

Abstract
Background
Lipocalin‐2 is a proinflammatory adipokine upregulated in obese humans and animals. A pathogenic role of lipocalin‐2 in hypertension has been suggested. Mice lacking lipocalin‐2 are protected from dietary obesity‐induced cardiovascular dysfunctions. Administration of lipocalin‐2 causes abnormal vasodilator responses in mice on a high‐fat diet (HFD).
Methods and Results
Wild‐type and lipocalin‐2 knockout mice were fed with standard chow or HFD. Immunoassays were performed for evaluating the circulating and tissue contents of lipocalin‐2. The relaxation and contraction of arteries were studied using a wire myograph. Blood pressure was monitored with implantable radio telemetry. Dietary obesity promoted the accumulation of lipocalin‐2 protein in blood and arteries. Deficiency of this adipokine protected mice from dietary obesity‐induced elevation of blood pressure. Mass spectrometry analysis revealed that human and murine lipocalin‐2 were modified by polyamination. Polyaminated lipocalin‐2 was rapidly cleared from the circulation. Adipose tissue was a major site for lipocalin‐2 deamidation. The circulating levels and the arterial accumulation of deamidated lipocalin‐2 were significantly enhanced by treatment with linoleic acid (18:2n−6), which bound to lipocalin‐2 with high affinity and prevented its interactions with matrix metalloproteinase 9 (MMP9). Combined administration of linoleic acid with lipocalin‐2 caused vascular inflammation and endothelial dysfunction and raised the blood pressure of mice receiving standard chow. A human lipocalin‐2 mutant with cysteine 87 replaced by alanine (C87A) contained less polyamines and exhibited a reduced capacity to form heterodimeric complexes with MMP9. After treatment, C87A remained in the circulation for a prolonged period of time and evoked endothelial dysfunction in the absence of linoleic acid.
Conclusions
Polyamination facilitates the clearance of lipocalin‐2, whereas the accumulation of deamidated lipocalin‐2 in arteries causes vascular inflammation, endothelial dysfunction, and hypertension.
Keywords: adipokine, endothelial dysfunction, inflammation, lipotoxicity, obesity
Introduction
Human lipocalin‐2, also called neutrophil gelatinase‐associated lipocalin, is a 25‐kDa protein originally purified from the exocytosed material of neutrophils and partly associated with the gelatinase, matrix metalloproteinase 9 (MMP9).1 It belongs to the functionally heterogeneous family of lipocalin proteins, which bind and carry small molecules to specific target cells.2 In obese human subjects and animals, the circulating concentration of lipocalin‐2 is augmented and positively correlated with body‐fat mass, arterial blood pressure (ABP), insulin resistance index, and abnormal lipid profiles.3 Because lipocalin‐2 is produced by adipose tissue, it is regarded as an adipokine.3–5 In mice, deficiency of this adipokine protects against the development of endothelial and cardiometabolic dysfunctions associated with genetic or dietary obesity.6–8 In mice fed a high‐fat diet (HFD), administration of recombinant lipocalin‐2 protein promotes endothelial dysfunction and induces adipose tissue inflammation.6–7 An augmented lipocalin‐2 level is likely to play a causative role in obesity‐related cardiovascular complications, including hypertension and heart disease.3,9 Despite this information, the mechanisms responsible for the elevated circulating level of lipocalin‐2 in obese subjects remain largely unknown. Moreover, it is not clear whether or not this adipokine exerts direct effects on the arterial wall leading to endothelial dysfunction.
