

Abstract
Thymosin-β4 (Tβ4) promotes cell survival, angiogenesis, and tissue regeneration and reduces inflammation. Cardiac rupture after myocardial infarction (MI) is mainly the consequence of excessive regional inflammation, whereas cardiac dysfunction after MI results from a massive cardiomyocyte loss and cardiac fibrosis. It is possible that Tβ4 reduces the incidence of cardiac rupture post-MI via anti-inflammatory actions and that it decreases adverse cardiac remodeling and improves cardiac function by promoting cardiac cell survival and cardiac repair. C57BL/6 mice were subjected to MI and treated with either vehicle or Tβ4 (1.6 mg·kg−1·day−1 ip via osmotic minipump) for 7 days or 5 wk. Mice were assessed for 1) cardiac remodeling and function by echocardiography; 2) inflammatory cell infiltration, capillary density, myocyte apoptosis, and interstitial collagen fraction histopathologically; 3) gelatinolytic activity by in situ zymography; and 4) expression of ICAM-1 and p53 by immunoblot analysis. Tβ4 reduced cardiac rupture that was associated with a decrease in the numbers of infiltrating inflammatory cells and apoptotic myocytes, a decrease in gelatinolytic activity and ICAM-1 and p53 expression, and an increase in the numbers of CD31-positive cells. Five-week treatment with Tβ4 ameliorated left ventricular dilation, improved cardiac function, markedly reduced interstitial collagen fraction, and increased capillary density. In a murine model of acute MI, Tβ4 not only decreased mortality rate as a result of cardiac rupture but also significantly improved cardiac function after MI. Thus, the use of Tβ4 could be explored as an alternative therapy in preventing cardiac rupture and restoring cardiac function in patients with MI.
Keywords: thymosin-β4, mice, myocardial infarction, cardiac rupture, cardiac function
myocardial infarction (MI) is a leading cause of death in the industrialized world. MI is associated with high rates of acute death caused by ventricular wall rupture, arrhythmias, or cardiogenic shock and long-term complications such as heart failure (17, 34). The percentage of autopsy-proven rupture of total autopsied patients who died of acute MI has increased from under 10% during the 1980s (1, 36) to 12–65% in recent decades (2, 14, 22, 34, 43). This increased percentage of autopsy-confirmed rupture deaths indicates the lack of effective strategies for rupture prevention post-MI. Both clinical and experimental studies have provided strong evidence that 1) wall rupture at an early stage of MI is mainly the consequence of excessive regional inflammation and degradation of the extracellular matrix (ECM) by proteinases, particularly matrix metalloproteinases (MMPs), resulting in infarct size extension, and 2) massive cardiomyocyte death associated with a significant decrease in capillary density causes cardiac dysfunction (6, 16, 24, 32, 47, 48).
Thymosin-β4 (Tβ4) is an endogenous 43-amino acid peptide found in the circulation and various organs, including the heart (29). Besides its known actin-sequestering activity (19), Tβ4 has numerous biological functions, including promotions of cell migration, angiogenesis, cell survival, and tissue regeneration and inhibition of inflammation (8). In permanently ligated mouse and ischemia-reperfusion pig models, Tβ4 stimulated myocardial cell migration, promoted angiogenesis and survival of cardiomyocytes, and decreased inflammation, thus improving cardiac function (4, 21). Therefore, we hypothesized that Tβ4 prevents cardiac rupture and improves cardiac function post-MI via its anti-inflammatory, proangiogenic, and antiapoptotic actions. The murine model of acute MI shares both clinical and pathological features of post-MI with changes found in human hearts, including cardiac rupture and dysfunction (17). Using C57BL/6 mice, we examined whether Tβ4 1) reduces the incidence of cardiac rupture during acute MI with a focus on its effects on inflammatory cell infiltration, gelatinolytic activity, neovascularization, and myocyte apoptosis in infarct border regions and 2) improves cardiac remodeling and function during the chronic phase of MI.
MATERIALS AND METHODS
Animals
All animal experiments conducted in this study were approved by the Institutional Animal Care and Use Committee of Henry Ford Hospital. Twelve-week-old male C57BL/6 mice (26–27 g, The Jackson Laboratory, Bar Harbor, ME) were housed in vented cages with a 12:12-h light-dark cycle and fed ad libitum. The dose and drug delivery route were based on previous studies of Tβ4 biodistribution (29) and the role of Tβ4 in cardioprotection and repair in a murine model of acute MI (4).
Experimental Design
The present study focused on the detrimental effects of MI on the heart and the protective cardiac effects of Tβ4 after MI. Due to the absence of any biological effect of Tβ4 when administered at a dose of 1.6 mg·kg−1·day−1 for 7 days or 5 wk in a sham-operated (sham) group (pilot study), we decided not to include a sham MI + Tβ4 group in the experimental design and statistical analysis. Thus, animals were allotted to the following three groups: 1) sham, 2) MI + vehicle, and 3) MI + Tβ4.
Protocol 1 (7-day protocol): effect of Tβ4 on cardiac rupture.
Mice were anesthetized with pentobarbital sodium (50 mg/kg ip), and an Alzet osmotic minipump (Durect, Cupertino, CA) containing either saline or Tβ4 (1.6 mg·kg−1·day−1, RegeneRx Biopharmaceuticals, Rockville, MD) was implanted intraperitoneally. To ensure that Tβ4 reached steady high circulating levels by the time of MI, 7 days after minipump implantation, mice were anesthetized with pentobarbital sodium (50 mg/kg ip), and MI was surgically induced by ligating the left anterior descending coronary artery as previously described (55). Animals were divided into the following three groups: 1) sham (n = 8), 2) MI + vehicle (n = 64), and 3) MI + Tβ4 (n = 22). Treatments were continued for 7 days. Mice were assessed for 1) cardiac remodeling and function by echocardiography and 2) changes in infiltrating neutrophils and macrophages, gelatinolytic activity, CD31 expression (angiogenesis), apoptotic myocytes, and ICAM-1 and p53 expression in the myocardium.
Protocol 2 (5-wk protocol): effect of Tβ4 on cardiac function.
Mice were anesthetized with pentobarbital sodium (50 mg/kg ip) and subjected to MI as described above in protocol 1. The osmotic minipump containing Tβ4 was implanted immediately after MI surgery as described above in protocol 1. The following three groups were used: 1) sham (n = 10), 2) MI + vehicle (n = 21), and 3) MI + Tβ4 (n = 24). Treatment lasted for 5 wk. Cardiac remodeling and function were evaluated by echocardiography and interstitial collagen, and capillary density measured in the left ventricle (LV) by histochemical staining.
Due to the small size of the mouse LV, some mice were used only for one or two analytic parameter(s) as indicated in the results.
