

Abstract
Background
Glutathionylation of endothelial nitric oxide synthase (eNOS) “uncouples” the enzyme, switching its function from nitric oxide (NO) to O2•− generation. We examined whether this reversible redox modification plays a role in angiotensin II (Ang II)‐induced endothelial dysfunction.
Methods and Results
Ang II increased eNOS glutathionylation in cultured human umbilical vein endothelial cells (HUVECs), rabbit aorta, and human arteries in vitro. This was associated with decreased NO bioavailability and eNOS activity as well as increased O2•− generation. Ang II‐induced decrease in eNOS activity was mediated by glutathionylation, as shown by restoration of function by glutaredoxin‐1. Moreover, Ang II‐induced increase in O2•− and decrease in NO were abolished in HUVECs transiently transfected, with mutant eNOS rendered resistant to glutathionylation. Ang II effects were nicotinamide adenine dinucleotide phosphate (NADPH) oxidase dependent because preincubation with gp 91ds‐tat, an inhibitor of NADPH oxidase, abolished the increase in eNOS glutathionylation and loss of eNOS activity. Functional significance of glutathionylation in intact vessels was supported by Ang II‐induced impairment of endothelium‐dependent vasorelaxation that was abolished by the disulfide reducing agent, dithiothreitol. Furthermore, attenuation of Ang II signaling in vivo by administration of an angiotensin converting enzyme (ACE) inhibitor reduced eNOS glutathionylation, increased NO, diminished O2•−, improved endothelium‐dependent vasorelaxation and reduced blood pressure.
Conclusions
Uncoupling of eNOS by glutathionylation is a key mediator of Ang II‐induced endothelial dysfunction, and its reversal is a mechanism for cardiovascular protection by ACE inhibition. We suggest that Ang II‐induced O2•− generation in endothelial cells, although dependent on NADPH oxidase, is amplified by glutathionylation‐dependent eNOS uncoupling.
Keywords: angiotensin II, endothelial nitric oxide synthase, glutathionylation, NADPH oxidase
Introduction
Endothelial nitric oxide synthase (eNOS), the predominant isoform of nitric oxide synthase in the vasculature, catalyzes the generation of nitric oxide (NO), a key regulator of vascular homeostasis. In its homodimeric form, eNOS oxidizes the substrate, l‐arginine, to l‐citrulline and NO. This involves the transfer of electrons from nicotinamide adenine dinucleotide phosphate (NADPH) in the reductase domain of one eNOS monomer to the heme in the oxidase domain of the opposite monomer1 in a tightly regulated process that requires the cofactor, tetrahydrobiopterin (BH4).2 In disease states with elevated “oxidative stress,” depletion of the redox‐sensitive BH4 leads to a disruption in the coupling of NADPH oxidation to NO synthesis, resulting in generation of superoxide (O2•−) from the oxidase domain3 in a process widely known as eNOS “uncoupling.” BH4 supplementation, however, only partially reverses eNOS uncoupling and endothelial dysfunction, suggesting that other redox‐related mechanisms may be involved in regulation of the eNOS function.4–5
The recent demonstration of modifications in eNOS activity through glutathionylation under oxidative4 and nitrosative6 stress has led to a paradigm shift in the understanding of the molecular mechanism of eNOS uncoupling. Under oxidative stress, cysteine (Cys) residues on eNOS are oxidized, leading to glutathionylation of eNOS6–7 as a result of the formation of a reversible disulfide bond between glutathione (GSH) and the reactive Cys residues of the enzyme. This GSH adduct has effects on tertiary structure and protein function akin to phosphorylation.8–10 Glutathionylation of eNOS leads to a loss of NO generation and, in contrast to BH4 depletion, production of O2•− primarily at the reductase domain.4 Increased eNOS glutathionylation in a hypertensive rat model, in which endothelial dysfunction was reversed by disulfide‐specific reducing agents and in streptozotocin‐induced diabetic rats, is supportive of the pathophysiological significance of eNOS glutathionylation.4,11 The precise signaling pathways leading to this oxidative modification in these disease states have not been elucidated.
Activation of the renin angiotensin system (RAS) is a key neurohormonal change in a multitude of disease states that are characterized by endothelial dysfunction (eg, hypertension, diabetes, atherosclerosis, and heart failure).12 Angiotensin II (Ang II) activates NADPH oxidase, a major source of vascular O2•−.13 Whereas O2•− from NADPH oxidase isoform 1 can trigger eNOS uncoupling indirectly through BH4 oxidation,14 the redox‐related mechanisms that directly modify eNOS and mediate Ang II‐induced eNOS uncoupling15 are not defined. We examined the hypothesis that glutathionylation mediates Ang II‐induced uncoupling of eNOS in an NADPH oxidase‐dependent manner. To complement in vitro data and establish in vivo significance, we examined the effects of attenuation of baseline Ang II receptor‐coupled redox signaling using the angiotensin converting enzyme (ACE) inhibitor, captopril, in rabbits, in which RAS is highly activated under basal conditions.16
Methods
Animals, Tissues, and Cells
Aortic segments were harvested from control male New Zealand White rabbits or rabbits treated with captopril (8 mg·kg−1·day−1) in their drinking water for 7 days. Surplus segments of human internal mammary or radial artery were obtained from patients undergoing coronary artery bypass graft operation. Pooled, multiple‐donor human umbilical vein endothelial cells (HUVECs; Lonza, Basel, Switzerland) were maintained at 37°C with 5% CO2 in MesoEndo Endothelial Cell Medium (Cell Applications, Inc., San Diego, CA), supplemented with 1% l‐glutamine, 1% penicillin/streptomycin, and 10% fetal bovine serum, and used for in vitro experiments. Protocols for animal and human experiments were approved by the appropriate research ethics committees.
Transfection Protocol
HUVECs were transfected using an Amaxa Nucleofection system (Lonza) as per the manufacturer's instructions. Briefly, 2×106 cells per cuvette were transfected, using 5‐μg DNA/transfection. Wild type (WT) and mutated eNOS lacking C689 and C90817 were kindly provided by Keith M. Channon's lab at Oxford University (Oxford, UK).
