

Abstract
Background
The pathogenesis of collateral growth (arteriogenesis) has been linked to both the innate and adaptive immune systems. While therapeutic approaches for the augmentation of arteriogenesis have focused on innate immunity, exploiting both innate and adaptive immune responses has not been examined. We hypothesized that tetanus toxoid (tt) immunization of mice followed by transplantation of monocytes (Mo) exposed ex vivo to tt augments arteriogenesis after ligation of the hind limb.
Methods and Results
Mo were generated from nonimmunized BALB/c mice, exposed ex vivo to tt for 24 hours and intravenously injected (ttMo, 2.5×106) into the tail veins of tt‐immunized syngeneic mice whose hind limbs had been ligated 24 hours prior to transplantation. Laser Doppler perfusion imaging was applied, and a perfusion index (PI) was calculated (ratio ligated/unligated). Twenty‐one days after ligation, the arteriogenesis of untreated BALB/c mice was limited (PI=0.49±0.09). Hind limb function was impaired in 80% of animals. Injection of non‐engineered Mo insignificantly increased the PI to 0.56±0.07. However, ttMo transplantation resulted in a strong increase of the PI to 0.82±0.08 (n=7; P<0.001), with no (0%) detectable functional impairment. ttMo injected into nonimmunized mice had no effect. The strong arteriogenic response of ttMo transplantation into immunized mice was prevented when mice had been depleted of T‐helper cells by CD4‐antibody pretreatment (PI=0.50±0.08; n=17; P<0.001), supporting the hypothesis that transplanted cells interact with recipient lymphocytes.
Conclusions
Transplantation of ttMo into pre‐immunized mice strongly promotes arteriogenesis. This therapeutic approach is feasible and highly attractive for the alleviation of morbidity associated with vascular occlusive disease.
Keywords: arteriogenesis, cell transplantation, innate and adaptive immune system, monocyte, tetanus toxoid
Introduction
The endogenous response of the body to a progressive vascular disease with consecutive luminal narrowing (eg, peripheral artery disease or coronary artery disease) is the active remodeling and growth of pre‐existing arterioles towards larger functional collateral arteries, a process termed arteriogenesis.1 Indeed, an association between collateral blood flow, myocardial viability and reduced long‐term mortality has been demonstrated in patients with coronary heart disease.2–4 However, only approximately one‐third of the maximal conductance of a normal artery is restored by endogenous collateral artery growth.5 Hence, therapeutic stimulation of arteriogenesis represents an appealing concept, especially considering that 20% of patients with vascular disease are not suitable for current treatments, such as percutaneous interventions or surgical bypass grafts.6
Collateral vessel growth is strongly driven by local inflammation. This inflammatory response includes endothelial activation and the local recruitment of leukocytes, mainly monocytes (Mo).7 Mo mature to macrophages and create a highly arteriogenic environment by secreting multiple growth factors that induce the remodeling of arterioles into functional collateral arteries.8–10 Accordingly, pharmacological reduction of blood Mo numbers or blockade of their local attraction by genetically targeting the chemokine (C‐C motif) receptor 2 (CCR2) leads to defective collateral growth.11–12 Vice versa, enhanced local attraction of Mo correlates with augmented arteriogenesis.13–14 However, not only innate immunity but also the adaptive immune system participates in the arteriogenic response.15 Blocking the T‐cell response (mice deficient in CD4/CD8 or MHC‐class‐II) resulted in a lack of collateral growth.16–18
The multifactorial nature of this process may explain why single‐factor approaches for the augmentation of arteriogenesis have generated mixed results (reviewed in1,19), which has thus led to the investigation of cell‐based therapies. Autologous bone marrow‐derived stem and progenitor cells have been identified as potential cells for transplantation, but only moderate clinical benefits were reported.20 However, in addition to dosage, isolation method, and administration route, further research is required to determine the optimal cell type. A disadvantage of autologous cell transplantation in particular could be the cells' inability to activate the innate and/or adaptive immune response, although either is of tremendous importance for arteriogenesis.
Our group has been intrigued by the idea that increasing local inflammation may represent the best stimulus for arteriogenesis. We have exploited the homing of Mo to areas of collateral growth for cell transplantation studies.21 Intriguingly, transplantation of allogeneic Mo resulted in the strongest arteriogenic response, implying that, indeed, targeting both the innate and the adaptive immune systems may emerge as the most efficient way to clinically augment collateralization.21–22
This observation led us to elaborate a clinical applicable immunotherapeutic approach. We hypothesized that tetanus toxoid (tt)‐pulsed syngeneic Mo vaccination into tt‐immunized mice whose hind limbs had been ligated elicits a strong arteriogenic immune response. As Mo isolation from humans and their ex vivo engineering are both easily performed and safe, translation of such an approach to clinical situations would be highly attractive.
Methods
Animal Model
Our study was performed with permission of the State of Saxony, Regierungspraesidium Dresden, according to Section 8 of the German Law for animal protection, and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health. We studied 8‐ to 13‐week‐old male BALB/c and C57Bl/6 mice from Charles River Laboratories. Mice were anesthetized by intraperitoneal injection of xylazine (20 mg/kg body weight) and ketamine (110 mg/kg body weight, MEDISTAR). The left femoral artery of each BALB/c mouse was occluded by ligation with 6‐0 sutures immediately distal to the origin of the deep femoral artery and proximal to the origin of any other distal branch.23–24 Hind limb perfusion was determined by laser Doppler perfusion imaging (LDPI) (PeriMed) as previously described.21,23 The mean values from the occluded and non‐occluded sides were divided to calculate a perfusion index (PI). LDPI was performed before, immediately after, and 7, 14, and 21 days following occlusion (n=7). Histological quantification of vessel density was performed on day 21 (n=3).
Isolation and Differentiation of Murine Bone Marrow‐Derived Monocytes (BMDMos)
Using a protocol previously published by our group,25 bone marrow cell suspensions were isolated by flushing femurs and tibias of 8‐ to 12‐week‐old BALB/c and C57Bl/6 mice (Charles River).
Mo were obtained by isolating CD115+, CD11b+, Gr‐1+, and F4/80 low cells as described previously.25 Cells were adjusted to a suspension of 106 cells/mL and were seeded on 6‐well ultra‐low‐attachment surface plates (Corning Costar).
FACS Analysis
For cell characterization, antibodies against the following antigens were used: CD4, CD8, CD11b, CD11c, CD25, CD45, CD45R, CD80, CD86, CD115, Gr‐1, CD154, the major histocompatibility complex (MHC) class II receptor (all from eBioscience), F4/80 (AbD Serotec), or CD117 (c‐kit) (Miltenyi Biotec). Samples were analyzed on a FACSCalibur (Becton Dickenson) using CellQuest software (Becton Dickenson). The phenotype of isolated Mo was further verified by detecting MHC class II receptor up‐regulation and antigen presentation. Functional phenotyping of isolated Mo was achieved via static and dynamic adhesion to endothelial cells in a fluid chamber as described previously.25
Ex Vivo Cell Engineering With Tetanus Toxoid
Mo were co‐stimulated with murine‐recombinant interferon‐γ (mrIFN‐γ) to increase MHC class II receptor up‐regulation and thereby improve CD4+ T cell activation via antigen presentation.26 Mo were stimulated with either 8 ng/mL mrIFN‐γ (eBioscience) and 100 μg/mL tt (ttMo) or with 8 ng/mL mrIFN‐γ alone for 24 hour (non‐engineered Mo; neMo). MHC class II receptor expression was quantified by FACS analysis. Mo were gated according to F4/80 expression.