Cardiac Rupture
In protocol 1, an autopsy was performed on each animal that was found dead after MI. The presence of a large amount of blood in the chest cavity and the perforation in the infarct-free wall indicated death due to cardiac rupture.
Cardiac Function by Echocardiography
Cardiac geometry and function were determined in awake mice at 7 days or 5 wk post-MI. LV diastolic dimensions (LVDd), LV diastolic areas (LVAd; obtained using two-dimensional echocardiography from the parasternal short-axis view), diastolic posterior wall thickness (PWTd), LV ejection fraction (EF), and shortening fraction (SF) were measured using a Doppler echocardiograph with a 15-MHz linear transducer (Acuson c256, Mountain View, CA) as previously described (54).
Histopathological Analysis
At the end of the experiments (7 days or 5 wk), mice were weighed and injected with an overdose of pentobarbital sodium (100 mg/kg ip). The heart was stopped at diastole by an intraventricular injection of 50 μl of 15% KCl, rapidly excised, and weighed. The LV, including the septum, was sectioned transversely into three slices from the apex to base. Slices were rapidly frozen in isopentane precooled in liquid nitrogen and then stored at −70°C until use. Cryosections of the LV (6 μm) were used for histological experiments. For the histology/morphology experiments detailed below, images of the sections were captured using a microscope (IX81, Olympus American, Melville, NY) equipped with a digital camera (DP70, Olympus American) and quantified with Microsuite Biological imaging software (Olympus).
Infarct size.
A 6-μm LV cryosection was cut from each slice and stained with Masson's trichrome. Whole heart images of the sections were captured at low magnification and processed with Microsuite Biological imaging software. Endocardial and epicardial circumferences as well as the length of the scar were measured for each slice with Microsuite Biological imaging software (Olympus). The ratio of scar length to ventricular circumference of the endocardium and epicardium of the three slices was determined, averaged, and expressed as a percentage to define infarct size. Mice with infarct size < 20% were excluded.
Immunohistochemical staining for CD68-positive macrophages, lymphocyte antigen 6B.2 alloantigen-positive neutrophils, and CD31-positive endothelial cells in infarct border regions.
LV cryosections were fixed in cold acetone for 10 min and rinsed in PBS for 5 min. Sections were incubated in 3% hydrogen peroxide in distilled water to quench endogenous peroxidase activity, rinsed in PBS, and preincubated with blocking serum (2% normal serum) for 30 min, after which sections were incubated at 4°C overnight with the following primary antibodies: 1) rat anti-mouse CD68 (a macrophage marker, 1:200, AbD Serotec, Raleigh, NC); 2) rat anti-mouse lymphocyte antigen 6B.2 alloantigen (a neutrophil marker, 1:200, AbD Serotec); and 3) rabbit polyclonal antibody against CD31 (a marker for endothelial cells, 1:200, Abcam, Cambrige, MA). Each section was then washed three times in PBS and assayed with a Vectastain ABC kit and 3-amino-9-ethylcarbazole substrate (Vector Laboratories, Burlingame, CA). Sections were counterstained with hematoxylin to show the nucleus in blue. Images of the sections were captured at high magnification. Positive staining was identified by a reddish-brown color. The number of positive cells in both border zone and remote areas and capillaries in the border zone were counted and expressed as numbers of cells or vessels per square millimeter of the myocardium, respectively. We identified the infarct border area according to the following histological features under the microscope: 1) intensified infiltrating leukocytes, 2) disarray of myofibrils, 3) necrotic myocytes and fiborotic tissue/scars, and 4) visible alive cardiomyocytes.
Gelatinolytic activity.
Gelatinolytic activity in the LV was determined in cryosections using in situ zymography (18). Briefly, DQ-gelatin (Molecular Probes, Eugene, OR) was dissolved at a concentration of 1 mg/ml in distilled water and then diluted 1:10 in 1% (wt/vol) low-gelling temperature agarose (Sigma, St. Louis, MO) in PBS. Subsequently, 25 μl of this mixture were placed on air-dried LV sections and incubated for 5 h at room temperature after placement of a 22 × 22-mm coverslip. The presence of autofluorescence in sections was tested by incubatation in the agarose-containing medium that lacks DQ-gelatin. Specific in situ zymography was tested by preincubation of the sections with 20 mM EDTA in PBS at room temperation for 1 h and then incubation of the sections with incubation medium. With appropriate background and autofluorescence correction, the remote areas showed no fluorescence. Images of the sections showing the borders of infarction (an area showing bright green fluorescence within the alive myocardium) were captured and processed with ImageJ (National Institutes of Health, http://rsbweb.nih.gov/ij/) to assess fluorescence intensity. The intensity was expressed as arbitrary units (mean tissue pixel intensity/tissue area).
In situ detection of cardiomyocyte apoptosis by TUNEL.
LV cryosections were double labeled by immunohistochemistry and TUNEL to identify α-actinin (sarcomeric, a marker of cardiomyocytes) and apoptotic cells, respectively. Briefly, 4% formaldehyde-fixed cryosections were pretreated with proteinase K (1:50) for 15 min at room temperature. Sections were incubated at 4°C overnight with monoclonal anti-α-actinin (sarcomeric) antibody (a marker of cardiomyocytes, EA-53, 1:800, Sigma) and then with Cy3-labeled goat anti-mouse IgG antibody (Jackson ImmunoResearch Laboratories, West Grove, PA). The TUNEL assay was performed using a TdT-Fluor in situ apoptosis detection kit (Trevigen, Gaithersburg, MD). Sections were counterstained with 4′,6-diamidino-2-phenylindole to show nuclei in blue. Images of the sections showing the myocardium were captured and processed with Microsuite Biological imaging software to assess the area of the myocardium. TUNEL-positive cardiomyocytes with bright green nuclei surrounded by red striations (α-actinin) were counted and expressed as numbers of TUNEL-positive myocytes per millimeter square.
Interstitial collagen fraction and capillary density.
LV sections were cut from each frozen slice and stained separately with fluorescein-labeled peanut agglutinin (Vector Laboratories) and rhodamine-labeled Griffonia simplicifolia lectin I (Vector Laboratories) to determine the interstitial collagen fraction (ICF) and capillary density in the heart, as we have previously described (25). Twelve fields were randomly chosen from each specific region, including noninfarct and infarct border areas.
Western Blot Analysis
About 20 mg of snap-frozen LV tissue from the base of the heart were thawed in 250 μl lysis buffer (Cell Signaling Technology, Danvers, MA) containing protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN) to which 1 mM PMSF was added right before use. Small pieces were disrupted and homogenized with a Polytron, with samples kept at 4°C throughout all procedures. Homogenized LV samples were centrifuged at 14,000 g for 10 min at 4°C, and supernatants containing total LV tissue lysates were collected. Protein in the supernatant was measured with Coomassie reagent (Thermo Scientific, Rockford, IL). The 60-μg aliquots of protein were kept at −72°C.
