

Abstract
Discoidin domain receptor 2 (DDR2) is a fibrillar collagen receptor that is expressed in mesenchymal cells throughout the body. In the heart, DDR2 is selectively expressed on cardiac fibroblasts. We generated a germline DDR2 knockout mouse and used this mouse to examine the role of DDR2 deletion on heart structure and function. Echocardiographic measurements from null mice were consistent with those from a smaller heart, with reduced left ventricular chamber dimensions and little change in wall thickness. Fractional shortening appeared normal. Left ventricular pressure measurements revealed mild inotropic and lusitropic abnormalities that were accentuated by dobutamine infusion. Both body and heart weights from 10-wk-old male mice were ∼20% smaller in null mice. The reduced heart size was not simply due to reduced body weight, since cardiomyocyte lengths were atypically shorter in null mice. Although normalized cardiac collagen mass (assayed by hydroxyproline content) was not different in null mice, the collagen area fraction was statistically higher, suggesting a reduced collagen density from altered collagen deposition and cross-linking. Cultured cardiac fibroblasts from null mice deposited collagen at a slower rate than wild-type littermates, possibly due to the expression of lower prolyl 4-hydroxylase α-isoform 1 enzyme levels. We conclude that genetic deletion of the DDR2 collagen receptor alters cardiac fibroblast function. The resulting perturbations in collagen deposition can influence the structure and function of mature cardiomyocytes.
Keywords: cardiac, fibroblast, collagen deposition, collagen receptor, development
discoidin domain receptor 2 (DDR2) is a receptor tyrosine kinase whose ligand is fibrillar (e.g., collagen types I and III) but not basement membrane collagen (e.g., collagen type IV). DDR2 has a requirement for triple helical collagen, since denatured collagen is not a ligand (23, 37). Activation of DDR2 has been shown to influence a variety of cell functions, including proliferation (16, 27), migration through collagen matrixes (1, 4, 27, 31), osteoblastic differentiation and chondrocyte maturation (24, 44), and integrin-mediated adhesion (39). The most commonly identified transcripts regulated by DDR2 are those of the matrix metalloproteinase (MMP) family, including MMP-1 (27, 43), MMP-2 (26, 27, 33, 43), MMP-8 (1), and MMP-13 (40, 41, 35). Other identified transcripts include osteocalcin and osteopontin (24), IL-6 (19), agrin (6), syndecan (Sdc)1 (6, 28), DDR2 itself (40), IL-12, and CD86 (22, 30). DDR2 gene deletion in immortalized skin fibroblasts reduces mRNA and protein levels for lysyl hydroxylase 1, lysyl oxidase, and osteonectin (28). Knockdown of DDR2 with short hairpin RNA has been shown to attenuate collagen-induced upregulation of lysyl oxidase protein in primary rodent osteoblast cultures (17). Collagen type I-α1 has also been reported to be upregulated by DDR2 in runt-related transcription factor 2-expressing cells (24) and skin fibroblasts (27). In contrast, DDR2 signaling appears to suppress collagen type I-α1 mRNA expression in vascular smooth muscle cells (7, 11), suggesting that the effect of DDR2 on transcript levels can be cell dependent.
Germline deletion of DDR2 has been previously shown to cause dwarfism in mice (35.3% lower body weight at 5 wk of age), which was believed to be caused by reduced chondrocyte proliferation and slower growth of long bones (21). Although the most pronounced developmental effects of DDR2 deletion or inhibition appear to be manifested in the skeletal system, DDR2 is expressed in many cells of mesenchymal origin, including interstitial fibroblasts of the heart (9, 10, 12), suggesting that DDR2 plays a role in determining cardiac structure and function and that its deletion or inhibition could result in a cardiac phenotype. Despite evidence showing that DDR2 appears to be involved in regulating extracellular matrix (ECM) deposition and composition, cardiovascular developmental effects have not been reported in DDR2-null mice to date.
During knockin of a MerCreMer gene into the DDR2 allele, we generated a mouse line that expressed little MerCreMer due to an unexpected frameshift mutation. This mouse, however, proved to be an ideal model of germline DDR2 deletion, which we used to determine whether the absence of DDR2 resulted in cardiac abnormalities. In the present study, we report that DDR2-null mice have smaller hearts, shorter cardiomyocytes, lower interstitial cardiac collagen density, and abnormalities in cardiac function, which support a role of this receptor in regulating cardiac structure and function. Cultured cardiac fibroblasts from knockout mice deposited insoluble collagen at a slower rate than wild-type (WT) mice, which could be caused by reduced expression of prolyl 4-hydroxylase α-isoform 1 (P4HA1) enzyme. Reduced levels of collagen synthesis could explain the smaller chamber sizes in null mice, providing a potential link between in vitro and in vivo observations.
EXPERIMENTAL PROCEDURES
Preparation of DDR2-null mice.
Homology arms were amplified by high-fidelity PCR from 129R1 genomic DNA using Pfu Ultra DNA polymerase (95°C for 2.5 min, 55°C for 30 s, 72°C for 5 min; 34 cycles of 95°C for 30 s, 65°C for 30 s, and 72°C for 5 min; 72°C for 10 min, held at 4°C; Agilent Technologies). The 4.0-kbp left homology arm (LHA) was amplified using the following primers: 5′-GTAAGGCTATCCAAGATCAGCTCGCACTCT-3′ and 5′-CCTAAGCTTCCCAGAGGATAGCGGCATATG-3′. The 3.1-kbp right homology arm (RHA) was amplified using the following primers: 5′-AACGGCCGAGGTACAGAGAGGTGTAGATTA-3′ and 5′-AGTTCACTCTCCACCCTTGAGCCAGTAGAA-3′. Arms were cloned into pCR-Blunt (Life Technologies), and the desired orientation was selected by restriction enzyme screening. The BamHI/HindIII fragment of the LHA and the EagI/NotI fragment of the RHA were purified from pCR-Blunt backbones. The MerCreMer-neo cassette was excised from the vector, pBKSII-MerCreMer-Mcl-Neo [kindly provided by Dr. Sylvia Evans, University of California-San Diego (UCSD)], by HindIII and NotI digestion. The targeting vector was then constructed in three sequential steps. In step 1, BamHI/HindIII LHA and HindIII/NotI MerCreMer-neo fragments were ligated into BamHI/NotI sites of the pCR2.1 cloning vector (Life Technologies). In step 2, the EagI/NotI RHA was ligated into the dephosphorylated NotI site of the result from step 1, ensuring that the preserved NotI site was distal to the MerCreMer. In step 3, to provide negative selection, a diphtheria toxin A coding cassette (PspOMI/NotI fragment, kindly provided by Dr. Ju Chen, UCSD) was then ligated into the dephosphorylated NotI site of the result from step 2, ensuring that the preserved NotI site was distal to the RHA. The resulting targeting vector was linearized with NotI and electroporated into 129R1 mouse embryonic stem cells, and individual G418-resistant clones were selected. Clones were screened by Southern blots using SpeI digestion and probe B (7.2-kbp WT and 5.2-kbp targeted; Fig. 1B). One targeted clone was karyotyped to verify the absence of chromosomal abnormalities, expanded, and microinjected into C57Bl/6 blastocysts. Injected blastocysts were implanted into pseudopregnant female hosts. Chimeric pups were bred, and agouti pups were screened for the targeted mutation. The neomycin cassette was removed from a positive mouse by breeding with a FLPe deleter mouse (kindly provided by Dr. Ju Chen, UCSD). The resulting mouse was backcrossed into C57Bl/6 for three to four generations. Generation of the targeted mice was made possible by the skilled assistance of the Transgenic Core and Embryonic Stem Cell Shared Resource at UCSD. All animal experiments were approved by the Institutional Animal Care and Use Committee of UCSD.
