

Abstract
The establishment of a functional vascular system requires multiple complex steps throughout embryogenesis, from endothelial cell (EC) specification to vascular patterning into venous and arterial hierarchies. Following the initial assembly of ECs into a network of cord-like structures, vascular expansion and remodeling occur rapidly through morphogenetic events including vessel sprouting, fusion, and pruning. In addition, vascular morphogenesis encompasses the process of lumen formation, critical for the transformation of cords into perfusable vascular tubes. Studies in mouse, zebrafish, frog, and human endothelial cells have begun to outline the cellular and molecular requirements underlying lumen formation. Although the lumen can be generated through diverse mechanisms, the coordinated participation of multiple conserved molecules including transcription factors, small GTPases, and adhesion and polarity proteins remains a fundamental principle, leading us closer to a more thorough understanding of this complex event.
Keywords: Casz1, Egfl7, lumen formation, morphogenesis, RhoA
Introduction
The cardiovascular system is one of the first organ systems to arise during embryogenesis. Its proper establishment during early development is critical for meeting the demands of a rapidly growing embryo in need of nutrients, oxygen, and waste removal. Vertebrate blood vessels develop via two distinct mechanisms known as vasculogenesis and angiogenesis. During vasculogenesis, the de novo assembly of new blood vessels, a subset of mesodermal cells differentiates into endothelial cell (EC) precursors called angioblasts. Angioblasts proliferate and migrate to specified positions in the embryo where they coalesce into cord-like structures to form the primary vascular plexus. These early EC behaviors are elicited through growth factor-mediated signaling including vascular endothelial growth factor (VEGF), fibroblast growth factor (FGF), and bone morphogenetic protein (BMP) [1]. Rapidly thereafter, endothelial cords transform into patent tubes and become further specified to contribute to either the venous, arterial, or lymphatic vasculature [2]. The primitive plexus undergoes dramatic remodeling during angiogenesis when new vessels sprout, elongate, and expand to form a complex network of arteries and veins. Henceforth, the majority of vascular development proceeds via angiogenesis, which can also be sub-divided into two processes. In sprouting angiogenesis, new vessels branch from preexisting vessels whereas in intussusceptive angiogenesis, existing vessels internally split to form new vessels [3, 4]. In later stages of vascular remodeling, mural cells including smooth muscle cells and pericytes are recruited to surround the EC-lined tubes and impart vessel stabilization and maturation [5].
In the adult, the vasculature is mostly quiescent, with little to no vessel growth or remodeling. However, in response to injury, reduced oxygen or hypoxic conditions, inflammation, tumor growth, or pregnancy, ECs are poised to react and form new blood vessels in a process known as neoangiogenesis. The ability of a cohort of ECs to dynamically assemble into a vascular network is remarkable, given the multitude of individual cellular behaviors that must first be coordinated, including migration, cell-cell and cell-extracellular matrix (ECM) adhesion, proliferation, ECM degradation, and sprouting. Therefore, it is not surprising that dysregulation of these behaviors can have detrimental effects on health and survival; e.g., during the pathogenesis of numerous vascular diseases, including atherosclerosis and myocardial ischemia, as well as during tumor progression [5, 6]. Understanding the cellular and molecular mechanisms underlying EC behavior will provide a basis for the development of therapeutics for these diseases.
Over the years, substantial contributions have been made toward understanding the mechanisms underlying sprouting angiogenesis that have significantly impacted the development of therapeutics promoting or inhibiting vessel growth in a wide range of diseases [7, 8]. Considerably less is known about the lumen formation process, critical for establishing a patent and functional vascular system during development as well as for efficient delivery of therapeutics to their targets during disease treatment. In this review, we will discuss the diverse mechanisms by which the lumens of various vascular beds arise. Although there is some debate as to which of these mechanisms predominate during development, the identification of several molecules and models of lumen formation has without a doubt shed light on this seemingly mysterious process, and has in recent years greatly expanded the number of groups currently studying lumen morphogenesis.
Vessel lumens arise by distinct mechanisms
The ability to culture primary mammalian ECs has significantly enabled the study of cellular behaviors associated with blood vessel assembly, such as proliferation, motility, and sprouting angiogenesis. The recent advent of three-dimensional models has further enhanced the ability to visualize EC morphogenesis into vascular tubes [9–12]. Specifically, in response to suspension in three-dimensional collagen or fibrin matrices, human umbilical vein endothelial cells (HUVECs) were shown to individually generate luminal spaces via a vacuolation mechanism known as cell hollowing. Live imaging revealed that cells use pinocytosis to internalize the plasma membrane and generate multiple vesicles that coalesce to form an intracellular lumen, defined as an ECM-free space surrounded by a single EC [13]. Contacts made between adjacent cells facilitate the fusion of vacuoles, resulting in a network of capillary-like tubes (Fig. 1A). This process depends on signaling between ECs and the ECM via integrins, particularly integrin α2β1 in collagen matrices, and integrins αvβ3 and α5β1 in fibrin matrices [14, 15]. Although these observations were made in cultured cells in a manipulated environment, the generation of an intracellular lumen via vacuolation was also observed in zebrafish ISVs [16].
Figure 1.

Mechanisms of blood vessel lumen formation. A: Intracellular lumens refer to hollow spaces enclosed by a single EC and devoid of junctions. Intracellular lumen formation can be sub-divided into two mechanisms. (i) During vacuole coalescence (top panel), individual ECs generate vacuole-like structures through membrane internalization. The vacuoles fuse to form a hollow compartment devoid of ECM. Therefore, when vacuolated cells assemble to form a network, contacts between adjacent cells are made (purple rectangles) however no junctions are found interrupting the cell-lumen boundary resulting in a unicellular tube. (ii) In apical membrane invagination (middle panel), once two distinct vessels (blue vs. pink) have established contact (purple oval), the luminal compartment is expanded by invagination of the membrane (light blue/pink). The membrane continues to extend through the point of contact resulting in one continuous lumen. Each cell now makes up a unicellular tube containing an intracellular lumen within the patent vessel. B: During extracellular lumen generation (bottom panel), ECs first form adherens junctions between their contact surfaces to generate cord-like structures. These junctions become redistributed from the center of the contact region to the periphery in order to allow a luminal compartment to take shape between the cells. Thus, multicellular tubes are generated in which the lumen is surrounded by two or more ECs that remain in contact with each other as well as with adjacent cells.