Tβ4 content in the LV.
LV lysates were separated by electrophoresis on 4–20% Tris-glycine gels (Invitrogen, Carlsbad, CA) and electrotransferred to nitrocellulose membranes (0.2-μm pore size) at a constant voltage of 100 V for 40 min at 4°C. Tβ4 was detected by immunoblot analysis of the membranes overnight at 4°C with a rabbit polyclonal antibody against Tβ4 (FL-44, 1:500, Santa Cruz Biotechnology, Santa Cruz, CA). Bound antibodies were visualized using a horseradish peroxidase-conjugated secondary antibody (Cell Signaling Technology) and enhanced chemiluminescence reagent (Amersham Biosciences, Piscataway, NJ). After the detection of Tβ4, the membrane was reblotted with a rabbit monoclonal antibody against GAPDH (1:3,000, Cell Signaling Technology). Band intensity was quantified by densitometry, Tβ4 was normalized to GAPDH, and the results are expressed as fold increases compared with sham groups.
ICAM-1 and p53 protein expression.
LV lysates were subjected to 10% SDS-PAGE under reducing conditions and electrotransferred to nitrocellulose membranes (0.45-μm pore size). ICAM-1 and the transcriptional factor p53 were detected by immunoblot analysis of the membranes overnight at 4°C with a mouse antibody against ICAM-1 (0.1 μg/ml, R&D Systems, Minneapolis, MN) and p53 (1C12, 1:1,000, Santa Cruz Biotechnology), respectively. The rest of the immunoblot procedure was the same as that described above for Tβ4 detection. Both ICAM-1 and p53 were normalized to GAPDH, and the results are expressed as fold increases compared with sham groups.
Statistical Analysis
Binary data (cardiac rupture) are expressed as proportions, and groups were compared using a χ2-test for two-by-two tables. Continuous data are expressed as means ± SE, and groups were compared using a two-sample Wilcoxon test. A nonparametric method was chosen as the variances differed substantially between groups. In all settings where multiple testing was used, Hochberg's method was used to determine significance. Adjusted P values of <0.05 were considered significant.
RESULTS
Protocol 1
Incidence of rupture after MI.
All 86 mice with MI (MI + vehicle group, n = 64, and MI + Tβ4 group, n = 22) survived 24 h after MI surgery. Seven days after MI, 56.3%of vehicle-treated mice (36 of 64 mice) and 22.7% of Tβ4-treated mice (5 of 22 mice) died due to LV rupture (Fig. 1A), and in Tβ4-treated animals, Tβ4 content was significantly higher compared with nontreated groups (P < 0.01; Fig. 1B). Thus, a significantly lower incidence of LV rupture was observed in the Tβ4-treated group. The time window of rupture was 3–6 days after MI in both groups.
Fig. 1.
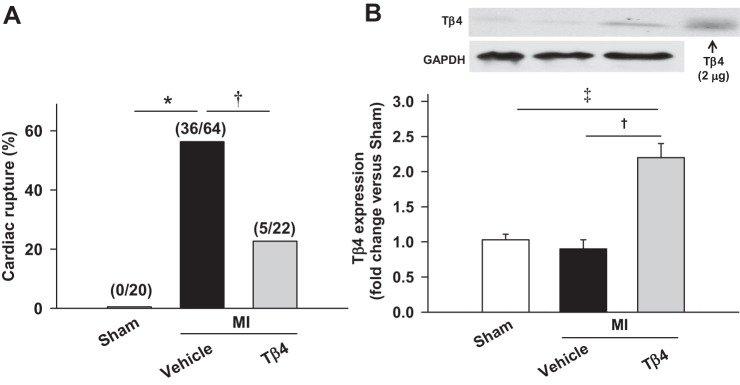
A: percentage of cardiac rupture deaths in C57BL/6 mice during the first week after myocardial infarction (MI). Thymosin-β4 (Tβ4) significantly reduced the incidence of cardiac rupture induced by MI. Values in parentheses are numbers of mice in the sham-operated (sham), MI + vehicle, and MI + Tβ4 groups. B: Tβ4 contents in mouse hearts, showing doubled cardiac Tβ4 in the Tβ4-treated group compared with nontreated groups. *P < 0.005; †P < 0.05; ‡P < 0.005.
Cardiac remodeling and function on day 7 after MI.
Echocardiographic data from surviving mice are shown in Table 1. MI caused LV chamber dilatation, as shown by increased LVDd and LVAd. Cardiac function as measured by EF and SF was markedly reduced by MI. Both MI-induced LV chamber dilation and cardiac dysfunction were not changed by 7-day Tβ4 treatment. Mice with MI had increased LV weight, which was not affected by 7-day treatment with Tβ4 (Table 1). No significant difference in infarct size was noted between vehicle- and Tβ4-treated groups at 7 days post-MI (Table 1).
Table 1.
Infarct size, LV weight, and echocardiographic measurements 7 days post-MI
| Parameter | Sham Group | MI + Vehicle Group | MI + Tβ4 Group |
|---|---|---|---|
| Infarct size, % | N/A | 37.4 ± 4.0 | 42.3 ± 4.0 |
| LV diastolic dimension, mm | 2.60 ± 0.08 | 5.10 ± 0.04* | 5.24 ± 0.50† |
| LV diastolic area, mm2 | 4.94 ± 0.58 | 23.06 ± 4.08* | 25.07 ± 4.31† |
| Diastolic posterior wall thickness, mm | 0.82 ± 0.02 | 0.82 ± 0.03 | 0.85 ± 0.01 |
| Ejection fraction, % | 75.16 ± 3.25 | 27.54 ± 3.87* | 30.64 ± 3.52† |
| Shortening fraction, % | 51.88 ± 2.12 | 18.61 ± 2.89* | 27.18 ± 4.31† |
| LV weight, mg/10 g body wt | 32.36 ± 1.35 | 49.26 ± 1.74* | 49.41 ± 2.95* |
| Body weight, g | 26.74 ± 0.83 | 25.35 ± 0.75 | 24.75 ± 0.84 |
Values are means ± SE; n = 8–10 animals/group. Animals were allotted to the following three groups: 1) sham operated (sham), 2) myocardial infarction (MI) + vehicle, and 3) MI + thymosin-β4 (Tβ4). LV, left ventricular; N/A, not applicable.
P < 0.005 and
P < 0.05 vs. the sham group.
Inflammatory cell infiltration in the LV.
