

A new idea is the most quickly acting antigen known to science.
—Wilfred Trotter
During the past few years, several distinguished senior colleagues who visited our department and inquired about the status of endogenous ouabain (EO) have asked, “How come I didn't know about these [published] data?” And, “Why isn't endogenous ouabain more widely accepted?” I usually simply shrug and express disappointment that EO is not yet a “mainstream hormone,” even though the seminal reports published more than two decades ago in leading journals (cited below) have been confirmed and supplemented (6, 7, 22). This has now happened often enough to give me pause.
Fierce competition for resources undoubtedly slowed the evolution of the EO story, with its several surprises and multiple components, making it difficult to sustain attention outside the immediate field. Research has also been stymied because the biosynthetic pathway has been only partly elucidated, and there are no commercial EO assays or clinically approved antagonists.
Upon further reflection, however, I have concluded that there is at least a partial explanation based on well-known human behavior to which we, as scientists, are particularly prone (28). We are all very wary of new, paradigm-shifting ideas that don't fit our preconceived notions. By profession, we are skeptics and we demand proof, but we are also often quick to express negative opinions without carefully assessing the data. We tend to ignore ideas and data that don't fit our preconceptions; in fact, we often don't even read (or digest) articles that fall outside our comfort zone. “Mea culpa!” As Darwin noted, established investigators are usually set in their ways; new ideas are for newcomers (9). And we are in good company (although this isn't a rationale): recall Einstein's famous put down of Heisenberg's uncertainty principle, “He [God] does not throw dice” (15).
Below I illustrate the problem with some examples, primarily from my own slow journey of recognition and acceptance of groundbreaking observations about which I was initially very skeptical. And, if I was so suspicious, how could I expect others to rapidly grasp and accept ideas and data that ostensibly don't fit existing dogma? Nevertheless, if the facts are inconsistent with the prevailing paradigm, progress requires a new one. This is “scientific revolution” (28).
In 1991, Hamlyn and colleagues (23) purified an endogenous cardiotonic steroid from human plasma and analytically identified it as ouabain. The following year, Doursout and coworkers (14) and, a year later, Yuan and colleagues (60) demonstrated that prolonged ouabain administration induces hypertension in normal rats. These remarkable observations have been replicated in a number of highly respected laboratories [reviewed in Blaustein et al. (7)]. The early studies were rapidly followed by reports that circulating EO is elevated in many patients with essential hypertension and mineralocorticoid hypertension (52) and in those with congestive heart failure (21). As in any research field, however, a few investigators failed to replicate the original findings, or reported false positives, and some have continued to question the validity of the original reports [e.g., Baecher et al. (2), Doris et al. (11), and Nicholls et al. (40). It is inappropriate to critique those studies here, but see, for example, Manunta et al. (35).] Thus it might seem understandable that individuals working outside the immediate field may be left in a quandary and would therefore simply ignore these data and arguments, but this is not the whole story.
Leenen's 1992 discovery of “brain ouabain,” including its key roles in salt-sensitive hypertension (25) and heart failure (31), was another seminal contribution. Numerous subsequent reports from that laboratory confirmed and extended those results by showing that brain ouabain was a distal component of a hypothalamic chronic renin-angiotensin II (ANG II)-aldosterone-epithelial Na+ channel-brain ouabain pathway (Fig. 1) (7, 18, 30). This slow brain pathway, which is activated by high salt and/or ANG II modulates central sympathoexcitatory neurons (i.e., the acute mechanisms) that are involved in both hypertension and heart failure (16, 29, 30, 43, 62). Note that high dietary salt/salt retention suppresses the peripheral renin-angiotensin-aldosterone system (RAAS) (1, 36), but, paradoxically, activates the brain RAAS (18, 43). The more proximal components of the slow pathway, ANG II and ANG II type 1 receptors, aldosterone and mineralocorticoid receptors, and epithelial Na+ channel (19, 20, 44), are often mentioned in the literature, but brain ouabain has been ignored. Indeed, even though I was aware of it, I, too, long disregarded brain ouabain; it didn't fit my biased, vasculocentric view of how EO works in hypertension. Finally, in 2010, John Hamlyn and I sat down with Frans Leenen and listened to each other's ideas. We agreed that the brain EO data and plasma EO data were both valid and needed to be reconciled. The outcome was the joint proposal of a new, comprehensive view of the pathogenesis of salt- and ANG II-dependent hypertension that involves both brain EO and circulating EO and links the kidneys, brain, and arteries (7). This led to collaboration and to the discovery that the chronic brain pathway regulates the plasma EO level (24).
