

Abstract
We tested the hypothesis that activation of inwardly rectifying potassium (KIR) channels and Na+-K+-ATPase, two pathways that lead to hyperpolarization of vascular cells, contributes to both the onset and steady-state hyperemic response to exercise. We also determined whether after inhibiting these pathways nitric oxide (NO) and prostaglandins (PGs) are involved in the hyperemic response. Forearm blood flow (FBF; Doppler ultrasound) was determined during rhythmic handgrip exercise at 10% maximal voluntary contraction for 5 min in the following conditions: control [saline; trial 1 (T1)]; with combined inhibition of KIR channels and Na+-K+-ATPase alone [via barium chloride (BaCl2) and ouabain, respectively; trial 2 (T2)]; and with additional combined nitric oxide synthase (NG-monomethyl-l-arginine) and cyclooxygenase inhibition [ketorolac; trial 3 (T3)]. In T2, the total hyperemic responses were attenuated ∼50% from control (P < 0.05) at exercise onset, and there was minimal further effect in T3 (protocol 1; n = 11). In protocol 2 (n = 8), steady-state FBF was significantly reduced during T2 vs. T1 (133 ± 15 vs. 167 ± 17 ml/min; Δ from control: −20 ± 3%; P < 0.05) and further reduced during T3 (120 ± 15 ml/min; −29 ± 3%; P < 0.05 vs. T2). In protocol 3 (n = 8), BaCl2 alone reduced FBF during onset (∼50%) and steady-state exercise (∼30%) as observed in protocols 1 and 2, respectively, and addition of ouabain had no further impact. Our data implicate activation of KIR channels as a novel contributing pathway to exercise hyperemia in humans.
Keywords: blood flow, contraction, skeletal muscle, vasodilation
the regulation of blood flow and oxygen delivery to contracting skeletal muscle is a complex response involving mechanical factors, the sympathetic nervous system, and local metabolic and endothelium-derived substances that influence vascular tone (47). In this context, prior studies in animal models established disruption of the endothelium attenuates the hyperemic response to muscle contraction (2, 44, 54) and subsequent studies have attempted to identify the endothelial-derived substances that may regulate blood flow during exercise. Local metabolic substances independent of the endothelium, such as K+, have also been suggested to be involved in exercise hyperemia (23, 29, 63), particularly at the onset of muscle contractions (1, 7, 40). However, to date, obligatory local vasodilators for the hyperemia at exercise onset and during steady-state conditions are still questioned in humans (28).
Endothelium-dependent vasodilation occurs via the synthesis of nitric oxide (NO) and vasodilating prostaglandins (PGs; i.e., prostacyclin) (19) or endothelium-derived hyperpolarization (18, 20). In young healthy humans, the contribution of NO and PGs is minimal-to-modest during mild- and moderate-intensity isolated muscle mass exercise (4, 10, 41, 51). Beyond NO and PGs, the endothelium is capable of stimulating vascular smooth muscle hyperpolarization leading to decreased calcium influx through voltage-gated calcium channels and thus vascular smooth muscle cell relaxation and subsequent vasodilation. Often, the endothelial cell layer itself also hyperpolarizes and this can stimulate the direct (through myoendothelial projections) or indirect (through efflux of a “factor”) hyperpolarization of vascular smooth muscle cells (36). In animals models, prevention of hyperpolarization (22, 39) or ascending vasodilation (54), to which hyperpolarization largely contributes (16), attenuates contraction-induced hyperemia. A few studies in humans have addressed candidate hyperpolarizing pathways including cytochrome P450 metabolites (25), calcium-activated potassium (KCa) channels (41), and adenosine triphosphate (ATP)-sensitive potassium channels (KATP) (50); however, the reductions in steady-state exercise hyperemia by these inhibitors have been minimal or nonexistent and the contribution of these pathways to the onset hyperemia has not been explored.
Recently, we have shown that K+-stimulated hyperpolarization of resistance vessels via the activation of inwardly rectifying potassium (KIR) channels and Na+-K+-ATPase contributes to a large portion (∼50%) of the hyperemic response following a single muscle contraction in the human forearm and, combined with NO and PGs, accounts for nearly all (∼80%) of the immediate total vasodilator response (7). In addition, activation of KIR channels underlies the majority of ATP-mediated vasodilation (6), a substance proposed to be involved in vascular regulation during exercise in humans (21, 32, 46). Lastly, inhibition of KIR channels significantly reduces the immediate peak reactive hyperemic response to temporary muscle ischemia and combined inhibition of KIR channels and Na+-K+-ATPase nearly abolishes the total [area under the curve (AUC)] response (12). Thus pathways capable of hyperpolarizing vascular smooth muscle and/or endothelial cells are attractive candidates for obligatory signaling at the onset and during continued muscle contractions.
With this collective information as a background, the purpose of the present experiment was to test the hypothesis that activation of KIR channels and Na+-K+-ATPase contributes to exercise hyperemia both at the onset of muscle contractions and during steady-state exercise in humans. Furthermore, we sought to determine whether there would be a combined role for NO and PGs after inhibiting activation of KIR channels and Na+-K+-ATPase. Finally, we performed a follow-up study based on recent observations from our laboratory to identify whether there was an independent role of KIR channel activation in this complex response in humans.
METHODS
Subjects
With Colorado State University Institutional Review Board approval and after written informed consent, a total of 27 young healthy adults [protocol 1: 7 men, 4 women; age = 23 ± 1 yr; weight = 71.6 ± 2.6 kg; height = 172 ± 3 cm; body mass index = 24.1 ± 0.7 kg/m2; forearm volume (FAV) = 848 ± 41 ml; protocol 2: 4 men, 4 women; age = 24 ± 2 yr; weight = 69.0 ± 3.0 kg; height = 173 ± 3 cm; body mass index = 23.1 ± 0.8 kg/m2; FAV = 811 ± 62 ml; protocol 3: 5 men, 3 women; age = 21 ± 1 yr; weight = 71.0 ± 4.2 kg; height = 176 ± 2 cm; body mass index = 23.0 ± 1.0 kg/m2; FAV = 945 ± 93 ml; means ± SE] participated in the present study. All subjects were sedentary to moderately active, nonsmokers, nonobese, normotensive (resting blood pressure <140/90 mmHg), and not taking any medications. Studies were performed after an overnight fast and 24-h abstention from caffeine and exercise with subjects in the supine position with the experimental arm abducted to 90° and slightly elevated above heart level upon a tilt-adjustable table. Female subjects were studied during the early follicular phase of their menstrual cycle or placebo phase of oral contraceptive use to minimize any potential cardiovascular effects of sex-specific hormones.
Arterial Catheterization, Arterial Blood Pressure, and Heart Rate
A 20-gauge, 7.6-cm catheter was placed in the brachial artery of the nondominant arm under aseptic conditions after local anesthesia (2% lidocaine) for local administration of study drugs and blood sampling. The catheter was connected to a three-port connector as well as a pressure transducer for mean arterial pressure (MAP) measurement and continuously flushed at 3 ml/h with heparinized saline. The two side ports were used for drug infusions (11, 34). Heart rate (HR) was determined using a three-lead electrocardiogram (ECG; Cardiocap/5, Datex-Ohmeda, Louisville, CO).
Forearm Blood Flow and Vascular Conductance
A 12-MHz linear-array ultrasound probe (Vivid 7; General Electric, Milwaukee, WI) was used to determine brachial artery mean blood velocity (MBV) and brachial artery diameter. The probe was securely fixed to the skin over the brachial artery proximal to the catheter insertion site as previously described (11). For blood velocity measurements, the probe insonation angle was maintained at <60° and the frequency used was 5 MHz. The Doppler shift frequency spectrum was analyzed via a Multigon 500M TCD (Multigon Industries, Mt. Vernon, NY) spectral analyzer from which mean velocity was determined as a weighted mean of the spectrum of Doppler shift frequencies. In protocols 1 and 3, brachial artery diameter measurements were made in duplex mode from images recorded on a VHS tape at end-diastole and between contractions (in triplicate) during rest and at 6, 12, 24, 60, 90, 120, 180, 240, and 300 s of exercise. All measurements were made by the same operator. An exponential line of best fit was generated for these data, and diameters were extrapolated from this function at relevant MBV time points. We utilized this approach in an attempt to minimize the effect of random diameter measurement error on blood flow (49). In protocol 2, diameter measurements were made in triplicate at rest and the end of exercise. Forearm blood flow (FBF) was then calculated as:
where the FBF is in ml/min, the MBV is in cm/s, the brachial diameter is in cm, and 60 was used to convert from ml/s to ml/min. Forearm vascular conductance (FVC) was calculated as (FBF/MAP) × 100 and expressed as ml·min−1·100 mmHg−1. All studies were performed in a cool (20–22°C) temperature-controlled environment with a fan directed toward the forearm to minimize the contribution of skin blood flow to forearm hemodynamics.