Immunohistochemical experiments showed markedly increased infiltration of both macrophages and neutrophils in the LV infarct border 7 days after MI (Fig. 2A). Numbers of macrophages were almost doubled compared with numbers of neutrophils in the MI + vehicle group. Tβ4 significantly reduced numbers of infiltrating macrophages (Fig. 2B) and neutrophils (Fig. 2C).
Fig. 2.

A: representative images of immunohistochemical staining for macrophages (top) and neutrophils (bottom) in the region of the infarct border at 7 days post-MI. B and C: quantitative analysis of CD68-positive cells (a marker for macrophage) and lymphocyte antigen (Ly) 6B.2-positive cells (a marker for neutrophils), respectively, showing that Tβ4 significantly reduced infiltrating macrophages and neutrophils. D: ICAM-1 (80 kDa) expression in the myocardium was significantly increased at 7 days post-MI, which was almost prevented by Tβ4. n = 5–7 animals/group. *P < 0.01; †P < 0.05; ‡P < 0.01.
Activation of ICAM-1 signaling plays an important role in the recruitment of inflammatory immune cells, including macrophages (12). Tβ4 exerts anti-inflammatory actions by inhibiting proinflammatory mediators in vivo (35, 46). Thus, we further examined whether inhibited macrophage infiltration by Tβ4 was associated with reduced expression of ICAM-1 in the LV. We found that ICAM-1 expression was significantly decreased in Tβ4-treated mice compared with vehicle-treated mice (Fig. 2D).
Gelatinolytic activity in the border of infarct area.
In situ zymography analysis revealed that gelatinolytic activity was not detectable in the normal myocardium (Fig. 3A). However, it increased markedly in the infarct border on day 7 post-MI, which was significantly inhibited in animals treated with Tβ4 (Fig. 3).
Fig. 3.

A: representative images of in situ zymography for gelatinolytic activity in the region near the infarct border. No clear gelatinolytic activity was detected in the sham heart. B: semiquantitative analysis of gelatinolytic activity showing that MI increased gelatinolytic activity, which was significantly blunted by Tβ4. n = 4–6 animals/group. *P < 0.005; †P < 0.05; ‡P < 0.01.
Angiogenesis.
Numbers of CD31-positive capillaries were markedly reduced in the infarct border in the MI + vehicle group compared with the sham group, whereas Tβ4 treatment significantly increased numbers of capillaries, indicating an enhanced angiogenic response (Fig. 4).
Fig. 4.

A: representative images of capillary vessels as indicated by positive staining for CD31 in the region of the infarct border. B: quantitative analysis of numbers of capillary vessels showing that capillary vessels were markedly reduced after MI but significantly increased with Tβ4 treatment. n = 5–6 animals/group. *P < 0.005; †P < 0.05; ‡P < 0.01.
TUNEL-positive cardiomyocytes.
TUNEL-positive cells were barely found in the myocardium of sham mice, as shown in Fig. 5A. Numbers of TUNEL-positive cardiomyocytes were remarkably increased post-MI, and positive myocytes were mainly located near the infarct area. Tβ4 treatment for 7 days markedly reduced TUNEL-positive cells (Fig. 5C).
Fig. 5.

A: representative images of TUNEL-positive myocytes in the left ventricular (LV) myocardium. TUNEL-positive nuclei are shown in bright green, cardiomyocytes in red (α-actinin), and nuclei in blue [4′,6-diamidino-2-phenylindole (DAPI)]. No clear TUNEL-positive myocytes were found in the sham heart. Numbers of positive cells increased significantly at 7 days post-MI, which were reduced by Tβ4. B: representative merged image showing TUNEL-positive myocytes (white arrow). C: quantitative analysis of the number of TUNEL-positive myocytes. D: p53 expression in the myocardium was markedly induced after MI, which was significantly downregulated by Tβ4 treatment. n = 6–8 animals/group. *P < 0.005; †P < 0.005; ‡P < 0.005.
The tumor suppressor protein p53 mediates cell apoptosis through its activity as a transcription factor (49). p53 expression in the myocardium was barely detected by Western blot analysis. MI markedly induced p53 protein expression at 7 days post-MI in the MI + vehicle group, and this expression was partially but significantly downregulated by Tβ4 treatment (Fig. 5D).
Protocol 2
Cardiac remodeling and function 5 wk after MI.
Echocardiographic data are shown in Figs. 6 and 7. MI caused LV chamber dilatation, as detected by increase in LVDd and LVAd. Cardiac function, as assessed by EF and SF, was markedly reduced by MI. However, by week 5, Tβ4 treatment significantly prevented LV dilation and improved cardiac function, as evidenced by increased EF and SF in the MI + Tβ4 group (Figs. 6, A–C, and 7). PWTd was similar among sham (0.83 ± 0.02 mm), MI + vehicle (0.85 ± 0.02 mm), and MI + Tβ4 groups (0.85 ± 0.02 mm). Untreated mice with MI had increased LV weight, which was not affected by Tβ4 with 5-wk treatment (Fig. 6D). Infarct size was similar between vehicle-treated (38.9 ± 3.1%) and Tβ4-treated (34.4 ± 4.0%) groups at 5 wk post-MI.
Fig. 6.

A: representative M-mode echocardiography performed on conscious mice given either vehicle or Tβ4 for 5 wk after MI. B and C: LV diastolic dimension (LVDd; B) and LV diastolic area (LVAd; C). LV chamber dilation in mice with MI was significantly prevented by Tβ4 treatment. D: LV weight-to-body weight ratio (LVW/BW) in mice with or without Tβ4 treatment after MI. n = 10–14 animals/group. *P < 0.005; †P < 0.05; ‡P < 0.005.
Fig. 7.
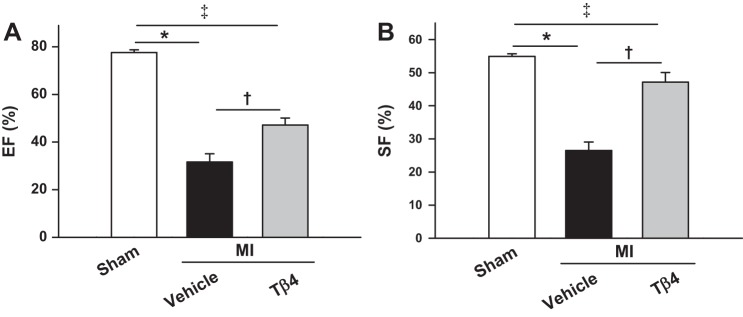
A and B: LV ejection fraction (EF; A) and shortening fraction (SF; B) in mice treated with or without Tβ4 post-MI, showing significantly increased cardiac function by five-wk Tβ4 treatment. n = 10–12 animals/group. *P = 0.0001; †P < 0.005; ‡P < 0.005.