Fig. 1.
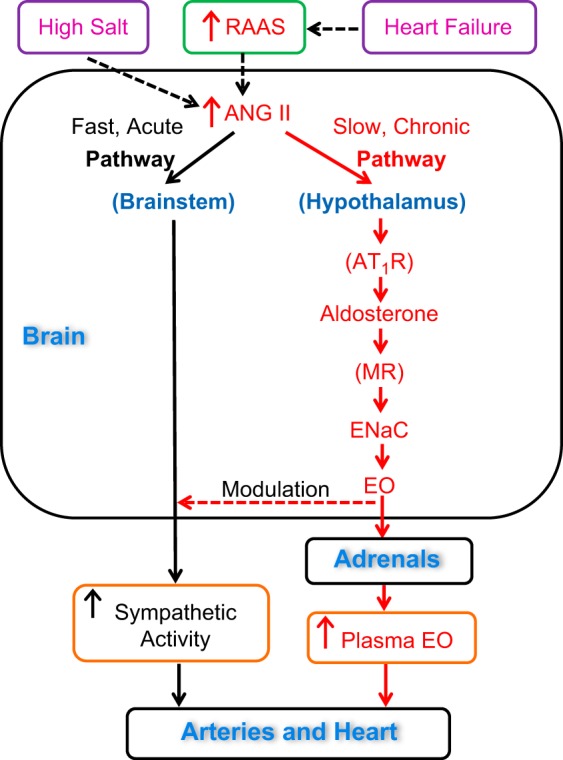
Diagram of the proposed fast (acute) and slow (chronic) brain pathways that regulate the cardiovascular system. They are located in, respectively, the brainstem (rostroventrolateral medulla) and hypothalamus. Both pathways are activated by excess dietary salt and/or increased angiotensin II (ANG II). The peripheral renin-angiotensin-aldosterone system (RAAS) is activated in heart failure; the increased circulating ANG II stimulates both pathways. High salt suppresses the peripheral RAAS but paradoxically activates the brain RAAS (see text for references). Activation of the slow brain pathway by either circulating or brain ANG II modulates the acute pathway and increases circulating endogenous ouabain (EO) by as yet unknown mechanisms. The effects of circulating EO on the arteries and heart are diagrammed in Fig. 2. AT1R, losartan-blockable ANG II type 1 receptors; MR, eplerenone-blockable mineralocorticoid receptors; ENaC, benzamil-blockable epithelial Na+ channels. “Brain EO” is locally synthesized and secreted; most circulating EO apparently comes from the adrenals. See text for further details.
Following the demonstration that ouabain induces hypertension (14, 60), Hamlyn, Manunta, and associates performed another key experiment: they infused digoxin into rats; after all, ouabain and digoxin are both cardiotonic steroids that indistinguishably inhibit Na+ pumps (5, 59). Astonishingly, digoxin did not induce hypertension; in fact, it normalized blood pressure in ouabain-infused rats. In direct contradiction to more than 60 years of pharmacological dogma, digoxin could also behave as an “ouabain antagonist.” These amazing observations appeared as an abstract in 1993 (38); the full paper wasn't published until 2000 (37), largely because of reviewer skepticism, but the results have been confirmed and extended to salt-sensitive hypertension (26, 63). These results were indisputable, but I didn't understand how digoxin and ouabain could have different effects, so I long ignored the digoxin data (55).
