1303–1309, 2008. [DOI] [PubMed] [Google Scholar]
- 40.Wappler EA, Institoris A, Dutta S, Katakam PV, Busija DW. Mitochondrial dynamics associated with oxygen-glucose deprivation in rat primary neuronal cultures. PLos One 8: e63206, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wojakowski W, Landmesser U, Bachowski R, Jadczyk T, Tendera M. Mobilization of stem and progenitor cells in cardiovascular diseases. Leukemia 26: 23–33, 2012. [DOI] [PubMed] [Google Scholar]
- 42.Xi Q, Cheranov SY, Jaggar JH. Mitochondria-derived reactive oxygen species dilate cerebral arteries by activating Ca2+ sparks. Circ Res 97: 354–362, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yin W, Signore AP, Iwai M, Cao G, Gao Y, Chen J. Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke 39: 3057–3063, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zamzami N, Kroemer G. The mitochondrion in apoptosis: how Pandora's box opens. Nat Rev Mol Cell Biol 2: 67–71, 2001. [DOI] [PubMed] [Google Scholar]