Cell Transplantation
Mo were harvested, resuspended in saline and injected into the tail veins of mice. Cell transplantation was performed 24 hours after ligation of the femoral artery. To control for possible contamination with remaining stem and progenitor cells, Mo for control and engineering experiments were taken from the same isolation pool. CD4+ T cell depletion: To deplete CD4+ cells, we intraperitoneally (i.p.) injected an anti‐CD4 antibody (100 μg/mouse, eBioscience) before and 24 hours after ligation of the femoral artery. CD4+ T cell depletion was verified by FACS analyses of mouse blood.
Immunization of Mice
Immunization and booster injections were performed with commercially available tt (Tetanol pur®; Novartis). To immunize the mice, 8 I.E. tt was used in a volume of 100 μL saline. tt was injected subcutaneously into the necks of the mice at day 0; a booster injection was given 3 weeks later by intraperitoneal injection of 8 I.E. tt. To determine the immunological response, 120 μL blood serum samples were taken before tt immunization, 2 weeks afterwards and 1 week following the booster injection. Blood samples were stored at −20°C for later analysis of tt‐antibody levels by enzyme‐linked immunosorbent assay (ELISA; Institute Virion/Serion). The measurements were validated with anti‐mouse‐IgG analysis (anti‐mouse‐IgG, Sigma Aldrich).
In Vitro Lymphocyte Stimulation, CFSE and MTT Proliferation Assay
Leukocytes were isolated from immunized and nonimmunized mice and were prepared according to standard protocols.27 Briefly, cell suspensions were adjusted to give densities of 5×106 cells/mL for cytokine secretion and 2.5×106 cells/mL for proliferation assays in complete DMEM high‐glucose medium (Sigma Aldrich). To each 24‐well of leukocytes, 7.5×106 Mo were added in different conditions: Mo+20 μg/mL tt, Mo+100 μg/mL tt, Mo+8 ng/mL IFN‐γ, Mo+20 μg/mL tt+8 ng/mL IFN‐γ, Mo+100 μg/mL tt+8 ng/mL IFN‐γ and non‐engineered Mo. The medium was supplemented with 20 ng/mL recombinant murine macrophage colony‐stimulation factor (rmM‐CSF, Miltenyi Biotec) for a 24‐hour pre‐stimulation period. Supernatant from the co‐cultures was stored at −80°C after a 48‐hour incubation period for quantification of cytokine production by ELISA (eBioscience). To determine CD4+ T cell proliferation, 2.5×106 cells/mL spleen‐derived leukocytes were incubated in co‐cultures with 7.5×106 Mo for 5 days and were supplemented with carboxyfluorescein‐diacetate‐succinimidyl‐ester (CFSE, Invitrogen). Cells were harvested after 5 days and counterstained with Alexa 647 anti‐CD4 (Sigma Aldrich) before quantification of CD4+ T cell proliferation by FACS analysis. Proliferating CD4+ T cells were quantified based on the dilution of CFSE fluorescence and CD4 co‐expression. The endogenous CD4+ T cell proliferation capacity was analyzed and termed “baseline proliferation.” To induce lymphocyte interaction above this baseline level, either pure tt (20 μg/mL) or ttMo were added to spleen‐derived T cells from tt pre‐immunized mice.
Proliferation was also determined in an MTT (3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide) assay. Cells were incubated with MTT, and cell numbers were determined by ELISA (Cell Growth Determination Kit; Sigma Aldrich). Clonal cell expansion was abolished by co‐incubation of the cultures with blocking functional grade anti‐MHC class II receptor antibodies (Rat IgG2b, eBioscience).
Cell Tracking of Mo Homing to Areas of Collateralization
Mo were stained with Vybrant‐DiI cell‐labeling solution (1,1′‐Dioctadecyl‐3,3,3′,3′‐Tetramethylindocarbocyanine, Invitrogen) for 24 hours. Histologic analysis was preformed 48 hours after systemic Mo transplantation for homing experiments. Fluorescein isothiocyanate (FITC) anti‐α‐actin staining was used to visualize and quantify collateral vessels. An anti‐F4/80 antibody (eBioscience) was used to identify Mo infiltration in areas of collateralization.
Immunofluorescence and Morphometric Analysis
Mice were sacrificed at indicated time points, and hind limb muscles were harvested. Frozen tissue sections were incubated with primary antibodies at recommended dilutions (rabbit anti‐CD31, Abcam; rat anti‐F4/80, eBioscience), followed by incubation with an appropriate cross‐absorbed secondary Cy3‐conjugated antibody. A FITC‐conjugated mouse anti α‐smooth muscle actin antibody (Sigma Aldrich) was used to identify growing collaterals, followed by nuclear counterstaining with 4′,6‐diamidino‐2‐phenylindole (DAPI). Sections were analyzed on an Axiovert S100 (Carl Zeiss) and an RT™ KE slider camera system (Diagnostic Instruments). Capillary density was measured in CD31‐stained sections, and vessel size and density were analyzed in α‐actin‐stained tissues; only vessels with media‐to‐lumen‐ratios typical for arteries were taken into consideration. Mo infiltration was assessed by counting F4/80‐positive cells near vessels identified as arteries by their α‐actin staining. Vessel density was calculated by automatically counting stained objects in cross‐section images. For this purpose, 3 sections per muscle were stained, and 10 representative images per slice were captured in 200‐fold magnification using a digital microscope and camera. Image analysis was performed using Image‐Pro plus 6 (Mediacybernatics).
Cytokine Quantification by Luminex Technology
Cytokines, chemokines, and growth factor levels in mouse serum were quantified at different time points (blood sample time line: before ligation and at 8 h, 24 h, 72 h, 7 days, 14 days, and 21 days after ligation). Serum was stored after a spin‐down procedure at −80°C, and mice were injected with 250 μL saline i.p. after each blood withdrawal. Cytokine concentrations of IL‐1α, IL‐1ß, IL‐2, IL‐3, IL‐4, IL‐5, IL‐6, IL‐9, IL‐10, IL‐12 (p40), IL‐12 (p70), IL‐13, IL‐17, Eotaxin, G‐CSF, GM‐CSF, IFN‐γ, KC, MCP‐1 (MCAF), MIP‐1α, MIP‐1ß, RANTES, TNF‐α, FGF basic, IL‐15, IL‐18, LIF, MCSF, MIG, MIP‐2, PDGF‐BB, and VEGF were determined using the multiplex pro‐mouse cytokine 23‐plex and 9‐plex panels and the cytokine reagent kit (Bio‐Rad Laboratories) as described previously.25 Samples were analyzed as triplets and standards in duplicates using a Luminex‐100 instrument with Bio‐Plex Manager 4.1 software (Bio‐Rad Laboratories).28
Statistical Analysis
The parameters analyzed are summarized as the means±std. The whiskers in the bar charts represent standard errors. Of special interest to the experiment are vessel proliferation (the main effect) and the time effect (after ligation) for developing from post‐ligation to 21 days post‐ligation. To analyze these effects, we performed a 2‐factorial ANOVA with cross effects using the GLM procedure in SAS®. In case of a significant global test, Tukey's test was used as a post hoc test for pairwise group comparisons. Significance levels were assessed using the adjusted F‐Test by Huynh‐Feldt.
All statistical decisions were 2‐tailed with a critical probability of α=5% without α‐adjustment, except for the Tukey's test for pairwise group comparisons. For this reason, additional statistical analyses to support the interpretations were performed in an exploratory manner. Significant results at P<0.05 are indicated with asterisks (*) to the figure legends.