The Na+ pump catalytic subunit-α, which contains the ouabain binding site, has four isoforms. All cells express pumps (αβ-dimers) with an α1-isoform and pumps with one other α-isoform (5, 33); cardiac and smooth muscles express α1 and α2 Na+ pumps, but the large majority, ∼75–80%, are α1 (13, 53, 61). Rodent α1 pumps are notoriously resistant to ouabain (EC50 ≈ 100 μM) (5, 41); they cannot be the physiological receptors for EO because plasma EO levels are normally subnanomolar (17, 36, 50). Accordingly, Lingrel and colleagues (12, 13) generated mice with mutant, ouabain-resistant, α2 Na+ pumps. These α2R/R mice do not develop hypertension when injected chronically with ouabain or adrenocorticotropic hormone, whereas wild-type (ouabain sensitive) α2 mice do (12, 13, 34). Mice in which α2 Na+ pumps are selectively knocked out in the cardiovascular system are also resistant to adrenocorticotropic hormone -induced hypertension (50). Additionally, recent studies indicate that either cardiac-specific overexpression (8) or knockout (51) of α2 Na+ pumps attenuates the development of pressure overload-induced cardiac hypertrophy and dysfunction. Clearly, the α2 Na+ pump and its endogenous ligand play a crucial role in at least some forms of hypertension and heart failure: additional remarkable, but largely ignored, data that are complementary to, but independent of, EO measurements.
In 2010, Golovina and colleagues (48) reported that expression of several Ca2+ transporter proteins is markedly increased in arterial smooth muscle (ASM) from rats with ouabain-induced hypertension. The proteins include Na+/Ca2+ exchanger-1 (NCX1); transient receptor potential cation channel, subfamily C, member 6 (TRPC6, component of some receptor-operated channels, ROCs); and sarcoplasmic reticulum Ca2+ ATPase pump-2 (SERCA2). Furthermore, they and others found that NCX1 and several other Ca2+ transporters are overexpressed in ASM in many common hypertension models [reviewed in Blaustein et al. (7) and Pulina et al. (47)]. These proteins are also upregulated in primary cultured normal rodent and human ASM cells incubated with nanomolar ouabain for 72–96 h (32, 48). In contrast, digoxin does not upregulate these proteins either in vivo or in vitro; in fact, digoxin antagonizes the effects of ouabain (Fig. 2) (63), consistent with the aforementioned blood pressure data (26, 37, 38). Ouabain-digoxin antagonism could no longer be overlooked simply because it did not fit the dogma that all cardiotonic steroids act only as Na+ pump inhibitors (5, 56). Also, another key discovery about ouabain that is inconsistent with this conventional wisdom could no longer be disregarded; namely, reports from Askari and colleagues (39, 45) that ouabain binding to Na+ pumps activates a C-Src-dependent protein kinase signaling cascade. In ASM, both in vivo and in vitro, this cascade is activated by ouabain, an effect antagonized by digoxin (Fig. 2) (63). This verifies ouabain-digoxin antagonism [and see Song et al. (55)] and implies that activation of the cascade and the protein upregulation do not depend on Na+ pump inhibition and NCX-mediated enhancement of Ca2+ signaling. Thus the Na+ pump is both an ion transporter and a hormone receptor (Fig. 2). The binding of EO and digoxin have similar short-term, but different long-term, effects.
Fig. 2.

Proposed effects of EO on the arteries (A) and heart (B). The well-documented acute action of EO (inhibition of α2 Na+ pumps), but not its chronic effect (α2 Na+ pump-mediated activation of the C-Src, ERK1/2, MAPK, and protein kinase cascade), is mimicked by digoxin. Sustained elevation of plasma EO leads, apparently via the protein kinase cascade, to increased expression of several arterial Ca2+ transporter proteins. These include Na+/Ca2+ exchanger-1 (NCX1), transient receptor potential cation channel, subfamily C, member 6 (TRPC6; component of receptor-operated channels), and the sarcoplasmic reticulum (SR) Ca2+ ATPase pump-2b (SERCA2b). These chronic effects of EO are blocked by digoxin (26, 37, 63), as indicated; digoxin may either mimic (not shown) or antagonize the acute effect of EO, depending upon the relative concentrations (55). (Note: ouabain-digoxin antagonism has been demonstrated in arteries and neurons, but not yet in heart, as indicated by the question marks in B.) Chronically elevated plasma EO may also increase NCX1 expression in the heart by a C-Src-activated protein kinase cascade (39, 45). Acute Na+ pump inhibition by EO raises the cytosolic Na+ concentration ([Na+]CYT). This reduction in the Na+ electrochemical gradient, in turn, promotes net Ca2+ gain via NCX1, and a rise in both cytosolic and SR calcium concentrations, [Ca2+]CYT and [Ca2+]SR, respectively, and, thus enhances Ca2+ signaling and contraction in both the heart and arteries [acute (green) pathways in A and B]. With increased NCX1 expression due to chronically elevated plasma EO, however, the heart and arteries apparently respond differently. The main role of NCX1 in the heart is to mediate Ca2+ extrusion and promote relaxation during diastole (3), whereas in arteries, its main role seems to be to mediate Ca2+ entry and maintain tone (61). Thus NCX1 upregulation in the heart should decrease contractility (B, chronic pathway), but in arteries it should increase contractility (A, chronic pathway). In other words, the acute and chronic effects of EO should be synergistic in arteries but antagonistic to one another in the heart. See text for further details.