Recent studies, however, have indicated that the lumen is more often generated extracellularly where it lies between two adjacent ECs connected by junctions, raising questions regarding whether cell vacuolation is the primary mechanism of lumen formation (Fig. 1B). Also known as cord hollowing, in the mouse dorsal aorta (DA) adjacent ECs are initially in contact with one another via adherens junctions [17]. The cells become polarized even before beginning to separate from each other with a number of molecules localizing specifically to the apical surface where the lumen will be generated. The establishment of apicobasal polarity is suggested to further promote cell separation by causing junctions to redistribute away from the cell-cell interface to the periphery. Furthermore, small GTPase signaling was also shown to enhance EC elongation in order to accommodate the luminal compartment in the center [17]. In addition to the mouse DA, extracellular lumens also arise in zebrafish ISVs. In contrast to the idea that intracellular vacuoles reside within the ISV ECs and fuse to form a seamless unicellular tube, labeling of multiple junction and polarity markers has in fact revealed that cells first establish apical polarity and form complex junctions that dynamically rearrange to generate a multicellular tube containing an extracellular lumen [18, 19]. However, ISV lumens are not generated strictly by cord hollowing as Wang et al. showed that the formation and fusion of vacuoles within ISVs also promote rapid expansion of the luminal compartment [19].
It is therefore increasingly clear that vessels arise in a heterogeneous manner and likely use a number of mechanisms to conveniently and efficiently generate lumens. Extracellular lumen formation may be the predominant mechanism by which major vessels such as the DA form, whereas small-caliber vessels like the ISVs and capillaries, which have also been demonstrated to resemble unicellular tubes in rats [20], may use a combination of both cord and cell hollowing. Nonetheless, analyses of the molecular determinants of extracellular and intracellular lumen formation have greatly contributed to our understanding of the cellular processes that must be coordinated in order to promote this complex step of blood vessel development (Table 1).
Table 1.
Molecular determinants that play diverse roles during lumen morphogenesis
| Molecule | Function/localization during lumen formation | Model system | Reference |
|---|---|---|---|
| VE-Cadherin | Localizes to adherens junctions in ECs; establishes apicobasal polarity | Mouse, zebrafish, cultured ECs | [17, 19, 26, 36–38] |
| Par complex (Par3-Par6-aPKC) | Promotes endothelial polarization; associates with tight and adherens junctions | Mouse, cultured ECs | [17, 31, 41, 87] |
| Moesin | Localizes to apical membrane; recruits F-actin to apical surface | Mouse, zebrafish | [17, 19] |
| CD34/PODXL | Localize to apical membrane; sialic acids promote cell-cell separation | Mouse, cultured ECs | [17, 26, 27] |
| Rac1/Cdc42 | Localize to vacuolar structures; mediate downstream signaling during intracellular lumen formation | Mouse, zebrafish, cultured ECs | [16, 31, 55, 56, 66, 67, 69] |
| RhoA | Positive and negative roles; modulates proper adhesion to ECM and ROCK-mediated shape changes | Mouse, cultured ECs | [17, 72–74, 76] |
| Rasip1 | Regulates RhoA activity with Arhgap29 to promote lumen formation | Mouse, cultured ECs | [73] |
| Casz1 | Binds and maintains Egfl7 expression in ECs | Xenopus, cultured ECs | [76] |
| Egfl7 | Secreted into ECM and required for initial establishment of lumens but specific function still unknown | Xenopus, zebrafish, cultured ECs | [76, 79] |
Vessels remodel lumens during branching and fusion
The processes described above reveal how the lumen forms de novo and independently of blood flow. More frequently, new vessels are formed during anastomosis, or fusion between two distinct perfused vessels that must combine luminal compartments to give rise to a patent network. The formation of the zebrafish dorsal longitudinal anastomosing vessel (DLAV) occurs via anastomosis between adjacent segmental arteries [21]. The DLAV is comprised of both multicellular and, in the majority of cases, unicellular tubes. Using a transgenic line expressing the tight junction molecule zonula occludens-1 (ZO-1) tagged to GFP, Herwig et al. found that multicellular lumens are generated by the rearrangement of junctions between three adjacent cells, resulting in the establishment of new contacts between the outer cells and the merging of local lumens into one continuous luminal compartment. Conversely in most instances, junctions between two cells are not as dynamic. Instead, a lumen is generated within a single cell by invagination of the apical membrane throughout the length of the cell resulting in a unicellular tube with an intracellular lumen. The luminal membrane of this single cell invaginates through the point of cell-cell contact to ultimately fuse with the neighboring lumen [21].
The presence of unicellular tubes is not unique to the DLAV and is conserved in other zebrafish vessels, such as the palatocerebral artery (PLA) in the cranial vasculature [22]. Using a similar fusion process, two distinct lumenized sprouts come into contact with each other, marked by junction markers such as ZO-1, and combine their luminal compartments by apical membrane invagination, resulting in two adjoining unicellular tubes comprised of intracellular lumens. However, later remodeling events reveal that these unicellular vessels are transient, and multicellular tubes take precedence within the PLA. The two cells adjacent to the unicellular tube are initially not in contact with each other but move and redistribute their junctions to form a new contact, thereby forcing the cell comprising the unicellular tube to split to one side of the tube (Fig. 2). Therefore, an extracellular lumen surrounded by multiple ECs is generated, analogous to the mouse DA lumen assembled via cord hollowing. In both the DLAV and PLA, apical membrane invagination and the subsequent generation of the lumen is dependent on blood flow, likely because of the force generated by increased blood pressure. However, polarity and junctional rearrangements still take place in the absence of flow [21, 22].
Figure 2.
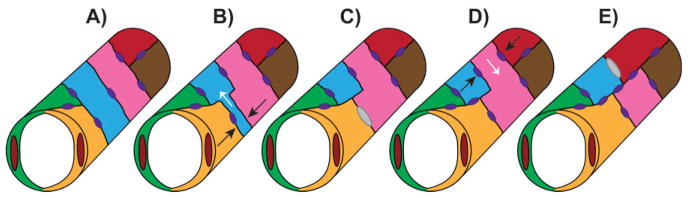
Lumen formation during vessel fusion. Unicellular tubes remodel into multicellular tubes by cell splitting. A: Two unicellular tubes (blue and pink) are flanked by multicellular tubes (green/yellow and red/brown) with junctions represented by purple ovals. B: Two cells flanking the blue unicellular tube (yellow and pink) rearrange their junctions and approach each other (black arrows) while the blue cell is forced to split to one side of the vessel (white arrow). C: The yellow and pink cells establish a new contact (gray oval) and the blue cell has narrowed to one side of the vessel incorporating into a multicellular tube. D: The second unicellular tube (pink) is now flanked by the blue and red cells which rearrange their junctions and approach each other (black arrows) forcing the pink cell to the opposite side (white arrow). E: The blue and red cells establish a new contact (gray oval) and the whole vessel is now comprised of multicellular tubes.
The presence of vacuole-like structures was not addressed in the above zebrafish studies and hence it is debatable whether vacuole coalescence and apical membrane invagination truly represent distinct mechanisms of lumen formation. However, because in both cases the lumen is completely surrounded by a single cell and devoid of any junctions throughout the unicellular region, vacuole coalescence, and apical membrane invagination can likely be annotated as sub-categories of intracellular lumen formation (Fig. 1A). It is also unknown what molecular forces govern lumen formation during vessel fusion. Because several distinct cellular behaviors are necessary for successful anastomosis, including migration, sprouting, and adhesion, it may be difficult to tease apart the specific molecular mechanism underlying apical membrane invagination and subsequent cell splitting. It is likely that multiple mechanisms are involved, and that different combinations of interacting factors and intersecting signaling pathways at each step of vascular morphogenesis may elicit more precise cellular responses.