Rhythmic Handgrip Exercise
Maximal voluntary contraction (MVC; all subjects mean 41 ± 2 kg, range 22–67 kg) was determined for the experimental arm as the average of three maximal squeezes of a handgrip dynamometer (Stoelting, Chicago, IL) that were within 3% of each other. Forearm exercise during the trials was performed with weight corresponding to 10% MVC (mean 4.1 ± 0.2 kg, range 2.2–6.7 kg) attached to a pulley system and lifted 4–5 cm over the pulley at a duty cycle of 1-s contraction-2-s relaxation (20 contractions/min) using both visual and auditory feedback to ensure the correct timing as described previously (11, 34). We chose this mild intensity rhythmic handgrip exercise to limit the contribution of systemic hemodynamics and reflex activation of the sympathetic nervous system on exercise hyperemia (42, 52). Under these conditions, changes in cardiac output are not necessary to achieve the forearm hyperemia observed (42), and thus our experimental model isolates the effects of muscle contractions on local vascular control mechanisms. Additionally, previous studies in our laboratory have determined that MVC is not affected by any of the vasoactive substances, particularly barium chloride and ouabain, administered in these studies (7).
Vasoactive Drug Infusion
All vasoactive drug infusions occurred via the brachial artery catheter to create a local effect in the forearm, and saline was utilized as a control infusate. Specific timing and duration of infusions are provided in Experimental Protocols.
To inhibit select hyperpolarizing vasodilator mechanisms (6, 12), ouabain octahydrate (Na+-K+-ATPase inhibitor; Sigma 03125, St. Louis, MO) was infused at 2.7 nmol/min in combination with barium chloride (BaCl2; KIR channel inhibitor; 10% wt/vol BDH3238; EMD Chemicals, Gibbstown, NJ) at 0.9 μmol·dl FAV−1·min−1 with a minimum dose of 8 μmol/min to a maximum dose of 10 μmol/min. This dose of BaCl2 is the same as we have previously utilized in hyperemic conditions and, importantly, did not impact forearm vasodilation to the endothelium-independent vasodilator sodium nitroprusside (12). Ouabain and BaCl2 were prepared in saline and confirmed sterile and free of fungus/endotoxin and particulate matter with a standard microbiology report (JCB-Analytical Research Labs, Wichita, KS) before use.
To block additional endothelium-dependent vasodilator pathways of interest, we administered NG-monomethyl-l-arginine [l-NMMA; NO synthase (NOS) inhibitor; Clinalfa/Bachem, Weil am Rhein, Germany] to inhibit the production of NO in combination with ketorolac [nonselective cyclooxygenase (COX) inhibitor; Hospira, Lake Forest, IL] to inhibit the synthesis of PGs. The doses of l-NMMA and ketorolac were 5 mg/min and 1,200 μg/min, respectively. We have previously demonstrated these doses to be effective during a similar exercise protocol (11, 14, 51). Forearm volume used for normalization for specific vasoactive drugs was determined from regional analysis of whole body dual-energy X-ray absorptiometry scans (QDR series software; Hologic, Bedford, MA).
Experimental Protocols
Protocol 1: onset exercise hyperemia.
Experimental protocols are presented in Fig. 1. The main purpose of protocol 1 was to determine the effect of inhibiting KIR channel and Na+-K+-ATPase activation (via BaCl2 and ouabain) alone, and in combination with NOS-COX blockade (via l-NMMA and ketorolac, respectively) on the hyperemia that occurs at the onset of repeated muscle contractions. To do this, subjects performed 10% MVC rhythmic handgrip exercise for 5 min in control conditions (trial 1; saline), following combined infusion of BaCl2 and ouabain (trial 2), and following combined infusion of BaCl2, ouabain, l-NMMA, and ketorolac (trial 3). Previously, we have observed acute fluctuations in resting vascular tone with BaCl2 and ouabain infusion (7); thus we infused BaCl2 and ouabain together for 3 min (preexercise) and then stopped infusion to obtain accurate resting blood flow for 30–60 s. After resting blood flow was obtained, subjects then began muscle contractions. This method of drug infusion was previously used by our laboratory and proven effective at significantly reducing the blood flow response to a single muscle contraction (7). This approach was performed for all trials in this protocol to appropriately quantify resting blood flow and the rapid hyperemia in response to exercise.
Fig. 1.
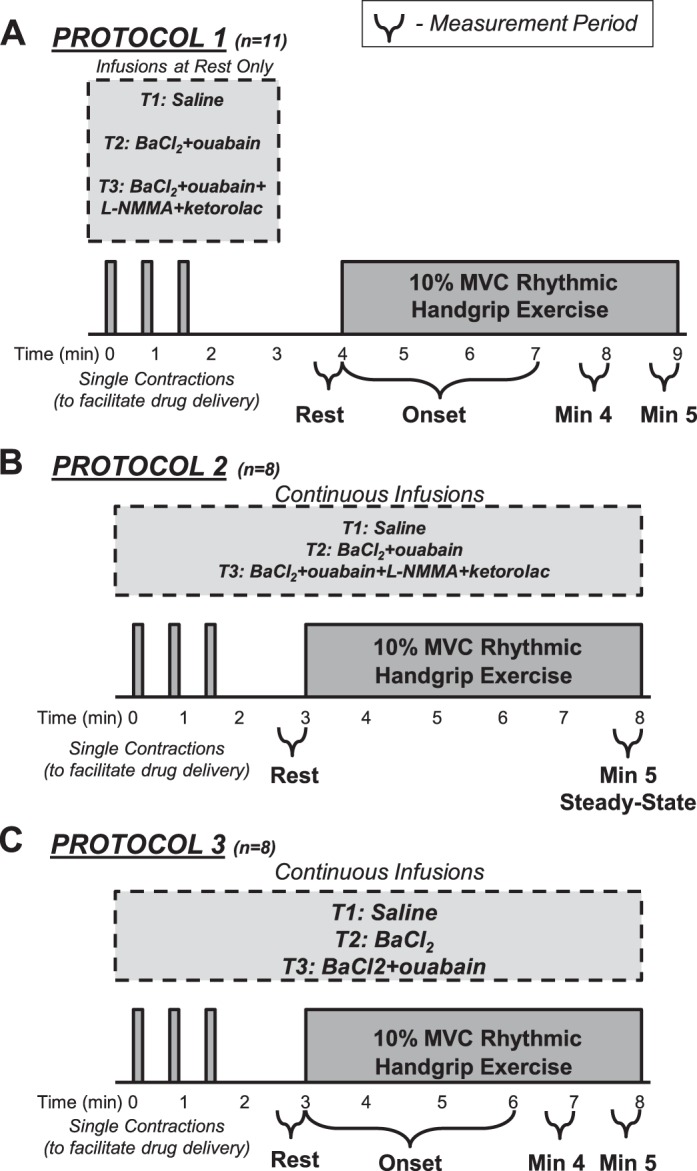
Experimental timeline. Three trials were performed in all protocols and trial 1 (T1) always consisted of saline infusion as a control condition. In protocols 1 (A) and 2 (B), trial 2 (T2) consisted of the combined infusion of barium chloride [BaCl2; inwardly rectifying potassium (KIR) channel inhibitor] and ouabain (Na+-K+-ATPase inhibitor) and trial 3 (T3) consisted of the combined infusion of BaCl2, ouabain, NG-monomethyl-l-arginine (l-NMMA; to inhibit nitric oxide synthesis) and ketorolac (to inhibit prostaglandin synthesis). During the loading period of drug infusions at rest, 3 single contractions were performed to facilitate drug delivery. In protocol 1, to avoid previously observed fluctuations in baseline hemodynamics, drugs were stopped before resting measures were made and exercise commenced. Measurements were made at rest, at the onset of 10% maximal voluntary contraction (MVC) rhythmic handgrip exercise, and throughout the 5-min bout of exercise. In protocol 2, drugs were continuously coinfused throughout rest and the entire handgrip exercise trial. C: in protocol 3, drugs were also continuously infused and to determine the independent effect of KIR channel inhibition in trial 2, BaCl2 alone was infused and in trial 3, combined BaCl2 and ouabain were administered.
In trial 1, saline was infused for 3 min before the start of resting blood flow measures. A single contraction (10% MVC) was performed at 30, 60, and 90 s of this infusion to facilitate delivery of the infusion to the vasculature of the muscle fibers recruited for this type of contraction (7). After saline infusion stopped, resting blood flow measures were made for 30–60 s and the subject was then instructed to begin the 5 min of contractions. Beat-to-beat blood flow was measured throughout the entire 5-min exercise protocol. Subjects rested comfortably for 20 min before the start of the next exercise trial. Trials 2 and 3 were similar; however, the infusions differed. In trial 2, ouabain was infused for 15 min and BaCl2 was added for the final 3 min before the start of resting blood flow measures. In trial 3, ouabain and BaCl2 were infused as in trial 2, and l-NMMA and ketorolac were added for the final 5 min before the start of resting blood flow measures.