In heart failure, the RAAS is activated (16, 29, 62) and ANG II-stimulated, EO-dependent mechanisms (Figs. 1 and 2) may also contribute to cardiac remodeling. In the heart, ouabain stimulates extracellular matrix formation (27, 49) and activates C-Src (39), and NCX1 overexpression is a common, but unexplained, feature of heart failure that, paradoxically, may impair cardiac contractility (42, 58).
The several aforementioned independent and seminal observations, together, reveal a new axis that links all these factors directly to hypertension (7) and heart failure (see above). The components include (Figs. 1 and 2) a stimulus (ANG II and/or high salt), the ANG II-stimulated central control system (the brain slow neurohumoral regulatory pathway), an hormonal messenger (EO), “biased” EO receptors (α2 Na+ pumps, which exhibit ouabain-digoxin antagonism), an EO-activated transducer (protein kinase cascade), protein kinase-modulated peripheral signal mechanisms (Ca2+ transporters and channels, e.g., NCX1, ROCs), a second messenger (Ca2+), and Ca2+-activated effectors (arterial and cardiac contractile apparatus). Because NCX1 promotes Ca2+ exit in heart (3) but Ca2+ entry in arteries (61), chronically elevated EO, which is pivotal, and upregulated NCX1 should make hearts hypocontractile and arteries hypercontractile (Fig. 2). Thus EO should promote both heart failure and hypertension. This is, of course, easy to see with hindsight but was certainly not anticipated when EO was discovered.
These considerations should provide novel insight into therapeutics. For example, plasma EO was not measured in the most widely cited, long-term trial of the therapeutic effectiveness of digoxin in heart failure (10). Yet, ouabain-digoxin antagonism implies that patients with low- and high-ambient EO are likely to have different outcomes when treated with digoxin. Thus a golden opportunity to predict who might benefit most from digoxin, the primary goal of the trial, was lost. Furthermore, it is now difficult to dismiss the striking correlation between the highest plasma EO levels and the worst morbidity and mortality statistics in patients with heart failure and related cardiovascular diseases (4, 21, 46, 54, 57). Surely, it is time to bring EO, a key neuroendocrine and cardiovascular hormone, in from the cold.
GRANTS
This study was supported by National Heart, Lung, and Blood Institute Grants R01-HL-045215 (to M. P. Blaustein and J. M. Hamlyn) and R01-HL-107555 (to M. P. Blaustein).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
M.P.B. conceived and designed research; analyzed data; interpreted results of experiments; prepared figures; and drafted, edited, revised, and approved final version of manuscript.
ACKNOWLEDGMENTS
I thank J. M. Hamlyn, F. H. H. Leenen, and W. G. Wier for helpful critiques of a preliminary version of this manuscript and H. Song for help with the figures.