Immunodetection of eNOS Glutathionylation
eNOS glutathionylation was examined in lysate or homogenate by coimmunoprecipitation. Glutathionylated proteins were immunoprecipitated with an anti‐GSH antibody (Ab; ViroGen, Watertown, MA), using protein A/G plus agarose beads,9 and immunoblotted with an Ab against eNOS (BD Biosciences [San Jose, CA] for human and Sigma‐Aldrich [St. Louis, MO] for rabbit samples) in Western blottings. In independent experiments, eNOS was immunoprecipitated from homogenates of rabbit aorta, followed by immunoblotting with anti‐GSH Ab, to control for the contribution of eNOS to glutathionylated protein immunoblottings. Gluathionylation of eNOS was also detected by the biotinylated GSH ethyl esther (BioGEE) technique in HUVECs, where glutathionylated subfraction was precipitated from HUVECs loaded with biotinylated GSH (500 μmol/L, 1 hour at room temperature) using streptavidin‐sepharose beads.9
Measurement of eNOS Activity
eNOS activity was measured by conversion of l‐[14C]arginine to l‐[14C] citrulline in cell lysate (Cayman Chemical, Ann Arbor, MI). To examine the specific role of glutathionylation in regulation of eNOS activity, cell lysates were incubated with recombinant human glutaredoxin 1 (hGrx1; 10 μg/mL), an enzyme that specifically catalyzes “deglutathionylation,” at 37°C for 30 minutes. Radioactivity was measured using a liquid scintillation counter.
Electron Paramagnetic Resonance Spin Trapping Measurement of NO Bioavailability
Electron paramagnetic resonance (EPR) measurement of NO in aortic segments (10×10 mm) was performed using the colloid spin‐trap Fe(DETC)2 (200 μmol/L), as previously described.18 The segments were incubated with Fe(DETC)2 at 37°C for 30 minutes, before snap‐freezing in liquid N2. Spectra were recorded at 77 K with a Bruker EMX X‐band spectrometer (Bruker Corporation, Billerica, MA) using the following parameters: microwave power, 2.5 mW; microwave frequency, 9.4 GHz; modulation amplitude, 0.2 mT; modulation frequency, 100 kHz; conversion time, 81.9 ms, time constant, 163.8 ms; and scan time, 83.9 seconds, with 12 scans averaged. The amplitude of NO signal was determined as the perpendicular height between the top of the first low‐field signal and the valley of the third high‐field signal.18
Confocal Fluorescence Microscopy
Immunostaining for subcellular localization of eNOS and glutathionylated protein was performed in control HUVECs and cells treated with Ang II (100 nmol/L, 1 hour). Subsequent to fixation and permeabilization, cells were incubated with mouse anti‐GSH and rabbit anti‐eNOS primary Abs, followed by secondary fluorescein‐isothiocyanate–conjugated anti‐mouse and Alexa Fluor 633–conjugated anti‐rabbit Abs.
Ang II‐induced changes in intracellular O2•− were examined in HUVECs loaded with the O2•−‐sensitive dye, dihydroethidium (DHE). To examine the contribution of eNOS glutathionylation to O2•− levels, after incubation with Ang II (100 nmol/L, 1 hour, 37°C), cells were incubated with the NOS inhibitor, l‐NG‐nitroarginine methyl ester (l‐NAME; 1 mmol/L), or the mixed disulfide‐reducing agent, dithiotreitol (DTT; 1 mmol/L), for 20 minutes at 37°C, washed with Krebs buffer containing diethylenetriaminepentacetic acid (100 μmol/L), and then loaded with DHE (2 μmol/L). Intracellular NO was examined in HUVECs loaded with NO‐sensitive fluorescent dye diaminofluorescein‐2 diacetate (DAF2DA; 5 μmol/L), with or without acetylcholine (ACh) treatment (1 μmol/L, 20 minutes).19 In transfected HUVECs, NO was detected by using 4‐amino‐5‐methylmino‐2′,7′‐difluorofluorescein (DAF‐FM) diacetate. Nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (DAPI; 1 μmol/L). To image O2•− or NO in aorta, cryosections (10 μm) of aorta in optimized cutting temperature medium were incubated with DHE (2 μmol/L) or DAF2DA (5 μmol/L), respectively, using adaptations of previously described methods.15,20 All cells and sections were mounted on glass slides with Vectashield mounting medium and viewed with a confocal microscope (Leica TCS SP5; Leica Microsystems, Heidelberg, Germany).
High Performance Liquid Chromatography Analysis of Dihydroethidium Oxidation Products
High performance liquid chromatography (HPLC) equipped with UV‐visible, as well as electrochemical detectors was used to separate the nonspecific ethidium (E+) and specific dihydroethidium (2‐OH‐E+) products of DHE oxidation in cells.21 Treatments of HUVECs were similar to fluorescence experiments, except for exposure to DHE at 50 μmol/L, for 30 minutes at 37°C. Cells were then lysed in methanol and the proteins precipitated by the addition of HClO4 (200 mmol/L) in methanol at 4°C and pelleted by centrifugation. The supernatant was stored at −80°C. Samples (50 μL) were separated by HPLC (Shimadzu Corporation, Kyoto, Japan) after injection onto a Synergi Polar RP C18 column (250×4.6 mm, 4 μmol/L, 80 Å; Phenomenex, Torrance, CA), equilibrated at 30°C with 60% mobile phase A (10% CH3CN in potassium phosphate buffer, 50 mmol/L, pH 2.6) and 40% mobile phase B (60% CH3CN in potassium phosphate buffer, 50 mmol/L, pH 2.6). Separation of products was performed by a linear increase to 100% mobile phase B over 30 minutes at a flow rate of 0.5 mL·min−1. Products were quantified by fluorescence (DHE and 2‐OH‐E+: λEX 358 nm, λEM 440 nm; E+ λEX 490 nm, λEM 565 nm) and electrochemical oxidation using a CoulArray detector (electrodes at 0, 200, 280, 365, 400, 450, 500, and 600 mV; ESA Biosciences, Inc., Chelmsford, MA ). The concentration of 2‐OH‐E+ was normalized to cell protein concentration.
Aortic Preparation and Functional Measurements
Aortic rings were excised from rabbits and mounted in organ chambers.22 Segments were incubated with Ang II (100 nmol/L, 1 hour) at 37°C before placement in the organ chambers, where indicated. This was followed by incubation with DTT (1 mmol/L) for 20 minutes in a group of experiments. Cumulative concentrations of ACh were applied to the rings precontracted with phenylephrine (PE; 300 nmol/L in experiments with DTT; 100 nmol/L in other experiments). Relaxation is expressed as percentage of PE‐induced contraction.