Protocol 2: steady-state exercise hyperemia.
Based on our observations in protocol 1 (see results), we believed it was necessary to perform a second protocol where we continued infusion of our inhibitors throughout the muscle contractions to determine whether we had underestimated the effect of our blockers on steady-state exercise hyperemia in protocol 1. The trials in protocol 2 mimicked those of protocol 1 (control, BaCl2 + ouabain, and BaCl2 + ouabain + l-NMMA + ketorolac) with the key experimental difference being that all inhibitors were infused for a similar duration at rest (preexercise) but then continued throughout the entire 5-min exercise duration. Forearm hemodynamics in this protocol were assessed in the final minutes of drug infusions at rest and in the final minute of exercise (minute 5).
Protocol 3: independent KIR channel inhibition.
Given that we have shown inhibition of KIR channels alone can have a significant effect on pharmacological- and ischemia-induced hyperemia and may explain the effects of combined BaCl2 and ouabain (6, 12), we designed a follow-up protocol to determine whether there was an independent contribution of KIR channels to onset and steady-state exercise hyperemia. In our previous studies, we did not observe fluctuations in resting vascular tone during BaCl2 infusions alone (6, 12), and thus here we continuously infused the inhibitors throughout rest and the duration of muscle contractions so as not to underestimate any steady-state effects. Protocol 3 consisted of three trials: control (saline), KIR channel inhibition (BaCl2), and combined KIR channel and Na+-K+-ATPase inhibition (BaCl2 + ouabain). Each trial consisted of a resting period followed by 5 min of rhythmic handgrip exercise at 10% MVC.
Data Acquisition and Analysis
Data were collected and stored on a computer at 250 Hz and were analyzed offline with signal-processing software (WinDaq; DATAQ Instruments, Akron, OH). MAP was determined from the arterial pressure waveform. For protocols 1 and 3, FBF and MAP were analyzed in 3-s bins that corresponded to each contraction:relaxation (1:2 s) cycle. This type of analysis was carried out for the first 3 min, and 30-s averages were used at minutes 4 and 5 of exercise. In the case that the MBV signal quality obtained during a 3-s cycle was altered due to operator error, a mathematical average of the preceding and the MBV of the subsequent bins was used. This occurred in <2% of all bins analyzed. HR was determined at rest and each minute of exercise thereafter.
In protocols 1 and 3, the total exercise-induced hyperemic and vasodilator responses were calculated as the sum of FBF and FVC, respectively, above baseline for 30, 60, 120, 180, 240, and 300 s of exercise (AUC). In protocol 2, the last 30 s of exercise was averaged to represent steady-state FBF and FVC.
To quantify the impact of vasoactive drugs in each protocol on FBF, the magnitude of inhibition of exercise hyperemia was calculated as:
Changes in FVC, as well as AUC, were calculated in a similar manner.
Statistics
Data are presented as means ± SE. In all protocols, two-way repeated-measures ANOVA (time point × drug condition) and post hoc pairwise comparisons were completed and Student-Newman-Keuls pairwise tests were used to analyze relevant time-averaged data. Paired Student's t-tests were used to analyze the calculated magnitude of inhibition (percent change) in steady-state hemodynamics between drug conditions. Significance was set a priori at P < 0.05.
RESULTS
For all protocols, systemic hemodynamics (HR and MAP) were largely unchanged throughout the trials and under all conditions. Some small (2–3 mmHg and 2–4 beats/min) but statistically significant differences in MAP and HR, respectively, were observed in the condition of prior BaCl2 + ouabain + l-NMMA + ketorolac infusion vs. control (Tables 1, 2, and 3).
Table 1.
Protocol 1: forearm and systemic hemodynamics
| Time Point/Condition | FVC, ml·min−1·100 mmHg−1 | MAP,‡ mmHg | HR,§ beats/min |
|---|---|---|---|
| Rest | |||
| Control | 36.4 ± 3.4 | 93 ± 2 | 56 ± 3 |
| BaCl2 + ouabain | 31.8 ± 2.3 | 94 ± 3 | 57 ± 4 |
| BaCl2 + ouabain + l-NMMA + ketorolac | 25.8 ± 2.5*† | 96 ± 2 | 53 ± 3 |
| Exercise minute 1 | |||
| Control | 140.2 ± 9.8 | 94 ± 3 | 61 ± 4 |
| BaCl2 + ouabain | 109.3 ± 7.6* | 96 ± 3 | 59 ± 4 |
| BaCl2 + ouabain + l-NMMA + ketorolac | 99.5 ± 5.7*† | 98 ± 2 | 57 ± 4 |
| Exercise minute 2 | |||
| Control | 151.3 ± 11.9 | 94 ± 2 | 59 ± 4 |
| BaCl2 + ouabain | 134.9 ± 9.6* | 96 ± 2 | 59 ± 3 |
| BaCl2 + ouabain + l-NMMA + ketorolac | 128.5 ± 7.9* | 97 ± 2 | 56 ± 4 |
| Exercise minute 3 | |||
| Control | 150.2 ± 9.8 | 94 ± 3 | 57 ± 5 |
| BaCl2 + ouabain | 139.6 ± 8.3* | 96 ± 3 | 59 ± 4 |
| BaCl2 + ouabain + l-NMMA + ketorolac | 134.7 ± 9.9* | 97 ± 2 | 57 ± 3 |
| Exercise minute 4 | |||
| Control | 155.6 ± 10.8 | 95 ± 2 | 60 ± 4 |
| BaCl2 + ouabain | 147.5 ± 8.7 | 95 ± 3 | 59 ± 3 |
| BaCl2 + ouabain + l-NMMA + ketorolac | 142.7 ± 10.3* | 97 ± 2 | 57 ± 3 |
| Exercise minute 5 | |||
| Control | 166.0 ± 15.3 | 95 ± 2 | 59 ± 4 |
| BaCl2 + ouabain | 157.1 ± 10.3 | 95 ± 2 | 59 ± 4 |
| BaCl2 + ouabain + l-NMMA + ketorolac | 145.4 ± 9.2* | 97 ± 2 | 58 ± 4 |
Values are means ± SE; n = 11 subjects. FVC, forearm vascular conductance; HR, heart rate; MAP, mean arterial pressure.
P < 0.05 vs. control.
P < 0.05 vs. BaCl2 + ouabain.
MAP: main effect of condition [BaCl2 + ouabain + NG-monomethyl-l-arginine (l-NMMA) + ketorolac vs. control; P < 0.05].
HR: main effect of condition (BaCl2 + ouabain + l-NMMA + ketorolac vs. control and vs. BaCl2 + ouabain, P < 0.05); main effect of time point (minutes 1–5 vs. rest; P < 0.05); no significant interaction (P = 0.35).
Table 2.
Protocol 2: systemic hemodynamics
| Time Point/Condition | MAP,* mmHg | HR,† beats/min |
|---|---|---|
| Rest | ||
| Control | 84 ± 2 | 56 ± 3 |
| BaCl2 + ouabain | 90 ± 3 | 54 ± 3 |
| BaCl2 + ouabain + l-NMMA + ketorolac | 94 ± 4 | 56 ± 3 |
| Steady-state exercise (minute 5) | ||
| Control | 86 ± 3 | 60 ± 3 |
| BaCl2 + ouabain | 95 ± 2 | 60 ± 3 |
| BaCl2 + ouabain + l-NMMA + ketorolac | 100 ± 3 | 59 ± 3 |
Values are means ± SE; n = 8 subjects.
MAP: main effect of time point (exercise vs. rest; P < 0.05) and condition (all conditions different; P < 0.05); no significant interaction (P = 0.07).
HR: main effect of time point (exercise vs. rest; P < 0.01); no significant main effect of condition (P = 0.32) or interaction (P = 0.47).
Table 3.