REFERENCES
- 1.Anderson DE, Gomez-Sanchez C, Dietz JR. Suppression of plasma renin and aldosterone in stress-salt hypertension in dogs. Am J Physiol Regul Integr Comp Physiol 251: R181–R186, 1986 [DOI] [PubMed] [Google Scholar]
- 2.Baecher S, Kroiss M, Fassnacht M, Vogeser M. No endogenous ouabain is detectable in human plasma by ultra-sensitive UPLC-MS/MS. Clin Chim Acta 431: 87–92, 2014 [DOI] [PubMed] [Google Scholar]
- 3.Bers DM, Despa S. Cardiac myocytes Ca2+ and Na+ regulation in normal and failing hearts. J Pharmacol Sci 100: 315–322, 2006 [DOI] [PubMed] [Google Scholar]
- 4.Bignami E, Casamassima N, Frati E, Lanzani C, Corno L, Alfieri O, Gottlieb S, Simonini M, Shah KB, Mizzi A, Messaggio E, Zangrillo A, Ferrandi M, Ferrari P, Bianchi G, Hamlyn JM, Manunta P. Preoperative endogenous ouabain predicts acute kidney injury in cardiac surgery patients. Crit Care Med 41: 744–755, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol Renal Physiol 275: F633–F650, 1998 [DOI] [PubMed] [Google Scholar]
- 6.Blaustein MP, Hamlyn JM. Signaling mechanisms that link salt retention to hypertension: Endogenous ouabain, the Na+ pump, the Na+/Ca2+ exchanger and TRPC proteins. Biochim Biophys Acta 1082: 1219–1229, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blaustein MP, Leenen FH, Chen L, Golovina VA, Hamlyn JM, Pallone TL, Van Huysse JW, Zhang J, Wier WG. How NaCl raises blood pressure: a new paradigm for the pathogenesis of salt-dependent hypertension. Am J Physiol Heart Circ Physiol 302: H1031–H1049, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Correll RN, Eder P, Burr AR, Despa S, Davis J, Bers DM, Molkentin JD. Overexpression of the Na+/K+ ATPase α2 but not α1 isoform attenuates pathological cardiac hypertrophy and remodeling. Circ Res 114: 249–256, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Darwin C. The Origin of Species by Means of Natural Selection; or, The Preservation of Favored Races in the Struggle for Life and the Descent of Man and Selection in Relation to Sex. New York: Modern Library, 1936, Chapt. XV, p. 368 [Google Scholar]
- 10.Digitalis Investigative Group. The effect of digoxin on mortality and morbidity in patients with heart failure. N Engl J Med 336: 525–533, 1997 [DOI] [PubMed] [Google Scholar]
- 11.Doris PA, Jenkins LA, Stocco DM. Is ouabain an authentic endogenous mammalian substance derived from the adrenal? Hypertension 23: 632–638, 1994 [DOI] [PubMed] [Google Scholar]
- 12.Dostanic-Larson I, Van Huysse JW, Lorenz JN, Lingrel JB. The highly conserved cardiac glycoside binding site of Na,K-ATPase plays a role in blood pressure regulation. Proc Natl Acad Sci USA 102: 15845–15850, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dostanic I, Paul RJ, Lorenz JN, Theriault S, Van Huysse JW, Lingrel JB. The α2-isoform of Na-K-ATPase mediates ouabain-induced hypertension in mice and increased vascular contractility in vitro. Am J Physiol Heart Circ Physiol 288: H477–H485, 2005 [DOI] [PubMed] [Google Scholar]
- 14.Doursout MF, Chelly JE, Liang YY, Buckley JP. The ouabain-dependent Na+-K+ pump and the brain renin-angiotensin system. Clin Exp Hypertens A 14: 393–411, 1992 [DOI] [PubMed] [Google Scholar]
- 15.Einstein A, Born M, Born H. A. Einstein letter to M. Born, 4 Dec. 1926. In: The Born-Einstein Letters: Correspondence Between Albert Einstein and Max and Hedwig Born from 1916 to 1955. New York: Walker, 1971, p. 91 [Google Scholar]
- 16.Felder RB, Francis J, Weiss RM, Zhang ZH, Wei SG, Johnson AK. Neurohumoral regulation in ischemia-induced heart failure. Role of the forebrain. Ann NY Acad Sci 940: 444–453, 2001 [DOI] [PubMed] [Google Scholar]