Statistical Analysis
Results are presented as mean±SEM. A paired (first panel in Figure 3B) or unpaired (remainder of Figures 3 and 5C and 5D) Student t test was used for comparison between 2 groups. A nonparametric test (Mann‐Whitney) was used for comparison between 2 groups where normal distribution could not be ascertained. For comparisons between more than 2 groups, a nonparametric ANOVA test (Kruskal‐Wallis) was used with Dunn's post‐hoc multiple comparisons. Vasorelaxation data were analyzed by 2‐way ANOVA with Tukey's post‐hoc analysis. P<0.05 was considered statistically significant.
Figure 3.

Ang II reduces NO bioavailability. A, Effect of Ang II on basal and ACh‐stimulated NO‐sensitive DAF2DA fluorescence in HUVECs. Cell nuclei were counterstained with DAPI (blue). Changes in the DAF2DA signal (df) were measured after stimulation with ACh (1 μmol/L) for 20 minutes (n=4; cells from 4 randomly chosen areas of interest in each group were included in the analysis). B, NO measurement by EPR with Fe(DETC)2 in control, in vitro Ang II‐treated (n=9), and in vivo captopril‐treated rabbit aortic segments (n=10 control and 6 captopril‐treated rabbits). Amplitude of NO‐Fe(DETC)2 signal was determined as the perpendicular height between the top of the first low‐field signal (g1=3297 G) and the valley of the third high‐field signal (g2=3323 G; arrows). Traces from the standard spermine NONOate in DMSO solution and endothelium‐denuded aorta are also shown. C, Effect of Ang II on NO‐sensitive DAF‐FM diacetate fluorescence in HUVECs overexpressing the WT or the double Cys mutant (Δ) eNOS in the presence of ACh. Statistical comparison was made in each cell type between conditions with and without Ang II (n=3). AU=arbitrary unit. Results are shown as means±SEM. Statistical significance (P<0.05) is indicated by asterisk (*). DAF indicates 4‐amino‐5‐methylmino‐2′,7′‐difluorofluorescein; DAPI, 4′,6‐diamidino‐2‐phenylindole; DHE, dihydroethidium; DTT, dithiotreitol; eNOS, endothelial nitric oxide synthase; EPR, electron paramagnetic resonance; HUVECs, human umbilical vein endothelial cells; l‐NAME, l‐NG‐nitroarginine methyl ester; NO, nitric oxide.
Figure 5.

ACE inhibition reduces eNOS glutathionylation and improves endothelium‐dependent vasorelaxation in vivo. A, eNOS expression in aortic homogenates of control and captopril‐treated rabbits. Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) immunoblot was used as the loading control (n=5 control and 4 captopril‐treated rabbits). B, eNOS glutathionylation in aorta of control and captopril‐treated rabbits (n=5 control and 4 captropril‐treated rabbits). C, O2•−‐sensitive DHE fluorescence in aortic cryosections. Intensity of the signal was measured within the endothelial layer of aorta from control and captopril‐treated rabbits (white arrows, magnified image of the endothelial layer is shown in white boxes; n=5 control and 4 captopril‐treated rabbits, with 5 randomly chosen regions of interest analyzed in each rabbit). D, NO detection in aortic cryosections by DAF2DA (5 μmol/L). Intensity of the signal was measured within the endothelial layer of aorta from control and captopril‐treated rabbits (white arrows, magnified image of the endothelial layer is shown in white boxes) (n=5 control and 4 captopril‐treated rabbits, with 5 randomly chosen regions of interest analyzed in each rabbit). E, Endothelium‐dependent vasorelaxation in aortic rings from control and captopril‐treated rabbits precontracted with PE (100 nmol/L). P value was <0.05 in the captopril‐treated group versus control. PE‐induced precontraction was 2.1±0.2 and 2.0±0.3 g in the control versus captopril group, respectively, and was not statistically different between groups. AU=arbitrary unit. Results are shown as means±SEM. Statistical significance (P<0.05) is indicated by asterisk (*). DHE indicates dihydroethidium; DTT, dithiotreitol; eNOS, endothelial nitric oxide synthase; NO, nitric oxide; PE, phenylephrine.
Results
Ang II Increases eNOS Glutathionylation and Inhibits Its Activity Through Glutathionylation in an NADPH Oxidase‐Dependent Manner
eNOS glutathionylation was detectable in serum‐starved HUVECs (Figure 1A). Ang II (100 nmol/L, 1 hour, 37°C) increased eNOS glutathionylation, compared to control (Figure 1A). Preincubation of cells with gp 91ds‐tat (5 μmol/L), a membrane‐permeable peptide inhibitor of NADPH oxidase subunit assembly, for 1 hour at 37°C had no effect on baseline eNOS glutathionylation, but abolished Ang II‐induced eNOS glutathionylation (Figure 1A). A lack of effect on baseline signal might be the result of low activity of the oxidase in serum‐free media and/or nonenzymatic oxidant generation under in vitro conditions.23
Figure 1.

Ang II increases eNOS glutathionylation and inhibits its activity through glutathionylation in an NADPH oxidase‐dependent manner. A, eNOS immunoblotting of GSH or IgG immunoprecipitate (IP) from HUVEC lysate with or without DTT (n=5). B, eNOS immunoblotting of total lysate and streptavidin precipitate (protein‐GSS) of HUVECs loaded with biotinylated GSH before Ang II exposure (n=5). C, eNOS immunoblotting of GSH IP from rabbit aorta (n=5). D, GSH immunoblotting of eNOS IP from rabbit aorta (n=5). E, eNOS immunoblotting of GSH IP from human arterial segments (n=4). F, Immunostaining of eNOS (first column, red fluorescence) and glutathionylation (second column, green fluorescence) in control HUVECs and cells treated with Ang II. The merged eNOS/glutathionylation image demonstrates in situ colocalization of glutathionylation and eNOS immunostaining (third column, yellow fluorescence, white arrows point to colocalization at membrane). Nuclei were stained with DAPI (blue). G, eNOS activity in HUVECs±Ang II, measured by the conversion of l‐[14C]arginine to l‐[14C]citrulline in cell lysates. Preincubation of cells with gp 91ds‐tat (5 μmol/L); or cell lysate with hGrx1 (10 μg/mL) is as indicated (n=6). H, eNOS glutathionylation in HUVECs preincubated with losartan (Los). Statistical comparison was made between the Ang II and Ang II+Los groups (n=4). Results are shown as means±SEM. Statistical significance (P<0.05) is indicated by asterisk. DTT indicates dithiotreitol; eNOS, endothelial nitric oxide synthase; GSH, glutathione; HUVECs, human umbilical vein endothelial cells.