Results
Cell Engineering and Characterization
Syngeneic Mo were generated from the bone marrows of nonimmunized BALB/c mice as described in the Methods section. To increase the expression of MHC class II and thereby enhance the capacity to activate CD4+ T cells, Mo were co‐stimulated with IFN‐γ. These cells were extensively phenotyped to verify their monocytic properties.25 Accordingly, we conducted all antigen uptake and presentation assays under IFN‐γ co‐stimulation. The Mo were then in vitro engineered to present tt antigen by exposing them to 100 μg/mL tt for 24 h.
To determine whether T cells from tt‐immunized mice recognized the ttMo, we quantified T cell expansion and cytokine secretion following co‐culture. Spleens were harvested from immunized and nonimmunized BALB/c mice to isolate leukocytes. Flow cytometry revealed that the cell suspension contained 27.9±5.4% CD4+ T cells, 14.2±3.6% CD8+ T cells, 46±4.4% B cells, and 3.2±0.8% CD4+/CD25+ T cells. Eight percent of the cells were positive for CD11b, which is a marker for Mo and polymorphonuclear leukocytes (PMN). Using an MTT proliferation assay, we determined the proliferation of T cells isolated from spleens of tt‐immunized mice after co‐incubation with either ttMo or neMo. The 2‐factor repeated measurement ANOVA indicated dependence on the T‐cell stimulation ratio (stimulation/baseline) for both proliferation (P=0.002) and blocking by anti‐MHC II antibodies (P=0.003). Baseline proliferation increased 1.28±0.06‐fold after adding neMo to the splenocyte culture paired t test, P=0.016). After adding ttMo, proliferation increased 2.64±0.16‐fold compared to baseline (P=0.003 and increased 1.28±0.06 baseline versus neMo, P=0.002, Figure 1A). Proliferation could be blocked almost completely when a specific antibody against MHC class II receptor had been added to the cell suspension, indicating that the proliferation was a specific response to the antigen presentation by ttMo (P=0.003, paired t test). The proliferation of T cells from immunized mice was not influenced by addition of tt to the medium (data not shown).
Figure 1.
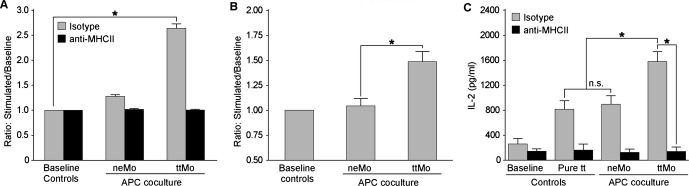
A, T cell proliferation assay (MTT) demonstrating augmentation of proliferation when ttMo were cultivated with APC in comparison to baseline (baseline=endogenous proliferation of non‐stimulated cells, P=0.002, ANOVA with 2 repeated factors). Proliferation could be blocked by supplementation of anti‐MHC II (P=0.003, ANOVA with 2 repeated factors). This result indicates the dependence of MHC II receptor‐mediated tt antigen presentation by ttMo. B, Quantitative analysis of CD4+ T‐cell proliferation after co‐cultivation with ttMo in an in vitro CFSE assay compared to baseline (repeated measurement ANOVA, P=0.022); (C) Quantitative analysis of IL‐2 production in spleen‐derived T cells from immunized mice after stimulation with ttMo, neMo or co‐cultivation with pure tt. IL‐2 production increased after adding ttMo to an APC co‐culture in comparison to co‐culturing with neMo or supplementation of pure tt (P=0.002, ANOVA). This augmentation of IL‐2 production could be blocked completely when anti‐MHC II was added to the cell suspension (P=0.023). No significant difference in IL‐2 production was observed between pure tt and neMo groups (P=0.153 paired t test). The bar charts represent the results as the means±standard errors. The asterisks indicate significant pairwise comparisons (P<0.05, without α‐adjustment). ANOVA indicates analysis of variance; APC, antigen‐presenting cell; CFSE, carboxyfluorescein‐diacetate‐succinimidyl‐ester; IL, interleukin; MHC, major histocompatibility complex; MTT, 3‐[4,5‐dimethylthiazol‐2‐yl]‐2,5‐diphenyltetrazolium bromide; neMo, non‐engineered monocytes; ttMo, tetanus toxoid monocytes.
The quantitative analysis for the CD‐4 T‐cell proliferation revealed an increase in the stimulation ratio of antigen‐presenting cell (APC) co‐cultures (repeated measurement ANOVA; P=0.022). Anti‐CD4 staining of CFSE‐labeled cells prior to FACS analysis revealed that CD4+ T cells expanded up to 1.43‐fold following incubation with ttMo compared with T cells exposed to neMo (Figure 1B). Additionally, we analyzed IL‐2 release of splenic T cells from pre‐immunized mice by ELISA. For the IL‐2 production of spleen‐derived T‐cells from immunized mice, we observed increasing levels of IL‐2 production (P=0.002) and a reduction in IL‐2 levels after co‐incubation with MHC II receptor antibody, (P=0.023, repeated measurement ANOVA). Upon co‐cultivation with ttMo, IL‐2 production increased dramatically from 262±230 pg/mL at baseline to 1577±425 pg/mL (P=0.003 Figure 1C). Stimulation of the T cells by adding pure tt directly to the cell culture medium resulted in an IL‐2 concentration of 816±363 pg/mL (P=0.001 versus baseline, paired t test). Remarkably, neMo also induced a significant increase in IL‐2 (897±358 pg/mL, P<0.0015 versus baseline), suggesting nonspecific lymphocyte (splenocyte) activation. Proliferation and cytokine secretion was abolished by incubation of co‐cultures with functional anti‐MHC class II receptor antibodies. Thus, Mo had been successfully engineered ex vivo to activate the adaptive immune system.
Transplantation of Tetanus Toxoid‐Engineered Monocytes Augments Blood Flow in the Ligated Hind Limbs of Immunized Mice
For this experiment, 2.5×106 neMo, 2.5×106 ttMo or normal saline were intravenously injected into the tail veins of non‐immunized BALB/c mice 24 hour after ligation of the femoral artery. Laser Doppler perfusion imaging of the hind limb allowed serial determination of the PI (Figure 2A). Endogenous collateralization was significant but insufficient. In these non‐immunized mice, the PI after saline injection only moderately increased over time (from 0.15±0.03 immediately post ligation to 0.30±0.08 1 week, 0.41±0.08 2 weeks and 0.49±0.09 3 weeks after ligation, ANOVA, P<0.001, Figure 2A), and 80% of these control mice suffered from hind limb necrosis and gangrene (Figure 2D). Transplantation of ttMo or neMo into nonimmunized mice induced a slight but insignificant further increase of arteriogenesis (PI after 3 weeks 0.58±0.11 versus 0.56±0.07 versus saline 0.49±0.09, ANOVA, n=25, P=0.105 t test, Figure 2A).
Figure 2.