- 17.Ferrandi M, Manunta P, Balzan S, Hamlyn JM, Bianchi G, Ferrari P. Ouabain-like factor quantification in mammalian tissues and plasma: comparison of two independent assays. Hypertension 30: 886–896, 1997 [DOI] [PubMed] [Google Scholar]
- 18.Gabor A, Leenen FH. Central neuromodulatory pathways regulating sympathetic activity in hypertension. J Appl Physiol 113: 1294–1303, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol Endocrinol Metab 262: E96–E99, 1992 [DOI] [PubMed] [Google Scholar]
- 20.Gomez-Sanchez EP, Gomez-Sanchez CE. Effect of central amiloride infusion on mineralocorticoid hypertension. Am J Physiol Endocrinol Metab 267: E754–E758, 1994 [DOI] [PubMed] [Google Scholar]
- 21.Gottlieb SS, Rogowski AC, Weinberg M, Krichten CM, Hamilton BP, Hamlyn JM. Elevated concentrations of endogenous ouabain in patients with congestive heart failure. Circulation 86: 420–425, 1992 [DOI] [PubMed] [Google Scholar]
- 22.Hamlyn JM, Blaustein MP. Salt sensitivity, endogenous ouabain and hypertension. Curr Opin Nephrol Hypertens 22: 51–58, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hamlyn JM, Blaustein MP, Bova S, DuCharme DW, Harris DW, Mandel F, Mathews WR, Ludens JH. Identification and characterization of a ouabain-like compound from human plasma. Proc Natl Acad Sci USA 88: 6259–6263, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hamlyn JM, Leenen FH, Golovina VA, Linde CI, Gao J, Huang B, Chen L, Li M, Blaustein MP. Central angiotensin II increases circulating endogenous ouabain, arterial myocyte Na/Ca exchanger expression and blood pressure via brain aldosterone and mineralocorticoid receptors. Hypertension 60: e419, 2012 [Google Scholar]
- 25.Huang BS, Harmsen E, Yu H, Leenen FH. Brain ouabain-like activity and the sympathoexcitatory and pressor effects of central sodium in rats. Circ Res 71: 1059–1066, 1992 [DOI] [PubMed] [Google Scholar]
- 26.Huang BS, Kudlac M, Kumarathasan R, Leenen FH. Digoxin prevents ouabain and high salt intake-induced hypertension in rats with sinoaortic denervation. Hypertension 34: 733–738, 1999 [DOI] [PubMed] [Google Scholar]
- 27.Jiang X, Ren YP, Lv ZR. Ouabain induces cardiac remodeling in rats independent of blood pressure. Acta Pharmacol Sin 28: 344–352, 2007 [DOI] [PubMed] [Google Scholar]
- 28.Kuhn TS. The Structure of Scientific Revolutions. Chicago: University of Chicago Press, 1996, p. 144–159 [Google Scholar]
- 29.Leenen FH. Actions of circulating angiotensin ii and aldosterone in the brain contributing to hypertension. Am J Hypertens. 17 Apr 2014. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 30.Leenen FH. The central role of the brain aldosterone-“ouabain” pathway in salt-sensitive hypertension. Biochim Biophys Acta 1802: 1132–1139, 2010 [DOI] [PubMed] [Google Scholar]
- 31.Leenen FH, Huang BS, Yu H, Yuan B. Brain ‘ouabain’ mediates sympathetic hyperactivity in congestive heart failure. Circ Res 77: 993–1000, 1995 [DOI] [PubMed] [Google Scholar]
- 32.Linde CI, Antos LK, Golovina VA, Blaustein MP. Nanomolar ouabain increases NCX1 expression and enhances Ca2+ signaling in human arterial myocytes: a mechanism that links salt to increased vascular resistance? Am J Physiol Heart Circ Physiol 303: H784–H794, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lingrel JB, Orlowski J, Shull MM, Price EM. Molecular genetics of Na,K-ATPase. Prog Nucleic Acid Res Mol Biol 38: 37–89, 1990 [DOI] [PubMed] [Google Scholar]