Fig. 1.
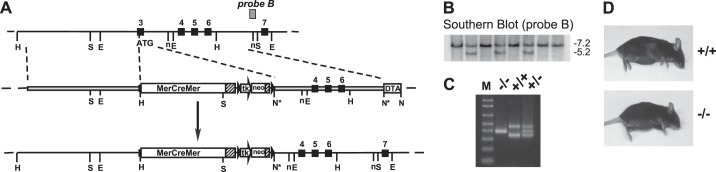
Production and screening of discoidin domain receptor 2 (DDR2) knockout embryonic stem (ES) cells and mice. A: diagrammatic representation of knockin of the MerCreMer cassette into exon 3 of the DDR2 allele to disrupt the DDR2 open reading frame. Top, wild-type (WT) allele; middle, targeting vector; bottom, targeted allele. Exons are shown as numbered solid boxes, polyadenylation sites are shown as hatched boxes, and Frt recombination sites are shown as black triangles surrounding the neomycin resistance gene (neo) cassette. This image is not drawn to scale. Restriction enzyme sites are as follows: HindIII (H), SpeI (S), EcoRI (E), NheI (n), NotI (N); and NotI site destroyed by ligation (N*). tk, thymidine kinase promoter; DTA, diphtheria toxin A expression cassette. B: Southern blot screening of ES cell clones. Genomic DNA that was isolated from a subset of cultured ES cells was digested with SpeI and subjected to Southern blot analysis with probe B (the hybridization region of the probe is shown in A). The WT band is ∼7.2 kbp, and the targeted band is ∼5.2 kbp. C: routine PCR screening of mice. DNA was isolated from tail clips or ear punches and subjected to PCR as described in experimental procedures. DDR2-null mice (−/−) generate a single band, WT mice (+/+) generate two bands, and heterozygous mice (+/−) generate three bands. M, Life Technologies 1-kb Plus DNA ladder from 100 to 1,000 bp. D: visual comparison of WT (top) and DDR-null (bottom) mice. Mice were 16-wk-old female littermates.
Routine genotyping of mice.
Genomic DNA was isolated from tail clips or ear punches (38). DNA (1 μl) in Tris-EDTA buffer was subjected to PCR using the FastStart Taq DNA Polymerase dNTPack kit (Roche Diagnostics) following manufacturer's instructions. Cycling conditions were as follows: 95°C for 4 min; 15 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 40 s; 17 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 40 s (with a 5-s increase in elongation time per cycle); 72°C for 7 min; and held at 4°C. Four primers were included in the reaction, namely 5′-GTGAGGTCCGGTAAAGCAAA-3′, 5′-CAGCCTCAAGTCAGTGGTCA-3′, 5′-GCCCAGTGGCATTTCAGATA-3′, and 5′-TAGTGAATTCCCGCGACTCT-3′. PCR products were electrophoresed on 2% agarose gels, poststained with Sybr Gold nucleic acid gel stain (Life Technologies), and visualized with a blue-light LED transilluminator (Spectronics). Under these conditions, a homozygous knockin mouse (i.e., a DDR2 knockout mouse) produces a single band, a WT mouse produces two bands, and a heterozygote mouse produces three bands, simplifying the genotyping process (Fig. 1C).
Hydroxyproline assays.
Cultured cells were scraped into, or minced tissue pieces were placed into, 4 N sodium hydroxide. Samples were subjected to alkaline hydrolysis followed by hydroxyproline and ninhydrin assays (32, 34). Standards were known masses of gelatin (for hydroxyproline) and BSA (for ninhydrin). Values are expressed as mass collagen per mass of noncollagenous protein.
High-fidelity RT-PCR of mutant DDR2 mRNA.
Total RNA was isolated from the skin of WT and DDR2-null mice with the RNeasy kit following manufacturer's instructions (Qiagen). RNA was quantified by absorbance at 260 nm. Reverse transcription was performed with random hexamers and Multiscribe Reverse Transcriptase (Life Technologies) followed by PCR using Pfu Ultra Hotstart DNA polymerase (Agilent Technologies). PCR primers were as follows: 5′-CACCCTCTGATACTCCAGATC-3′ with 5′-GTTGGTCAATAAGCCCATCATT-3′ for DDR2-MerCreMer fusion mRNA and 5′-TCTGAAATGATCCCGATTCC-3′ with 5′-GACTGGGATAAGGCGAACAA-3′ for DDR2 mRNA (from exon 2 to exon 5). Cycling conditions were as follows: 95°C for 2.5 min; 30 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 1 min; 72°C for 10 min; and held at 4°C. After visualization by agarose gel electrophoresis, PCR products were cloned into the vector, PCR Blunt II-TOPO, following the manufacturer's recommendations (Life Technologies). Inserts were then sequenced from both directions using T7 promoter and M13 reverse primers by Eton Bioscience (San Diego, CA).
Transthoracic echocardiography and cardiac catheterization.
Animals were anesthetized with 5% isoflurane for 1 min and then maintained at 1% throughout the examination. The anterior chest wall was shaved, and Nair was applied to remove the remaining hair. Small needle electrodes for simultaneous electrocardiogram were inserted into one upper limb and one lower limb. Transthoracic echocardiography (M-mode and two-dimensional) was performed using the VisualSonics (FUJIFILM) Vevo 2100 ultrasound system with a linear transducer 32–55 MHz. Measurements of left ventricular (LV) end-diastolic dimension, LV end-systolic dimension, end-diastolic interventricular septal thickness, and LV posterior wall thickness were determined from the LV M-mode tracing. Fractional shortening (in %) was used as an indicator of systolic cardiac function. The following day, mice were anesthetized with an intraperitoneal injection of ketamine (100 mg/kg) and xylazine (10 mg/kg). Mice were then intubated, placed on a ventilator (100–110 strokes/min, stroke volume: 0.4–0.5 ml), and the right carotid artery was exposed. The femoral vein was cannulated with stretched polyethylene-50 tubing for drug administration. A 1.4-Fr high-fidelity catheter-tip micromanometer (Millar Instruments, Houston, TX) was inserted retrograde into the aorta via the left carotid artery and advanced into the LV. Pressure was recorded on LabChart (ADinstruments). Both sides of the vagal nerve were cut, pressure was allowed to stabilize, and baseline pressure was recorded. After baseline pressures were obtained, dobutamine was given through the femoral vein at a dosage of 0.75, 2, 4, 6, and 8 μg·kg−1·min−1. Pressure data were analyzed at the end of 3 min for each dose.
Preparation of heart tissue.
Mice were weighed, anesthetized by an intraperitoneal injection of 100 mg/kg ketamine and 10 mg/kg xylazine, and euthanized by cervical dislocation. Hearts were excised and placed into room temperature PBS with 60 mM potassium chloride until they ceased to beat. Hearts were then weighed, and atria were removed. For hydroxyproline assays, a section of the LV was fixed in 4% buffered paraformaldehyde for 1 day and then stored at −80°C until needed. For microscopy, a section of the heart was fixed in 4% buffered paraformaldehyde for 2 days and then embedded in paraffin, cross-sectioned, and mounted onto slides by the UCSD Histology and Immunohistochemistry Shared Resource. Approximately one-half of the LV distal to the apex was used for microscopy, whereas the proximal half was used for biochemical assays.
Collagen area fraction.