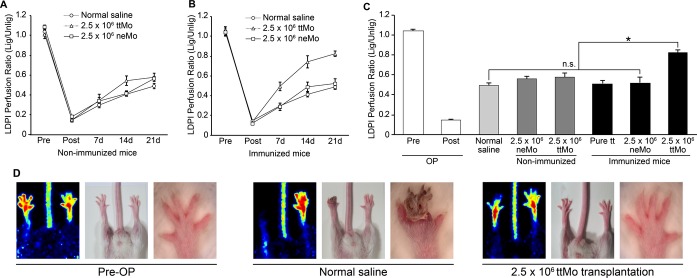
A+B, Hind limb perfusion assayed by laser Doppler measurement, shown as the ratio of ligated to unligated hind limbs at various time points after femoral artery ligation. The 2‐factor ANOVA for treatment groups as the main factor and the restoration of hind limb perfusion from the time point post‐ligation up to 21 days as the repeated factor shows significance between these 2 factors and in cross effects of these factors (P<0.001). A, Cell transplantation into non‐immunized mice: the P values among ttMo, neMo and saline injection are P<0.001 for the time effect and P<0.001 for cross‐effect stimulation×time P=0.019. Tukey's post hoc test at day 21 for the LDPI measurement shows only a difference between ttMo and normal saline (P<0.05). B, Cell transplantation into pre‐immunized mice with tt: the P values of all the 3 groups are P<0.001. Tukey's post hoc test at day 21 indicates a difference between transplantation and ttMo, neMo and normal saline (P<0.05). C, Comparison of the LDPI perfusion ratios between all experimental groups 3 weeks after ligation of the femoral artery, the time point of maximal collateralization: Tukey's post hoc test of ANOVA indicates significant differences (P<0.05) between transplantation of ttMo into neMo‐immunized mice and transplantation of ttMo into non‐immunized mice and between pure tt injection and transplantation of neMo into pre‐immunized mice. Between these groups, no significant differences were noted (ANOVA, P=0.245, n=39). D, Representative pictures of the clinical status of hind limbs pre‐operatively and 3 weeks after ligation. ANOVA indicates analysis of variance; LDPI, laser Doppler perfusion imaging; lig, ligated; neMo, non‐engineered monocytes; ttMo, tetanus toxoid monocytes; Unlig, unligated.
However, transplantation of 2.5×106 ttMo into mice that had been pre‐immunized with tt tremendously increased collateralization compared with normal saline injection or transplantation of neMo into pre‐immunized mice (PI 0.82±0.08 versus 0.49±0.09 and 0.52±0.14, respectively, ANOVA, n=25, P<0.001, Figure 2B). None of the ttMo‐injected mice developed ischemic symptoms, such as necrosis or gangrene (Figure 2D). Notably, the strong response of tt pre‐immunized mice was comparable to the results that we obtained when we transplanted allogeneic Mo (generated from C57Bl/6 mice) into nonimmunized BALB/c mice, a condition that was used to recapitulate our initial observation in rabbits.22,25
Additionally, when tt was injected subcutaneously into pre‐immunized mice, no additional arteriogenic responses were detected (PI 0.51±0.09 versus normal saline injection, 0.49±0.09, P<0.585, Figure 2C). Figure 2C summarizes the results of all experimental groups 3 weeks after ligation of the femoral artery, the time point of maximal collateralization. These results indicate that the tt‐antigen presentation capacity of the ttMo and their homing to sites of arteriogenesis is tremendously important for augmentation of collateral vessel growth. Indeed, their homing to areas of collateralization was visualized by transplantation of ex vivo DiI‐labeled ttMo and subsequent immunohistochemical analysis of the upper limbs (Figure 5).
Figure 5.
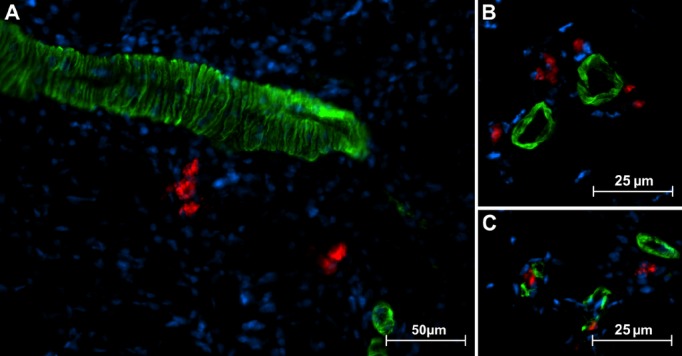
Representative immunostaining of adductor muscle sections. non‐engineered monocytes (neMo) were stained with DiI prior to transplantation. DiI‐positive (red) syngeneic Mo near α‐actin‐positive (green) vessels 48 hour after transplantation. DiI‐positive cells were rarely found near large vessels (A) but typically accumulated near small arterioles (B and C).
To test whether transplantation of ttMo augments collateralization through a CD4+ T‐cell‐dependent response, BALB/c mice were depleted of CD4+ T cells by antibody treatment, which reduced peripheral blood CD4+ T cells by 90.2% after 2 weeks. Quantitative analysis of peripheral blood leukocyte concentrations up to 21 days after anti‐CD4 antibody injection revealed clear time dependence (Figure 3A, left panel). The quantitative analysis of peripheral blood leukocyte concentrations indicates a pre‐post effect after anti‐CD4 antibody injection (P=0.040, paired t test). The proportions of B cells and CD4+ T cells in lymphocyte subfractions changed from day 1 to 3 weeks (ANOVA, P=0.002), and a pre‐post effect after antibody injection was evident (P=0.006, paired t test) (Figure 3A, right panel). When ttMo were injected after CD4+ T cell depletion, the arteriogenic response was completely abolished (PI 0.82±0.08 versus PI 0.49±0.09, ANOVA; n=17; P<0.001; Figure 3B). These data suggest that the immunological response of the host to the graft is CD4+ T cell‐dependent.
Figure 3.
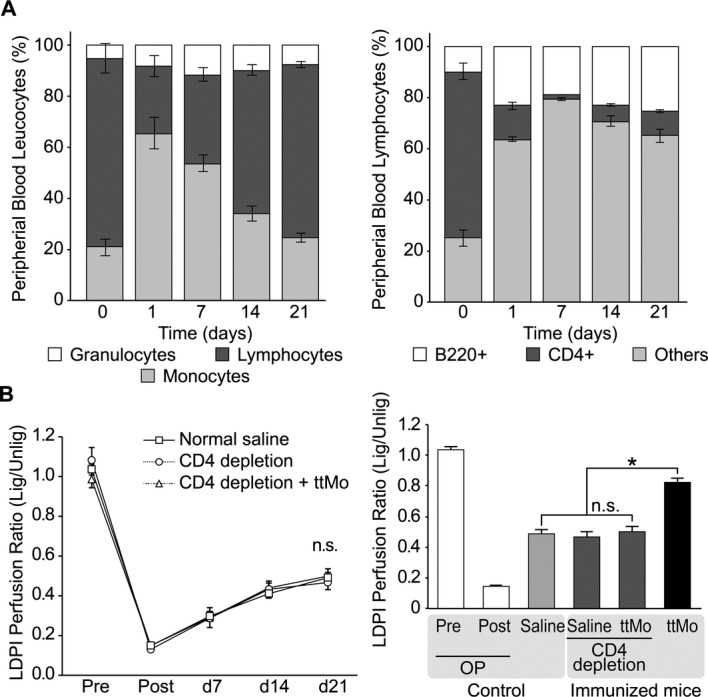
A, Quantitative analysis of peripheral blood leukocyte concentrations up to 21 days after anti‐CD4 antibody injection. Left panel: peripheral blood leukocytes, P=0.023 for the change in lymphocyte proportions between day 1 and 3 weeks (ANOVA); right panel: sub‐fractionation of lymphocytes into B cells and CD4+ T cells demonstrated the changing proportions from day 1 to 3 weeks (P=0.0021, ANOVA) and the pre‐post effect (P=0.006, paired t test). B, Left panel: kinetics of the LDPI perfusion ratio of pre‐immunized mice (normal saline vs CD4‐depletion vs CD4‐depletion plus ttMo transplantation, P<0.189, ANOVA); right panel: LDPI perfusion ratio 3 weeks after ligation of the femoral artery, the time point of maximal collateralization (P<0.001, t test). The transplantation of ttMo into non‐CD4+ T‐cell‐depleted mice is shown for comparison (black bar). ANOVA indicates analysis of variance; LDPI, laser Doppler perfusion imaging; lig, ligated; ttMo, tetanus toxoid monocytes; Unlig, unligated.