Protocol 3: forearm and systemic hemodynamics
| Time Point/Condition | FVC, ml·min−1·100 mmHg−1 | MAP,‡ mmHg | HR,§ beats/min |
|---|---|---|---|
| Rest | |||
| Control | 36.5 ± 5.2 | 93 ± 4 | 62 ± 5 |
| BaCl2 | 27.6 ± 3.7 | 95 ± 3 | 61 ± 5 |
| BaCl2 + ouabain | 31.8 ± 3.7 | 97 ± 3 | 59 ± 5 |
| Exercise minute 1 | |||
| Control | 135.4 ± 20.7 | 93 ± 3 | 64 ± 5 |
| BaCl2 | 73.2 ± 16.2* | 94 ± 3 | 62 ± 6 |
| BaCl2 + ouabain | 74.9 ± 11.9* | 97 ± 3 | 62 ± 6 |
| Exercise minute 2 | |||
| Control | 157.6 ± 20.3 | 94 ± 3 | 64 ± 6 |
| BaCl2 | 99.9 ± 18.1* | 96 ± 3 | 63 ± 5 |
| BaCl2 + ouabain | 97.3 ± 12.4* | 98 ± 3 | 62 ± 5 |
| Exercise minute 3 | |||
| Control | 162.6 ± 17.6 | 94 ± 3 | 65 ± 6 |
| BaCl2 | 110.9 ± 17.1* | 97 ± 2 | 64 ± 6 |
| BaCl2 + ouabain | 103.6 ± 12.2* | 100 ± 2 | 64 ± 5 |
| Exercise minute 4 | |||
| Control | 169.3 ± 18.3 | 94 ± 3 | 66 ± 6 |
| BaCl2 | 119.1 ± 15.9* | 94 ± 3 | 64 ± 6 |
| BaCl2 + ouabain | 112.3 ± 12.1* | 100 ± 2 | 64 ± 6 |
| Exercise minute 5 | |||
| Control | 168.0 ± 15.5 | 94 ± 3 | 64 ± 6 |
| BaCl2 | 119.0 ± 16.8* | 95 ± 3 | 64 ± 6 |
| BaCl2 + ouabain | 111.2 ± 11.8* | 101 ± 2 | 65 ± 6 |
Values are means ± SE; n = 8 subjects.
P < 0.05 vs. control. †P < 0.05 vs. BaCl2 + ouabain.
MAP: main effect of condition (BaCl2 + ouabain vs. both control and BaCl2; P < 0.05); main effect of time point (minute 5 vs. rest and minute 1; P < 0.05); no significant interaction [(P = 0.23); 0.05]; main effect of time point (minutes 1–5 vs. rest; P < 0.05); no significant interaction (P = 0.39).
Protocol 1: Onset Exercise Hyperemia
In control (saline) conditions, FBF increased rapidly from rest (34 ± 3 ml/min) in response to muscle contractions (Fig. 2A). In trial 2, combined inhibition of KIR channels and Na+-K+-ATPase (via infusion of BaCl2 + ouabain that was stopped before resting measures and the start of exercise) had no effect on resting FBF (30 ± 3 ml/min) but significantly attenuated FBF for the first 2 min of exercise (Fig. 2B). FBF at minutes 3, 4, and 5 was not different compared with control. Additionally, in trial 2, the total hyperemic response was significantly attenuated from control and this effect was more pronounced earlier in the exercise bout (30 s: −49 ± 5%; 60 s: −34 ± 5%; 120 s: −20 ± 5%; 180 s: −13 ± 4%; 240 s: −10 ± 4%; 300 s: −8 ± 3%). In trial 3, FBF for the first 15 s after exercise onset was reduced compared with trial 2; however, FBF was not different thereafter until minute 5 of exercise where FBF was reduced ∼8% from control (Fig. 2B). When the total hyperemic response was quantified, l-NMMA and ketorolac only had an additional effect at 30 and 300 s (%Δ from control: 30 s: −65 ± 2%; 60 s: −40 ± 3%; 120 s: −23 ± 4%; 180 s: −15 ± 4%; 240 s: −14 ± 4%; 300 s: −14 ± 4%). Changes in FVC paralleled those observed in FBF (Table 1).
Fig. 2.
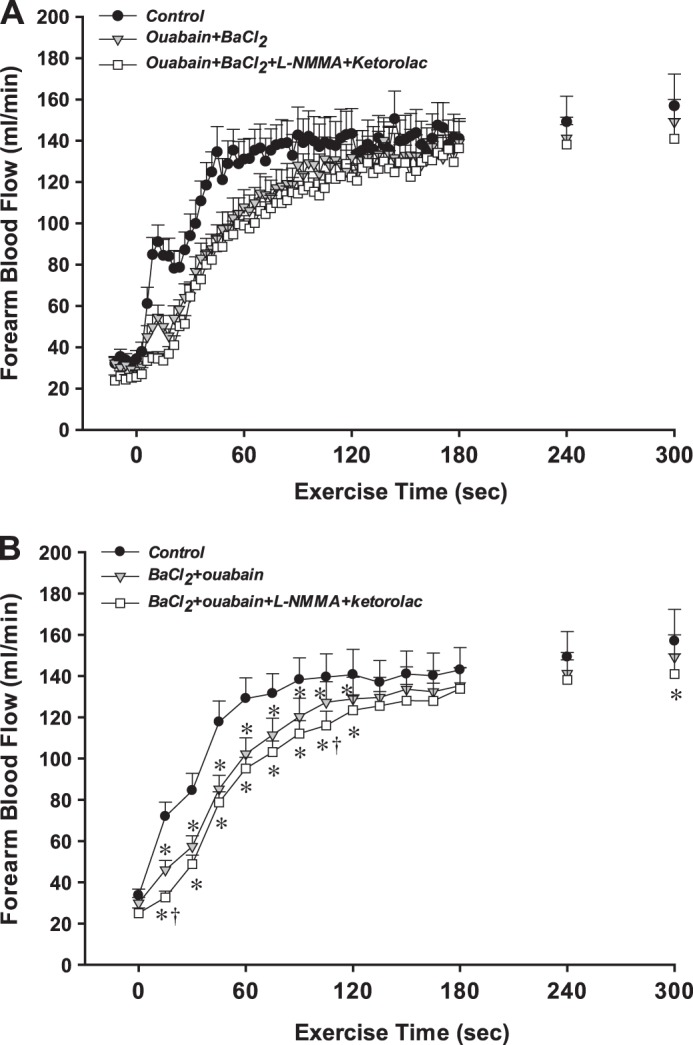
Protocol 1: effect of inhibition of hyperpolarizing and endothelium-dependent vasodilator mechanisms on the onset of exercise hyperemia. A: forearm blood flow (FBF) across the 3 experimental conditions is shown at rest and during 10% MVC rhythmic handgrip exercise. Data are in 3-s bins corresponding to a contraction:relaxation cycle during exercise. B: statistical analysis was performed on 15-s averages of blood flow data (5 3-s bins). BaCl2 + ouabain significantly reduced the onset of exercise hyperemia, and l-NMMA + ketorolac had an additional effect within the first 15 s of exercise. *P < 0.05 vs. control; †P < 0.05 vs. BaCl2 + ouabain; n = 11 subjects.
Protocol 2: Steady-State Exercise Hyperemia
After 5 min of exercise, steady-state FBF (Fig. 3A) and FVC (Fig. 3B) were significantly lower with continuous infusion of BaCl2 and ouabain throughout muscle contractions (trial 2: 133 ± 15 ml/min; 140 ± 15 ml·min−1·100 mmHg−1) than in control conditions (trial 1: 167 ± 17 ml/min; 194 ± 18 ml·min−1·100 mmHg−1). In trial 3, inhibiting NO and PG synthesis in addition to KIR channels and Na+-K+-ATPase resulted in a further attenuation of exercise hyperemia and vasodilation compared with control conditions (Fig. 3, A and B). Compared with control, BaCl2 and ouabain reduced steady-state FBF and FVC −20 ± 3 and −28 ± 2%, respectively, and the addition of l-NMMA and ketorolac had a total impact of −29 ± 3 and −40 ± 3% on FBF and FVC, respectively.
Fig. 3.
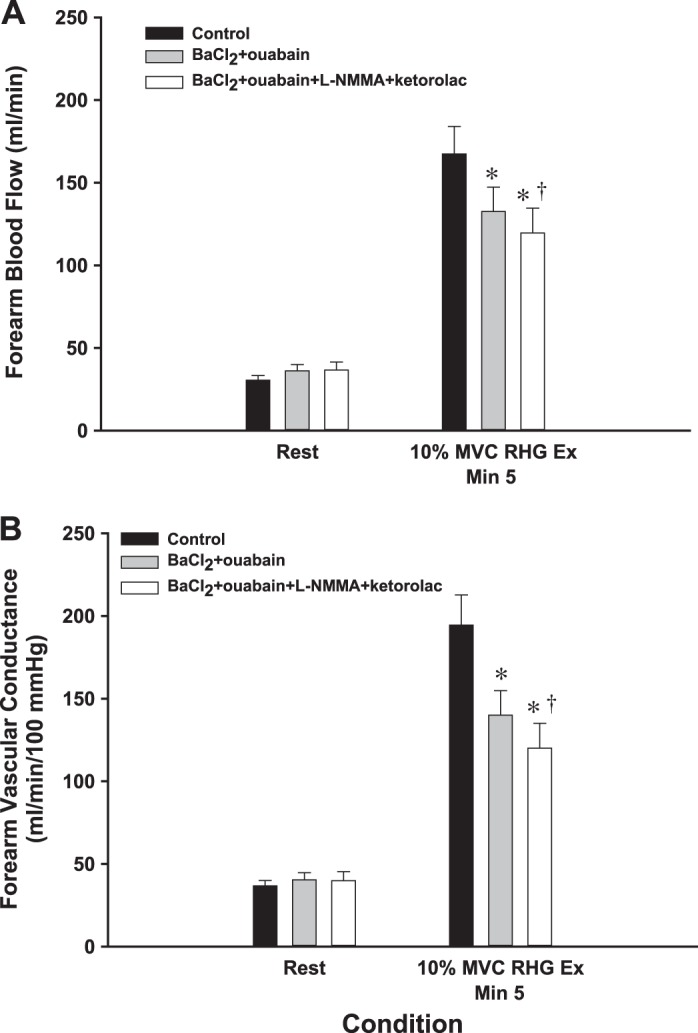
Protocol 2: effect of inhibition of hyperpolarizing and endothelium-dependent vasodilator mechanisms on steady-state exercise hyperemia. In protocol 2, with continuous infusion of the inhibitors, a significant reduction in FBF (A) and forearm vascular conductance (FVC; B) was observed with combined BaCl2 + ouabain infusion. Here, the addition of l-NMMA + ketorolac to BaCl2 + ouabain further reduced both steady-state FBF and FVC. RHG Ex, rhythmic handgrip exercise. *P < 0.05 vs. control; †P < 0.05 vs. BaCl2 + ouabain; n = 8 subjects.