The establishment of polarity initiates lumen formation
Distinct surface markers define the apical and basal membranes
Despite the distinction between intracellular and extracellular lumens, a common theme reconciling these differences is that in all cases, cells must first establish polarity to define the apical/luminal (“inside”) and basal/abluminal (“outside”) vessel surface [23]. This specialized distinction between the inside and outside of the tube is not unique to ECs. Multiple other tubular organs such as the kidney and intestine also display apicobasal polarity, and many of the molecular players currently being investigated in vessel lumen formation unsurprisingly have reported roles in epithelial tubulogenesis as well [24].
A number of markers have been established that have preferential localization to either the apical or basal side of EC-lined tubes. Notably, cell-surface transmembrane proteins such as the CD34 sialomucins, consisting of CD34 and podocalyxin-like proteins 1 and 2 (PODXL/PODXL2), and the Ezrin-Radixin-Moesin (ERM) protein Moesin are enriched at the apical membrane [17, 19, 25, 26]. However, the function of these proteins extends beyond expression at the apical surface. The negatively charged sialic acids coating the extracellular domain of the CD34 sialomucins are required for the electrostatic repulsion needed to separate the adjoining ECs from each other to accommodate the central luminal compartment [27]. Furthermore, in PODXL-null mice, Moesin no longer localizes to the apical membrane of the early DA, and only 40% of lumens fully develop [17]. ERM proteins modulate cytoskeletal dynamics and signal transduction by anchoring the actin cytoskeleton to various transmembrane proteins [28, 29]. Accordingly, Moesin depletion was associated with significantly diminished F-actin enrichment at the apical surface of the developing mouse DA and zebrafish ISVs, ultimately resulting in reduced or failed lumen formation [17, 19]. In addition, the partition-defective (Par) polarity complex (Par3/Par6/atypical protein kinase C [aPKC]) has also been implicated in apical polarity in epithelial and ECs, and disruption of the Par complex is strongly associated with failed lumen formation [30, 87]. Depletion of Par3 and Par6 in HUVECs embedded in collagen matrices results in failed intracellular lumen formation, whereas chemical inhibition of PKC in the mouse DA prevents Moesin phosphorylation and subsequent lumen generation [17, 31].
On the basal surface of the vessel, ECs contact various constituents of the ECM that facilitate interactions with mural cells as well as influence whether vessels will become stable and quiescent or “activated” to sprout [32]. Basement membrane markers include fibronectin, laminin, collagen IV, and integrins. A recent study suggests that this specialized separation of apical and basal membranes in vertebrate blood vessels is a consequence of EC evolution. Unlike vertebrate vessels, the lumen of the aorta in the invertebrate cephalochordate amphioxus, which lacks ECs, is directly lined with the laminin-containing basement membrane implying that ECs serve to separate basal surfaces from the luminal compartment [33, 34].
VE-Cadherin promotes proper localization of polarity markers
What is the basis for initiating polarity in the first place? In the case of extracellular lumen formation, ECs first coalesce into cords where they adhere to each other via adherens junctions. Numerous markers, such as β-catenin and N-cadherin, constitute adherens junctions in multiple cell types. Vascular endothelial (VE)-cadherin (also known as Cdh5), however, specifically marks junctions in ECs [35]. Far from simply labeling cell-cell junctions, VE-cadherin plays a critical role in vascular morphogenesis. VE-cadherin deficiency in mice results in early embryonic lethality due to severe vascular defects, including failed establishment of yolk sac vasculature, disorganized and disconnected embryonic vessels, and notably, minimal or absent lumens [36, 37]. A functional vascular network also fails to form in VE-cadherin-depleted zebrafish embryos in which vessel lumens are small (or absent) and fail to connect to form a complete circulatory loop [38]. In the adult, VE-cadherin is essential for maintaining vascular homeostasis and endothelial barrier integrity [39, 40].
Subsequent studies in cultured ECs as well as in vivo have determined that the molecular basis for aberrant lumen morphogenesis in VE-cadherin-deficient animals is associated with impaired establishment of cell polarity. In VE-cadherin null mouse DAs, apical markers such as CD34, PODXL, and Moesin fail to localize to the cell-cell contact, and consequently lumens do not form [17]. These results were confirmed in detailed analyses of VE-cadherin-depleted zebrafish where it was noted that Moesin was no longer enriched at the luminal surface of the ISVs [19]. In addition, the distribution of ZO-1-containing junctions was disorganized in VE-cadherin-depleted zebrafish, indicating a role for VE-cadherin in maintaining adherens junctions as well as tight junctions. Tubular HUVEC networks in collagen matrices also display specific localization of apical and basal markers [26]. However, in the absence of VE-cadherin, PODXL and collagen IV are aberrantly localized with little distinction between apical and basal membranes. Consequently, there are multiple small lumens instead of one central compartment (Fig. 3). In addition, members of the Par complex are also absent from cell-cell contacts in VE-cadherin-depleted cells [26]. Moreover, VE-cadherin has been shown to directly associate with Par3 and Par6 in cultured ECs, strongly suggesting that the interplay between junctional and polarity molecules promotes lumen formation [41]. Interestingly, VE-cadherin is not required for polarity during PLA anastomosis. Instead, it is needed to stabilize the initial contact made between fusing sprouts, implying diverse roles for this junctional marker during various steps of vascular assembly [22].
Figure 3.
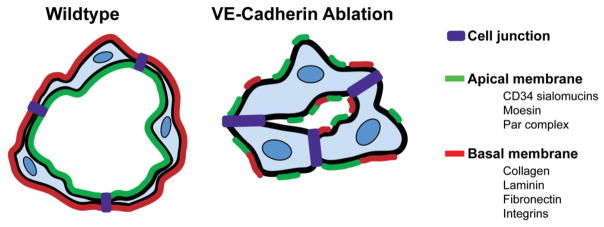
Role of VE-Cadherin in establishment of polarity. In the presence of VE-Cadherin (left panel), ECs establish proper polarity with a specified apical surface, as marked by proteins such as the CD34 sialomucins, the ERM protein Moesin, and members of the Par polarity complex, and a basal surface marked by ECM constituents and integrins. However, when VE-Cadherin is absent (right panel), apical and basal markers are disorganized and fail to localize to the appropriate surface resulting in smaller or absent vessel lumens.
Small GTPases modulate the cytoskeleton to facilitate lumen formation
In addition to cell-cell contact and the establishment of polarity, lumen morphogenesis requires cells to make dynamic contacts with the underlying ECM as well as rearrange their internal actin cytoskeleton to adopt a suitable shape conducive to expansion of the luminal compartment. Directly involved in regulating cell motility and cytoskeletal organization, the Rho family of small GTPases has been at the forefront of cell biology for decades. The number of studies on their downstream effectors, roles in diverse biological processes such as gene expression, proliferation, and membrane trafficking, and implications for human health is continually growing [42–44]. The most well-known GTPases, RhoA, Rac1, and Cdc42, have been extensively characterized in fibroblasts, where they have critical roles in organizing the actin cytoskeleton during cell migration and mitosis [45–48]. It is therefore unsurprising that ECs also depend on GTPase activity for a multitude of cellular behaviors. However, the functions of the Rho GTPases in vascular development and disease are only just beginning to be uncovered, including their roles in lumen formation.