Tetanus Toxoid‐Engineered Syngeneic Monocyte Transplantation Reduces Ischemia Within the Lower Limbs
Because capillary density within the lower limb indicates hypoxia‐driven angiogenesis, we histologically quantified their numbers within the calf muscle prior to and after ligation. The capillary density increased from 280±41 capillaries/mm2 in the hind limbs of unligated mice to 1336±198 1 week, 1570±90 2 weeks and 1682±348 capillaries/mm2 3 weeks after ligation (ANOVA, P=0.003, Figure 4A), suggesting an insufficient proximal endogenous collateralization. Animals were sacrificed at the 3‐week time point for histological analysis of the effects of cell transplantation. ANOVA between the 5 treatment groups (Figure 4B) indicated that the differences were significant (P=0.005), and Tukey's post hoc test showed that ttMo resulted in less distal capillary formation in comparison to the other treatment groups (ttMo 1095±181 capillaries/mm2 versus 2.5×106 neMo in immunized mice 1478±109 capillaries/mm2 versus 2.5×106 neMo in nonimmunized mice 1891±139 capillaries/mm2 P<0.05). These results imply that collateralization resulted in sufficient blood supply to the distal limb.
Figure 4.
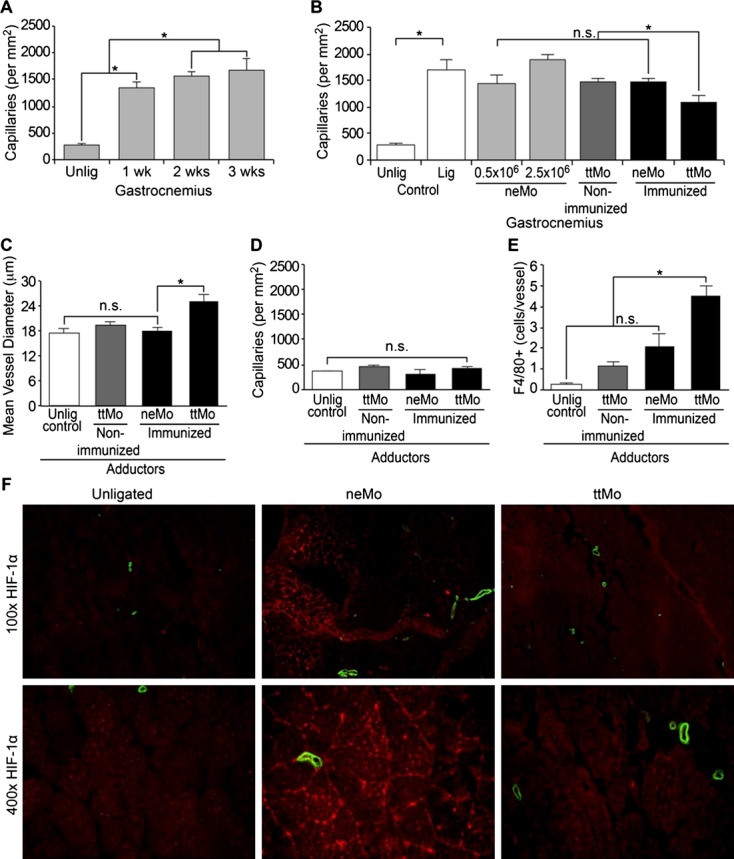
A, Histologic workup of capillary density within the gastrocnemius muscle before and up to 3 weeks after ligation in untreated control animals with increasing capillary density after ligation (P=0.003, ANOVA); (B) Quantitative comparison of capillary density between the experimental groups 3 weeks after ligation; (C) Determination of mean vessel diameter within the adductor muscle, increasing vessel diameter in immunized mice after transplantation of ttMo (P=0.005, ANOVA; P<0.005, Tukey's post hoc test); (D) Quantitative analysis of capillary density in the adductor muscle (P=0.181, ANOVA); (E) Quantification of perivascular Mo infiltration after systemic cell transplantation (P=0.001, ANOVA); (F) Immunofluorescence staining of HIF‐1α (Cy3, red) and smooth muscle‐α‐actin (FITC, green) demonstrate low levels of HIF‐1α within the distal limbs of animals that had received ttMo, while normal saline controls and neMo transplantation exhibited pronounced HIF‐1α stabilization and high capillary density. ANOVA indicates analysis of variance; FITC, fluorescein isothiocyanate; HIF, hypoxia‐inducible factor; lig, ligated; neMo, non‐engineered monocytes; ttMo, tetanus toxoid monocytes; Unlig, unligated.
ANOVA was used to determine changes in vessel diameter within the adductor muscle. Increased blood supply by therapeutic arteriogenesis was further evidenced by a 12% increase in arteriolar diameter in areas of collateralization (upper limb) when ttMo had been transplanted to tt pre‐immunized mice. Tukey's post hoc test indicated that there were no differences in vessel diameter after neMo transplantation, injection of normal saline or transplantation of ttMo to non‐pre‐immunized mice (Figure 4C).
No changes in capillary density within the adductor muscle were evident in any of the groups (Figure 4D, ANOVA; P=0.181).
A global comparison (ANOVA; P=0.001) demonstrated significantly more perivascular cell infiltration in the ttMo group versus the neMo and ttMo nonimmunized mouse groups. Perivascular Mo/macrophage infiltration within the adductor muscle, where collateralization is best visualized, was examined. Compared to the unligated leg, ttMo transplantation into nonimmunized mice already resulted in an increase of local Mo/macrophage accumulation (0.3±0.1 versus 1.1±0.4 cells/vessel, respectively). Infiltration was comparable to transplantation of neMo into immunized animals (2.1±1.1 cells/vessel). However, transplantation of ttMo into immunized mice led to a pronounced Mo/macrophage accumulation (4.5±0.8 cells/vessel, Tukey's post hoc test demonstrated that most cell infiltration was in the ttMo group; P<0.05; Figure 4E). Perivascular infiltration of Mo was visualized in representative pictures (Figure 5).
Finally, immunofluorescence visualization of HIF‐1α was used as an additional marker of ischemia within the distal gastrocnemius muscle. Three weeks after ligation of the hind limbs of nontreated BALB/c mice, HIF‐1α expression was still detectable, implying persistent local ischemia triggering angiogenesis. However, when ttMo had been transplanted, HIF‐1α expression was almost completely abolished, suggesting normoxic conditions in the lower limb (Figure 4F, right panel). Transplantation of neMo was unable to alleviate hypoxia‐induced HIF‐1α expression. Remarkably, no functional impairment was detected when ttMo had been transplanted.
Immunization of Mice
BALB/c mice were immunized with tt. Antibody titers were determined by serum titration and reached 1:1000±100 after 14 days and 1:9500±2100 after 28 days (n=3, repeated measurement ANOVA, P=0.016), indicating an appropriate immune response against tt (Figure 6).
Figure 6.
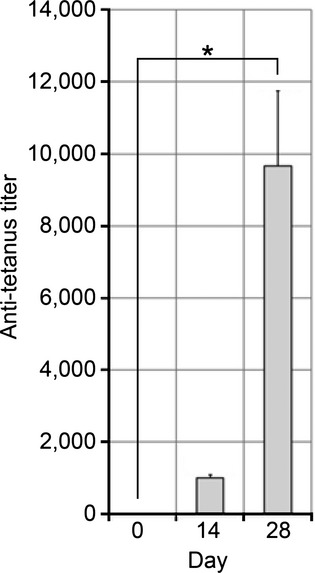
Mouse tt antibody titers; the results are presented as mean±std., n=3, *P<0.05 ANOVA. ANOVA indicates analysis of variance; tt, tetanus toxoid.