The interstitial collagen area fraction was measured on Masson's trichrome-stained sections using ImageJ software (National Institutes of Health) and expressed as the percentage of collagen area to total tissue area, excluding large vessels and perivascular fibrosis. The trichrome stain was purchased as a kit (HT15–1KT, Sigma-Aldrich), and the provided instructions were followed. Randomly selected fields (10–20 fields total) at ×400 magnification were quantified per heart in a blinded fashion.
Myocyte cross-sectional area and length.
Myocyte cross-sectional area was measured in hematoxylin and eosin-stained sections visualized at ×400 and photographed using a digital camera under a microscope as previously described (13, 18). At least 100 myocytes across several sections were scored per heart. Myocyte lengths were measured after immunohistochemical staining of gap junctions with a pan-cadherin antibody (no. 4068, Cell Signaling Technology) using a similar technique to that previously described (3, 18). At least 25 myocytes across several sections were scored per heart. All measurements were performed by blinded investigators. Myocyte volumes were estimated as the product of length and cross-sectional area.
Quantifying pyridinoline cross-links in collagen.
Isolation of the nonreducible hydroxypyridinium cross-link was accomplished by reverse-phase HPLC, similar to that previously described (5). Lyophilized tissue (2–3 mg) was hydrolyzed in 6 N HCl at 108°C for 24 h. The liberated hydroxypyridinium cross-link was then isolated on a reverse-phase C-18 ODS HPLC column (4.6 × 250 mm, Beckman Instruments). Elution was performed with an acetonitrile gradient in the presence of 0.1 M heptafluorobutyric acid as the ion-pairing agent. Quantitation of hydroxypyridinium was simultaneously achieved by measuring in-line fluorescence (excitation: 297 nm and emission: 390 nm) with a filter fluorometer (model 121, Gilson). Pyridoxamine was used as a standard.
Isolation and culture of cardiac fibroblasts.
Mouse cardiac fibroblasts were isolated from mixed-sex adult littermates similarly to that previously described (14). Cells were cultured in high-glucose DMEM (no. 11965-092, Life Technologies) supplemented with 10% FBS and antibiotic-antimycotic, unless otherwise noted. Second-passage cells were routinely used for experiments.
Real-time RT-PCR.
Confluent cardiac fibroblasts were cultured for 10 days with daily changes of DMEM + 10% FBS + antibiotic-antimycotic + 25 μg/ml ascorbic acid. Total RNA was isolated using the RNeasy kit following the manufacturer's instructions (Qiagen). Contaminating DNA was removed by DNase I digestion before the absorbance was quantified at 260 nm. Total RNA was reverse transcribed with qScript cDNA SuperMix according to the manufacturer's instructions (Quanta Biosciences). TaqMan qPCR was performed on 50 ng cDNA (in duplicate). This work was performed with the support of the Genomics and Sequencing Core at the UCSC Center for AIDS Research (P30 AI036214), the VA San Diego Healthcare System, and the Veterans Medical Research Foundation. FAM/MGB primers/probes were purchased from Life Technologies and were as follows: collagen type I-α1 (Mm00801666_g1), collagen type I-α2 (Mm00483888_m1), collagen type III-α1 (Mm01254476_m1), MMP-2 (Mm00439498_m1), lysyl oxidase (Mm00495386_m1), a disintegrin and metalloproteinase with thrombospondin motifs 2 (Adamts2; Mm00805170_m1), bone morphogenetic protein 1 (Bmp1; Mm00802220_m1), P4HA1 (Mm00803137_m1), Sdc1 (Mm00448920_g1), and Gapdh (Mm99999915_g1). Gapdh was used as a normalizer.
Western blot analysis.
Adherent cells were lysed with RIPA buffer [50 mM Tris·HCl (pH 8.0), 150 mM NaCl, 1.0% (vol/vol) IGEPAL CA-630, 0.5% (wt/vol) sodium deoxycholate, and 0.1% (wt/vol) SDS containing protease inhibitors] on ice. Heart tissue was homogenized on ice in the same RIPA buffer with a tissue homogenizer (Fisher TissueMiser, Fisher Scientific). Extracts were cleared by centrifugation, and total protein was quantified using the Bio-Rad Protein Assay (catalog no. 500-0006). Equal amounts of protein were electrophoresed on discontinuous SDS-PAGE and electrophoretically transferred to polyvinylidene difluoride membranes. Primary antibodies were as follows: DDR2 (polyclonal goat antibody, AF2538, R&D Systems), GAPDH (AM4300 mouse monoclonal 6C5, Ambion, or no. 2118 rabbit monoclonal antibody 14C10, Cell Signaling), P4HA1 (goat polyclonal antibody NB100-57852, Novus Biologicals), lysyl oxidase (rabbit polyclonal antibody no. 17958-1-AP, Proteintech Group). Secondary antibodies were species-matched horse radish peroxidase conjugates. Signals were visualized using ECL Plus (GE Healthcare) and X-ray film, a Storm 860 fluorescent scanner (GE Healthcare), or a C-DiGit Blot Scanner (LI-COR Biosciences).
RESULTS
Generation and initial observations of DDR2-null mice.
During knockin of a MerCreMer gene into the DDR2 allele, we generated two mouse lines, one line targeting exon 2 and one line targeting exon 3 (see Fig. 1A for exon 3 targeting), due to uncertainties at the time regarding the start of protein translation. Exon 2 was later found to contain the actual start codon and signal peptide, so our targeting of the downstream ATG in exon 3 expressed only low levels of MerCreMer protein (data not shown). This mouse, however, proved to be a good model of germline DDR2 deletion. Two mRNA were found to be transcribed from the targeted DDR2 allele: the out-of-frame MerCreMer fusion (PCR product of 391 bp; Fig. 2A) and an exon 2-exon 4 splice variant (PCR product of 410 bp for the null mouse vs. 513 bp for the WT mouse; Fig. 2B). Based on primary sequence data, both mRNA were predicted to produce no DDR2 protein, and this was confirmed by immunoblot of cultured cardiac fibroblast extracts (Fig. 2C). Reprobing with an anti-GAPDH antibody confirmed equivalent protein loading (Fig. 2C). Our mice were 20% smaller than WT littermates by weight (Table 2), with shorter limbs and skull shape abnormalities highlighted by blunted snouts (Fig. 1D). These characteristics are believed to be the result of skeletal growth defects that have been noted in all DDR2-null models to date (15, 21, 41). Our mice were fertile when young, but fecundity dropped dramatically with age in both male and female mice, so that the mice were essentially sterile by 4 mo of age. In addition, pregnancies in young female knockout mice could prove fatal, with some mice dying either during or after pregnancy (data not shown). The reason for this mortality is not known. Routine breeding of mice used heterozygote crosses to avoid these reproductive problems.
Fig. 2.