Rac1 and Cdc42 positively regulate lumen formation
Rac1 and Cdc42 are classically known to promote lamellipodial and filopodial protrusions, respectively, at the leading edge of migrating cells including fibroblasts, ECs, and smooth muscle cells [46, 49, 50]. Early cell culture studies first showed that Rac1 and Cdc42 have distinct functions in ECs, such as stabilizing cell-cell junctions to regulate vascular permeability and mediating downstream signaling in response to shear stress [51–57]. Although global Rac1 inactivation in mice results in defects far preceding vascular development, endothelial-specific ablation of Rac1 results in embryonic lethality at slightly later stages with severe defects in angiogenic sprouting and remodeling of secondary vessels [58, 59]. Genetic inactivation of Rac1 in cultured ECs reinforced the requirement for Rac1 in adhesion, migration, and tube assembly [59]. Similarly, global ablation of Cdc42 also results in early developmental abnormalities prior to vessel development [60]; however, mouse embryonic stem cells devoid of Cdc42 fail to assemble into vascular networks during embryoid body differentiation due to impaired directional migration [61]. Very recently, an endothelial-specific Cdc42-null mouse has been generated and displays severe defects in overall vasculogenesis likely due to increased EC apoptosis [62].
Rac1 and Cdc42 have been strongly implicated in promoting formation of epithelial tubes and therefore, have recently been the targets of study during vascular tubulogenesis [63–65]. Indeed, inhibiting Rac1 or Cdc42 activity or expression in HUVECs suspended in collagen matrices prevents cell vacuolation and subsequent lumen formation [31, 66]. Furthermore, signaling downstream of Rac1 and Cdc42 that is associated with polarity (Par complex) and cytoskeletal dynamics (Pak2/Pak4) is also required for intracellular lumen formation in collagen matrices [31, 67, 68]. In vivo, Cdc42 localizes to intracellular vacuolar structures in zebrafish ISVs; however, studies to examine whether depleting Cdc42 in these vessels hinders lumen formation have yet to be performed [16]. In a mouse skin model of angiogenesis, retroviral delivery of VEGF and a dominant negative form of Cdc42 decreased the size and number of perfusable vessels whereas the constitutively active form promoted formation of lumens, thereby supporting a positive role for Cdc42 in lumen generation [56]. Rac1 has also been linked to in vivo lumen formation through its interaction with cerebral cavernous malformation-1 (CCM1) protein. In CCM1-depleted zebrafish embryos, although lumens of major vessels form normally, the lumens of the smaller ISVs fail to form, most likely because intracellular vacuoles are not generated [69]. In addition, depleting CCM1 in HUVECs inhibits Rac1 activity, implying that GTPase signaling downstream of CCM1 is necessary for lumen morphogenesis in microvessels. Taken together, these studies support a function for Rac1 and Cdc42 in lumen generation, additionally potentiating the hypothesis that intracellular lumen formation may indeed be a mechanism by which small-caliber vessels open and expand.
RhoA activity must be tightly controlled during lumen formation
The major physiological outputs of RhoA-mediated signaling in various cell types include the formation of focal adhesions between cells and their underlying substrates as well as formation of stress fibers that promote actomyosin contraction [70, 71]. The role for RhoA in lumen morphogenesis has been controversial. Early studies showed that disrupted RhoA function had no effect on intracellular vacuole formation in collagen matrices [31, 66]. However, later studies demonstrated that disruption of microtubules using chemical agents, an event known to activate RhoA, results in the collapse of lumens in three-dimensional assays. This is prevented when RhoA activity is inhibited, implying that increased RhoA activity in fact antagonizes the stability of tubular networks [72]. Furthermore, Xu et al. recently showed that negative regulation of RhoA signaling via interaction between Ras-interacting protein 1 (RASIP1) and the GTPase activating protein (GAP) Arhgap29 is required for lumen formation [73]. In the absence of either interacting protein, RhoA activity increases but Rac1 and Cdc42 levels are decreased, in agreement with studies highlighting the requirement for the latter two GTPases in lumen formation. Consequently, cultured and mouse embryonic ECs are unable to maintain adhesion to the underlying ECM and display polarity defects with improper localization of apical and basal markers [73]. These data strongly suggest that de-regulation of RhoA activity prevents proper cell behaviors associated with lumen formation.
However, results from a number of studies conflict with the hypothesis that RhoA suppresses lumen formation and instead, propose a positive role for RhoA during this process. Introduction of VEGF and a dominant negative form of RhoA in mouse skin resulted in fewer new blood vessels, and of vessels that did form, most did not contain lumens [74]. Further evidence highlighting a requirement for RhoA signaling was demonstrated in the mouse DA, where normal lumen formation proceeds when F-actin and non-muscle myosin II first localize to the apical surface between the two opposing ECs [17]. In the presence of a Rho kinase (ROCK) inhibitor, F-actin correctly positions at the cell-cell contact; however, myosin II fails to colocalize with F-actin, thus resulting in defective lumen formation. These results indicate that RhoA downstream signaling through ROCK is required for the proper EC shape changes that facilitate opening of the luminal compartment.
More recently, a study demonstrated that RhoA lies downstream of a transcriptional cascade responsible for promoting proper EC adhesion, shape, and proliferation to facilitate vessel assembly [75, 76]. The transcription factor CASTOR (CASZ1), previously implicated in cardiac development [77] and genetically linked to hypertension and high blood pressure [78], and its direct target Epidermal Growth Factor Like-Domain 7 (Egfl7) are required for vessel branching and lumen formation in Xenopus embryos [76]. EGFL7 is an ECM-associated protein exclusively secreted by ECs during embryonic development that has been demonstrated to play a role in vessel morphogenesis [79, 80]. In CASZ1 and EGFL7-depleted HUVECs, RhoA expression levels, and consequently activity, are significantly diminished, resulting in reduced stress fiber and focal adhesion formation associated with contractility defects and loss of adhesion between cells and their underlying substrate. These defects can be rescued by reintroduction of EGFL7 strongly suggesting that the CASZ1/Egfl7 transcriptional hierarchy is required for proper expression of RhoA and the cellular outputs associated with its activation (Fig. 4) [76]. Therefore, these results would favor a model by which RhoA is necessary for eliciting the proper EC behaviors (i.e. cell shape and adhesion) for vascular morphogenesis to proceed. Whether reduced RhoA levels explain the impaired lumen formation in CASZ1 and EGFL7-depleted Xenopus embryos remains to be established. However, because these small GTPases likely have roles in numerous processes during vascular development, this is a situation where small animal models can be particularly valuable. Pharmacological inhibition or the use of caged morpholinos to inhibit GTPase activity in specific tissues during a tightly controlled temporal window can be useful in teasing apart these diverse roles [81–83]. Nonetheless, it is evident that too much or too little GTPase signaling, particularly RhoA, can be detrimental to vascular morphogenesis.