Serum Cytokine Quantification
Serum was taken prior and directly post‐ligation and at 8 h, 24 h, 72 h, 7 days, 14 days, and 21 days for cytokine quantification.
Significant increases of IL‐4 and IL‐5 levels in all different groups (immunized mice treated with pure tt only and with either neMo or ttMo) pointed towards an acute phase reaction (Table). IL‐5 levels only rose in immunized mice treated with pure tt or ttMo, suggesting a T‐cell‐dependent immune reaction with consecutive B‐cell stimulation (Table), which finally resulted in an increase in tt antibody titers (see above).
Table 1.
Peak Interleukin and Cytokine Increases After Transplantation of neMo, ttMo or Injection of Pure Tetanol in Immunized Mice Depicted as Ratio of Pre to Post Treatment Levels
| Interleukin/Cytokine | neMo | ttMo | Pure Tetanol | ANOVA |
|---|---|---|---|---|
| Mean Values±Std | Mean Values±Std | Mean Values±Std | P Value | |
| IL‐1β | 2.886±0.281 | 2.004±0.523 | 1.856±0.753 | 0.123 |
| IL‐4 | 1.260±0.275 | 1.506±0.288 | 2.187±0.344 | 0.049 |
| IL‐5 | 1.843±0.199 | 5.569±0.750 | 8.605±1.762 | 0.001 |
| IL‐6 | 5.291±1319 | 7.435±2.263 | 9.589±4.237 | 0.264 |
| IL‐10 | 2.658±2.010 | 3.407±3.235 | 13.461±14177 | 0.263 |
| G‐CSF | 8.601±1.569 | 84.194±16.410 | 25.482±5.986 | <0.001 |
Note that peak occurred at different time points within the 21 day period. ANOVA with main factor transplantation group (quotient between peak of cytokine or interleukin pre‐operation and post transplantation) (n=9). ANOVA indicates analysis of variance; G‐CSF, granulocyte colony‐stimulating factor; IL, interleukin; neMo, non‐engineered monocytes; ttMo, tetanus toxoid monocytes.
G‐CSF levels significantly differed in immunized mice treated with ttMo compared with mice given pure tt or neMo (Table). G‐CSF levels rose from 30±3 pg/mL (prior to ligation) to 211±55 pg/mL after ligation and further increased up to 2020±329 pg/mL within 8 hours when ttMo had been transplanted compared with injection with neMo (728±138 pg/mL) or pure tt (554±235 pg/mL, P<0.05 for either comparison). This G‐CSF peak declined within the next 24 hours and reached control levels 3 days after transplantation (ttMo: 34±3 pg/mL; neMo 36±6 pg/mL; pure tt 123±6 pg/mL).
Discussion
Collateral vessels develop in response to arterial stenosis and occlusion. These vessels have a significant protective effect on organ function and survival. Recently, a meta‐analysis of Seiler and coworkers came to the conclusion that good collateralization in patients with coronary artery disease reduced mortality by 36% in comparison with limited or no collateralization.4 Still, only one‐third of the maximal conductance of a normal artery is restored by endogenous collateral artery growth, and two‐thirds of patients do not have sufficient collateral flow to prevent ischemia during vascular occlusion,29 which illustrates the enormous importance of therapeutic efforts.
Collateral vessels remodel from small interconnecting arterioles into conductance vessels under 2 main conditions: (1) fluid shear stress due to the pressure gradient along the stenosis/occlusion and (2) attraction of mononuclear cells that attach to activated endothelium or enter the vessel adventitia to secrete the optimal repertoire of cytokines and growth factors needed for arteriogenesis to occur.30 In addition to this innate immune response, arteriogenesis is hampered in T‐lymphocyte‐deficient nude mice and CD4−/− mice,16–18,31 suggesting an additional role for the adaptive immune system.
As single‐factor approaches have generated mixed results, we hypothesized that targeting both the innate and adaptive immune systems may emerge as the most efficient way to clinically augment collateralization. This joint targeting was achieved by transplantation of syngeneic Mo, which had been loaded ex vivo with tt, into tt‐immunized mice. We hypothesized that antigen presentation of transplanted Mo to primed T cells would result in their activation and the further stimulation of Mo. An augmentation of endogenous collateral growth based on the principle of activated innate and adaptive immune systems was expected.
First, antigen presentation and the in vivo behavior of the engineered Mo were studied. Indeed, the Mo took up the antigen in vitro and stimulated specific T cells through an MHCII‐restricted mechanism. The adaptive transfer of these cells into immunized mice resulted in an additional increase of the antibody titer, resembling a boost‐immunization, implying that the transplanted Mo were functionally active and accepted in vivo as APCs.
Transplantation of these immunologically active cells into immunized mice resulted in a distinct stimulation of endogenous arteriogenesis. This effect was as potent as previously shown for allogeneic Mo transplantation.21 However, the effect was absent in both non‐immunized mice and immunized mice injected only with pure tt. These control experiments underscored that the arteriogenic effect was based on re‐stimulation of primed T cells with antigen‐presenting Mo and excluded sole activation of the immune system through a boost with tt. Additionally, the absence of collateral growth after CD4+ T‐cell depletion demonstrated that the effect was dependent on the interaction of the transplanted ttMo with specific CD4+ T cells.32–33
We propose that the combination of both stimulation of the adaptive immune system and the homing of transplanted Mo to areas of collateralization and their local activation accounts for the augmentation of arteriogenesis. Indeed, we were able to visualize the homing of fluorescently marked Mo to areas of collateralization. According to,34 incorporation into the vessel wall was never observed. Rather, transplanted cells seemed to be incorporated into the regular Mo pool, as unloaded cells and tt‐loaded cells were locally visualized.
In addition to the local effects caused by antigen‐loaded and activated Mo that had homed to areas of collateralization, we examined the systemic cytokine profile. We detected a rather nonspecific acute‐phase reaction and a TH2 response under both conditions (pure tt immunization and ttMo transplantation). Only G‐CSF was distinguishably induced after transplantation of ttMo. These results imply that transplanted Mo are actively involved in chemotaxis and recruitment of other leukocyte populations and, thus, could further modulate the arteriogenic response. Interestingly, the arteriogenic potential of G‐CSF has recently been shown.35
Preliminary evidence has established the safety and feasibility of autologous cell transplantation.36–37 Bone marrow‐derived stem and progenitor cells in particular have been identified as potential new therapeutic options to augment angiogenesis and collateralization (reviewed in20). Encouraging results of preclinical studies have rapidly led to several small clinical trials. For example, implantation of bone marrow‐derived mononuclear cells in patients with severe peripheral artery diseases resulted in a moderate improvement of clinical parameters.37–38 Nonetheless, large, randomized, placebo‐controlled, double‐blind studies are both necessary and currently ongoing.20 Notably, all cells used thus far share a monocytic phenotype. As Mo are crucial in arteriogenesis, it seems that cell therapies in vascular diseases based on the application of whole monocytic cells are more successful than therapies using sub‐fractionated cell preparations. Additionally, Mo are easily obtainable. However, to date, peripheral blood autologous Mo have not been able to elicit any effect.38 These data are confirmed in our study. Similarly, in another study, PBMCs depleted of CD34+ cells were not able to increase capillary density or to decrease limb loss.39 When examined closely, all approaches, independent of the cell type, have been designed to minimize immunologic and inflammatory reactions against the transplanted cells (autologous cells were used, or stem cells were delivered to syngeneic mice or rats or xenogeneic stem cells were used in severely immunosuppressed mice, such as nude or SCID mice). However, in our hands, transplantation of allogeneic PBMCs was able to elicit the strongest arteriogenic response.21–22 This observation led us to place the emphasis on the inflammatory and immunologic aspects of collateral growth and to engineer autologous Mo locally serving both entities. We strongly believe that this approach represents a new avenue for future successful clinical trials.