DTT (1 mmol/L) abolished the signal for eNOS glutathionylation when incubated with lysate protein for 15 minutes preceding immunoprecipitation (Figure 1A), consistent with a mixed disulfide bond between eNOS and GSH.24 Similar data were obtained in independent experiments using the BioGEE technique (Figure 1B).
Consistent with the results in HUVECs, Ang II (100 nmol/L, 1 hour) increased eNOS glutathionylation in rabbit aorta (ex vivo) in an NADPH oxidase‐dependent manner (Figure 1C and 1D). To determine the relevance of our data to human vascular physiology and pathophysiology, we examined the effect of Ang II (100 nmol/L) on eNOS glutathionylation in intact segments of human arteries ex vivo. eNOS glutathionylation was present in human vessels at baseline and was significantly increased by exposure to Ang II (Figure 1E).
In immunofluorescence experiments, immunostaining for colocalization of GSH and eNOS that reflects eNOS glutathionylation in situ was observed particularly at the membrane, and this was increased from baseline by incubating HUVECs with Ang II (Figure 1F).
In functional studies, Ang II induced a reduction in eNOS‐mediated conversion of l‐[14C]arginine to l‐[14C]citrulline in HUVEC lysates (Figure 1G). This effect on eNOS function appeared relatively smaller compared to effects on eNOS glutathionylation detected by Western blotting (Figure 1A through 1D). Whereas Western blotting is a nonquantitative method and linearity in detected signal for eNOS glutathionylation by Western blotting with the actual glutathionylation levels and eNOS activity have not been shown, we speculate that such apparent discrepancy might be a result of the presence of reducing agents in the commercial kit used for functional measurements. The effect on eNOS activity was blocked by preincubation of cells with gp91ds‐tat, indicating NADPH oxidase‐dependence of the effect and was reversed by incubation of lysates with hGrx1 (10 μg/mL; Figure 1G). To examine the type of Ang II receptor involved in mediating the effects, HUVECs were preincubated with losartan (5 μmol/L, 1 hour, 37°C), a specific Ang II type 1 receptor (AT1R) blocker, before incubation with Ang II (100 nmol/L, 1 hour, 37°C). Abolition of Ang II effects by losartan (Figure 1H) was supportive of AT1R mediating Ang II‐induced eNOS glutathionylation. Taken together, these results indicate that Ang II induces an NADPH oxidase‐dependent increase in eNOS glutathionylation, through AT1R, resulting in a decrease in eNOS activity.
Ang II Uncouples eNOS in HUVECs Through Glutathionylation
Exposure of HUVECS to Ang II (100 nmol/L, 1 hour) increased O2•−‐sensitive DHE fluorescence (Figure 2A). Addition of l‐NAME increased DHE fluorescence in control cells, suggesting that a proportion of basal O2•− is tonically quenched by NO (Figure 2A). In contrast, l‐NAME added to cells pre‐exposed to Ang II had no significant effect on DHE fluorescence. A lack of effect on DHE signal by l‐NAME, in contrast to control cells, suggested that Ang II had induced uncoupling of eNOS. Because l‐NAME does not inhibit O2•− generation from the reductase domain of uncoupled eNOS, the known site of glutathionylation,4 we examined the effect of disulfide reduction by DTT. A decrease in DHE fluorescence by DTT (Figure 2A) in Ang II‐treated cells supported the role of glutathionylation. Similar to l‐NAME, DTT increased the DHE signal in control cells (Figure 2A).
Figure 2.
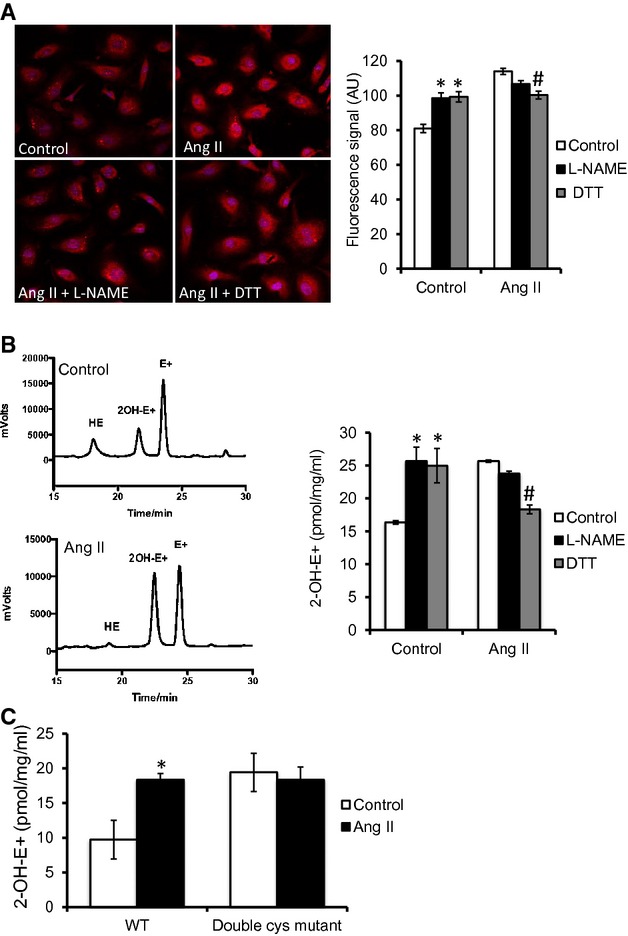
Ang II induces eNOS‐derived O2•− generation through glutathionylation. A, Effect of Ang II on O2•−‐sensitive DHE fluorescence. Cell nuclei were counterstained with DAPI (blue). Preincubation with l‐NAME (1 mmol/L) or DTT (1 mmol/L) is as indicated (n=9 control, 9 Ang II, and 6 in other groups; cells from 4 randomly chosen regions of interest in each group were included in the analysis). B, HPLC analysis of DHE oxidation products to separate the specific product (2‐OH‐E+) from the nonspecific product (E+) of DHE oxidation. [2‐OH‐E+] is calculated as the mean of values determined by fluorescence and electrochemical detectors, normalized to total protein concentration (pmol·mg−1·mL−1). C, Ang II‐induced O2•− as detected by HPLC analysis of specific DHE oxidation product (2‐OH‐E+) in HUVECs transfected with WT eNOS (WT) and double Cys mutant eNOS (double Cys mutant). Statistical comparison was made in each cell type between conditions of with and without Ang II (n=3). AU=arbitrary unit. Results are shown as means±SEM. Statistical significance, compared to control (P<0.05), is indicated by asterisk (*). In Figure 2A and 2B, statistical significance, compared to Ang II (P<0.05), is indicated by pound sign (#).DHE indicates dihydroethidium; DTT, dithiotreitol; eNOS, endothelial nitric oxide synthase; GSH, glutathione; HPLC, high performance liquid chromatography; HUVECs, human umbilical vein endothelial cells; PE, phenylephrine; WT, wild type.