Capillary density and collagen deposition.
Capillary density was significantly lowered in the MI + vehicle group compared with the sham group; 5-wk treatment with Tβ4 led to a significant increase in capillaries in the myocardium (Fig. 8, A, top, and B). MI also caused cardiac fibrosis, as indicated by increased myocardial ICF in the MI + vehicle group. Tβ4 significantly inhibited collagen deposition in the heart after MI (Fig. 8, A, bottom, and C).
Fig. 8.

A: representative images of capillaries (red stained, top) and interstitial collagen (green stained, bottom) in the LV 5 wk after MI. B and C: quantitative analysis of capillary density (B) and interstitial collagen fraction (ICF; C) showing that MI caused a decrease in capillary density and an increase in ICF and that these adverse effects were significantly prevented by Tβ4. n = 5–8 animals/group. *P < 0.005, vehicle-treated vs. sham groups; †P < 0.01; ‡P < 0.05.
DISCUSSION
The major findings of the present study are as follows: 1) mice treated with Tβ4 exhibited a reduced mortality rate as a result of decreased cardiac rupture during the acute MI; 2) MI was associated with an excessive inflammatory response, as demonstrated by the increases in neutrophil and macrophage infiltration and ICAM-1 expression, enhanced gelatinolytic activity, reduced numbers of capillaries, and increased cardiomyocyte apoptotsis in the heart; 3) these excessive inflammatory responses were partially prevented by Tβ4; and 4) Tβ4 treatment for 5 wk ameliorated LV dilatation and improved cardiac function, and it reduced interstitial fibrosis and increased capillary density in the myocardium.
MI evokes intense inflammatory responses both systemically and within the infarcted myocardium. Although inflammatory responses are essential for post-MI wound healing and scar formation, excessive inflammatory responses and ECM remodeling can lead to harmful consequences (15, 16), such as cardiac rupture, maladaptive LV remodeling, and cardiac dysfunction. Clinical studies have shown that hearts from patients with cardiac rupture revealed a higher density of inflammatory cells and more abundant MMP-9 expression in the infarct myocardium compared with MI patients without cardiac rupture (23, 48). We and others (6, 51, 56) have reported that in murine models of acute MI, cardiac rupture mainly occurs at 3–5 days post-MI and that neutrophils constitute the majority of of infiltrating cells within the first 1–2 days after MI, which are followed by a marked accumulation of macrophages on days 3–4. The peak times of rupture occurrence and macrophage infiltration in the infarcted myocardium overlap (at days 3–5 post-MI), indicating macrophages as the major inflammatory cell population correlated to the risk of cardiac rupture. Here, we report, for the first time, that Tβ4 significantly reduced the incidence of cardiac rupture, which can be attributed to its anti-inflammatory action, as evidenced by the reduced numbers of infiltrating macrophages and neutrophils in the infarct border.
Pronounced inflammatory cell infiltration is always associated with upregulated expression of proinflammatory cytokines and mediators and with enhanced expression and activities of gelatinolytic proteinases (13). MMP-2 and MMP-9 are the typical gelatinolytic proteinases. As demonstrated by Heymans et al. (20), deletion of MMP-9 significantly lowered cardiac rupture in mouse models of MI. Similarly, MMP-2 expression in the heart was also elevated after MI; disruption of MMP-2 decreased mortality caused by post-MI cardiac rupture compared with wild-type mice (27). Together, these clinical and animal experimental results suggest that excessive leukocyte infiltration and gelatinolytic proteinase expression are closely associated with cardiac rupture. Since Tβ4 has been shown to reduce inflammatory cell infiltration and downregulate inflammatory mediators (26, 45), these activities have been linked to its ability to decrease gelatinolytic proteinases and gene transcript levels of a variety of proinflammatory cytokines and chemokines. We tested whether Tβ4 prevented cardiac rupture after MI due to its anti-inflammatory actions. Indeed, Tβ4-treated mice had a low incidence of cardiac rupture and reduced inflammatory responses, as indicated by a decrease in inflammatory cell infiltration and gelatinolytic activity in the infarct border region. Our findings are consistent with both clinical and experimental results from other studies showing that excessive cardiac inflammation constitutes one of the central mechanisms of cardiac rupture pathogenesis. ICAM-1 is expressed on the membranes of leukocytes and endothelial cells, and it binds to integrins of type CD11a/CD18 (lymphocytes) or CD11b/CD18 (macrophages) (38, 53). ICAM-1 signaling can mediate the recruitment of inflammatory immune cells (12). In our murine model of MI, ICAM-1 expression in the myocardium was significantly increased, which was consistent with the clinical findings showing that serum levels of ICAM-1 in patients within the first week of MI were significantly higher than those in normal control subjects (11); this enhanced ICAM-1 expression was inhibited by Tβ4. The changes in ICAM-1 expression in our study corresponded to changes in macrophage infiltration, indicating the role of ICAM-1 in macrophage recruitment. Thus, we believe that Tβ4 may have decreased the numbers of infiltrating macrophages in part by decreasing local ICAM-1 levels post-MI. In clinical studies, proinflammatory cytokines, including IL-8, have been shown to be induced during MI through the NF-κB pathway in the myocardium (9) and to predict the development of heart failure after MI (10, 30). Importantly, Tβ4 inhibits NF-κB activation and thus the expression of the downstream IL-8 gene as well (35). Therefore, we can speculate that the anti-inflammatory effects of Tβ4 in the heart after MI could be due to its inhibition on NF-κB activation and thus the expression of proinflammatory cytokines/chemokines.
Heart angiogenesis/neovascularization is implicated to play a role in improving prognosis post-MI (41, 42). Tβ4 has been shown to promote coronary vessel development and collateral growth not only during embryonic development but also in the adult epicardium by stimulating epicardial vascular progenitors, which migrate and differentiate into smooth muscle and endothelial cells (44). Indeed, in the present study, we showed that Tβ4 exerted a proangiogenic effect at an early stage of MI, as evidenced by the increased numbers of CD31-postive capillaries in the infarct border. Since newly formed capillaries are an important component of tissue granulation and repair, the enhanced angiogenic/vasculogenic response may contribute greatly to the reduced incidence of cardiac rupture in the acute phase of MI and also later on to the improved cardiac remodeling and function.