Figure 4.
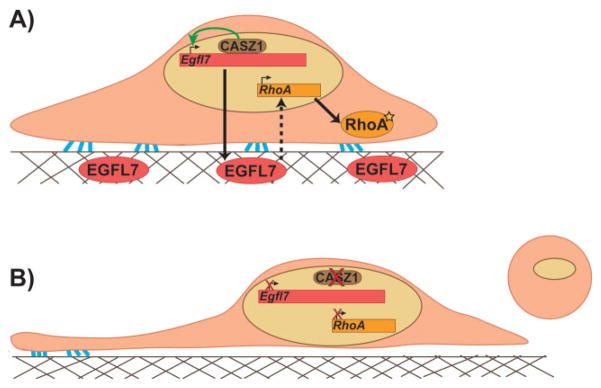
RhoA lies downstream of the CASZ1/Egfl7 transcriptional network. A: The zinc-finger transcription factor CASZ1 binds to and activates expression of Egfl7 in ECs (top panel). EGFL7 is then secreted to the ECM and by a yet unknown mechanism, promotes RhoA expression and subsequent GTPase activity (star) leading to proper EC shape and adhesion to the substrate (as indicated by blue lines). B: In the absence of CASZ1 (bottom panel), transcriptional regulation of Egfl7 is dysregulated and RhoA expression is diminished resulting in thin, elongated cells that no longer maintain adhesion to the substrate and eventually round up and detach.
Endothelial cells enclose around a central lumen via a novel mechanism
The molecular players and pathways discussed above have been deciphered based on two primary mechanisms of lumen morphogenesis: intracellular lumen formation via vacuole coalescence or membrane invagination and the formation of extracellular lumens via cell-cell separation. Nonetheless, characterization of these mechanisms has been confined to limited, albeit informative, vascular contexts, such as cultured ECs suspended in ECM matrices, the mouse DA, and zebrafish ISVs. Recent evidence has elucidated a new mechanism by which lumens form. Helker et al. demonstrated that the zebrafish common cardinal veins (CCVs), which bifurcate from the major posterior cardinal vein, develop a central compartment via a novel lumen ensheathment mechanism [84]. In this model, specified angioblasts destined to become ECs of the CCV are first positioned in a monolayer on top of the yolk syncytial layer (YSL). These cells then detach from the YSL and migrate to the epidermal side, joining together and shaping a new lumen-containing tube located between the YSL and the epidermis, prior to the commencement of circulation. The cells continue to migrate as a sheet to extend the tube and connect it to the heart inflow tract. Interestingly, although VE-cadherin was shown to be required for detachment and migration of ECs during this process, establishment of apicobasal polarity does not appear to be a prerequisite for lumen formation, based on the absence of the apical marker PODXL2. Because the ECs are not initially in contact with each other during the detachment and ensheathment steps, perhaps defining the apical surface between adjacent cells is superfluous. However, it would be interesting to determine whether CCVs form properly in Moesin-depleted fish or other polarity mutants.
Conclusions and Implications
With the amount of heterogeneity that occurs in different vascular beds throughout embryonic and neonatal development, it would not be surprising to identify additional mechanisms of lumen formation. The assembly of the vasculature through distinct processes such as de novo EC coalescence into cords, angiogenesis, anastomosis, or in response to mechanical forces such as blood flow, may directly influence how the lumen is generated, expanded, and maintained. With advances in imaging technology and more detailed understanding of the spatial and temporal requirements of critical molecules during vascular morphogenesis, the discovery of novel lumen formation mechanisms or events that remodel the vessel architecture is very likely (Table 1). In addition to examining how lumens of embryonic vessels develop, it would be interesting to investigate how lumens are re-established postnatally, such as in neoangiogenesis after injury or during vascular regeneration in the heart following myocardial ischemia. Furthermore, the need for effective cancer therapies continues to sustain drug development industries, and trials testing the efficacy of anti-angiogenic therapies in controlling tumor vasculature growth are ongoing [85]. Although the majority of current treatments are aimed at inhibiting vessel growth and expansion in order to starve tumors of oxygen and induce shrinkage, a seemingly contradictory idea know as “vascular normalization” has emerged [86]. The tumor vasculature is highly abnormal; vessels are tortuous, leaky, uneven, and disorganized. Inducing tumor vessels and ECs to become more “normal” by stabilizing junctions and recruiting mural cells to provide cohesive coverage of EC-lined tubes may lead to improved perfusion of drugs and soluble factors directly to the tumor site as well as reduce the chance of metastasis. Obviously, these outcomes are concomitant with the presence of an impermeable, stable lumen. Thus, a greater understanding of lumen morphogenesis, particularly the molecular players involved, may contribute to new approaches for normalizing tumor vessels.