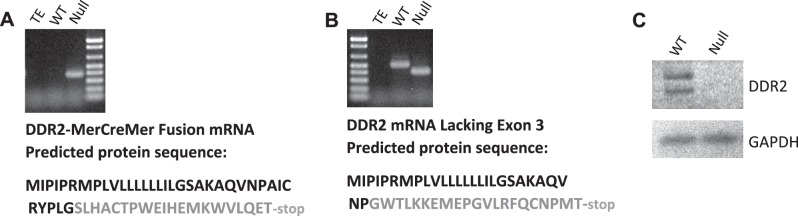
Verification that DDR2-null mice produce no DDR2 protein. A: DDR2-null mice transcribe a nonsense DDR2-MerCreMer fusion mRNA. Total RNA was isolated from the skin of either WT or DDR2-null mice and subjected to high-fidelity RT-PCR for the DDR2-MerCreMer fusion mRNA, as described in experimental procedures. Tris-EDTA (TE) buffer was amplified as a negative control. The stained agarose gel is shown on top. The PCR product was fully sequenced, and the expected, truncated translation product is shown underneath. B: DDR2-null mice also transcribe a nonsense DDR2 splice variant that lacks exon 3. Total RNA was isolated from the skin of either WT or null mice and subjected to high-fidelity RT-PCR for DDR2 mRNA, as described in experimental procedures. TE buffer was amplified as a negative control. The stained agarose gel is shown on top. The PCR product was fully sequenced, and the expected, truncated translation product is shown underneath. Protein sequences shown in black are correct for DDR2, whereas those shown in gray are missense. C: cultured cardiac fibroblasts express no detectable DDR2 protein. Cardiac fibroblasts from WT and null mice were isolated and cultured. Western blots for DDR2 (molecular mass: 120–140 kDa) and GAPDH (molecular mass: 37 kDa) were performed as indicated in experimental procedures. Note that the DNA ladder shown on both agarose gel images (A and B) is the same as shown in Fig. 1C.
Table 2.
Comparison of heart measurements between germline DDR2-null and WT mice
| Parameter | WT Mice | DDR2-Null Mice | n | P Value |
|---|---|---|---|---|
| Body weight, g | 27.1 ± 2.1 | 21.6 ± 2.5* | 10 | 0.00004 |
| Heart weight, g | 0.155 ± 0.017 | 0.123 ± 0.014* | 10 | 0.0002 |
| Tibia length, mm | 17.1 ± 0.3 | 15.4 ± 0.5* | 10 | 4.00 e−7 |
| Heart weight/body weight, % | 0.573 ± 0.039 | 0.572 ± 0.032 | 10 | 0.977 |
| Heart weight/tibia length, % | 0.910 ± 0.097 | 0.801 ± 0.089* | 10 | 0.017 |
| Myocyte cross-sectional area, μm2 | 109 ± 15 | 101 ± 5 | 6 | 0.27 |
| Myocyte length, μm | 75 ± 4 | 63 ± 6* | 6 | 0.002 |
| Myocyte volume, μm3 | 8,191 ± 1,490 | 6,382 ± 451* | 6 | 0.017 |
| Collagen area fraction, % | 2.1 ± 0.6 | 3.4 ± 1.0* | 6 | 0.019 |
| Nanograms of collagen/microgram of protein | 16.65 ± 1.29 | 17.14 ± 2.58 | 6 | 0.688 |
| Pyridinoline cross-links, pmol/mg tissue | 273 ± 74 | 245 ± 65 | 5 | 0.535 |
| Lox protein, densitometric units | 58.6 ± 15.8 | 58.3 ± 16.0 | 5 | 0.79 |
Values are means ± SE; n, number of 10-wk-old male mice per genotype. Lox, lysyl oxidase.
P < 0.05 vs. WT.
In vivo heart physiology of DDR2-null mice.
Despite evidence of DDR2 expression in cardiac fibroblasts (9, 10, 12) and phenotypic alterations of other mesenchymal cells that could be extrapolated to affect the cardiac interstitium, to our knowledge, there have been no studies on the hearts of DDR2-null mice. Echocardiographic measurements of heart dimensions demonstrated trends toward reductions in LV end-diastolic dimension, LV end-systolic dimension, LV end-diastolic dimension/LV posterior wall thickness, and estimated LV mass in null mice, and these would likely have reached significance with a larger sample size (Table 1). Indeed, echocardiography of an independent group of mice (5–6 mo old, mixed sex and strain) did show statistically lower chamber dimensions and estimated LV masses in null mice (data not shown).
Table 1.
Comparison of echocardiographic heart measurements between germline DDR2-null and WT mice
| Parameter | WT Mice | DDR2-Null Mice | P Value |
|---|---|---|---|
| LVIDd, mm | 3.70 ± 0.29 | 3.17 ± 0.58 | 0.114 |
| LVIDs, mm | 2.05 ± 0.47 | 1.66 ± 0.27 | 1.56 |
| IVSd, mm | 0.64 ± 0.04 | 0.61 ± 0.06 | 0.386 |
| IVSs, mm | 1.10 ± 0.09 | 1.07 ± 0.06 | 0.576 |
| LVPWd, mm | 0.64 ± 0.04 | 0.64 ± 0.08 | 0.963 |
| LVPWs, mm | 1.24 ± 0.13 | 1.19 ± 0.09 | 0.484 |
| LVIDd/LVPWd | 5.80 ± 0.274 | 4.59 ± 1.50 | 0.146 |
| Estimated LV mass, mg | 79.0 ± 16.0 | 57.9 ± 14.4 | 0.061 |
| LV mass/body weight | 28.5 ± 4.6 | 26.5 ± 6.0 | 0.566 |
| LVIDd/body weight | 0.134 ± 0.008 | 0.145 ± 0.025 | 0.379 |
| Fractional shortening, % | 44.8 ± 10.7 | 47.1 ± 4.8 | 0.673 |
Values are means ± SD; n = 5 male mice (10 wk old) per genotype. WT, wild type; DDR2, discoidin domain receptor 2; LVIDd, left ventricular (LV) end-diastolic dimension; LVIDs, LV end-systolic dimension; IVSd, end-diastolic interventricular septal thickness; IVSs, end-systolic interventricular septal thickness; LVPWd, end-diastolic LV posterior wall thickness; LVPWs, end-systolic LV posterior wall thickness.
To further assess cardiac function, LV hemodynamic variables were measured at baseline and after inotropic stimulation with increasing doses of dobutamine (Fig. 3). Heart rate in null mice remained lower across all dobutamine doses, although the difference was not quite significant at P = 0.078 (Fig. 3A). With the exception of end-diastolic pressure (where there was no difference between genotypes or dobutamine dose), null mice recorded values that reflected reduced contractility (maximum pressure and maximum dP/dt) and slower relaxation (minimum dP/dt and time constant of relaxation), indicating the presence of mild systolic and diastolic abnormalities brought on by stress.
Fig. 3.

Changes in left ventricular (LV) pressure values with dobutamine. Hemodynamic LV measurements were obtained as described in experimental procedures. A–F: calculated values for heart rate [in beats/min (bpm); A], maximum LV pressure (B), maximum dP/dt (C), minimum dP/dt (D), end-diastolic pressure (EDP; E) and the time constant for relaxation (τ; F) for both 10-wk-old WT mice (solid line; n = 5) and DDR2-null mice (dashed line; n = 5). Plotted points represent means ± SE. Dobutamine doses are given in micrograms per kilogram per minute in each abscissa. *Significantly different between WT and null mice by t-test (P < 0.05). In addition, repeated measures two-way ANOVA was performed for each data set. The effect of dobutamine dose was found to be statistically significant in all measurements except EDP. The P values for the effect of genotype and the influence of genotype on dobutamine response (interaction) are shown for each graph.
Ex vivo analyses of DDR2-null hearts.