Both experimental (3, 39) and human (31, 40) studies have detected increased numbers of TUNEL-positive cardiomyocytes in post-MI hearts, as early as the initiation of acute MI, and as well as up to 6 mo post-MI, along with LV dilation and contractile dysfunction. This implies that myocyte death may have an important pathogenic role in early and late LV remodeling and dysfunction after MI (39, 50). The tumor suppressor protein p53 regulates cell cycle activity and apoptosis through its activity as a transcription factor (49). Matsusaka et al. (28) has shown that p53−/− mice with MI exhibited improved survival entirely due to a reduced rupture death rate, associated with increased infarct wall thickness and markedly reduced numbers of apoptotic myocytes in the infarct region. Notably, these mice also had better preservation of LV function on day 3 after MI. These studies demonstrated that p53-dependent myocyte apoptosis is involved not only in early cardiac rupture (28) but also in late remodeling and dysfunction (39) after MI. Importantly, Tβ4 markedly decreased cardiomyocyte apoptosis, as assessed by TUNEL assay 24 h after ligation in an acute MI mouse model (4). Consistent with other studies, we observed notable apoptotic myocytes and high levels of p53 expression in the myocardium of mice 7 days after MI, which were partially prevented by Tβ4. The reduced incidence of cardiac rupture and restored cardiac function by Tβ4 in MI models in the present study might be due to negative regulation of p53 by Tβ4 and thus reduced myocyte apoptosis mediated by Tβ4.
Tβ4 treatment for 5 wk significantly ameliorated LV dilatation and improved cardiac function, as evidenced by the increase in SF and EF post-MI, which were associated with significantly increased numbers of capillary and decreased cardiac fibrosis in the LV. Our results are consistent with previously published data showing improved cardiac function as evaluated by echocardiography after 2- and 4-wk treatment with Tβ4 using the same model (4). As found in the present study and pilot studies, Tβ4 limited excessive inflammation and promoted cardiomyocyte survival and angiogenesis after ischemic heart injury (4, 7, 21). These studies demonstrated a strong rationale for the use of Tβ4 to prevent cardiac damage and promote regeneration after an infarction. Since N-acetyl-seryl-aspartyl-lysyl-proline, an anti-inflammatory and anti-fibrotic tetrapeptide (33, 37, 52), is released by endoproteinase cleavage of the NH2-terminal sequence of Tβ4 (5), Tβ4-induced antifibrotic effect on the heart could be mediated by increased N-acetyl-seryl-aspartyl-lysyl-proline and/or indirectly due to its negative role on inflammation. Together, these findings show that Tβ4 has a clear, positive role in cardioprotection and repair. Tβ4 is currently under a phase II clinical trial for the treatment of patients with acute MI (http://clinicaltrials.gov/ct2/show/NCT01311518).
In conclusion, the present study shows, for the first time, that 1) Tβ4 is able to reduce incidence of cardiac rupture post-MI and markedly improve cardiac function at 5 wk after MI and 2) the cardiac protective effects of Tβ4 after MI were associated with a significant reduction of the excessive inflammatory response and cardiomyocyte apoptosis as well as with an enhancement of angiogenesis. In the clinical point of view, Tβ4 could potentially be a therapeutic candidate for patients with acute MI.
GRANTS
This work was supported by the Henry Ford Health System institutional fund (to N.-E. Rhaleb) and in part by National Heart, Lung, and Blood Institute Grant HL-028982 (to O. A. Carretero).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: H.P., J.X., and X.D. performed experiments; H.P. and E.L.P. analyzed data; H.P., X.-P.Y., and N.-E.R. interpreted results of experiments; H.P. and N.-E.R. prepared figures; H.P. and N.-E.R. drafted manuscript; X.-P.Y., O.A.C., and N.-E.R. edited and revised manuscript; O.A.C. and N.-E.R. conception and design of research; N.-E.R. approved final version of manuscript.
ACKNOWLEDGMENTS
The author thank RegeneRx Biopharmaceuticals for providing Tβ4 and are grateful to the scientific editorial work of Branislava Janic.
REFERENCES
- 1.Batts KP, Ackermann DM, Edwards WD. Postinfarction rupture of the left ventricular free wall: clinicopathologic correlates in 100 consecutive autopsy cases. Hum Pathol 21: 530–535, 1990 [DOI] [PubMed] [Google Scholar]
- 2.Becker RC, Hochman JS, Cannon CP, Spencer FA, Ball SP, Rizzo MJ, Antman EM. Fatal cardiac rupture among patients treated with thrombolytic agents and adjunctive thrombin antagonists: observations from the Thrombolysis and Thrombin Inhibition in Myocardial Infarction 9 Study. J Am Coll Cardiol 33: 479–487, 1999 [DOI] [PubMed] [Google Scholar]
- 3.Bialik S, Geenen DL, Sasson IE, Cheng R, Horner JW, Evans SM, Lord EM, Koch CJ, Kitsis RN. Myocyte apoptosis during myocardial infarction in the mouse localizes to hypoxic regions but occurs independently of p53. J Clin Invest 100: 1363–1372, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bock-Marquette I, Saxena A, White MD, DiMaio JM, Srivastava D. Thymosin β4 activates integrin-linked kinase and promotes cardiac cell migration, survival and cardiac repair. Nature 432: 466–472, 2004 [DOI] [PubMed] [Google Scholar]
- 5.Cavasin MA. Therapeutic potential of thymosin-beta4 and its derivative N-acetyl-seryl-aspartyl-lysyl-proline (Ac-SDKP) in cardiac healing after infarction. Am J Cardiovasc Drugs 6: 305–311, 2006 [DOI] [PubMed] [Google Scholar]
- 6.Cavasin MA, Tao Z, Menon S, Yang XP. Gender differences in cardiac function during early remodeling after acute myocardial infarction in mice. Life Sci 75: 2181–2192, 2004 [DOI] [PubMed] [Google Scholar]
- 7.Crockford D. Development of thymosin β4 for treatment of patients with ischemic heart disease. Ann NY Acad Sci 1112: 385–395, 2007 [DOI] [PubMed] [Google Scholar]