Protocol 3: Independent KIR Channel Inhibition
In control (saline) conditions, FBF increased rapidly from rest (34 ± 5 ml/min) in response to muscle contractions (Fig. 4A). In trial 2, continuous infusion of BaCl2 throughout the trial did not alter resting FBF (26 ± 3 ml/min) but significantly attenuated FBF for all 5 min of exercise (Fig. 4B). The total hyperemic response was significantly attenuated with BaCl2 (%Δ from control: 30 s: −64 ± 6%; 60 s: −60 ± 7%; 120 s: −48 ± 7%; 180 s: −42 ± 7%; 240 s: −38 ± 6%; 300 s: −36 ± 6%). In trial 3, continuous infusion of BaCl2 + ouabain did not affect resting FBF (31 ± 3 ml/min). Absolute FBF at the onset of exercise, although significantly lower than control conditions, was not different than the BaCl2 alone condition (Fig. 4B). A similar pattern was evident when the total hyperemic response was quantified and the reduction from control conditions was calculated (30 s: −54 ± 10%; 60 s: −56 ± 8%; 120 s: −47 ± 5%; 180 s: −42 ± 5%, 240 s: −45 ± 4%; 300 s: −46 ± 5%). Again, changes in FVC paralleled those observed in FBF (Table 3). The percent reductions in FBF and FVC from control steady-state exercise levels (minute 5) were similar in magnitude (∼30%) to those observed in protocol 2 when both BaCl2 + ouabain were continuously infused throughout exercise.
Fig. 4.
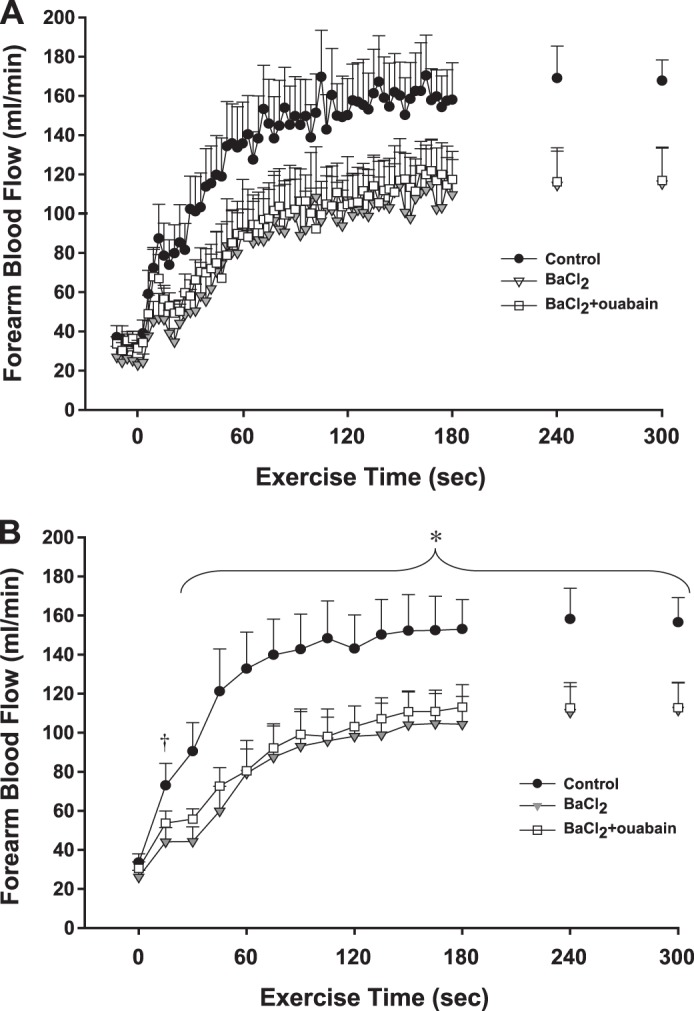
Protocol 3: effect of independent inhibition of KIR channels on exercise hyperemia. A: FBF across the 3 experimental conditions is shown at rest and during 10% MVC rhythmic handgrip exercise. Data are in 3-s bins corresponding to a contraction:relaxation cycle during exercise. B: statistical analysis was performed on 15-s averages of blood flow data (5 3-s bins). In all inhibited conditions, exercise hyperemia was lower from 30 to 300 s. *P < 0.05 vs. BaCl2; vs. BaCl2 + ouabain. After 15 s, there was no statistically significant reduction with infusion of BaCl2 + ouabain. †P < 0.05 vs. BaCl2; n = 8 subjects.
DISCUSSION
The primary purpose of the present study was to determine the contribution of activation of KIR channels and Na+-K+-ATPase to exercise hyperemia at the onset of muscle contractions and during the steady-state portion of the blood flow response and to determine whether prior inhibition of these pathways revealed a role for NO and PGs. We then followed up our initial findings and further sought to identify if there was an independent role for KIR channel activation in exercise hyperemia. The primary novel findings of the present study are as follows. First, combined inhibition of KIR channels and Na+-K+−ATPase significantly attenuates onset of exercise hyperemia following repeated muscle contractions (protocol 1) and this is largely due to KIR channel activation (protocol 3). Additionally, in line with prior findings in the human forearm, NO and PGs do not play a significant role in the onset of the response except potentially for the initial 15 s (protocol 1). Second, we demonstrate for the first time in humans a significant attenuation of steady-state exercise hyperemia with inhibition of activation of KIR channels and Na+-K+-ATPase and similar to the onset, this is primarily due to activation of KIR channels (protocols 2 and 3). Interestingly, prior inhibition of KIR channels and Na+-K+-ATPase reveals a significant role for NO and PGs in mediating steady-state exercise hyperemia that has not been previously observed in this model of mild-to-moderate intensity rhythmic handgrip exercise (protocol 2). Our collective findings indicate a novel role for KIR channel activation in exercise hyperemia in humans, and this contribution is greater at exercise onset (∼50%) compared with steady-state conditions (∼30%).
Onset Exercise Hyperemia: Contributing Signaling Pathways (Protocols 1 and 3)
At the onset of exercise, there is a rapid increase in blood flow that occurs immediately upon muscular relaxation, and recent evidence indicates this is largely due to rapid local vasodilation (5, 7, 31). With repeated muscle contractions, this increase in blood flow continues in a biphasic manner comprised of an initial rapid increase that plateaus by ∼5–7 s and a second, slower phase that begins ∼20 s after the onset of exercise and continues in an intensity-dependent manner until steady-state hyperemia has been achieved (49, 60). In the present study, we observe similar dynamic characteristics in the hyperemic response to mild intensity handgrip exercise in control conditions (Fig. 2A). Previous studies in the human forearm demonstrated that inhibition of NO or PGs does not impact the rapid onset of exercise hyperemia (56, 57) nor does antagonism of muscarinic receptors (56) that would be bound by acetylcholine either from an endothelial source (38) or spillover from the motor neuron junction (61). Studies in animals suggest that during continuous muscle contractions distal resistance arterioles within the active tissue relax (62) and this dilation is conducted upstream to the level of the feed arteries (53). Hyperpolarization of endothelial and vascular smooth muscle cells is known to contribute more robustly to distal vessel vasodilation as opposed to vasodilator autocoids such as NO and PGs (48, 55) and is capable of stimulating conducted vasodilation, whereas, in general, NO and PGs do not (16, 26). Recently, we demonstrated that activation of KIR channels and Na+-K+-ATPase contributes to the total vasodilation following a single muscle contraction (7), the first such observation in humans. In the present study, we determined what role these pathways have in the onset of hyperemia in response to repeated muscle contractions.