Endothelial dysfunction occurs at the early stages of obesity and promotes the development of cardiometabolic complications.3,10–11 Obesity impairs endothelium‐dependent vasodilator responses to shear stress, insulin, and other neurohumoral mediators.12 By contrast, weight loss in obese human subjects substantially improves endothelial function.13–14 The endothelium of obese subjects is exposed directly to elevated lipid levels (including free fatty acids, triglycerides, and low‐density lipoproteins [LDLs]). In particular, circulating nonesterified free fatty acids (NEFAs) play a key pathogenic role in obesity‐induced endothelial dysfunction.15–19 It is not known whether or not lipocalin‐2‐evoked endothelial dysfunction is related to NEFA‐mediated lipotoxicity under obese conditions. The present study demonstrates that lipocalin‐2 is polyaminated and rapidly cleared from the circulation. NEFAs upregulate and activate lipocalin‐2 by promoting its deamidation. The deamidated lipocalin‐2 accumulates in arteries, inducing endothelial dysfunction and causing elevated ABP. These lines of evidence collectively reveal a novel mechanism underlying obesity‐induced endothelial dysfunction and hypertension, which involves lipid‐mediated activation of an inflammatory adipokine, lipocalin‐2.
Materials and Methods
Experimental Design
The present study investigated the mechanisms underlying upregulated lipocalin‐2 and the cause‐effect relationships with endothelial dysfunction and hypertension in obese animals. Data comparisons were performed between mice fed with standard chow and HFD, or between mice with and without lipocalin‐2 expressions, or among different treatment conditions (vehicle, lipocalin‐2 protein, linoleic acid [LA], and lipocalin‐2 plus LA) in age‐matched mice under the same type of diet.
Animal Studies
All the animal experimental procedures were approved by the Committee on the Use of Live Animals for Teaching and Research of the University of Hong Kong and carried out in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (Publication No. 85‐23, revised 1996). Male wild‐type (WT) and lipocalin‐2 knockout (Lcn2‐KO) mice were maintained in a C57BL/6J background.6–8,20 Mice were housed in a room under controlled temperature (23±1°C) and 12‐hour light‐dark cycle, with free access to water and standard chow (LabDiet 5053; Purina Mills, Richmond, IN) or HFD (composed of 21.3% protein, 23.6% fat [primarily lard; 45% of the caloric intake], 5.8% fiber, and 41.2% carbohydrate; 4.65 kcal·g−1; D12451; Research Diet, New Brunswick, NJ). For NEFA administration, mice under standard chow were fasted (food removal) for 16 hours and then administered intraperitoneally with a total volume of 30 μL of solution containing 3 mg of LA, oleic acid (OA), palmitic acid (PA), docosahexaenoic acid (DHA), or eicosapentaenoic acid (EPA). A lipocalin‐2 protein solution (4 μg/μL in phosphate buffer; 800 μg per mouse) was subsequently injected intraperitoneally. MMP9 Inhibitor I (Cat No. 444278; Calbiochem, EMD Chemicals, Inc, San Diego, CA) was administered by tail vein injection (2 μg per mouse). Unless specified otherwise, tissues were collected 6 hours after treatment.
Production of Lipocalin‐2 Protein
Recombinant murine (mlipocalin‐2) and human (hlipocalin‐2) lipocalin‐2 was expressed, purified, and endotoxin was removed as previously described.5–7 Purity of the protein was confirmed by SDS‐PAGE and mass spectrometry (MS) analysis.
Site‐Directed Mutagenesis
The pPRO‐His‐hLCN2 vector5 was used to generate a construct pPRO‐His‐hLCN2‐C87A encoding a mutant form of hlipocalin‐2 with the cysteine 87 residue replaced by alanine. Primers used for mutagenesis were 5′‐AGGACTTTTGTTCCAGGTGCCCAGCCCGGCGAGTTCACG‐3′ (forward) and 5′‐CGTGAACTCGCCGGGCTGGGCACCTGGAACAAAAGTCCT‐3′ (reverse). The mutation was verified by DNA sequencing.
Quantitative Reverse‐Transcription Polymerase Chain Reaction
Quantitation of target genes was performed using SYBR Green polymerase chain reaction (PCR) Master Mix (Qiagen, Hamburg, Germany) and an ABI PRISM 7900 HT Sequence Detection System (Applied Biosystems, Foster City, CA). The sequences of the primers used are listed in Table 1. Data were calculated and are presented as relative expression of transcripts normalized to GAPDH.