Because total DHE fluorescence has limitations in specificity for detection of O2•− in biological systems,21 we used HPLC to separate the specific product of DHE oxidation by O2•− (2‐OH‐E+) from the nonspecific, but fluorescent product, E+. Consistent with the fluorescence studies, Ang II induced an increase in 2‐OH‐E+ that was reduced by DTT, but was insenstitive to l‐NAME (Figure 2B). Similar results were observed for the ratio of 2‐OH‐E+/HE, which accounts for any potential differences in the amount of HE that entered the cells25 (data not shown).
We next examined the effect of mutating the reactive Cys residues located in the eNOS reductase domain (C689 and C908).4,17 There was a nonsignificant decrease in 2‐OH‐E+ levels in cells transfected with WT eNOS compared with the cells transfected with double Cys mutant (Figure 2C), which had a similar 2‐OH‐E+ level to the previous nontrasnfected control cells (Figure 2B). Ang II exposure increased O2•− in HUVECs overexpressing WT eNOS (Figure 2C). However, this Ang II‐induced increased in O2•− was abolished in cells expressing the double Cys mutant eNOS compared to their no‐Ang II control, pointing to a major contribution of eNOS glutathionylation to Ang II‐induced O2•− production in endothelial cells (ECs).
Because uncoupling of eNOS is expected to result in reduced NO bioavailability, we quantified NO formation. Ang II reduced both baseline and ACh‐induced increases in NO‐sensitive DAF2DA fluorescence in HUVECs (Figure 3A). Quantitative measurement of NO in EPR spin‐trapping experiments by Fe(DETC)2 demonstrated that exposing rabbit aortic segments to Ang II in vitro causes a reduction in NO level (Figure 3B). Consistent with O2•− measurements, the effect of Ang II on NO‐sensitive DAF‐FM diacetate fluorescence was also abolished in cells expressing the double Cys mutant eNOS (Figure 3C), indicating a critical role for these reactive Cys residues in Ang II‐induced eNOS uncoupling.
Ang II Reduces Endothelium‐Dependent Relaxation in Aorta Through Reversible Thiol Oxidation
To examine whether Ang II‐induced eNOS glutathionylation affects vascular endothelial function, aortic rings were treated with Ang II for 1 hour at 37°C before transfer to organ chambers. Ang II pre‐exposure significantly reduced ACh‐induced vasorelaxation (Figure 4A). To investigate the role of glutathionylation, we exposed aortic rings to DTT (1 mmol/L, 20 minutes).4 Because DTT shifted the PE concentration‐contraction curve (Figure 4B), these experiments were performed at a concentration with minimal differences in PE‐induced precontraction (ie, 300 nmol/L; Figure 4B). DTT abolished the Ang II‐induced impairment of endothelium‐dependent relaxation (Figure 4C), supporting a role for glutathionylation in mediating the Ang II effect on ACh‐induced vasorelaxation.
Figure 4.
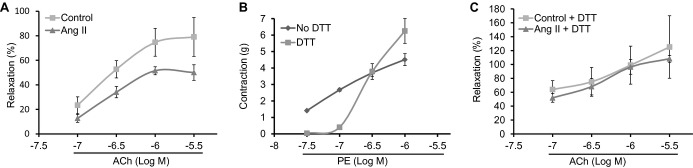
Ang II impairs ACh‐induced vasorelaxation through thiol oxidation. A, Endothelium‐dependent vasorelaxation in control and Ang II (100 nmol/L)‐treated rabbit aortic rings precontracted with PE (300 nmol/L). (n=5 rabbits in each group with 3 rings studied in each rabbit). P value was <0.05 in the Ang II‐treated group versus control on 2‐way ANOVA. PE‐induced precontraction was 2.4±0.6 and 2.6±0.5 g in control versus Ang II‐treated rings, respectively, and was not statistically different in 2 groups. B, Effect of DTT on PE‐induced contraction in rabbit aorta (n=5; with 2 rings studied in each rabbit). C, ACh‐induced relaxation in rings with and without DTT (1 mmol/L) added after Ang II exposure (n=4 control and 5 Ang II groups with 3 rings studied in each rabbit). PE‐induced precontraction was 2.3±0.2 and 2.5±0.3 g in control versus Ang II‐treated rings, respectively, and was not statistically different between groups. Aortic relaxation is expressed as percentage of PE‐induced contraction (at 300 nmol/L). DTT indicates dithiotreitol; PE, phenylephrine; WT, wild type.
Attenuation of Ang II Signaling by ACE Inhibition In Vivo Reduces eNOS Glutathionylation and Improves Endothelium‐Dependent Vasorelaxation
Because Ang II‐induced eNOS glutathionylation impaired endothelium‐dependent vasorelaxation in aortic rings ex vivo, we examined whether attenuation of Ang II signaling by ACE inhibition could reduce baseline redox signaling within critical microdomains by reversing eNOS glutathionylation, thereby improving endothelial function in an in vivo setting. Treatment of rabbits with the ACE inhibitor captopril had no effect on eNOS expression in aorta, but reduced baseline eNOS glutathionylation (Figure 5A and 5B). This was associated with a decrease in endothelial O2•− formation, an increase in endothelial NO levels, and improvement in endothelium‐dependent vasorelaxation (Figures 3B and 5C through 5E). Parallel to the decrease in eNOS glutathionylation, both systolic and diastolic blood pressures were significantly reduced in captopril‐treated rabbits, without an effect on heart rate (Table).
Table 1.
Hemodynamic Effect of Captopril in Rabbits
| Control | Captopril | |
|---|---|---|
| Heart rate | 202±13 | 214±9 |
| Systolic blood pressure | 92±1 | 65±6* |
| Diastolic blood pressure | 82±1 | 57±6* |
| Mean arterial pressure | 86±1 | 59±6* |
Heart rate, systolic, diastolic and mean arterial pressure measured in control (n=5) and captopril‐treated (n=3) rabbits. Results are shown as means±SEM. Statistical significance *P<0.05).
Discussion
Extensive evidence exists for Ang II‐mediated increase in endothelial oxidative stress, with subsequent adverse effects on vascular function12 and eNOS activity.15 Here, we show that NADPH oxidase‐dependent glutathionylation of eNOS is a key molecular mechanism for this phenomenon. Our data demonstrate, for the first time, the major quantitative contribution that glutathionylation‐mediated eNOS uncoupling makes to Ang II‐induced endothelial O2•− generation.