- 34.Lorenz JN, Loreaux EL, Dostanic-Larson I, Lasko V, Schnetzer JR, Paul RJ, Lingrel JB. ACTH-induced hypertension is dependent on the ouabain-binding site of the α2-Na+-K+-ATPase subunit. Am J Physiol Heart Circ Physiol 295: H273–H280, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Manunta P, Ferrandi M, Bianchi G, Hamlyn JM. Endogenous ouabain in cardiovascular function and disease. J Hypertens 27: 9–18, 2009 [DOI] [PubMed] [Google Scholar]
- 36.Manunta P, Hamilton BP, Hamlyn JM. Salt intake and depletion increase circulating levels of endogenous ouabain in normal men. Am J Physiol Regul Integr Comp Physiol 290: R553–R559, 2006 [DOI] [PubMed] [Google Scholar]
- 37.Manunta P, Hamilton J, Rogowski AC, Hamilton BP, Hamlyn JM. Chronic hypertension induced by ouabain but not digoxin in the rat: antihypertensive effect of digoxin and digitoxin. Hypertens Res 23 Suppl: S77–S85, 2000 [DOI] [PubMed] [Google Scholar]
- 38.Manunta P, Tyzack J, Hamilton BP, Hamlyn JM. Augmentation and antagonism of ouabain-induced hypertension. Hypertension 22: 432, 1993 [Google Scholar]
- 39.Mohammadi K, Kometiani P, Xie Z, Askari A. Role of protein kinase C in the signal pathways that link Na+/K+-ATPase to ERK1/2. J Biol Chem 276: 42050–42056, 2001 [DOI] [PubMed] [Google Scholar]
- 40.Nicholls MG, Lewis LK, Yandle TG, Lord G, McKinnon W, Hilton PJ. Ouabain, a circulating hormone secreted by the adrenals, is pivotal in cardiovascular disease. Fact or fantasy? J Hypertens 27: 3–8, 2009 [DOI] [PubMed] [Google Scholar]
- 41.O'Brien WJ, Lingrel JB, Wallick ET. Ouabain binding kinetics of the rat alpha two and alpha three isoforms of the sodium-potassium adenosine triphosphate. Arch Biochem Biophys 310: 32–39, 1994 [DOI] [PubMed] [Google Scholar]
- 42.O'Rourke B, Kass DA, Tomaselli GF, Kaab S, Tunin R, Marban E. Mechanisms of altered excitation-contraction coupling in canine tachycardia-induced heart failure, I: experimental studies. Circ Res 84: 562–570, 1999 [DOI] [PubMed] [Google Scholar]
- 43.Osborn JW, Fink GD, Sved AF, Toney GM, Raizada MK. Circulating angiotensin II and dietary salt: converging signals for neurogenic hypertension. Curr Hypertens Rep 9: 228–235, 2007 [DOI] [PubMed] [Google Scholar]
- 44.Pare MC, Maltais S, Escher E. The neurogenic origin of hypertension in SHR may be mediated by angiotensin II through a receptor different from AT1 and AT2. Regul Pept 47: 81–86, 1993 [DOI] [PubMed] [Google Scholar]
- 45.Peng M, Huang L, Xie Z, Huang WH, Askari A. Partial inhibition of Na+/K+-ATPase by ouabain induces the Ca2+-dependent expressions of early-response genes in cardiac myocytes. J Biol Chem 271: 10372–10378, 1996 [DOI] [PubMed] [Google Scholar]
- 46.Pitzalis MV, Hamlyn JM, Messaggio E, Iacoviello M, Forleo C, Romito R, de Tommasi E, Rizzon P, Bianchi G, Manunta P. Independent and incremental prognostic value of endogenous ouabain in idiopathic dilated cardiomyopathy. Eur J Heart Fail 8: 179–186, 2006 [DOI] [PubMed] [Google Scholar]
- 47.Pulina MV, Zulian A, Baryshnikov SG, Linde CI, Karashima E, Hamlyn JM, Ferrari P, Blaustein MP, Golovina VA. Cross talk between plasma membrane Na+/Ca2+ exchanger-1 and TRPC/Orai-containing channels: key players in arterial hypertension. Adv Exp Med Biol 961: 365–374, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pulina MV, Zulian A, Berra-Romani R, Beskina O, Mazzocco-Spezzia A, Baryshnikov SG, Papparella I, Hamlyn JM, Blaustein MP, Golovina VA. Upregulation of Na+ and Ca2+ transporters in arterial smooth muscle from ouabain-induced hypertensive rats. Am J Physiol Heart Circ Physiol 298: H263–H274, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Quintas LE, Pierre SV, Liu L, Bai Y, Liu X, Xie ZJ. Alterations of Na+/K+-ATPase function in caveolin-1 knockout cardiac fibroblasts. J Mol Cell Cardiol 49: 525–531, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rindler