Table 1.
Sequences of Primers Used for QPCR Analysis
| Gene Name (symbol) | Accession ID | Sequence Range | Product Size (bp) | Primer Sequences |
|---|---|---|---|---|
| GAPDH (GAPD) |
GI:126012538 | 654 to 836 | 183 | Forward 5′CAGAACATCATCCCTGCATC3′ Reverse 5′CTGCTTCACCACCTTCTTGA3′ |
| ICAM‐1 (Icam1) |
GI:30172560 | 149 to 341 | 213 | Forward 5′GTGATGCTCAGGTATCCATCCA3′ Reverse 5′CACAGTTCTCAAAGCACAGCG3′ |
| E‐selectin (Sele) |
GI:118130193 | 141 to 268 | 128 | Forward 5′ATGCCTCGCGCTTTCTCTC3′ Reverse 5′GTAGTCCCGCTGACAGTATGC3′ |
| P‐selectin (Selp) |
GI:327412298 | 1345 to 1445 | 101 | Forward 5′TGAACTGAAGGGATCAAGAAGACT3′ Reverse 5′GCCGAGGGACATCATCACAT3′ |
| MCP‐1 (Ccl2) |
GI:141803162 | 111 to 250 | 140 | Forward 5′GCCTGCTGTTCACAGTTGC3′ Reverse 5′GGTGATCCTCTTGTAGCTCTCC3′ |
| TNFα (Tnf) |
GI:518831586 | 440 to 646 | 207 | Forward 5′TGGTGCCTGGTCTGATGATG3′ Reverse 5′GTGGTAACCGCTCAGGTGTTG3′ |
| NGALR (Lrp2) |
GI:124487371 | 3101 to 3208 | 127 | Forward 5′ CCACCCATCCCAACGGCGAC3′ Reverse 5′ CGGGCTGGGTCTCCCTCACA3′ |
| 24p3R (Slc22a) |
GI:270341383 | 752 to 918 | 186 | Forward 5′ CGGCCAGGTTTGGCCGTCG3′ Reverse 5′ GGGTTGGGTCGCACAGCTCC3′ |
| Lipocalin‐2 (Lcn‐2) |
GI:34328048 | 216 to 383 | 168 | Forward 5′ GGTTTGGTGGCAGGCTATTA3′ Reverse 5′ CAGAGTGGCTTTCCCCATAA3′ |
GAPDH indicates glyceraldehyde 3‐phosphate dehydrogenase; ICAM‐1, intercellular adhesion molecule‐1; MCP‐1, monocyte chemotactic protein‐1; NGALR, neutrophil gelatinase‐associated lipocalin receptor; QPCR, quantitative reverse‐transcription polymerase chain reaction; TNF‐α, tumor necrosis factor alpha.
SDS‐PAGE, Western Blotting, and ELISA
Protein samples, serum (1.5 μL), or tissue lysates (30 or 50 μg) were incubated with either a Laemmli sample buffer or a nonreducing buffer (1% SDS, 5% glycerol, and 10 mmol/L of Tris‐HCl; pH 6.8) at room temperature for 10 minutes and then separated by SDS‐PAGE.21 Protein bands were visualized by Coomassie Brilliant Blue staining. After transferring to polyvinylidene difluoride (PVDF) membranes, Western blotting was performed by incubating with antibodies (Abs) against mlipocalin‐2 or hlipocalin‐2,5 spermidine, spermine, and cytochrome P4502C (Abcam, Cambridge, MA), MMP9 (BD Transduction Laboratories, San Jose, CA) and beta‐actin (Cell Signaling, Beverly, MA), respectively. After incubation with secondary Ab, the immune complexes were detected with enhanced chemiluminescence (ECL) reagents from GE Healthcare (Uppsala, Sweden). Albumin detected with Ponceau S staining, or immunoglobulin G probed by Western blotting, was used as a loading control for serum samples. Beta‐actin was probed as a loading control for tissue samples. Immune complexes were detected with ECL reagents from GE Healthcare. Total serum lipocalin‐2 levels were measured using an in‐house ELISA.5
Immunohistochemistry
Paraffin sections (5 μm) were prepared for hematoxylin staining and immunostaining using in‐house Abs against mlipocalin‐2 or hlipocalin‐2.5–6 Images were captured and analyzed under a microscope (Leica Microsystems, Bensheim, Germany).