Ang II increased eNOS glutathionylation and resulted in a reduction in NO as well as an increase in eNOS‐derived O2•−. A lack of effect on O2•− levels in experiments with eNOS inhibition by l‐NAME (Figure 2A and 2B), which blocks O2•− generation from the oxidase domain of the enzyme, suggests that the electron leak and O2•− are largely derived from the reductase domain. This is in agreement with a previous report, where l‐NAME had partial or no effect on O2•− generation from glutathionylated or alkylated human eNOS (heNOS).4 A nonsignificant trend for reduction in O2•− by l‐NAME in Ang II‐treated cells in our study (Figure 2A and 2B) may be consistent with a partial reduction in glutathionylated heNOS‐derived O2•−,4 or indicate the coexistence of other uncoupling mechanisms, such as BH4 depletion, that primarily affect the oxidase domain. Interplay of these 2 distinct mechanisms of eNOS uncoupling has been shown, where eNOS uncoupling by BH4 oxidation can induce uncoupling of the enzyme by glutathionylation of Cys residues at reductase domain and vice versa.17
In contrast to l‐NAME, the disulfide reductant DTT significantly decreased O2•− levels (Figure 2A and 2B). This supports a contribution of reversible thiol oxidation to eNOS‐derived O2•− generation. Interestingly, DTT increased O2•− levels in control cells, as opposed to the reduction it caused in Ang II‐treated cells. Because eNOS is expected to be mostly in the coupled state in control cells, we speculate that the increase in signal at baseline might be a result of the effects of DTT on the NADPH oxidase complex, a major source of O2•− under basal physiological conditions in membrane signaling microdomains. In particular, the p47phox subunit has Cys residues that are critical for enzyme activity,26 which might be susceptible to, and regulated by, glutathionylation.
In addition to the effects of DTT on O2•− formation, a direct role for eNOS glutathionylation in mediating the effects of Ang II is strongly supported by restoration of eNOS activity by hGrx1, which specifically catalyzes deglutathionylation. Although mechanisms regulating the cellular activity of Grx1 are poorly understood,27 our report of a functional interaction between Grx1 and eNOS (Figure 1G) is in agreement with a recent study.28 This highlights the importance of thiol reductase enzymes as potential targets in treatment of many disease states where the RAS plays a central role in endothelial dysfunction.
Abolishment of Ang II‐induced increase in O2•− in HUVECs overexpressing eNOS without the reactive Cys residues (Figure 2C) not only provides the most definitive evidence for glutathionylation mediating Ang II‐induced eNOS uncoupling, but also suggests that the majority of O2•− under Ang II stimulation in ECs is generated by uncoupled eNOS. NADPH‐derived O2•− appears to act as a trigger to initiate the uncoupling through glutathionylation of eNOS. This scheme (as shown in Figure 6) strongly supports the role of NADPH oxidase and its derived O2•− as the “kindling” and uncoupled eNOS as the “bonfire,” as previously proposed,29 but adds glutathionylation as the critical switch.
Figure 6.
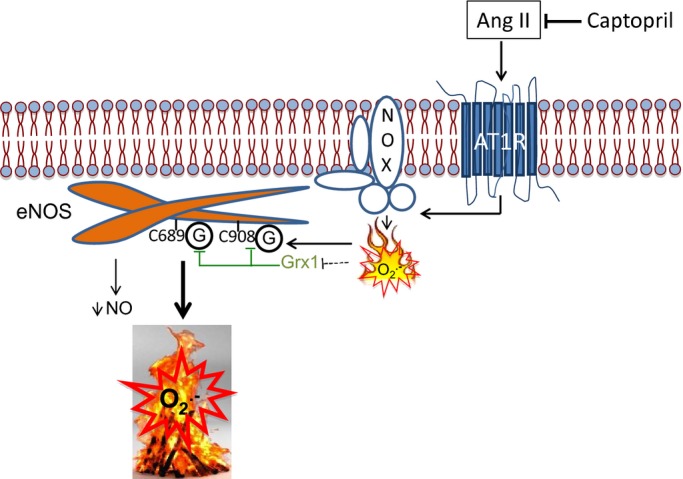
Schematic representation of Ang II‐induced, NADPH oxidase‐dependent eNOS uncoupling through glutathionylation in endothelial cells. NADPH oxidase (NOX)‐derived O2•−, under Ang II type 1 receptor (AT1R)‐coupled activation,12 initiates eNOS uncoupling by glutathionylation of critical Cys residues (C689 and C908) in the reductase domain (shown as G), leading to a decrease in NO and amplification of O2•− production. This scheme strongly supports the previously proposed paradigm of NOX and derived O2•− acting as the “kindling” and uncoupled eNOS the “bonfire,” but adds eNOS glutathionylation as the critical switch. ACE inhibition by captopril blocks these effects. Grx1 reverses glutathionylation, and its activity is redox‐sensitive by yet to be identified mechanisms (dotted line). ACE indicates angiotensin converting enzyme; eNOS, endothelial nitric oxide synthase; NO, nitric oxide.
eNOS modifications induced by Ang II are expected to affect the downstream NO‐activated signaling pathways that regulate vascular tone. Indeed, Ang II decreased endothelium‐dependent relaxation in intact aortic rings in a DTT‐sensitive manner (Figure 4), supporting the significance of eNOS glutathionylation in Ang II‐induced endothelial dysfunction in intact vessels. These effects are similar to effects of DTT to restore the endothelium‐dependent vasorelaxation in spontaneously hypertensive rats (SHRs) reported previously.4 Of note, we show that DTT caused a shift in PE concentration‐contraction curve. This effect of DTT on PE precontraction has not been previously discussed4 and may be a result of effects of DTT on glutathionylation of other cellular redox‐sensitive molecules, such as ryanodine receptor30 and sarco/endoplasmic reticulum Ca2+‐ATPase,31 that are key determinants of intracellular Ca2+ and hence vascular tone.
Our findings provide insights into the signaling cascade that leads to the reported increase in glutathionylated eNOS4 and eNOS uncoupling32 in SHR and streptozotocin‐induced diabetic rats.11 Ang II plays an important role in etiology of hypertension in the SHR and endothelial dysfunction in diabetes, at least in part, through an increase in oxidative stress.33 Ang II‐induced, NADPH oxidase‐dependent uncoupling of eNOS through glutathionylation may provide a mechanistic rationale for these observations and may implicate a similar pathway in the pathogenesis of hypertension and diabetes in humans. Consistent with the clinical relevance of captopril‐induced decrease in eNOS glutathionylation, the improved eNOS coupling and endothelium‐dependent vasorelaxation was also associated with a reduction in blood pressure in rabbits (Table).