- 8.Crockford D, Turjman N, Allan C, Angel J. Thymosin β4: structure, function, and biological properties supporting current and future clinical applications. Ann NY Acad Sci 1194: 179–189, 2010 [DOI] [PubMed] [Google Scholar]
- 9.Damas JK, Eiken HG, Oie E, Bjerkeli V, Yndestad A, Ueland T, Tonnessen T, Geiran OR, Aass H, Simonsen S, Christensen G, Froland SS, Attramadal H, Gullestad L, Aukrust P. Myocardial expression of CC- and CXC-chemokines and their receptors in human end-stage heart failure. Cardiovasc Res 47: 778–787, 2000 [DOI] [PubMed] [Google Scholar]
- 10.De GL, Brunetti ND, Montrone D, De RF, Cuculo A, Di BM. Subacute inflammatory activation in subjects with acute coronary syndrome and left ventricular dysfunction. Inflammation 35: 363–370, 2012 [DOI] [PubMed] [Google Scholar]
- 11.Elahi AW, Vijayakumar AN, Lichstein E, Mokhtarian F. Interplay of antibody and T cell responses in acute myocardial infarction. J Lab Clin Med 138: 112–118, 2001 [DOI] [PubMed] [Google Scholar]
- 12.Etienne-Manneville S, Chaverot N, Strosberg AD, Couraud PO. ICAM-1-coupled signaling pathways in astrocytes converge to cyclic AMP response element-binding protein phosphorylation and TNF-α secretion. J Immunol 163: 668–674, 1999 [PubMed] [Google Scholar]
- 13.Fang L, Gao XM, Samuel CS, Su Y, Lim YL, Dart AM, Du XJ. Higher levels of collagen and facilitated healing protect against ventricular rupture following myocardial infarction. Clin Sci (Lond) 115: 99–106, 2008 [DOI] [PubMed] [Google Scholar]
- 14.Figueras J, Curos A, Cortadellas J, Sans M, Soler-Soler J. Relevance of electrocardiographic findings, heart failure, and infarct site in assessing risk and timing of left ventricular free wall rupture during acute myocardial infarction. Am J Cardiol 76: 543–547, 1995 [DOI] [PubMed] [Google Scholar]
- 15.Frangogiannis NG. The immune system and cardiac repair. Pharmacol Res 58: 88–111, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res 53: 31–47, 2002 [DOI] [PubMed] [Google Scholar]
- 17.Gao XM, White DA, Dart AM, Du XJ. Post-infarct cardiac rupture: recent insights on pathogenesis and therapeutic interventions. Pharmacol Ther 134: 156–179, 2012 [DOI] [PubMed] [Google Scholar]
- 18.Gkantidis N, Katsaros C, Chiquet M. Detection of gelatinolytic activity in developing basement membranes of the mouse embryo head by combining sensitive in situ zymography with immunolabeling. Histochem Cell Biol 138: 557–571, 2012 [DOI] [PubMed] [Google Scholar]
- 19.Hannappel E. β-Thymosins. Ann NY Acad Sci 1112: 21–37, 2007 [DOI] [PubMed] [Google Scholar]
- 20.Heymans S, Luttun A, Nuyens D, Theilmeier G, Creemers E, Moons L, Dyspersin GD, Cleutjens JP, Shipley M, Angellilo A, Levi M, Nübe O, Baker A, Keshet E, Lupu F, Herbert JM, Smits JF, Shapiro SD, Baes M, Borgers M, Collen D, Daemen MJ, Carmeliet P. Inhibition of plasminogen activators or matrix metalloproteinases prevents cardiac rupture but impairs therapeutic angiogenesis and causes cardiac failure. Nat Med 5: 1135–1142, 1999 [DOI] [PubMed] [Google Scholar]
- 21.Hinkel R, El-Aouni C, Olson T, Horstkotte J, Mayer S, Muller S, Willhauck M, Spitzweg C, Gildehaus FJ, Munzing W, Hannappel E, Bock-Marquette I, DiMaio JM, Hatzopoulos AK, Boekstegers P, Kupatt C. Thymosin beta4 is an essential paracrine factor of embryonic endothelial progenitor cell-mediated cardioprotection. Circulation 117: 2232–2240, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hutchins KD, Skurnick J, Lavenhar M, Natarajan GA. Cardiac rupture in acute myocardial infarction: a reassessment. Am J Forensic Med Pathol 23: 78–82, 2002 [DOI] [PubMed] [Google Scholar]
- 23.Kameda K, Matsunaga T, Abe N, Fujiwara T, Hanada H, Fukui K, Fukuda I, Osanai T, Okumura K. Increased pericardial fluid level of matrix metalloproteinase-9 activity in patients with acute myocardial infarction: possible role in the development of cardiac rupture. Circ J 70: 673–678, 2006 [DOI] [PubMed] [Google Scholar]
- 24.Kawano H, Miyauchi K, Okada R, Daida H, Yokoi H, Miyano H, Takaya J, Satoh H, Yamaguchi H, Suda K, Shirai T. Histopathological study of cardiac rupture following myocardial infarction with and without thrombolytic therapy. J Cardiol 24: 249–255, 1994 [PubMed] [Google Scholar]
- 25.Liu YH, Yang XP, Sharov VG, Nass O, Sabbah HN, Peterson E, Carretero OA. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure. Role of kinins and angiotensin II type 2 receptors. J Clin Invest 99: 1926–1935, 1997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Malinda KM, Sidhu GS, Mani H, Banaudha K, Maheshwari RK, Goldstein AL, Kleinman HK. Thymosin β4 accelerates wound healing. J Invest Dermatol 113: 364–368, 1999 [DOI] [PubMed] [Google Scholar]
- 27.Matsumura S, Iwanaga S, Mochizuki S, Okamoto H, Ogawa S, Okada Y. Targeted deletion or pharmacological inhibition of MMP-2 prevents cardiac rupture after myocardial infarction in mice. J Clin Invest 115: 599–609, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Matsusaka H, Ide T, Matsushima S, Ikeuchi M, Kubota T, Sunagawa K, Kinugawa S, Tsutsui H. Targeted deletion of p53 prevents cardiac rupture after myocardial infarction in mice. Cardiovasc Res 70: 457–465, 2006 [DOI] [PubMed] [Google Scholar]
- 29.Mora CA, Baumann CA, Paino JE, Goldstein AL, Badamchian M. Biodistribution of synthetic thymosin β4 in the serum, urine, and major organs of mice. Int J Immunopharmacol 19: 1–8, 1997 [DOI] [PubMed] [Google Scholar]
- 30.Nymo SH, Hulthe J, Ueland T, McMurray J, Wikstrand J, Askevold ET, Yndestad A, Gullestad L, Aukrust P. Inflammatory cytokines in chronic heart failure: interleukin-8 is associated with adverse outcome. Results from CORONA. Eur J Heart Fail 16: 68–75, 2014 [DOI] [PubMed] [Google Scholar]
- 31.Olivetti G, Quaini F, Sala R, Lagrasta C, Corradi D, Bonacina E, Gambert SR, Cigola E, Anversa P. Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J Mol Cell Cardiol 28: 2005–2016, 1996 [DOI] [PubMed] [Google Scholar]