Adoptive cell therapy and immunotherapy also continue to play increasing roles in the treatment of many hematologic malignancies.40 Investigators have initiated clinical trials of established cancer with tumor antigen‐loaded dendritic cells.40 They have established proof of principle that properly activated dendritic cells, loaded with the proper form and dose of antigen and properly activated, along with properly migrating to lymph nodes, can initiate and expand tumor‐specific CD4+ and CD8+ T‐cell responses to induce meaningful therapeutic responses.41–43 Thus far, despite the induction of a robust tumor‐specific T‐cell response in many patients and occasional spectacular complete tumor regression, particularly in patients with melanoma, this hope has not been fulfilled.
Translation of our approach to the clinical situation seems attractive, as Mo isolation can be performed easily in humans, and injection of in vitro‐grown cells has been performed already without adverse effects. Furthermore, immunization against a recommended antigen, such as tt, confers a lifelong immunity against the disease and adoptive cell transplantation. Furthermore, a lifelong protective immunity would also be ideal for repetitive cell transplantations for a more effective augmentation of collateralization. Still, although the safety of bone marrow mononuclear cell transplantation has been observed up to 3.2 years,44 especially in pro‐arteriogenic therapies, safety issues regarding stimulation of pathophysiological angiogenesis (ie, tumor angiogenesis, diabetic retinopathy, and intra‐plaque angiogenesis) need to be considered. However, prior to human studies, further insight into the mechanism of cell transplantation would be needed to assure safety in human use. Here, we demonstrated the efficiency of an adoptive Mo‐transplantation in a murine model of acute hind‐limb ischemia in healthy, young, wild‐type mice. Further studies should address the influence in chronic ischemia models and the use in genetic‐disease models (ie, apo‐e‐ or eNOS‐deficient mice).
Sources of Funding
This work was supported by the DFG (Deutsche Forschungsgemeinschaft, German Research Foundation) SFB 854 (Sonderforschungsbereich, collaborative research center).
Disclosures
None.
References
- 1.van Royen N, Piek JJ, Schaper W, Fulton WF. A critical review of clinical arteriogenesis research. J Am Coll Cardiol. 2010; 55:17-25 [DOI] [PubMed] [Google Scholar]
- 2.Sabia PJ, Powers ER, Ragosta M, Sarembock IJ, Burwell LR, Kaul S. An association between collateral blood flow and myocardial viability in patients with recent myocardial infarction. N Engl J Med. 1992; 327:1825-1831 [DOI] [PubMed] [Google Scholar]
- 3.Meier P, Gloekler S, Zbinden R, Beckh S, de Marchi SF, Zbinden S, Wustmann K, Billinger M, Vogel R, Cook S, Wenaweser P, Togni M, Windecker S, Meier B, Seiler C. Beneficial effect of recruitable collaterals: a 10‐year follow‐up study in patients with stable coronary artery disease undergoing quantitative collateral measurements. Circulation. 2007; 116:975-983 [DOI] [PubMed] [Google Scholar]
- 4.Meier P, Hemingway H, Lansky AJ, Knapp G, Pitt B, Seiler C. The impact of the coronary collateral circulation on mortality: a meta‐analysis. Eur Heart J. 2012; 33:614-621 [DOI] [PubMed] [Google Scholar]
- 5.Schaper W, Schaper J. Collateral Circulation: Heart, Brain, Kidney, Limbs. 1993Boston, MA: Kluwer Academic Publishers [Google Scholar]
- 6.Seiler C. The human coronary collateral circulation. Heart. 2003; 89:1352-1357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arras M, Mollnau H, Strasser R, Wenz R, Ito WD, Schaper J, Schaper W. The delivery of angiogenic factors to the heart by microsphere therapy. Nat Biotechnol. 1998; 16:159-162 [DOI] [PubMed] [Google Scholar]
- 8.Wu S, Wu X, Zhu W, Cai WJ, Schaper J, Schaper W. Immunohistochemical study of the growth factors, aFGF, bFGF, PDGF‐AB, VEGF‐A and its receptor (Flk‐1) during arteriogenesis. Mol Cell Biochem. 2010; 343:223-229 [DOI] [PubMed] [Google Scholar]
- 9.Limbourg A, Korff T, Napp LC, Schaper W, Drexler H, Limbourg FP. Evaluation of postnatal arteriogenesis and angiogenesis in a mouse model of hind‐limb ischemia. Nat Protoc. 2009; 4:1737-1746 [DOI] [PubMed] [Google Scholar]
- 10.Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000; 6:389-395 [DOI] [PubMed] [Google Scholar]
- 11.Heil M, Ziegelhoeffer T, Pipp F, Kostin S, Martin S, Clauss M, Schaper W. Blood monocyte concentration is critical for enhancement of collateral artery growth. Am J Physiol. 2002; 283:H2411-H2419 [DOI] [PubMed] [Google Scholar]
- 12.Heil M, Ziegelhoeffer T, Wagner S, Fernandez B, Helisch A, Martin S, Tribulova S, Kuziel WA, Bachmann G, Schaper W. Collateral artery growth (arteriogenesis) after experimental arterial occlusion is impaired in mice lacking CC‐chemokine receptor‐2. Circ Res. 2004; 94:671-677 [DOI] [PubMed] [Google Scholar]
- 13.Buschmann IR, Hoefer IE, van Royen N, Katzer E, Braun‐Dullaeus RC, Heil M, Kostin S, Bode C, Schaper W. GM‐CSF: a strong arteriogenic factor acting by amplification of monocyte function. Atherosclerosis. 2001; 159:343-356 [DOI] [PubMed] [Google Scholar]
- 14.Ito WD, Arras M, Winkler B, Scholz D, Schaper J, Schaper W. Monocyte chemotactic protein‐1 increases collateral and peripheral conductance after femoral artery occlusion. Circ Res. 1997; 80:829-837 [DOI] [PubMed] [Google Scholar]
- 15.Weinert S, Poitz DM, Auffermann‐Gretzinger S, Eger L, Herold J, Medunjanin S, Schmeisser A, Strasser RH, Braun‐Dullaeus RC. The lysosomal transfer of LDL/cholesterol from macrophages into vascular smooth muscle cells induces their phenotypic alteration. Cardiovasc Res. 2013; 97:544-552 [DOI] [PubMed] [Google Scholar]
- 16.van Weel V, Toes RE, Seghers L, Deckers MM, de Vries MR, Eilers PH, Sipkens J, Schepers A, Eefting D, van Hinsbergh VW, van Bockel JH, Quax PH. Natural killer cells and CD4+ T‐cells modulate collateral artery development. Arterioscler Thromb Vasc Biol. 2007; 27:2310-2318 [DOI] [PubMed] [Google Scholar]
- 17.Stabile E, Kinnaird T, la Sala A, Hanson SK, Watkins C, Campia U, Shou M, Zbinden S, Fuchs S, Kornfeld H, Epstein SE, Burnett MS. CD8+ T lymphocytes regulate the arteriogenic response to ischemia by infiltrating the site of collateral vessel development and recruiting CD4+ mononuclear cells through the expression of interleukin‐16. Circulation. 2006; 113:118-124 [DOI] [PubMed] [Google Scholar]