Interactions between Ang II‐induced, NADPH oxidase‐generated O2•− and eNOS occur in spatially distinct compartments within the endothelial rafts/caveolae, with the structural protein caveolin‐1 playing a critical role in the assembly of caveolar signalosomes as well as mediating Ang II‐induced eNOS uncoupling.34 Consistent with this, blocking assembly of NADPH oxidase complex with gp91ds‐tat abolished Ang II‐induced eNOS glutathionylation (Figure 1A through 1D) and prevented loss of eNOS enzymatic activity (Figure 1G), suggesting NADPH oxidase as the prime source of O2•− mediating the effects of Ang II on eNOS, rather than other putative cellular sources, such as mitochondria or xanthine oxidase. Because gp91ds‐tat prevents the docking of the cytosolic regulatory p47phox subunit to the membranous gp91phox, the isoforms activated by Ang II are likely to be NOX1 or NOX2, the activity of which requires p47phox translocation to the membrane.35
ACE inhibitors and angiotensin receptor blockers (ARBs) are known to protect against major cardiovascular events beyond what might be expected from their antihypertensive effects.36 Improvement in endothelial function has been implicated.37 Our findings that ACE inhibition by captopril reduces eNOS glutathionylation and results in increased NO and diminished O2•− levels, as well as improvement in endothelium‐dependent relaxation, may rationalize analogous effects of the ARB, telmisartan, in nitrate‐tolerant rats38 and provide molecular insights into the protective effect of these agents. Indeed, the detected increase in glutathionylation of eNOS in intact human vessels by Ang II supports the direct relevance of our findings to human vascular pathophysiology.
Sources of Funding
The work was supported by Heart Research Australia. Liu was supported by a fellowship from the National Heart Foundation of Australia, Gentile by a Marcus Blackmore fellowship (Heart Research Institute), and Figtree by the University of Sydney Medical Foundation.
Disclosures
None.
References
- 1.Palmer RM, Ashton DS, Moncada S. Vascular endothelial cells synthesize nitric oxide from l‐arginine. Nature. 1988; 333:664-666 [DOI] [PubMed] [Google Scholar]
- 2.Bredt DS, Hwang PM, Glatt CE, Lowenstein C, Reed RR, Snyder SH. Cloned and expressed nitric oxide synthase structurally resembles cytochrome P‐450 reductase. Nature. 1991; 351:714-718 [DOI] [PubMed] [Google Scholar]
- 3.Wever RM, van Dam T, van Rijn HJ, de Groot F, Rabelink TJ. Tetrahydrobiopterin regulates superoxide and nitric oxide generation by recombinant endothelial nitric oxide synthase. Biochem Biophys Res Commun. 1997; 237:340-344 [DOI] [PubMed] [Google Scholar]
- 4.Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen YR, Druhan LJ, Zweier JL. S‐glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010; 468:1115-1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Talib J, Kwan J, Suryo Rahmanto A, Witting PK, Davies MJ. The smoking‐associated oxidant hypothiocyanous acid induces endothelial nitric oxide synthase dysfunction. Biochem J. 2014; 457:89-97 [DOI] [PubMed] [Google Scholar]
- 6.Manevich Y, Townsend DM, Hutchens S, Tew KD. Diazeniumdiolate mediated nitrosative stress alters nitric oxide homeostasis through intracellular calcium and S‐glutathionylation of nitric oxide synthetase. PLoS ONE. 2010; 5:e141517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen CA, Lin CH, Druhan LJ, Wang TY, Chen YR, Zweier JL. Superoxide induces endothelial nitric‐oxide synthase protein thiyl radical formation, a novel mechanism regulating eNOS function and coupling. J Biol Chem. 2011; 286:29098-29107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zweier JL, Chen CA, Druhan LJ. S‐glutathionylation reshapes our understanding of endothelial nitric oxide synthase uncoupling and nitric oxide/reactive oxygen species‐mediated signaling. Antioxid Redox Signal. 2011; 14:1769-1775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Figtree GA, Liu CC, Bibert S, Hamilton EJ, Garcia A, White CN, Chia KKM, Cornelius F, Geering K, Rasmussen HH. Reversible oxidative modification: a key mechanism of Na+–K+ pump regulation. Circ Res. 2009; 105:185-193 [DOI] [PubMed] [Google Scholar]
- 10.Liu CC, Karimi Galougahi K, Weisbrod RM, Hansen T, Ravaie R, Nunez A, Liu YB Garcia A, Fry NAS, Hamilton EJ, Sweadner KJ, Cohen RA, Figtree GA. Oxidative inhibition of the vascular Na+–K+ pump via NADPH oxidase‐dependent β1 subuit glutathionylation: implications for angiotensin II‐induced vascular dysfunction. Free Radic Biol Med. 2013; 65C:563-572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schuhmacher S, Oelze M, Bollmann F, Kleinert H, Otto C, Heeren T, Steven S, Hausding M, Knorr M, Pautz A, Reifenberg K, Schulz E, Gori T, Wenzel P, Münzel T, Daiber A. Vascular dysfunction in experimental diabetes is improved by pentaerythrityl tetranitrate but not isosorbide‐5‐mononitrate therapy. Diabetes. 2011; 60:2608-2616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Toda N, Ayajiki K, Okamura T. Interaction of endothelial nitric oxide and angiotensin in the circulation. Pharmacol Rev. 2007; 59:54-87 [DOI] [PubMed] [Google Scholar]
- 13.Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II‐mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996; 97:1916-1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Youn YJ, Gao L, Cai H. The p47phox‐ and NADPH oxidase organiser 1 (NOXO1)‐dependent activation of NADPH oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS) uncoupling and endothelial dysfunction in a streptozotocin‐induced murine model of diabetes. Diabetologia. 2012; 55:2069-2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mollnau H, Wendt M, Szocs K, Lassegue B, Schulz E, Oelze M, Li H, Bodenschatz M, August M, Kleschyov AL, Tsilimingas N, Walter U, Forstermann U, Meinertz T, Griendling K, Munzel T. Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling. Circ Res. 2002; 90:E58-E65 [DOI] [PubMed] [Google Scholar]