TN, Dostanic I, Lasko VM, Nieman ML, Neumann JC, Lorenz JN, Lingrel JB. Knockout of the Na,K-ATPase α2-isoform in the cardiovascular system does not alter basal blood pressure but prevents ACTH-induced hypertension. Am J Physiol Heart Circ Physiol 301: H1396–H1404, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rindler TN, Lasko VM, Nieman ML, Okada M, Lorenz JN, Lingrel JB. Knockout of the Na,K-ATPase α2-isoform in cardiac myocytes delays pressure overload-induced cardiac dysfunction. Am J Physiol Heart Circ Physiol 304: H1147–H1158, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rossi G, Manunta P, Hamlyn JM, Pavan E, De Toni R, Semplicini A, Pessina AC. Immunoreactive endogenous ouabain in primary aldosteronism and essential hypertension: relationship with plasma renin, aldosterone and blood pressure levels. J Hypertens 13: 1181–1191, 1995 [DOI] [PubMed] [Google Scholar]
- 53.Shelly DA, He S, Moseley A, Weber C, Stegemeyer M, Lynch RM, Lingrel J, Paul RJ. Na+ pump α2-isoform specifically couples to contractility in vascular smooth muscle: evidence from gene-targeted neonatal mice. Am J Physiol Cell Physiol 286: C813–C820, 2004 [DOI] [PubMed] [Google Scholar]
- 54.Simonini M, Lanzani C, Bignami E, Casamassima N, Frati E, Meroni R, Messaggio E, Alfieri O, Hamlyn J, Body SC, Collard CD, Zangrillo A, Manunta P. A new clinical multivariable model that predicts postoperative acute kidney injury: impact of endogenous ouabain. Nephrol Dial Transplant. 11 Jun 2014. pii: gfu200. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Song H, Karashima E, Hamlyn JM, Blaustein MP. Ouabain-digoxin antagonism in rat arteries and neurones. J Physiol 592: 941–969, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Song H, Thompson SM, Blaustein MP. Nanomolar ouabain augments Ca2+ signalling in rat hippocampal neurones and glia. J Physiol 591: 1671–1689, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stella P, Manunta P, Mallamaci F, Melandri M, Spotti D, Tripepi G, Hamlyn JM, Malatino LS, Bianchi G, Zoccali C. Endogenous ouabain and cardiomyopathy in dialysis patients. J Intern Med 263: 274–280, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Studer R, Reinecke H, Bilger J, Eschenhagen T, Bohm M, Hasenfuss G, Just H, Holtz J, Drexler H. Gene expression of the cardiac Na+-Ca2+ exchanger in end-stage human heart failure. Circ Res 75: 443–453, 1994 [DOI] [PubMed] [Google Scholar]
- 59.Yoda A. Association and dissociation rate constants of the complexes between various cardiac monoglycosides and Na, K-ATPase. Ann NY Acad Sci 242: 598–616, 1974 [DOI] [PubMed] [Google Scholar]
- 60.Yuan CM, Manunta P, Hamlyn JM, Chen S, Bohen E, Yeun J, Haddy FJ, Pamnani MB. Long-term ouabain administration produces hypertension in rats. Hypertension 22: 178–187, 1993 [DOI] [PubMed] [Google Scholar]
- 61.Zhang J, Lee MY, Cavalli M, Chen L, Berra-Romani R, Balke CW, Bianchi G, Ferrari P, Hamlyn JM, Iwamoto T, Lingrel JB, Matteson DR, Wier WG, Blaustein MP. Sodium pump α2 subunits control myogenic tone and blood pressure in mice. J Physiol 569: 243–256, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zucker IH, Xiao L, Haack KK. The central renin-angiotensin system and sympathetic nerve activity in chronic heart failure. Clin Sci (Lond) 126: 695–706, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zulian A, Linde CI, Pulina MV, Baryshnikov SG, Papparella I, Hamlyn JM, Golovina VA. Activation of c-SRC underlies the differential effects of ouabain and digoxin on Ca2+ signaling in arterial smooth muscle cells. Am J Physiol Cell Physiol 304: C324–C333, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]