Ex vivo analyses revealed that null hearts weighed significantly less than WT hearts (Table 2). Statistical significance was maintained if heart weights were normalized to tibial lengths but not to body weights (Table 2). Both body weights and tibial lengths were significantly lower in null mice (Table 2), however, making the validity of these normalized comparisons uncertain. To assess whether heart weights were lower because the mice were smaller, histological sections from potassium-arrested WT hearts of 10-wk-old male mice were quantified by blinded investigators for myocyte cross-sectional area, myocyte length, and estimated myocyte volume. Linear regression analyses revealed that cross-sectional area correlated well with body weight (r2 = 0.72), volume correlation was weaker, albeit still reasonable (r2 = 0.5), but length correlated poorly (r2 = 0.078), suggesting that myocyte length was independent of body weight in 10-wk-old mice. When WT hearts were compared with null hearts, both myocyte length and volume were statistically lower in null hearts (with lengths demonstrating the best statistics), but there was no significant difference between WT and null cross-sectional area (Table 2). Moreover, estimated myocyte volume in null hearts was 22% lower than that of WT hearts, which agrees very well with the measured 21% heart weight reduction. These values indicated that DDR2-null mice had smaller hearts, with most of the effect attributable to a reduction in cardiomyocyte lengths that could not be explained by a lower body weight.
There was no statistical difference in normalized collagen mass in LVs (estimated by hydroxyproline) between null and WT hearts (Table 2). However, collagen area fraction in the null group was 67% higher, and this increase was statistically significant (Table 2 and Fig. 4). Thus, absolute mass of cardiac collagen in the null group was lower since hearts were smaller, but total cardiac protein was proportionally lower so that normalized collagen mass was similar between null and WT groups. However, the collagen that was present in the null group was occupying a larger volume in the myocardium, suggesting that it was less densely packed. Analysis of stable collagen cross-links by HPLC revealed no statistical differences between genotypes (Table 2). There was also no difference in the level of lysyl oxidase, a protein that plays an important role in collagen cross-linking, in the myocardium, as assessed by immunoblot (Table 2). The reason for lower density of collagen in the hearts of the knockout mice remains uncertain.
Fig. 4.
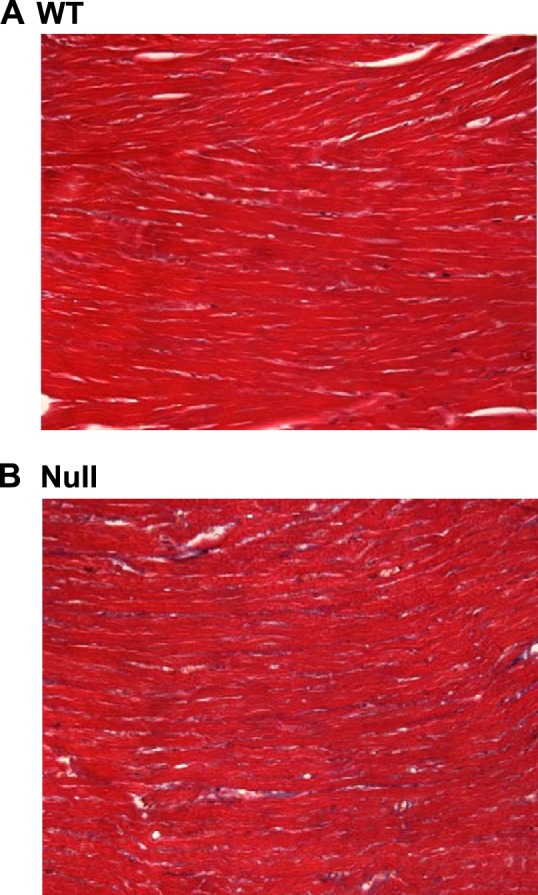
Representative trichrome stains for the assessment of collagen area fraction. Fixed and mounted tissue sections were stained with Masson's trichrome stain and quantified as described in experimental procedures. Photographs of representative microscopic images (×400 magnification) are shown for both WT (A) and DDR2-null (B) mice.
Analyses of cultured cardiac fibroblasts from DDR2-null mice.
To further assess how DDR2 affects collagen deposition in the heart, cardiac fibroblasts from adult mice were isolated and cultured. After 24 h of [3H]proline radiolabeling, there were no significant differences between genotypes in normalized collagenase-sensitive radiolabel that was secreted into the media in a soluble form [59,994 ± 853 disintegrations/min (dpm) in WT mice vs. 58,567 ± 1,378 dpm in null mice, n = 3, P = 0.216]. Cultured cardiac fibroblasts also slowly accumulate collagen in an insoluble, cell-associated form. DDR2-null fibroblasts were found to synthesize insoluble, cell-associated collagen at a slower rate than WT fibroblasts (8.75 vs. 5.98 ng normalized collagen/day for WT vs. null fibroblasts; Fig. 5). To better understand the reduced deposition of insoluble collagen, quantitative RT-PCR was performed on total RNA from cultured cardiac fibroblasts (Table 3). Transcripts that influence the deposition of mature collagen were selected in addition to two genes that have been previously shown to be regulated by DDR2 activation [i.e., MMP-2 and Sdc1 (6, 26–28, 33, 43)]. When multiple isoforms of a protein existed, only the major isoform expressed in mouse cardiac fibroblasts was assayed (e.g., Adamts2, Bmp1, and P4HA1; data not shown). Cardiac fibroblasts were cultured with ascorbate for 10 days before RNA isolation to ensure the synthesis of triple-helical collagen and subsequent stimulation of DDR2. Although all mRNA species assayed were expressed at slightly lower levels in null fibroblasts, the differences did not achieve statistical significance in most of the tested mRNA (Table 3). In fact, both MMP-2 and Sdc1, which have been shown to be substantially reduced in the absence of DDR2 activation in other cell types (6, 26–28, 33, 43), were not significantly altered by DDR2 deletion in cardiac fibroblasts (Table 3). However, P4HA1 mRNA was consistently and significantly downregulated in the null group.
Fig. 5.
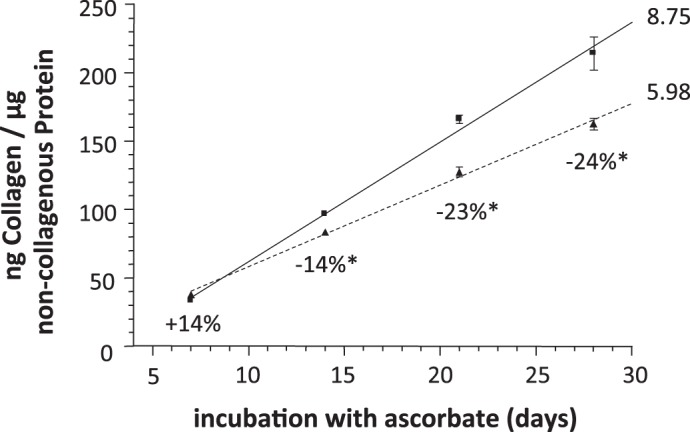
Cultured cardiac fibroblasts from DDR2-null mice synthesize insoluble collagen more slowly than those from WT littermates. Cardiac fibroblasts were isolated from the hearts of DDR2-null mice and WT littermates. Cells were grown to confluence in complete media and then cultured for the indicated number of days in complete media + 25 μg/ml ascorbic acid. Media were changed daily. At the indicated times, cells were assayed for collagen content (by hydroxyproline) and total protein (by ninhydrin). Details are provided in experimental procedures. The percent differences between null and WT mice are indicated below each time point. *P < 0.05 at this time point, r2 of both linear regression lines > 0.995. WT, squares with solid line; null, triangles with dashed line. Values are means ± SE; n = 3. The slope (in ng collagen·μg protein−1·day−1) is shown immediately to the right of each line.
Table 3.