When BaCl2 and ouabain were infused before contractions to inhibit KIR channels and Na+-K+-ATPase, respectively, there was a significant attenuation of the initial hyperemic response for the first 2 min of exercise (Fig. 2B) and the findings of protocol 3 suggest this is due to activation of KIR channels (Fig. 4). In protocol 1, beyond 2 min, there was no longer a significant effect on the absolute level of blood flow; however, the initial impact on the hyperemia was of large enough magnitude to affect the total (AUC) hyperemic response through 4 min of exercise. In this protocol, due to fluctuations in resting blood flow that we have observed during infusion of BaCl2 and ouabain in our laboratory (7), we stopped drug infusions at rest then obtained blood flow measures for 30–60 s before the start of muscle contractions. Utilizing this method allowed us to appropriately quantify resting FBF and the rapid blood flow responses to muscle contraction; however, because drugs were not coinfused throughout muscle contractions, we may have underestimated the impact of inhibition on steady-state hyperemia. Given our possible underestimation, we sought to more directly address a role for activation of KIR channels and Na+-K+-ATPase during steady-state hyperemia in protocol 2 where all inhibitors were coinfused during muscle contractions (see section below).
Several lines of evidence suggest that there is crossover between the various vasodilator pathways and that, in the presence of inhibition of one pathway, another may compensate to preserve blood flow and oxygen delivery, particularly during muscle activation or decreased arterial oxygen content (25, 37, 59). Thus we investigated whether in the presence of inhibition of KIR channels and Na+-K+-ATPase, there would be a role for NO and PGs, despite prior evidence suggesting they do not contribute to initial exercise hyperemia in the human forearm (56, 57). Combined infusion of l-NMMA and ketorolac to enzymatically inhibit the production of NO and PGs, respectively, reduced FBF beyond that which occurred with BaCl2 + ouabain at the start of forearm contractions (Fig. 2B). However, this further impairment diminished rapidly over time, and by 30 s there was no longer a significant reduction in FBF from the BaCl2 + ouabain condition. Similarly, there was minimal additional effect of NO and PG inhibition on the total hyperemic response. It is possible that the reduction in resting blood flow during combined NO and PG inhibition attributed to the initial and brief reduction in exercise hyperemia, similar to prior observations of lower resting blood flow and slower forearm blood flow response times to handgrip exercise when the arm was positioned above vs. below heart level (57). In the presence of BaCl2 + ouabain + l-NMMA + ketorolac, muscle blood flow was still able to increase rapidly and achieve levels significantly greater than rest and near steady-state levels by 3 min of exercise, indicating redundant vasodilator signals, even from the onset of muscle contractions.
Steady-State Exercise Hyperemia: Contributing Signaling Pathways (Protocols 2 and 3)
To date, multiple studies have attempted to identify the various vasodilator pathways involved in exercise hyperemia and most have focused on the steady-state portion of the response (13, 28, 45). Steady-state muscle blood flow is thought to reflect a homeostatic balance between oxygen delivery via increases in blood flow and oxygen demand of the contracting muscle (43). In the human forearm, the results of previous studies have been rather unimpressive in terms of the magnitude of reduction in muscle blood flow consequent to vasodilator inhibition. In fact, combined inhibition of NO and PGs, two important endothelium-dependent dilators, does not impact the rest to steady-state muscle blood flow response (10, 51). Given animal studies that suggested an important role for the endothelium (2, 44) and particularly for conducted hyperpolarizing stimuli (39, 54) in exercise hyperemia and the lack of involvement of NO and PGs, we wanted to address the role of resistance vessel hyperpolarization in muscle blood flow control during exercise in humans.
One issue with attempting to address hyperpolarizing pathways in humans is a lack of specific antagonists and agonists of the proposed pathways involved in initiating and conducting hyperpolarization in the vasculature. Additionally, in human studies, it is not possible to directly measure membrane potential or identify the cell type (endothelial and vascular smooth muscle) that may hyperpolarize. Previously, we have demonstrated that combined administration of ouabain and BaCl2 can abolish KCl-mediated vasodilation, a stimulus for direct vascular smooth muscle cell hyperpolarization (6). In the present study we used this established pharmacology (6, 7, 12) to test the role of vascular hyperpolarization in muscle blood flow regulation during continued muscle contractions.
Due to the observed fluctuations in resting blood flow in the present studies and in our past experience with ouabain and BaCl2 (6, 7), in protocol 1 where we were focused on capturing the onset of exercise hyperemia, we did not coinfuse these inhibitors during contractions. However, when we did not observe a significant effect during steady-state exercise (Fig. 1), we questioned whether there was “wash-out” of our inhibitors as blood flow increased or a reduction in effectiveness over time. Subsequently, we performed studies in protocol 2 in which we coinfused our inhibitors throughout muscle contractions as is more typical of our approach for these types of exercise studies (10, 11). Furthermore, given these fluctuations appeared to be due to ouabain, in protocol 3 where we assessed an independent impact of BaCl2 we also continuously infused our pharmacology throughout the entire rest and exercise period. Under these conditions of continuous inhibition, we do in fact observe a significant contribution of KIR channels and Na+-K+-ATPase to steady-state exercise hyperemia (Fig. 3) and this is specifically due to a role of KIR channel activation alone (Fig. 4; range from both protocols 20–30%).
Interestingly, combined inhibition of NO and PGs in addition to hyperpolarization mediated by KIR channel and Na+-K+-ATPase activation resulted in a statistically significant further attenuation of steady-state muscle blood flow and vascular conductance in protocol 2 (Fig. 3). This finding is in contrast with the lack of a combined role for NO and PGs in the human forearm in previous studies (10, 15, 51) but highlights the ability of vasodilator pathways to compensate for one another, a phenomenon that has been observed in a variety of previous studies (25, 37, 59). It is important to emphasize the magnitude of the effect of combined inhibition of KIR channels, Na+-K+-ATPase, and NO and PGs on FBF and FVC during steady-state exercise. A 40% reduction in forearm vasodilation (FVC) is profound, particularly in a small muscle mass such as the forearm that typically is minimally affected following pharmacological inhibition of vasodilator pathways. Furthermore, this change approaches the observed effects of completely inhibiting conducted vasodilation (∼45% reduction in contraction-induced hyperemia) in animal models (54) providing an additional context for our findings.
Potential Stimuli for Signaling Through KIR channels, Na+-K+-ATPase, and NO and PGs During Exercise
As previously stated, there are a variety of substances that can stimulate hyperpolarization of the vasculature, both endothelium dependent and independent. One candidate substance is the K+ ion, as interstitial K+ increases as a result of muscle contractions (1, 18, 24, 30) and can also be released as an endothelial-derived hyperpolarizing factor (18). Agonist binding to receptors on endothelial cells can cause intracellular calcium changes that stimulate small- and intermediate-conductance KCa channels to open and K+ to efflux into the interstitial space or microdomain signaling complexes at the physical intersection of vascular smooth muscle and endothelial cells (35). In animal models where these small- and intermediate-conductance KCa channels can be specifically inhibited, a significant attenuation in contraction-induced hyperemia is observed (39). While the K+ ion would not account for the compensatory involvement of NO and PG during steady-state exercise (6), it could be involved in directly stimulating both KIR channels and Na+-K+-ATPase (6, 18) ultimately resulting in vascular smooth muscle cell hyperpolarization, thus contributing to both the onset and steady-state levels of exercise hyperemia.
Another potential stimulus for hyperpolarization of endothelial and vascular smooth cells is ATP, a potent vasoactive molecule released from endothelial and red blood cells in response to deoxygenation, hypercapnia, and mechanical stresses that increases in plasma samples of blood draining contracting skeletal muscle tissue (3, 8, 21, 32, 58). ATP causes profound vasodilation that is endothelium dependent (17), and we have determined in humans the primary underlying signaling mechanism is activation of KIR channels (6). Additionally, we have demonstrated a modest component of ATP-mediated vasodilation occurs through NO and PG synthesis (9) and thus particularly during steady-state exercise where a significant role for NO and PGs is revealed when hyperpolarization is inhibited, ATP may also serve as a potential stimulus for these pathways.
Experimental Considerations
Our group has recently utilized the pharmacological approach of local BaCl2 and ouabain infusions to inhibit hyperpolarization stimulated by KIR channel and Na+-K+-ATPase activation, respectively (6, 7, 12). While these inhibitors are safe for use, given that they are not used clinically, we attempt to limit the total number of subjects exposed to these agents and the total dose given to any one subject. Therefore, we have not repeated control experiments within this protocol to demonstrate effective inhibition of hyperpolarization-mediated (KCl) vasodilation, preserved vasodilator capacity [to endothelium-dependent (acetylcholine) and endothelium-independent (sodium nitroprusside) agonists], and preserved maximal force production (6, 7). Similarly, given our extensive use of l-NMMA and ketorolac (6, 7, 9–12) and various tests of efficacy (14, 51), we have not repeated those control studies within this experiment. When all four inhibitors (BaCl2, ouabain, l-NMMA, and ketorolac) are administered, vasodilation to sodium nitroprusside remains preserved (12). It is possible that our dose of BaCl2 may not be selective for KIR channels and inhibits other potassium channels, specifically KATP channels (27). However, given that previously there was no significant effect of inhibiting KATP channels during steady-state exercise in an identical experimental model (50), we do not feel this changes our primary conclusions. Furthermore, inhibition of other potential pathways leading eventually to vascular smooth muscle cell hyperpolarization such as cytochrome P450 metabolites (25) and large-conductance KCa channels (41) has no impact on exercise hyperemia, adding to the support for our specific impact of BaCl2 on KIR channels.