Abbreviations
- BMP
bone morphogenetic protein
- CCV
common cardinal vein
- DA
dorsal aorta
- DLAV
dorsal longitudinal anastomosing vessel
- EC
endothelial cell
- ECM
extracellular matrix
- FGF
fibroblast growth factor
- HUVEC
human umbilical vein endothelial cell
- ISV
intersegmental vessels
- PLA
palatocerebral artery
- VEGF
vascular endothelial growth factor
- YSL
yolk syncytial layer
References
- 1.Patel-Hett S, D’Amore PA. Signal transduction in vasculogenesis and developmental angiogenesis. Int J Dev Biol. 2011;55:353–63. doi: 10.1387/ijdb.103213sp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Swift MR, Weinstein BM. Arterial-venous specification during development. Circ Res. 2009;104:576–88. doi: 10.1161/CIRCRESAHA.108.188805. [DOI] [PubMed] [Google Scholar]
- 3.Djonov V, Schmid M, Tschanz SA, Burri PH. Intussusceptive angiogenesis: its role in embryonic vascular network formation. Circ Res. 2000;86:286–92. doi: 10.1161/01.res.86.3.286. [DOI] [PubMed] [Google Scholar]
- 4.Eilken HM, Adams RH. Dynamics of endothelial cell behavior in sprouting angiogenesis. Curr Opin Cell Biol. 2010;22:617–25. doi: 10.1016/j.ceb.2010.08.010. [DOI] [PubMed] [Google Scholar]
- 5.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. 2011;146:873–87. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 6.Chappell JC, Bautch VL. Vascular development: genetic mechanisms and links to vascular disease. Curr Top Dev Biol. 2010;90:43–72. doi: 10.1016/S0070-2153(10)90002-1. [DOI] [PubMed] [Google Scholar]
- 7.Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. 2011;473:298–307. doi: 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Said SS, Pickering JG, Mequanint K. Advances in growth factor delivery for therapeutic angiogenesis. J Vasc Res. 2013;50:35–51. doi: 10.1159/000345108. [DOI] [PubMed] [Google Scholar]
- 9.Nakatsu MN, Sainson RC, Aoto JN, Taylor KL, et al. Angiogenic sprouting and capillary lumen formation modeled by human umbilical vein endothelial cells (HUVEC) in fibrin gels: the role of fibroblasts and Angiopoietin-1. Microvasc Res. 2003;66:102–12. doi: 10.1016/s0026-2862(03)00045-1. [DOI] [PubMed] [Google Scholar]
- 10.Davis GE, Black SM, Bayless KJ. Capillary morphogenesis during human endothelial cell invasion of three-dimensional collagen matrices. In Vitro Cell Dev Biol Anim. 2000;36:513–9. doi: 10.1290/1071-2690(2000)036<0513:CMDHEC>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 11.Nehls V, Drenckhahn D. A novel, microcarrier-based in vitro assay for rapid and reliable quantification of three-dimensional cell migration and angiogenesis. Microvasc Res. 1995;50:311–22. doi: 10.1006/mvre.1995.1061. [DOI] [PubMed] [Google Scholar]
- 12.Kleinman HK, Martin GR. Matrigel: basement membrane matrix with biological activity. Semin Cancer Biol. 2005;15:378–86. doi: 10.1016/j.semcancer.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 13.Davis GE, Bayless KJ. An integrin and Rho GTPase-dependent pinocytic vacuole mechanism controls capillary lumen formation in collagen and fibrin matrices. Microcirculation. 2003;10:27–44. doi: 10.1038/sj.mn.7800175. [DOI] [PubMed] [Google Scholar]
- 14.Davis GE, Camarillo CW. An alpha 2 beta 1 integrin-dependent pinocytic mechanism involving intracellular vacuole formation and coalescence regulates capillary lumen and tube formation in three-dimensional collagen matrix. Exp Cell Res. 1996;224:39–51. doi: 10.1006/excr.1996.0109. [DOI] [PubMed] [Google Scholar]
- 15.Bayless KJ, Salazar R, Davis GE. RGD-dependent vacuolation and lumen formation observed during endothelial cell morphogenesis in three-dimensional fibrin matrices involves the alpha(v)beta(3) and alpha(5)beta(1) integrins. Am J Pathol. 2000;156:1673–83. doi: 10.1016/s0002-9440(10)65038-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kamei M, Saunders WB, Bayless KJ, Dye L, et al. Endothelial tubes assemble from intracellular vacuoles in vivo. Nature. 2006;442:453–6. doi: 10.1038/nature04923. [DOI] [PubMed] [Google Scholar]
- 17.Strilic B, Kucera T, Eglinger J, Hughes MR, et al. The molecular basis of vascular lumen formation in the developing mouse aorta. Dev Cell. 2009;17:505–15. doi: 10.1016/j.devcel.2009.08.011. [DOI] [PubMed] [Google Scholar]
- 18.Blum Y, Belting HG, Ellertsdottir E, Herwig L, et al. Complex cell rearrangements during intersegmental vessel sprouting and vessel fusion in the zebrafish embryo. Dev Biol. 2008;316:312–22. doi: 10.1016/j.ydbio.2008.01.038. [DOI] [PubMed] [Google Scholar]
- 19.Wang Y, Kaiser MS, Larson JD, Nasevicius A, et al. Moesin1 and Ve-cadherin are required in endothelial cells during in vivo tubulogenesis. Development. 2010;137:3119–28. doi: 10.1242/dev.048785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoshida Y, Yamada M, Wakabayashi K, Ikuta F, et al. Endothelial basement membrane and seamless-type endothelium in the repair process of cerebral infarction in rats. Virchows Arch A Pathol Anat Histopathol. 1989;414:385–92. doi: 10.1007/BF00718621. [DOI] [PubMed] [Google Scholar]
- 21.Herwig L, Blum Y, Krudewig A, Ellertsdottir E, et al. Distinct cellular mechanisms of blood vessel fusion in the zebrafish embryo. Curr Biol. 2011;21:1942–8. doi: 10.1016/j.cub.2011.10.016. [DOI] [PubMed] [Google Scholar]
- 22.Lenard A, Ellertsdottir E, Herwig L, Krudewig A, et al. In Vivo analysis reveals a highly stereotypic morphogenetic pathway of vascular anastomosis. Dev Cell. 2013;25:492–506. doi: 10.1016/j.devcel.2013.05.010. [DOI] [PubMed] [Google Scholar]
- 23.Lizama CO, Zovein AC. Polarizing pathways: balancing endothelial polarity, permeability, and lumen formation. Exp Cell Res. 2013;319:1247–54. doi: 10.1016/j.yexcr.2013.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodriguez-Fraticelli AE, Galvez-Santisteban M, Martin-Belmonte F. Divide and polarize: recent advances in the molecular mechanism regulating epithelial tubulogenesis. Curr Opin Cell Biol. 2011;23:638–46. doi: 10.1016/j.ceb.2011.07.002. [DOI] [PubMed] [Google Scholar]
- 25.Nielsen JS, McNagny KM. Novel functions of the CD34 family. J Cell Sci. 2008;121:3683–92. doi: 10.1242/jcs.037507. [DOI] [PubMed] [Google Scholar]
- 26.Lampugnani MG, Orsenigo F, Rudini N, Maddaluno L, et al. CCM1 regulates vascular-lumen organization by inducing endothelial polarity. J Cell Sci. 2010;123:1073–80. doi: 10.1242/jcs.059329. [DOI] [PubMed] [Google Scholar]
- 27.Strilic B, Eglinger J, Krieg M, Zeeb M, et al. Electrostatic cell-surface repulsion initiates lumen formation in developing blood vessels. Curr Biol. 2010;20:2003–9. doi: 10.1016/j.cub.2010.09.061. [DOI] [PubMed] [Google Scholar]