TaqMan real-time RT-PCR of total RNA from cultured cardiac fibroblasts of DDR2-null and WT mice
| mRNA | Function | WT Mice | DDR2-Null Mice | P Value |
|---|---|---|---|---|
| Collagen type I-α1 | Collagen type I subunit | 1.02 ± 0.06 | 0.95 ± 0.26 | 0.527 |
| Collagen type I-α2 | Collagen type I subunit | 1.02 ± 0.08 | 0.93 ± 0.24 | 0.401 |
| Collagen type III-α1 | Collagen type III subunit | 1.03 ± 0.08 | 0.75 ± 0.33 | 0.090 |
| Lox | Initiates cross-linking | 1.04 ± 0.05 | 0.89 ± 0.18 | 0.092 |
| Adamts2 | N-propeptidase | 0.99 ± 0.05 | 0.85 ± 0.23 | 0.187 |
| Bone morphogenetic protein 1 | C-propeptidase | 1.00 ± 0.09 | 0.86 ± 0.24 | 0.241 |
| Prolyl 4-hydroxylase α-isoform 1 | Triple helix stability | 0.98 ± 0.05 | 0.86 ± 0.04* | 0.002 |
| Matrix metalloproteinase-2 | 0.98 ± 0.05 | 0.81 ± 0.30 | 0.222 | |
| Syndecan 1 | 1.02 ± 0.04 | 0.91 ± 0.13 | 0.091 |
Data are means ± SD of 2(−ΔΔCT) values (where CT is threshold cycle); n = 6 per genotype.
P < 0.05 vs. WT.
To further examine the possibility that lowered P4HA1 levels may have resulted in the reduced deposition of cell-associated collagen shown in Fig. 5, cardiac fibroblasts were cultured for 10 days in the presence of ascorbate to ensure the production of triple-helical collagen and DDR2 receptor activation. Immunoblot analysis revealed that mean P4HA1 protein levels were 27% lower in null mice (Fig. 6). Although the observed reduction in P4HA1 protein was small, it was reproducible in independent experiments and was of similar magnitude as the ∼30% reduction in collagen deposition rate shown in Fig. 5. Culture in the absence of ascorbate abrogated the reduction (data not shown), suggesting that DDR2 activation was able to increase P4HA1 levels in cardiac fibroblasts.
Fig. 6.
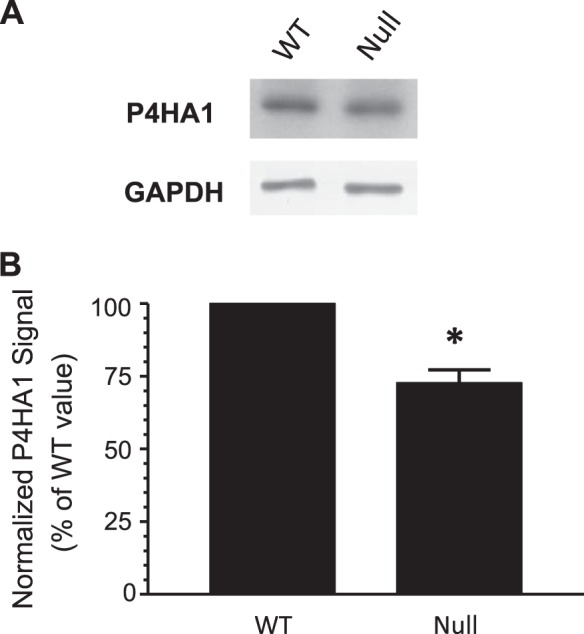
Cultured cardiac fibroblasts from DDR2-null mice have lower levels of prolyl 4-hydroxylase α-isoform 1 (P4HA1) protein. Cardiac fibroblasts were isolated from the hearts of DDR2-null mice and WT littermates. Cells were grown to confluence in complete media and then cultured for 10 days in complete media + 25 μg/ml ascorbic acid. Media were changed daily. Cell lysates were prepared, and equal amounts of protein were subjected to immunoblot analysis for P4HA1 (molecular mass: 61 kDa), as detailed in experimental procedures. Blots were stripped and then reprobed for GAPDH (molecular mass: 37 kDa) to ensure equal loading. A: representative images. Band intensities were quantified by ImageJ software (for scanned X-ray film) or C-DiGit Blot Scanner software (LI-COR Biosciences). Each P4HA1 signal was normalized to the corresponding GAPDH signal and expressed as a percentage of the WT value (arbitrarily set to 100%). B: graph of normalized P4HA1 signals. Values are means ± SE; n = 4. *P = 0.009 by Student's t-test.
DISCUSSION
In a DDR2 knockout mouse generated by insertion of a MerCreMer cassette into exon 3 of the DDR2 gene, we found evidence of abnormal cardiac structure and function. Ten-week-old DDR-null mice had hearts that were ∼21% smaller by weight, which did not appear to be due solely to a smaller body size, since myocyte lengths were atypically shorter in null mice. Fractional shortening appeared normal by echocardiography, but catheter measurements revealed mild inotropic and lusitropic abnormalities that were accentuated by dobutamine infusion. Although normalized cardiac collagen mass was similar in null mice, collagen area fraction (i.e., the volume that it occupied) was significantly higher, suggesting abnormalities in collagen deposition and cross-linking. We also found that cultured cardiac fibroblasts from null mice deposited collagen at a slower rate than WT littermates, possibly due to the expression of lower P4HA1 enzyme levels in null mice.
Although overall heart size appeared proportional to body size in DDR2-null mice, the reduction appeared to be caused by shorter cardiomyocytes that were not typical of WT animals. The mechanism for reduced cardiomyocyte lengths is not certain. It is noteworthy, however, that the shorter cardiomyocytes are presumed to possess fewer sarcomeric units in series along their length (8). Others have shown that cultured cardiomyocytes can rearrange their sarcomeres and conform to the shape of micropatterned ECM surfaces (2, 29). It is possible that the reduced rate of collagen deposition by DDR2-null fibroblasts reduces the size of the cardiac ECM scaffold and limits the growth of cardiomyocytes during development. The fact that cultured cardiac fibroblasts from DDR2-null mice deposit insoluble collagen at a slower rate than those from WT littermates supports this hypothesis. Our data suggest that, if true, this growth conformation occurs mostly along the long axis of the cardiomyocytes, restricting their eccentric growth.
Although the mechanism through which DDR2 affects collagen deposition in these cells was not identified, the receptor could directly affect deposition via its extracellular collagen-binding domain (25) in a manner analogous to that shown for integrins (36, 42). Another possibility is that DDR2 activation by triple-helical collagen produces a sustained increase in P4HA1 levels that directly influences the secretion and deposition of collagen. Indeed, the ∼30% reduction in the rate of synthesis of insoluble collagen observed in null mice (Fig. 5) mimics the ∼27% reduction in P4HA1 protein levels by immunoblot analysis (Fig. 6).
Evaluation of mRNA expression for proteins involved in collagen deposition in the myocardium in WT and null mice shown in Table 3 was largely unrevealing, with greater variability demonstrated between cell preparations than between genotypes. Also, two mRNA species reported to be regulated greatly by DDR2 in other cell types, namely, MMP-2 and Sdc1 (6, 26–28, 33, 43), were not significantly different in cardiac fibroblasts. Potential explanations include 1) tissue differences; 2) our analysis of stable phenotypic alterations that manifest over longer periods of time, rather than transient stimulation of DDR2; and 3) our analysis of primary cells, which could differ from the immortalized cells used in other published works (21, 27, 28).
Dobutamine infusion was used to help elucidate how DDR2 gene deletion affected cardiac function when the heart was stressed by catecholamines. We found mild reductions in contractility and diastolic relaxation consistent with reduced cardiac reserve. Although the mechanisms for the abnormalities in both systolic and diastolic function are not fully understood, both force transmission during systole and recoil during diastole could have been impeded by reduced collagen density (20). It is also possible that the reduction in cardiomyocyte lengths contributed to these functional abnormalities, as could intrinsic abnormalities in cardiomyocyte functional properties. We cannot rule out the possibility that the anesthetic used to sedate the mice differentially affected cardiac function (e.g., heart rate) between knockout and WT mice. Future mechanistic studies are required to clarify these possibilities.