- 16.Oster P, Bauknecht H, Hackenthal E. Active and passive immunization against angiotensin II in the rat and rabbit. Evidence for a normal regulation of the renin‐angiotensin system. Circ Res. 1975; 37:607-614 [DOI] [PubMed] [Google Scholar]
- 17.Crabtree MJ, Brixey R, Batchelor H, Hale AB, Channon KM. Integrated redox sensor and effector functions for tetrahydrobiopterin‐ and glutathionylation‐dependent endothelial nitric‐oxide synthase uncoupling. J Biol Chem. 2013; 288:561-569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kleschyov AL, Mollnau H, Oelze M, Meinertz T, Huang Y, Harrison DG, Munzel T. Spin trapping of vascular nitric oxide using colloid Fe(II)‐diethyldithiocarbamate. Biochem Biophys Res Commun. 2000; 275:672-677 [DOI] [PubMed] [Google Scholar]
- 19.Sugimoto K, Fujii S, Takemasa T, Yamashita K. Detection of intracellular nitric oxide using a combination of aldehyde fixatives with 4,5‐diaminofluorescein diacetate. Histochem Cell Biol. 2000; 113:341-347 [DOI] [PubMed] [Google Scholar]
- 20.Heydemann A, Huber JM, Kakkar R, Wheeler MT, McNally EM. Functional nitric oxide synthase mislocalization in cardiomyopathy. J Mol Cell Cardiol. 2004; 36:213-223 [DOI] [PubMed] [Google Scholar]
- 21.Zielonka J, Vasquez‐Vivar J, Kalyanaraman B. Detection of 2‐hydroxyethidium in cellular systems: a unique marker product of superoxide and hydroethidine. Nat Protoc. 2008; 3:8-21 [DOI] [PubMed] [Google Scholar]
- 22.Verbeuren TJ, Coene MC, Jordaens FH, Van Hove CE, Zonnekeyn LL, Herman AG. Effect of hypercholesterolemia on vascular reactivity in the rabbit. II. Influence of treatment with dipyridamole on endothelium‐dependent and endothelium‐independent responses in isolated aortas of control and hypercholesterolemic rabbits. Circ Res. 1986; 59:496-504 [DOI] [PubMed] [Google Scholar]
- 23.Liochev SI, Fridovich I. The effects of superoxide dismutase on H2O2 formation. Free Radic Biol Med. 2007; 42:1465-1469 [DOI] [PubMed] [Google Scholar]
- 24.Hansen RE, Winther JR. An introduction to methods for analyzing thiols and disulfides: Reactions, reagents, and practical considerations. Anal Biochem. 2009; 394:147-158 [DOI] [PubMed] [Google Scholar]
- 25.Maghzal GJ, Stocker R. Improved analysis of hydroethidine and 2‐hydroxyethidium by HPLC and electrochemical detection. Free Radic Biol Med. 2007; 43:1095-1096 [DOI] [PubMed] [Google Scholar]
- 26.Babior BM. The activity of leukocyte NADPH oxidase: regulation by p47PHOX cysteine and serine residues. Antioxid Redox Signal. 2002; 4:35-38 [DOI] [PubMed] [Google Scholar]
- 27.Gallogly MM, Mieyal JJ. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr Opin Pharmacol. 2007; 7:381-391 [DOI] [PubMed] [Google Scholar]
- 28.Chen CA, Pascali F, Basye A, Hemann C, Zweier JL. Redox modulation of endothelial nitric oxide synthase by glutaredoxin‐1 through reversible oxidative post‐translational modification. Biochemistry. 2013; 52:6712-6723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gongora MC, Harrison DG. Sad heart from no SOD. Hypertension. 2008; 51:28-30 [DOI] [PubMed] [Google Scholar]
- 30.Sanchez G, Pedrozo Z, Domenech RJ, Hidalgo C, Donoso P. Tachycardia increases NADPH oxidase activity and RyR2 S‐glutathionylation in ventricular muscle. J Mol Cell Cardiol. 2005; 39:982-991 [DOI] [PubMed] [Google Scholar]
- 31.Adachi T, Weisbrod RM, Pimentel DR, Ying J, Sharov VS, Schoneich C, Cohen RA. S‐glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat Med. 2004; 10:1200-1207 [DOI] [PubMed] [Google Scholar]
- 32.Li H, Witte K, August M, Brausch I, Godtel‐Armbrust U, Habermeier A, Closs EI, Oelze M, Munzel T, Forstermann U. Reversal of endothelial nitric oxide synthase uncoupling and up‐regulation of endothelial nitric oxide synthase expression lowers blood pressure in hypertensive rats. J Am Coll Cardiol. 2006; 47:2536-2544 [DOI] [PubMed] [Google Scholar]
- 33.Bolterman RJ, Manriquez MC, Ortiz Ruiz MC, Juncos LA, Romero JC. Effects of captopril on the renin angiotensin system, oxidative stress, and endothelin in normal and hypertensive rats. Hypertension. 2005; 46:943-947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lobysheva I, Rath G, Sekkali B, Bouzin C, Feron O, Gallez B, Dessy C, Balligand JL. Moderate caveolin‐1 downregulation prevents NADPH oxidase‐dependent endothelial nitric oxide synthase uncoupling by angiotensin II in endothelial cells. Arterioscler Thromb Vasc Biol. 2011; 31:2098-2105 [DOI] [PubMed] [Google Scholar]
- 35.Guzik TJ, Harrison DG. Vascular NADPH oxidases as drug targets for novel antioxidant strategies. Drug Discov Today. 2006; 11:524-533 [DOI] [PubMed] [Google Scholar]
- 36.Probstfield JL, O'Brien KD. Progression of cardiovascular damage: the role of renin‐angiotensin system blockade. Am J Cardiol. 2010; 105:10A-20A [DOI] [PubMed] [Google Scholar]
- 37.Shahin Y, Khan JA, Samuel N, Chetter I. Angiotensin converting enzyme inhibitors effect on endothelial dysfunction: a meta‐analysis of randomised controlled trials. Atherosclerosis. 2011; 216:7-16 [DOI] [PubMed] [Google Scholar]
- 38.Knorr M, Hausding M, Kroller‐Schuhmacher S, Steven S, Oelze M, Heeren T, Scholz A, Gori T, Wenzel P, Schulz E, Daiber A, Munzel T. Nitroglycerin‐induced endothelial dysfunction and tolerance involve adverse phosphorylation and S‐glutathionylation of endothelial nitric oxide synthase: beneficial effects of therapy with the AT1 receptor blocker telmisartan. Arterioscler Thromb Vasc Biol. 2011; 31:2223-2231 [DOI] [PubMed] [Google Scholar]