- 28.Fehon RG, McClatchey AI, Bretscher A. Organizing the cell cortex: the role of ERM proteins. Nat Rev Mol Cell Biol. 2010;11:276–87. doi: 10.1038/nrm2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Speck O, Hughes SC, Noren NK, Kulikauskas RM, et al. Moesin functions antagonistically to the Rho pathway to maintain epithelial integrity. Nature. 2003;421:83–7. doi: 10.1038/nature01295. [DOI] [PubMed] [Google Scholar]
- 30.Joberty G, Petersen C, Gao L, Macara IG. The cell-polarity protein Par6 links Par3 and atypical protein kinase C to Cdc42. Nat Cell Biol. 2000;2:531–9. doi: 10.1038/35019573. [DOI] [PubMed] [Google Scholar]
- 31.Koh W, Mahan RD, Davis GE. Cdc42- and Rac1-mediated endothelial lumen formation requires Pak2, Pak4 and Par3, and PKC-dependent signaling. J Cell Sci. 2008;121:989–1001. doi: 10.1242/jcs.020693. [DOI] [PubMed] [Google Scholar]
- 32.Hynes RO. Cell-matrix adhesion in vascular development. J Thromb Haemost. 2007;5:32–40. doi: 10.1111/j.1538-7836.2007.02569.x. [DOI] [PubMed] [Google Scholar]
- 33.Kucera T, Strilic B, Regener K, Schubert M, et al. Ancestral vascular lumen formation via basal cell surfaces. PLoS One. 2009;4:e4132. doi: 10.1371/journal.pone.0004132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Strilic B, Kucera T, Lammert E. Formation of cardiovascular tubes in invertebrates and vertebrates. Cell Mol Life Sci. 2010;67:3209–18. doi: 10.1007/s00018-010-0400-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lampugnani MG. Endothelial cell-to-cell junctions: adhesion and signaling in physiology and pathology. Cold Spring Harb Perspect Med. 2012;2:a006528. doi: 10.1101/cshperspect.a006528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gory-Faure S, Prandini MH, Pointu H, Roullot V, et al. Role of vascular endothelial-cadherin in vascular morphogenesis. Development. 1999;126:2093–102. doi: 10.1242/dev.126.10.2093. [DOI] [PubMed] [Google Scholar]
- 37.Carmeliet P, Lampugnani MG, Moons L, Breviario F, et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98:147–57. doi: 10.1016/s0092-8674(00)81010-7. [DOI] [PubMed] [Google Scholar]
- 38.Montero-Balaguer M, Swirsding K, Orsenigo F, Cotelli F, et al. Stable vascular connections and remodeling require full expression of VE-cadherin in zebrafish embryos. PLoS One. 2009;4:e5772. doi: 10.1371/journal.pone.0005772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dejana E, Orsenigo F, Lampugnani MG. The role of adherens junctions and VE-cadherin in the control of vascular permeability. J Cell Sci. 2008;121:2115–22. doi: 10.1242/jcs.017897. [DOI] [PubMed] [Google Scholar]
- 40.Dejana E, Vestweber D. The role of VE-cadherin in vascular morphogenesis and permeability control. Prog Mol Biol Transl Sci. 2013;116:119–44. doi: 10.1016/B978-0-12-394311-8.00006-6. [DOI] [PubMed] [Google Scholar]
- 41.Iden S, Rehder D, August B, Suzuki A, et al. A distinct PAR complex associates physically with VE-cadherin in vertebrate endothelial cells. EMBO Rep. 2006;7:1239–46. doi: 10.1038/sj.embor.7400819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parsons JT, Horwitz AR, Schwartz MA. Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol. 2010;11:633–43. doi: 10.1038/nrm2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–79. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- 44.Karlsson R, Pedersen ED, Wang Z, Brakebusch C. Rho GTPase function in tumorigenesis. Biochim Biophys Acta. 2009;1796:91–8. doi: 10.1016/j.bbcan.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 45.Nobes CD, Hall A. Rho, rac and cdc42 GTPases: regulators of actin structures, cell adhesion and motility. Biochem Soc Trans. 1995;23:456–9. doi: 10.1042/bst0230456. [DOI] [PubMed] [Google Scholar]
- 46.Nobes CD, Hall A. Rho, rac, and cdc42 GTPases regulate the assembly of multimolecular focal complexes associated with actin stress fibers, lamellipodia, and filopodia. Cell. 1995;81:53–62. doi: 10.1016/0092-8674(95)90370-4. [DOI] [PubMed] [Google Scholar]
- 47.Maddox AS, Burridge K. RhoA is required for cortical retraction and rigidity during mitotic cell rounding. J Cell Biol. 2003;160:255–65. doi: 10.1083/jcb.200207130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Olson MF, Ashworth A, Hall A. An essential role for Rho, Rac, and Cdc42 GTPases in cell cycle progression through G1. Science. 1995;269:1270–2. doi: 10.1126/science.7652575. [DOI] [PubMed] [Google Scholar]
- 49.Ridley AJ, Paterson HF, Johnston CL, Diekmann D, et al. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401–10. doi: 10.1016/0092-8674(92)90164-8. [DOI] [PubMed] [Google Scholar]
- 50.Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006;16:522–9. doi: 10.1016/j.tcb.2006.08.006. [DOI] [PubMed] [Google Scholar]
- 51.Beckers CM, van Hinsbergh VW, van Nieuw Amerongen GP. Driving Rho GTPase activity in endothelial cells regulates barrier integrity. Thromb Haemost. 2010;103:40–55. doi: 10.1160/TH09-06-0403. [DOI] [PubMed] [Google Scholar]
- 52.Broman MT, Kouklis P, Gao X, Ramchandran R, et al. Cdc42 regulates adherens junction stability and endothelial permeability by inducing alpha-catenin interaction with the vascular endothelial cadherin complex. Circ Res. 2006;98:73–80. doi: 10.1161/01.RES.0000198387.44395.e9. [DOI] [PubMed] [Google Scholar]
- 53.Spindler V, Schlegel N, Waschke J. Role of GTPases in control of microvascular permeability. Cardiovasc Res. 2010;87:243–53. doi: 10.1093/cvr/cvq086. [DOI] [PubMed] [Google Scholar]
- 54.Wojciak-Stothard B, Potempa S, Eichholtz T, Ridley AJ. Rho and Rac but not Cdc42 regulate endothelial cell permeability. J Cell Sci. 2001;114:1343–55. doi: 10.1242/jcs.114.7.1343. [DOI] [PubMed] [Google Scholar]
- 55.Hoang MV, Nagy JA, Senger DR. Active Rac1 improves pathologic VEGF neovessel architecture and reduces vascular leak: mechanistic similarities with angiopoietin-1. Blood. 2011;117:1751–60. doi: 10.1182/blood-2010-05-286831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hoang MV, Nagy JA, Senger DR. Cdc42-mediated inhibition of GSK-3 beta improves angio-architecture and lumen formation during VEGF-driven pathological angiogenesis. Microvascular Research. 2011;81:34–43. doi: 10.1016/j.mvr.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tzima E, Del Pozo MA, Kiosses WB, Mohamed SA, et al. Activation of Rac1 by shear stress in endothelial cells mediates both cytoskeletal reorganization and effects on gene expression. EMBO J. 2002;21:6791–800. doi: 10.1093/emboj/cdf688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sugihara K, Nakatsuji N, Nakamura K, Nakao K, et al. Rac1 is required for the formation of three germ layers during gastrulation. Oncogene. 1998;17:3427–33. doi: 10.1038/sj.onc.1202595. [DOI] [PubMed] [Google Scholar]
- 59.Tan W, Palmby TR, Gavard J, Amornphimoltham P, et al. An essential role for Rac1 in endothelial cell function and vascular development. FASEB J. 2008;22:1829–38. doi: 10.1096/fj.07-096438. [DOI] [PubMed] [Google Scholar]