- 32.Ortiz-Perez JT, Riera M, Bosch X, De Caralt TM, Perea RJ, Pascual J, Soler MJ. Role of circulating angiotensin converting enzyme 2 in left ventricular remodeling following myocardial infarction: a prospective controlled study. PLoS One 8: e61695, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Peng H, Carretero OA, Liao TD, Peterson EL, Rhaleb NE. Role of N-acetyl-seryl-aspartyl-lysyl-proline in the antifibrotic and anti-inflammatory effects of the angiotensin-converting enzyme inhibitor captopril in hypertension. Hypertension 49: 695–703, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pouleur AC, Barkoudah E, Uno H, Skali H, Finn PV, Zelenkofske SL, Belenkov YN, Mareev V, Velazquez EJ, Rouleau JL, Maggioni AP, Kober L, Califf RM, McMurray JJ, Pfeffer MA, Solomon SD. Pathogenesis of sudden unexpected death in a clinical trial of patients with myocardial infarction and left ventricular dysfunction, heart failure, or both. Circulation 122: 597–602, 2010 [DOI] [PubMed] [Google Scholar]
- 35.Qiu P, Wheater MK, Qiu Y, Sosne G. Thymosin β4 inhibits TNF-α-induced NF-κB activation, IL-8 expression, and the sensitizing effects by its partners PINCH-1 and ILK. FASEB J 25: 1815–1826, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reddy SG, Roberts WC. Frequency of rupture of the left ventricular free wall or ventricular septum among necropsy cases of fatal acute myocardial infarction since introduction of coronary care units. Am J Cardiol 63: 906–911, 1989 [DOI] [PubMed] [Google Scholar]
- 37.Rhaleb NE, Peng H, Yang XP, Liu YH, Mehta D, Ezan E, Carretero OA. Long-term effect of N-acetyl-seryl-aspartyl-lysyl-proline on left ventricular collagen deposition in rats with 2-kidney, 1-clip hypertension. Circulation 103: 3136–3141, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rothlein R, Dustin ML, Marlin SD, Springer TA. A human intercellular adhesion molecule (ICAM-1) distinct from LFA-1. J Immunol 137: 1270–1274, 1986 [PubMed] [Google Scholar]
- 39.Sam F, Sawyer DB, Chang DL, Eberli FR, Ngoy S, Jain M, Amin J, Apstein CS, Colucci WS. Progressive left ventricular remodeling and apoptosis late after myocardial infarction in mouse heart. Am J Physiol Heart Circ Physiol 279: H422–H428, 2000 [DOI] [PubMed] [Google Scholar]
- 40.Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M, Voipio-Pulkki LM. Apoptosis in human acute myocardial infarction. Circulation 95: 320–323, 1997 [DOI] [PubMed] [Google Scholar]
- 41.Sellke FW, Simons M. Angiogenesis in cardiovascular disease. Current status and therapeutic potential. Drugs 58: 391–396, 1999 [DOI] [PubMed] [Google Scholar]
- 42.Semenza GL. Angiogenesis in ischemic and neoplastic disorders. Annu Rev Med 54: 17–28, 2003 [DOI] [PubMed] [Google Scholar]
- 43.Shamshad F, Kenchaiah S, Finn PV, Soler-Soler J, McMurray JJ, Velazquez EJ, Maggioni AP, Califf RM, Swedberg K, Kober L, Belenkov Y, Varshavsky S, Pfeffer MA, Solomon SD. Fatal myocardial rupture after acute myocardial infarction complicated by heart failure, left ventricular dysfunction, or both: the Valsartan In Acute Myocardial Infarction Trial (VALIANT). Am Heart J 160: 145–151, 2010 [DOI] [PubMed] [Google Scholar]
- 44.Smart N, Risebro CA, Melville AA, Moses K, Schwartz RJ, Chien KR, Riley PR. Thymosin β4 induces adult epicardial progenitor mobilization and neovascularization. Nature 445: 177–182, 2007 [DOI] [PubMed] [Google Scholar]
- 45.Sosne G, Christopherson PL, Barrett RP, Fridman R. Thymosin-β4 modulates corneal matrix metalloproteinase levels and polymorphonuclear cell infiltration after alkali injury. Invest Ophthalmol Vis Sci 46: 2388–2395, 2005 [DOI] [PubMed] [Google Scholar]
- 46.Sosne G, Szliter EA, Barrett R, Kernacki KA, Kleinman H, Hazlett LD. Thymosin β4 promotes corneal wound healing and decreases inflammation in vivo following alkali injury. Exp Eye Res 74: 293–299, 2002 [DOI] [PubMed] [Google Scholar]
- 47.Tao ZY, Cavasin MA, Yang F, Liu YH, Yang XP. Temporal changes in matrix metalloproteinase expression and inflammatory response associated with cardiac rupture after myocardial infarction in mice. Life Sci 74: 1561–1572, 2004 [DOI] [PubMed] [Google Scholar]
- 48.van den Borne SW, Cleutjens JP, Hanemaaijer R, Creemers EE, Smits JF, Daemen MJ, Blankesteijn WM. Increased matrix metalloproteinase-8 and -9 activity in patients with infarct rupture after myocardial infarction. Cardiovasc Pathol 18: 37–43, 2009 [DOI] [PubMed] [Google Scholar]
- 49.Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer 2: 594–604, 2002 [DOI] [PubMed] [Google Scholar]
- 50.Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest 111: 1497–1504, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yang F, Liu YH, Yang XP, Xu J, Kapke A, Carretero OA. Myocardial infarction and cardiac remodelling in mice. Exp Physiol 87: 547–555, 2002 [DOI] [PubMed] [Google Scholar]
- 52.Yang F, Yang XP, Liu YH, Xu J, Cingolani O, Rhaleb NE, Carretero OA. Ac-SDKP reverses inflammation and fibrosis in rats with heart failure after myocardial infarction. Hypertension 43: 229–236, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yang L, Froio RM, Sciuto TE, Dvorak AM, Alon R, Luscinskas FW. ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-α-activated vascular endothelium under flow. Blood 106: 584–592, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang XP, Liu YH, Rhaleb NE, Kurihara N, Kim HE, Carretero OA. Echocardiographic assessment of cardiac function in conscious and anesthetized mice. Am J Physiol Heart Circ Physiol 277: H1967–H1974, 1999 [DOI] [PubMed] [Google Scholar]
- 55.Yang XP, Liu YH, Shesely EG, Bulagannawar M, Liu F, Carretero OA. Endothelial nitric oxide gene knockout mice. Cardiac phenotypes and the effect of angiotensin-converting enzyme inhibitor on myocardial ischemia/reperfusion injury. Hypertension 34: 24–30, 1999 [DOI] [PubMed] [Google Scholar]
- 56.Yang Y, Ma Y, Han W, Li J, Xiang Y, Liu F, Ma X, Zhang J, Fu Z, Su YD, Du XJ, Gao XM. Age-related differences in postinfarct left ventricular rupture and remodeling. Am J Physiol Heart Circ Physiol 294: H1815–H1822, 2008 [DOI] [PubMed] [Google Scholar]