In summary, DDR2-null mice had structural and functional abnormalities that were associated with differences in heart size, heart rate, contractility, and relaxation. Since DDR2 is selectively expressed on fibroblasts in the heart (9, 10, 12), these findings are consistent with the hypothesis that abnormalities in the cardiac matrix that are present throughout development can result in abnormalities in cardiac structure and function. Whether the effects of the loss of DDR2 on cardiac function are due to an impact of the ability of the matrix to transduce contractile and relaxation forces of cardiomyocytes or to a direct influence of the interstitium on intrinsic properties of cardiomyocytes is uncertain. Elucidation of this question, however, could provide novel insights into cardiac development, the interaction of cell types within the heart, the interplay of contractile myocytes with the ECM, and the development and treatment of heart failure.
GRANTS
This work was funded in part by National Heart, Lung, and Blood Institute Grant R21-HL-080358 (to R. T. Cowling).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: R.T.C., Y.G., N.D.D., K.L.P., and B.H.G. conception and design of research; R.T.C., S.J.Y., I.J.K., J.I.P., Y.G., and N.D.D. performed experiments; R.T.C., S.J.Y., I.J.K., J.I.P., Y.G., N.D.D., K.L.P., and B.H.G. analyzed data; R.T.C., S.J.Y., I.J.K., J.I.P., Y.G., N.D.D., K.L.P., and B.H.G. interpreted results of experiments; R.T.C. prepared figures; R.T.C. drafted manuscript; R.T.C. and B.H.G. edited and revised manuscript; R.T.C., S.J.Y., I.J.K., J.I.P., Y.G., N.D.D., K.L.P., and B.H.G. approved final version of manuscript.
ACKNOWLEDGEMENTS
The authors thank Dr. Ju Chen (University of California-San Diego) and Dr. Edie Goldsmith (University of South Carolina) for the helpful discussions. The authors also thank Dr. David Amiel and Fred Harwood at the University of California-San Diego for the skilled assistance in performing collagen cross-link assays, Jonathan Hu for help with DDR2 immunoblots, and Erika Alvarez for help in analyzing the echocardiographic and hemodynamic data.
Present address of J. I. Park: Seoul Veterans Healthcare Medical Center, Seoul, South Korea.
Present address of I. J. Kim: Dept. of Cardiology, CHA Bundang Medical Ctr., CHA Univ., Seongnam-si, Gyunggi-do, Republic of Korea.
REFERENCES
- 1.Afonso PV, McCann CP, Kapnick SM, Parent CA. Discoidin domain receptor 2 regulates neutrophil chemotaxis in 3D collagen matrices. Blood 121: 1644–1650, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bray MA, Sheehy SP, Parker KK. Sarcomere alignment is regulated by myocyte shape. Cell Motil Cytoskeleton 65: 641–651, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chaudhry HW, Dashoush NH, Tang H, Zhang L, Wang X, Wu EX, Wolgemuth DJ. Cyclin A2 mediates cardiomyocyte mitosis in the postmitotic myocardium. J Biol Chem 279: 35858–35866, 2004 [DOI] [PubMed] [Google Scholar]
- 4.Chen SC, Wang BW, Wang DL, Shyu KG. Hypoxia induces discoidin domain receptor-2 expression via the p38 pathway in vascular smooth muscle cells to increase their migration. Biochem Biophys Res Commun 374: 662–667, 2008 [DOI] [PubMed] [Google Scholar]
- 5.Eyre DR, Koob TJ, Van Ness KP. Quantitation of hydroxypyridinium crosslinks in collagen by high-performance liquid chromatography. Anal Biochem 137: 380–388, 1984 [DOI] [PubMed] [Google Scholar]
- 6.Faraci E, Eck M, Gerstmayer B, Bosio A, Vogel WF. An extracellular matrix-specific microarray allowed the identification of target genes downstream of discoidin domain receptors. Matrix Biol 22: 373–381, 2003 [DOI] [PubMed] [Google Scholar]
- 7.Ferri N, Carragher NO, Raines EW. Role of discoidin domain receptors 1 and 2 in human smooth muscle cell-mediated collagen remodeling: potential implications in atherosclerosis and lymphangioleiomyomatosis. Am J Pathol 164: 1575–1585, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Goktepe S, Abilez OJ, Parker KK, Kuhl E. A multiscale model for eccentric and concentric cardiac growth through sarcomerogenesis. J Theor Biol 265: 433–442, 2010 [DOI] [PubMed] [Google Scholar]
- 9.Goldsmith EC, Hoffman A, Morales MO, Potts JD, Price RL, McFadden A, Rice M, Borg TK. Organization of fibroblasts in the heart. Dev Dyn 230: 787–794, 2004 [DOI] [PubMed] [Google Scholar]
- 10.Hartner A, Cordasic N, Rascher W, Hilgers KF. Deletion of the alpha8 integrin gene does not protect mice from myocardial fibrosis in DOCA hypertension. Am J Hypertens 22: 92–99, 2009 [DOI] [PubMed] [Google Scholar]
- 11.Hou G, Wang D, Bendeck MP. Deletion of discoidin domain receptor 2 does not affect smooth muscle cell adhesion, migration, or proliferation in response to type I collagen. Cardiovasc Pathol 21: 214–218, 2012 [DOI] [PubMed] [Google Scholar]
- 12.Hudon-David F, Bouzeghrane F, Couture P, Thibault G. Thy-1 expression by cardiac fibroblasts: lack of association with myofibroblast contractile markers. J Mol Cell Cardiol 42: 991–1000, 2007 [DOI] [PubMed] [Google Scholar]
- 13.Inada T, Fujiwara H, Hasegawa K, Araki M, Yamauchi-Kohno R, Yabana H, Fujiwara T, Tanaka M, Sasayama S. Upregulated expression of cardiac endothelin-1 participates in myocardial cell growth in Bio14.6 Syrian cardiomyopathic hamsters. J Am Coll Cardiol 33: 565–571, 1999 [DOI] [PubMed] [Google Scholar]
- 14.Iwata M, Cowling RT, Gurantz D, Moore C, Zhang S, Yuan JX, Greenberg BH. Angiotensin-(1–7) binds to specific receptors on cardiac fibroblasts to initiate antifibrotic and antitrophic effects. Am J Physiol Heart Circ Physiol 289: H2356–H2363, 2005 [DOI] [PubMed] [Google Scholar]
- 15.Kano K, Marin dE, Young J, Wnek C, Maddatu TP, Nishina PM, Naggert JK. A novel dwarfism with gonadal dysfunction due to loss-of-function allele of the collagen receptor gene, Ddr2, in the mouse. Mol Endocrinol 22: 1866–1880, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawai I, Hisaki T, Sugiura K, Naito K, Kano K. Discoidin domain receptor 2 (DDR2) regulates proliferation of endochondral cells in mice. Biochem Biophys Res Commun 427: 611–617, 2012 [DOI] [PubMed] [Google Scholar]