- 60.Chen F, Ma L, Parrini MC, Mao X, et al. Cdc42 is required for PIP(2)-induced actin polymerization and early development but not for cell viability. Curr Biol. 2000;10:758–65. doi: 10.1016/s0960-9822(00)00571-6. [DOI] [PubMed] [Google Scholar]
- 61.Qi Y, Liu J, Wu X, Brakebusch C, et al. Cdc42 controls vascular network assembly through protein kinase Ciota during embryonic vasculogenesis. Arterioscler Thromb Vasc Biol. 2011;31:1861–70. doi: 10.1161/ATVBAHA.111.230144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jin Y, Liu Y, Lin Q, Li J, et al. Deletion of Cdc42 enhances ADAM17-mediated vascular endothelial growth factor receptor 2 shedding and impairs vascular endothelial cell survival and vasculogenesis. Mol Cell Biol. 2013;33:4181–97. doi: 10.1128/MCB.00650-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jaffe AB, Kaji N, Durgan J, Hall A. Cdc42 controls spindle orientation to position the apical surface during epithelial morphogenesis. J Cell Biol. 2008;183:625–33. doi: 10.1083/jcb.200807121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu KD, Datta A, Yu W, Brakeman PR, et al. Rac1 is required for reorientation of polarity and lumen formation through a PI 3-kinase-dependent pathway. Am J Physiol Renal Physiol. 2007;293:F1633–40. doi: 10.1152/ajprenal.00053.2007. [DOI] [PubMed] [Google Scholar]
- 65.Myllymaki SM, Teravainen TP, Manninen A. Two distinct integrin-mediated mechanisms contribute to apical lumen formation in epithelial cells. PLoS One. 2011;6:e19453. doi: 10.1371/journal.pone.0019453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bayless KJ, Davis GE. The Cdc42 and Rac1 GTPases are required for capillary lumen formation in three-dimensional extracellular matrices. J Cell Sci. 2002;115:1123–36. doi: 10.1242/jcs.115.6.1123. [DOI] [PubMed] [Google Scholar]
- 67.Koh W, Sachidanandam K, Stratman AN, Sacharidou A, et al. Formation of endothelial lumens requires a coordinated PKCepsilon-, Src-, Pak- and Raf-kinase-dependent signaling cascade downstream of Cdc42 activation. J Cell Sci. 2009;122:1812–22. doi: 10.1242/jcs.045799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sacharidou A, Koh W, Stratman AN, Mayo AM, et al. Endothelial lumen signaling complexes control 3D matrix-specific tubulogenesis through interdependent Cdc42- and MT1-MMP-mediated events. Blood. 2010;115:5259–69. doi: 10.1182/blood-2009-11-252692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu H, Rigamonti D, Badr A, Zhang J. Ccm1 regulates microvascular morphogenesis during angiogenesis. J Vasc Res. 2011;48:130–40. doi: 10.1159/000316851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389–99. doi: 10.1016/0092-8674(92)90163-7. [DOI] [PubMed] [Google Scholar]
- 71.Chrzanowska-Wodnicka M, Burridge K. Rho-stimulated contractility drives the formation of stress fibers and focal adhesions. J Cell Biol. 1996;133:1403–15. doi: 10.1083/jcb.133.6.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bayless KJ, Davis GE. Microtubule depolymerization rapidly collapses capillary tube networks in vitro and angiogenic vessels in vivo through the small GTPase Rho. J Biol Chem. 2004;279:11686–95. doi: 10.1074/jbc.M308373200. [DOI] [PubMed] [Google Scholar]
- 73.Xu K, Sacharidou A, Fu S, Chong DC, et al. Blood Vessel Tubulogenesis Requires Rasip1 Regulation of GTPase Signaling. Dev Cell. 2011;20:526–39. doi: 10.1016/j.devcel.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hoang MV, Whelan MC, Senger DR. Rho activity critically and selectively regulates endothelial cell organization during angiogenesis. Proc Natl Acad Sci USA. 2004;101:1874–9. doi: 10.1073/pnas.0308525100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Charpentier MS, Dorr KM, Conlon FL. Transcriptional regulation of blood vessel formation: The role of the CASZ1/Egfl7/RhoA pathway. Cell Cycle. 2013;12:2165–6. doi: 10.4161/cc.25539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Charpentier MS, Christine KS, Amin NM, Dorr KM, et al. CASZ1 promotes vascular assembly and morphogenesis through the direct regulation of an EGFL7/RhoA-mediated pathway. Dev Cell. 2013;25:132–43. doi: 10.1016/j.devcel.2013.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Christine KS, Conlon FL. Vertebrate CASTOR is required for differentiation of cardiac precursor cells at the ventral midline. Dev Cell. 2008;14:616–23. doi: 10.1016/j.devcel.2008.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Takeuchi F, Isono M, Katsuya T, Yamamoto K, et al. Blood pressure and hypertension are associated with 7 loci in the Japanese population. Circulation. 2010;121:2302–9. doi: 10.1161/CIRCULATIONAHA.109.904664. [DOI] [PubMed] [Google Scholar]
- 79.Parker LH, Schmidt M, Jin SW, Gray AM, et al. The endothelial-cell-derived secreted factor Egfl7 regulates vascular tube formation. Nature. 2004;428:754–8. doi: 10.1038/nature02416. [DOI] [PubMed] [Google Scholar]
- 80.Fitch MJ, Campagnolo L, Kuhnert F, Stuhlmann H. Egfl7, a novel epidermal growth factor-domain gene expressed in endothelial cells. Dev Dyn. 2004;230:316–24. doi: 10.1002/dvdy.20063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Langdon Y, Tandon P, Paden E, Duddy J, et al. SHP-2 acts via ROCK to regulate the cardiac actin cytoskeleton. Development. 2012;139:948–57. doi: 10.1242/dev.067579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Deiters A, Garner RA, Lusic H, Govan JM, et al. Photocaged morpholino oligomers for the light-regulation of gene function in zebrafish and Xenopus embryos. J Am Chem Soc. 2010;132:15644–50. doi: 10.1021/ja1053863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morckel AR, Lusic H, Farzana L, Yoder JA, et al. A photo-activatable small-molecule inhibitor for light-controlled spatiotemporal regulation of Rho kinase in live embryos. Development. 2012;139:437–42. doi: 10.1242/dev.072165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Helker CS, Schuermann A, Karpanen T, Zeuschner D, et al. The zebrafish common cardinal veins develop by a novel mechanism: lumen ensheathment. Development. 2013;140:2776–86. doi: 10.1242/dev.091876. [DOI] [PubMed] [Google Scholar]
- 85.Meadows KL, Hurwitz HI. Anti-VEGF therapies in the clinic. Cold Spring Harb Perspect Med. 2012;2:a006577. doi: 10.1101/cshperspect.a006577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Carmeliet P, Jain RK. Principles and mechanisms of vessel normalization for cancer and other angiogenic diseases. Nat Rev Drug Discov. 2011;10:417–27. doi: 10.1038/nrd3455. [DOI] [PubMed] [Google Scholar]
- 87.Zovein AC, Luque A, Turlo KA, Hofmann JJ, et al. Beta1 integrin establishes endothelial cell polarity and arteriolar lumen formation via a Par3-dependent mechanism. Dev Cell. 2010;18:39–51. doi: 10.1016/j.devcel.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]