The forearm model, while advantageous for many reasons, also has some inherent limitations. We acknowledge that the muscle mass engaged in rhythmic handgrip exercise is relatively small and extrapolating our results to more traditional modes of whole body exercise would need to be done cautiously. It may be desirable to test ideas related to the role of hyperpolarization via KIR channel activation in regulating vascular control during larger muscle mass exercise that engage the sympathetic nervous system to assess the influence of these pathways on the interaction with sympathetic vasoconstriction and overall blood pressure regulation; however, safety concerns of systemic administration of BaCl2 in doses suitable to appropriately inhibit KIR channels preclude this at the present time.
Conclusions
Continuous skeletal muscle contractions result in a rapid and significant rise in muscle blood flow to deliver oxygen to meet the increased metabolic need of the tissue. The regulation of muscle blood flow during exercise is a complex response and given the strong relation among muscle blood flow, oxygen delivery, and exercise capacity in health and disease, there is great interest in understanding the various vasodilator signals that contribute to this response. Here, we demonstrate for the first time in humans that KIR channel activation significantly contributes to the hyperemia observed at the onset of and during steady-state muscle contractions. Additionally, NO and PGs have at most a modest contribution to the initial (10–15 s) rise in muscle blood flow, and interestingly, in the presence of inhibition of KIR channels and Na+-K+-ATPase, we reveal a role for these dilators during steady-state exercise hyperemia, an observation that was not previously made in this experimental model. The primary stimuli for activation of KIR channels during muscle contractions remain unclear at this time, but the K+ ion and circulating ATP are two likely candidates. Future work will be needed to definitively identify these stimuli and, furthermore, to determine what role these pathways may play in the reduced muscle perfusion often observed with aging and disease.
GRANTS
This research was supported by National Heart, Lung, and Blood Institute Grant HL-102720 (to F. A. Dinenno).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: A.R.C. and F.A.D. conception and design of research; A.R.C., G.J.L., D.G.L., and F.A.D. performed experiments; A.R.C. analyzed data; A.R.C. and F.A.D. interpreted results of experiments; A.R.C. prepared figures; A.R.C. and F.A.D. drafted manuscript; A.R.C. and F.A.D. edited and revised manuscript; A.R.C., G.J.L., D.G.L., and F.A.D. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the subjects who volunteered to participate and Dr. Brett S. Kirby, Dr. Jennifer C. Richards, and Leora J. Garcia and for contributions to this project.
Present address of A. R Crecelius: Dept. of Health and Sport Science, Univ, of Dayton, Dayton, OH.
REFERENCES
- 1.Armstrong ML, Dua AK, Murrant CL. Potassium initiates vasodilatation induced by a single skeletal muscle contraction in hamster cremaster muscle. J Physiol 581: 841–852, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berdeaux A, Ghaleh B, Dubois-Rande JL, Vigue B, Drieu La Rochelle C, Hittinger L, Giudicelli JF. Role of vascular endothelium in exercise-induced dilation of large epicardial coronary arteries in conscious dogs. Circulation 89: 2799–2808, 1994 [DOI] [PubMed] [Google Scholar]
- 3.Bodin P, Bailey D, Burnstock G. Increased flow-induced ATP release from isolated vascular endothelial cells but not smooth muscle cells. Br J Pharmacol 103: 1203–1205, 1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boushel R, Langberg H, Gemmer C, Olesen J, Crameri R, Scheede C, Sander M, Kjaer M. Combined inhibition of nitric oxide and prostaglandins reduces human skeletal muscle blood flow during exercise. J Physiol 543: 691–698, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clifford PS. Skeletal muscle vasodilatation at the onset of exercise. J Physiol 583: 825–833, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crecelius AR, Kirby BS, Luckasen GJ, Larson DG, Dinenno FA. ATP-mediated vasodilatation occurs via activation of inwardly-rectifying potassium channels in humans. J Physiol 590: 5349–5359, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Crecelius AR, Kirby BS, Luckasen GJ, Larson DG, Dinenno FA. Mechanisms of rapid vasodilation after a brief contraction in human skeletal muscle. Am J Physiol Heart Circ Physiol 305: H29–H40, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crecelius AR, Kirby BS, Richards JC, Dinenno FA. Mechanical effects of muscle contraction increase intravascular ATP draining quiescent and active skeletal muscle in humans. J Appl Physiol 114: 1085–1093, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crecelius AR, Kirby BS, Richards JC, Garcia LJ, Voyles WF, Larson DG, Luckasen GJ, Dinenno FA. Mechanisms of ATP-mediated vasodilation in humans: modest role for nitric oxide and vasodilating prostaglandins. Am J Physiol Heart Circ Physiol 301: H1302–H1310, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crecelius AR, Kirby BS, Voyles WF, Dinenno FA. Augmented skeletal muscle hyperaemia during hypoxic exercise in humans is blunted by combined inhibition of nitric oxide and vasodilating prostaglandins. J Physiol 589: 3671–3683, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crecelius AR, Kirby BS, Voyles WF, Dinenno FA. Nitric oxide, but not vasodilating prostaglandins, contributes to the improvement of exercise hyperemia via ascorbic acid in healthy older adults. Am J Physiol Heart Circ Physiol 299: H1633–H1641, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crecelius AR, Richards JC, Luckasen GJ, Larson DG, Dinenno FA. Reactive hyperemia occurs via activation of inwardly rectifying potassium channels and Na+/K+-ATPase in humans. Circ Res 113: 1023–1032, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delp MD, Laughlin MH. Regulation of skeletal muscle perfusion during exercise. Acta Physiol Scand 162: 411–419, 1998 [DOI] [PubMed] [Google Scholar]
- 14.Dinenno FA, Joyner MJ. Blunted sympathetic vasoconstriction in contracting skeletal muscle of healthy humans: is nitric oxide obligatory? J Physiol 553: 281–292, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dinenno FA, Joyner MJ. Combined NO and PG inhibition augments α-adrenergic vasoconstriction in contracting human skeletal muscle. Am J Physiol Heart Circ Physiol 287: H2576–H2584, 2004 [DOI] [PubMed] [Google Scholar]
- 16.Domeier TL, Segal SS. Electromechanical and pharmacomechanical signalling pathways for conducted vasodilatation along endothelium of hamster feed arteries. J Physiol 579: 175–186, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duza T, Sarelius IH. Conducted dilations initiated by purines in arterioles are endothelium dependent and require endothelial Ca2+. Am J Physiol Heart Circ Physiol 285: H26–H37, 2003 [DOI] [PubMed] [Google Scholar]
- 18.Edwards G, Dora KA, Gardener MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature 396: 269–272, 1998 [DOI] [PubMed] [Google Scholar]
- 19.Feletou M, Vanhoutte PM. Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture). Am J Physiol Heart Circ Physiol 291: H985–H1002, 2006 [DOI] [PubMed] [Google Scholar]
- 20.Garland CJ, Plane F, Kemp BK, Cocks TM. Endothelium-dependent hyperpolarization: a role in the control of vascular tone. Trends Pharmacol Sci 16: 23–30, 1995 [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez-Alonso J, Olsen DB, Saltin B. Erythrocyte and the regulation of human skeletal muscle blood flow and oxygen delivery: role of circulating ATP. Circ Res 91: 1046–1055, 2002 [DOI] [PubMed] [Google Scholar]
- 22.Hamann JJ, Buckwalter JB, Clifford PS. Vasodilatation is obligatory for contraction-induced hyperaemia in canine skeletal muscle. J Physiol 557: 1013–1020, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hazeyama Y, Sparks HV. Exercise hyperemia in potassium-depleted dogs. Am J Physiol Heart Circ Physiol 236: H480–H486, 1979 [DOI] [PubMed] [Google Scholar]
- 24.Hazeyama Y, Sparks HV. A model of potassium ion efflux during exercise of skeletal muscle. Am J Physiol Regul Integr Comp Physiol 236: R83–R90, 1979 [DOI] [PubMed] [Google Scholar]