- 18.Stabile E, Burnett MS, Watkins C, Kinnaird T, Bachis A, la Sala A, Miller JM, Shou M, Epstein SE, Fuchs S. Impaired arteriogenic response to acute hindlimb ischemia in CD4‐knockout mice. Circulation. 2003; 108:205-210 [DOI] [PubMed] [Google Scholar]
- 19.Sneider EB, Nowicki PT, Messina LM. Regenerative medicine in the treatment of peripheral arterial disease. J Cell Biochem. 2009; 108:753-761 [DOI] [PubMed] [Google Scholar]
- 20.Lawall H, Bramlage P, Amann B. Stem cell and progenitor cell therapy in peripheral artery disease. A critical appraisal. Thromb Haemost. 2010; 103:696-709 [DOI] [PubMed] [Google Scholar]
- 21.Herold J, Pipp F, Fernandez B, Xing Z, Heil M, Tillmanns H, Braun‐Dullaeus RC. Transplantation of monocytes: a novel strategy for in vivo augmentation of collateral vessel growth. Hum Gene Ther. 2004; 15:1-12 [DOI] [PubMed] [Google Scholar]
- 22.Francke A, Weinert S, Strasser RH, Braun‐Dullaeus RC, Herold J. Transplantation of bone marrow derived monocytes: a novel approach for augmentation of arteriogenesis in a murine model of femoral artery ligation. Am J Transl Res. 2013; 5:155-169 [PMC free article] [PubMed] [Google Scholar]
- 23.Wuestenfeld JC, Herold J, Niese U, Kappert U, Schmeisser A, Strasser RH, Braun‐Dullaeus RC. Indocyanine green angiography: a new method to quantify collateral flow in mice. J Vasc Surg. 2008; 48:1315-1321 [DOI] [PubMed] [Google Scholar]
- 24.Scholz D, Cai WJ, Schaper W. Arteriogenesis, a new concept of vascular adaptation in occlusive disease. Angiogenesis. 2001; 4:247-257 [DOI] [PubMed] [Google Scholar]
- 25.Francke A, Herold J, Weinert S, Strasser RH, Braun‐Dullaeus RC. Generation of mature murine monocytes from heterogeneous bone marrow and description of their properties. J Histochem Cytochem. 2011; 59:813-825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poncet P, Arock M, David B. MHC class II‐dependent activation of CD4+ T cell hybridomas by human mast cells through superantigen presentation. J Leukoc Biol. 1999; 66:105-112 [DOI] [PubMed] [Google Scholar]
- 27.Alatery A, Basta S. An efficient culture method for generating large quantities of mature mouse splenic macrophages. J Immunol Methods. 2008; 338:47-57 [DOI] [PubMed] [Google Scholar]
- 28.Yao Q, Doan LX, Zhang R, Bharadwaj U, Li M, Chen C. Thymosin‐alpha1 modulates dendritic cell differentiation and functional maturation from human peripheral blood CD14+ monocytes. Immunol Lett. 2007; 110:110-120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.van Royen N, Piek JJ, Schaper W, Fulton WF. A critical review of clinical arteriogenesis research. J Am Coll Cardiol. 2009; 55:17-25 [DOI] [PubMed] [Google Scholar]
- 30.Arras M, Ito D, Scholz D, Winkler B, Schaper J, Schaper W. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J Clin Invest. 1998; 101:40-50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.la Sala A, Pontecorvo L, Agresta A, Rosano G, Stabile E. Regulation of collateral blood vessel development by the innate and adaptive immune system. Trends Mol Med. 2012; 18:494-501 [DOI] [PubMed] [Google Scholar]
- 32.Tang C, Inman MD, van Rooijen N, Yang P, Shen H, Matsumoto K, O'Byrne PM. Th type 1‐stimulating activity of lung macrophages inhibits Th2‐mediated allergic airway inflammation by an IFN‐gamma‐dependent mechanism. J Immunol. 2001; 166:1471-1481 [DOI] [PubMed] [Google Scholar]
- 33.Randolph GJ, Jakubzick C, Qu C. Antigen presentation by monocytes and monocyte‐derived cells. Curr Opin Immunol. 2008; 20:52-60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ziegelhoeffer T, Fernandez B, Kostin S, Heil M, Voswinckel R, Helisch A, Schaper W. Bone marrow‐derived cells do not incorporate into the adult growing vasculature. Circ Res. 2004; 94:230-238 [DOI] [PubMed] [Google Scholar]
- 35.Sugiyama Y, Yagita Y, Oyama N, Terasaki Y, Omura‐Matsuoka E, Sasaki T, Kitagawa K. Granulocyte colony‐stimulating factor enhances arteriogenesis and ameliorates cerebral damage in a mouse model of ischemic stroke. Stroke. 2011; 42:770-775 [DOI] [PubMed] [Google Scholar]
- 36.Amann B, Ludemann C, Ruckert R, Lawall H, Liesenfeld B, Schneider M, Schmidt‐Lucke J. Design and rationale of a randomized, double‐blind, placebo‐controlled phase III study for autologous bone marrow cell transplantation in critical limb ischemia: the bone marrow outcomes trial in critical limb ischemia (BONMOT‐CLI). Vasa. 2008; 37:319-325 [DOI] [PubMed] [Google Scholar]
- 37.Amann B, Luedemann C, Ratei R, Schmidt‐Lucke JA. Autologous bone marrow cell transplantation increases leg perfusion and reduces amputations in patients with advanced critical limb ischemia due to peripheral artery disease. Cell Transplant. 2009; 18:371-380 [DOI] [PubMed] [Google Scholar]
- 38.Tateishi‐Yuyama E, Matsubara H, Murohara T, Ikeda U, Shintani S, Masaki H, Amano K, Kishimoto Y, Yoshimoto K, Akashi H, Shimada K, Iwasaka T, Imaizumi T. Therapeutic angiogenesis for patients with limb ischaemia by autologous transplantation of bone‐marrow cells: a pilot study and a randomised controlled trial. Lancet. 2002; 360:427-435 [DOI] [PubMed] [Google Scholar]
- 39.Li S, Zhou B, Han ZC. Therapeutic neovascularization by transplantation of mobilized peripheral blood mononuclear cells for limb ischemia. A comparison between CD34+ and CD34− mononuclear cells. Thromb Haemost. 2006; 95:301-311 [DOI] [PubMed] [Google Scholar]
- 40.Melief CJ. Cancer immunotherapy by dendritic cells. Immunity. 2008; 29:372-383 [DOI] [PubMed] [Google Scholar]
- 41.Timmerman JM, Czerwinski DK, Davis TA, Hsu FJ, Benike C, Hao ZM, Taidi B, Rajapaksa R, Caspar CB, Okada CY, van Beckhoven A, Liles TM, Engleman EG, Levy R. Idiotype‐pulsed dendritic cell vaccination for B‐cell lymphoma: clinical and immune responses in 35 patients. Blood. 2002; 99:1517-1526 [DOI] [PubMed] [Google Scholar]
- 42.Timmerman JM, Levy R. Dendritic cell vaccines for cancer immunotherapy. Annu Rev Med. 1999; 50:507-529 [DOI] [PubMed] [Google Scholar]
- 43.Timmerman JM, Levy R. Linkage of foreign carrier protein to a self‐tumor antigen enhances the immunogenicity of a pulsed dendritic cell vaccine. J Immunol. 2000; 164:4797-4803 [DOI] [PubMed] [Google Scholar]
- 44.Matoba S, Tatsumi T, Murohara T, Imaizumi T, Katsuda Y, Ito M, Saito Y, Uemura S, Suzuki H, Fukumoto S, Yamamoto Y, Onodera R, Teramukai S, Fukushima M, Matsubara H. Long‐term clinical outcome after intramuscular implantation of bone marrow mononuclear cells (therapeutic angiogenesis by cell transplantation [TACT] trial) in patients with chronic limb ischemia. Am Heart J. 2008; 156:1010-1018 [DOI] [PubMed] [Google Scholar]