- 17.Khosravi R, Sodek KL, Faibish M, Trackman PC. Collagen advanced glycation inhibits its discoidin domain receptor 2 (DDR2)-mediated induction of lysyl oxidase in osteoblasts. Bone 58: 33–41, 2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim MA, Yang D, Kida K, Molotkova N, Yeo SJ, Varki N, Iwata M, Dalton ND, Peterson KL, Siems WE, Walther T, Cowling RT, Kjekshus J, Greenberg B. Effects of ACE2 inhibition in the post-myocardial infarction heart. J Card Fail 16: 777–785, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klatt AR, Zech D, Kuhn G, Paul-Klausch B, Klinger G, Renno JH, Schmidt J, Malchau G, Wielckens K. Discoidin domain receptor 2 mediates the collagen II-dependent release of interleukin-6 in primary human chondrocytes. J Pathol 218: 241–247, 2009 [DOI] [PubMed] [Google Scholar]
- 20.Krams R, Janssen M, Van der Lee C, Van MJ, De Jong JW, Slager CJ, Verdouw PD. Loss of elastic recoil in postischemic myocardium induces rightward shift of the systolic pressure-volume relationship. Am J Physiol Heart Circ Physiol 267: H1557–H1564, 1994 [DOI] [PubMed] [Google Scholar]
- 21.Labrador JP, Azcoitia V, Tuckermann J, Lin C, Olaso E, Manes S, Bruckner K, Goergen JL, Lemke G, Yancopoulos G, Angel P, Martinez C, Klein R. The collagen receptor DDR2 regulates proliferation and its elimination leads to dwarfism. EMBO Rep 2: 446–452, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee JE, Kang CS, Guan XY, Kim BT, Kim SH, Lee YM, Moon WS, Kim DK. Discoidin domain receptor 2 is involved in the activation of bone marrow-derived dendritic cells caused by type I collagen. Biochem Biophys Res Commun 352: 244–250, 2007 [DOI] [PubMed] [Google Scholar]
- 23.Leitinger B, Hohenester E. Mammalian collagen receptors. Matrix Biol 26: 146–155, 2007 [DOI] [PubMed] [Google Scholar]
- 24.Lin KL, Chou CH, Hsieh SC, Hwa SY, Lee MT, Wang FF. Transcriptional upregulation of DDR2 by ATF4 facilitates osteoblastic differentiation through p38 MAPK-mediated Runx2 activation. J Bone Miner Res 25: 2489–2503, 2010 [DOI] [PubMed] [Google Scholar]
- 25.Mihai C, Iscru DF, Druhan LJ, Elton TS, Agarwal G. Discoidin domain receptor 2 inhibits fibrillogenesis of collagen type 1. J Mol Biol 361: 864–876, 2006 [DOI] [PubMed] [Google Scholar]
- 26.Olaso E, Ikeda K, Eng FJ, Xu L, Wang LH, Lin HC, Friedman SL. DDR2 receptor promotes MMP-2-mediated proliferation and invasion by hepatic stellate cells. J Clin Invest 108: 1369–1378, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Olaso E, Labrador JP, Wang L, Ikeda K, Eng FJ, Klein R, Lovett DH, Lin HC, Friedman SL. Discoidin domain receptor 2 regulates fibroblast proliferation and migration through the extracellular matrix in association with transcriptional activation of matrix metalloproteinase-2. J Biol Chem 277: 3606–3613, 2002 [DOI] [PubMed] [Google Scholar]
- 28.Olaso E, Lin HC, Wang LH, Friedman SL. Impaired dermal wound healing in discoidin domain receptor 2-deficient mice associated with defective extracellular matrix remodeling. Fibrogenesis Tissue Repair 4: 5, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parker KK, Tan J, Chen CS, Tung L. Myofibrillar architecture in engineered cardiac myocytes. Circ Res 103: 340–342, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Poudel B, Ki HH, Lee YM, Kim DK. Induction of IL-12 production by the activation of discoidin domain receptor 2 via NF-κB and JNK pathway. Biochem Biophys Res Commun 434: 584–588, 2013 [DOI] [PubMed] [Google Scholar]
- 31.Ruiz PA, Jarai G. Discoidin domain receptors regulate the migration of primary human lung fibroblasts through collagen matrices. Fibrogenesis Tissue Repair 5: 3, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwartz DE, Choi Y, Sandell LJ, Hanson WR. Quantitative analysis of collagen, protein and DNA in fixed, paraffin-embedded and sectioned tissue. Histochem J 17: 655–663, 1985 [DOI] [PubMed] [Google Scholar]
- 33.Shyu KG, Wang BW, Chang H. Hyperbaric oxygen activates discoidin domain receptor 2 via tumour necrosis factor-α and the p38 MAPK pathway to increase vascular smooth muscle cell migration through matrix metalloproteinase 2. Clin Sci (Lond) 116: 575–583, 2009 [DOI] [PubMed] [Google Scholar]
- 34.Starcher B. A ninhydrin-based assay to quantitate the total protein content of tissue samples. Anal Biochem 292: 125–129, 2001 [DOI] [PubMed] [Google Scholar]
- 35.Su J, Yu J, Ren T, Zhang W, Zhang Y, Liu X, Sun T, Lu H, Miyazawa K, Yao L. Discoidin domain receptor 2 is associated with the increased expression of matrix metalloproteinase-13 in synovial fibroblasts of rheumatoid arthritis. Mol Cell Biochem 330: 141–152, 2009 [DOI] [PubMed] [Google Scholar]
- 36.Velling T, Risteli J, Wennerberg K, Mosher DF, Johansson S. Polymerization of type I and III collagens is dependent on fibronectin and enhanced by integrins α11β1 and α2β1. J Biol Chem 277: 37377–37381, 2002 [DOI] [PubMed] [Google Scholar]
- 37.Vogel WF, Abdulhussein R, Ford CE. Sensing extracellular matrix: an update on discoidin domain receptor function. Cell Signal 18: 1108–1116, 2006 [DOI] [PubMed] [Google Scholar]
- 38.Wang Z, Storm DR. Extraction of DNA from mouse tails. Biotechniques 41: 410–412, 2006 [DOI] [PubMed] [Google Scholar]
- 39.Xu H, Bihan D, Chang F, Huang PH, Farndale RW, Leitinger B. Discoidin domain receptors promote α1β1- and α2β1-integrin mediated cell adhesion to collagen by enhancing integrin activation. PLOS ONE 7: e52209, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xu L, Peng H, Wu D, Hu K, Goldring MB, Olsen BR, Li Y. Activation of the discoidin domain receptor 2 induces expression of matrix metalloproteinase 13 associated with osteoarthritis in mice. J Biol Chem 280: 548–555, 2005 [DOI] [PubMed] [Google Scholar]
- 41.Xu L, Servais J, Polur I, Kim D, Lee PL, Chung K, Li Y. Attenuation of osteoarthritis progression by reduction of discoidin domain receptor 2 in mice. Arthritis Rheum 62: 2736–2744, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Young EF, Marcantonio EE. A novel subcellular collagen organization process visualized by total internal reflection fluorescence microscopy. Cell Commun Adhes 14: 169–180, 2007 [DOI] [PubMed] [Google Scholar]
- 43.Zhang W, Ding T, Zhang J, Su J, Li F, Liu X, Ma W, Yao L. Expression of discoidin domain receptor 2 (DDR2) extracellular domain in pichia pastoris and functional analysis in synovial fibroblasts and NIT3T3 cells. Mol Cell Biochem 290: 43–53, 2006 [DOI] [PubMed] [Google Scholar]
- 44.Zhang Y, Su J, Yu J, Bu X, Ren T, Liu X, Yao L. An essential role of discoidin domain receptor 2 (DDR2) in osteoblast differentiation and chondrocyte maturation via modulation of Runx2 activation. J Bone Miner Res 26: 604–617, 2011 [DOI] [PubMed] [Google Scholar]