- 25.Hillig T, Krustrup P, Fleming I, Osada T, Saltin B, Hellsten Y. Cytochrome P450 2C9 plays an important role in the regulation of exercise-induced skeletal muscle blood flow and oxygen uptake in humans. J Physiol 546: 307–314, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoepfl B, Rodenwaldt B, Pohl U, De Wit C. EDHF, but not NO or prostaglandins, is critical to evoke a conducted dilation upon ACh in hamster arterioles. Am J Physiol Heart Circ Physiol 283: H996–H1004, 2002 [DOI] [PubMed] [Google Scholar]
- 27.Jackson WF. Potassium channels in the peripheral microcirculation. Microcirculation 12: 113–127, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Joyner MJ, Wilkins BW. Exercise hyperaemia: is anything obligatory but the hyperaemia? J Physiol 583: 855–860, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Juel C, Olsen S, Rentsch RL, Gonzalez-Alonso J, Rosenmeier JB. K+ as a vasodilator in resting human muscle: implications for exercise hyperaemia. Acta Physiol (Oxf) 190: 311–318, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Kirby BS, Carlson RE. Potassium, contracting myocytes and rapid vasodilatation: peaking more than just our interest? J Physiol 586: 315–317, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kirby BS, Carlson RE, Markwald RR, Voyles WF, Dinenno FA. Mechanical influences on skeletal muscle vascular tone in humans: insight into contraction-induced rapid vasodilatation. J Physiol 583: 861–874, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kirby BS, Crecelius AR, Voyles WF, Dinenno FA. Impaired skeletal muscle blood flow control with advancing age in humans: attenuated ATP release and local vasodilation during erythrocyte deoxygenation. Circ Res 111: 220–230, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kirby BS, Voyles WF, Carlson RE, Dinenno FA. Graded sympatholytic effect of exogenous ATP on postjunctional alpha-adrenergic vasoconstriction in the human forearm: implications for vascular control in contracting muscle. J Physiol 586: 4305–4316, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ledoux J, Taylor MS, Bonev AD, Hannah RM, Solodushko V, Shui B, Tallini Y, Kotlikoff MI, Nelson MT. Functional architecture of inositol 1,4,5-trisphosphate signaling in restricted spaces of myoendothelial projections. Proc Natl Acad Sci USA 105: 9627–9632, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ledoux J, Werner ME, Brayden JE, Nelson MT. Calcium-activated potassium channels and the regulation of vascular tone. Physiology (Bethesda) 21: 69–78, 2006 [DOI] [PubMed] [Google Scholar]
- 37.Markwald RR, Kirby BS, Crecelius AR, Carlson RE, Voyles WF, Dinenno FA. Combined inhibition of nitric oxide and vasodilating prostaglandins abolishes forearm vasodilatation to systemic hypoxia in healthy humans. J Physiol 589: 1979–1990, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martin CM, Beltran-Del-Rio A, Albrecht A, Lorenz RR, Joyner MJ. Local cholinergic mechanisms mediate nitric oxide-dependent flow-induced vasorelaxation in vitro. Am J Physiol Heart Circ Physiol 270: H442–H446, 1996 [DOI] [PubMed] [Google Scholar]
- 39.Milkau M, Kohler R, de Wit C. Crucial importance of the endothelial K+ channel SK3 and connexin40 in arteriolar dilations during skeletal muscle contraction. FASEB J 24: 3572–3579, 2010 [DOI] [PubMed] [Google Scholar]
- 40.Mohrman DE, Sparks HV. Role of potassium ions in the vascular response to a brief tetanus. Circ Res 35: 384–390, 1974 [DOI] [PubMed] [Google Scholar]
- 41.Mortensen SP, Gonzalez-Alonso J, Damsgaard R, Saltin B, Hellsten Y. Inhibition of nitric oxide and prostaglandins, but not endothelial-derived hyperpolarizing factors, reduces blood flow and aerobic energy turnover in the exercising human leg. J Physiol 581: 853–861, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Richards JC, Crecelius AR, Kirby BS, Larson DG, Dinenno FA. Muscle contraction duration and fibre recruitment influence blood flow and oxygen consumption independent of contractile work during steady-state exercise in humans. Exp Physiol 97: 750–761, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rowell LB. Human Cardiovascular Control. New York: Oxford Univ Press, 1993 [Google Scholar]
- 44.Sagach VF, Kindybalyuk AM, Kovalenko TN. Functional hyperemia of skeletal muscle: role of endothelium. J Cardiovasc Pharmacol 20, Suppl 12: S170–175, 1992 [DOI] [PubMed] [Google Scholar]
- 45.Saltin B. Exercise hyperaemia: magnitude and aspects on regulation in humans. J Physiol 583: 819–823, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saltin B. In search of a vasodilator: is ATP the answer? J Physiol 590: 5261–5262, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Saltin B, Radegran G, Koskolou MD, Roach RC. Skeletal muscle blood flow in humans and its regulation during exercise. Acta Physiol Scand 162: 421–436, 1998 [DOI] [PubMed] [Google Scholar]
- 48.Sandow SL, Tare M, Coleman HA, Hill CE, Parkington HC. Involvement of myoendothelial gap junctions in the actions of endothelium-derived hyperpolarizing factor. Circ Res 90: 1108–1113, 2002 [DOI] [PubMed] [Google Scholar]
- 49.Saunders NR, Pyke KE, Tschakovsky ME. Dynamic response characteristics of local muscle blood flow regulatory mechanisms in human forearm exercise. J Appl Physiol 98: 1286–1296, 2005 [DOI] [PubMed] [Google Scholar]
- 50.Schrage WG, Dietz NM, Joyner MJ. Effects of combined inhibition of ATP-sensitive potassium channels, nitric oxide, and prostaglandins on hyperemia during moderate exercise. J Appl Physiol 100: 1506–1512, 2006 [DOI] [PubMed] [Google Scholar]
- 51.Schrage WG, Joyner MJ, Dinenno FA. Local inhibition of nitric oxide and prostaglandins independently reduces forearm exercise hyperaemia in humans. J Physiol 557: 599–611, 2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Seals DR, Victor RG. Regulation of muscle sympathetic nerve activity during exercise in humans. Exerc Sport Sci Rev 19: 313–349, 1991 [PubMed] [Google Scholar]
- 53.Segal SS. Regulation of blood flow in the microcirculation. Microcirculation 12: 33–45, 2005 [DOI] [PubMed] [Google Scholar]
- 54.Segal SS, Jacobs TL. Role for endothelial cell conduction in ascending vasodilatation and exercise hyperaemia in hamster skeletal muscle. J Physiol 536: 937–946, 2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shimokawa H, Yasutake H, Fujii K, Owada MK, Nakaike R, Fukumoto Y, Takayanagi T, Nagao T, Egashira K, Fujishima M, Takeshita A. The importance of the hyperpolarizing mechanism increases as the vessel size decreases in endothelium-dependent relaxations in rat mesenteric circulation. J Cardiovasc Pharmacol 28: 703–711, 1996 [DOI] [PubMed] [Google Scholar]
- 56.Shoemaker JK, Halliwill JR, Hughson RL, Joyner MJ. Contributions of acetylcholine and nitric oxide to forearm blood flow at exercise onset and recovery. Am J Physiol Heart Circ Physiol 273: H2388–H2395, 1997 [DOI] [PubMed] [Google Scholar]
- 57.Shoemaker JK, Naylor HL, Pozeg ZI, Hughson RL. Failure of prostaglandins to modulate the time course of blood flow during dynamic forearm exercise in humans. J Appl Physiol 81: 1516–1521, 1996 [DOI] [PubMed] [Google Scholar]
- 58.Sprague RS, Ellsworth ML, Stephenson AH, Lonigro AJ. ATP: the red blood cell link to NO and local control of the pulmonary circulation. Am J Physiol Heart Circ Physiol 271: H2717–H2722, 1996 [DOI] [PubMed] [Google Scholar]
- 59.Taddei S, Ghiadoni L, Virdis A, Buralli S, Salvetti A. Vasodilation to bradykinin is mediated by an ouabain-sensitive pathway as a compensatory mechanism for impaired nitric oxide availability in essential hypertensive patients. Circulation 100: 1400–1405, 1999 [DOI] [PubMed] [Google Scholar]
- 60.Tschakovsky ME, Sheriff DD. Immediate exercise hyperemia: contributions of the muscle pump vs. rapid vasodilation. J Appl Physiol 97: 739–747, 2004 [DOI] [PubMed] [Google Scholar]
- 61.Welsh DG, Segal SS. Coactivation of resistance vessels and muscle fibers with acetylcholine release from motor nerves. Am J Physiol Heart Circ Physiol 273: H156–H163, 1997 [DOI] [PubMed] [Google Scholar]
- 62.Welsh DG, Segal SS. Endothelial and smooth muscle cell conduction in arterioles controlling blood flow. Am J Physiol Heart Circ Physiol 274: H178–H186, 1998 [DOI] [PubMed] [Google Scholar]
- 63.Wilson JR, Kapoor SC, Krishna GG. Contribution of potassium to exercise-induced vasodilation in humans. J Appl Physiol 77: 2552–2557, 1994 [DOI] [PubMed] [Google Scholar]