Peptide Mass Finger Printing by Matrix‐Assisted Laser Desorption/Ionization Time‐of‐Flight MS Analysis
Proteins of interest separated by SDS‐PAGE were excised and digested in gel with trypsin.22 Tryptic peptide mixtures were then mixed with an equal amount of α‐cyano‐4‐hydroxycinnamic acid matrix, spotted on the sample plates, and air‐dried for analysis on a 4800 matrix‐assisted laser desorption/ionization dual time‐of‐flight (MALDI TOF/TOF)™ system (Applied Biosystems). All ion spectra were recorded in the positive mode with an acceleration voltage of 20.0 kV. The spectrometer was externally calibrated by use of Cal Mix 2 standard mixture.
Isometric Force Measurement
Mice were anesthetized with pentobarbital sodium (30 mg·mL−1·kg−1, IP). The thoracic aortae and carotid arteries were isolated and cut into rings (≈2 mm length), which were suspended in a Mulvany‐Halpern wire myograph (Model 610M; Danish Myo Technology A/S, Aarhus, Denmark) for recording of isometric force (PowerLab 4SP; ADInstrruments, Inc., Colorado Springs, CO). The myograph chambers contained modified Krebs‐Ringer bicarbonate (KRB) solution (in mmol/L: 118 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 25 NaHCO3, and 11.1 d‐glucose [control solution]). After equilibration, the aortae were contracted with phenylephrine (10−5 mol/L) before being exposed to increasing concentrations of acetylcholine (ACh; 10−10 to 10−4.5 mol/L; Sigma‐Aldrich, St. Louis, MO). Decreases in tension are expressed as the percentage of the contraction to phenylephrine. In addition, the aortae were contracted with thromboxane‐prostanoid receptor agonist U46619 (1 to 3×10−8 mol/L; Biomol, Plymouth Meeting, PA) before being exposed to increasing concentrations of insulin (10−10 to 3×10−6 mol/L; Actrapid HM, Novo Nordisk, Bagsvaerd, Denmark). Decreases in tension are expressed as the percentage of the contraction to U46619. Carotid arteries were preincubated with N‐nitro‐l‐arginine methyl ester (l‐NAME; nitric oxide synthase [NOS] inhibitor; 10−4 mol/L) for 40 minutes and then exposed to increasing concentrations of ACh (10−10 to 3×10−4 mol/L; Sigma Aldrich, St. Louis, MO). Increases in tension are expressed as the percentage of a reference contraction to KCl (60 mmol/L) obtained at the beginning of the experiment.
Blood Pressure Measurement
Blood pressure was measured using an implantable telemetry system (Data Sciences International, St. Paul, MN). In brief, WT and Lcn2‐KO mice were anesthetized with a Hypnorm/Dormicum mixture (Hypnorm: 0.315 mg/mL of fentanyl and 10 mg/mL of fluanisone [VetaPharma Ltd, Leeds, UK]; Dormicum: 5 mg/mL of midazolam [Roche A/S, Hvidovre, Denmark]). Telemetry catheters (HD‐X11 or PA‐C10; Data Sciences International, St. Paul, MN) were inserted into the left common carotid artery with the transmitter implanted subcutaneously. Mice were housed individually in a temperature‐ and humidity‐controlled facility and maintained under a 12‐hour dark (lights off 7:00 pm) and 12‐hour light (lights on 7:00 am) cycle. Blood pressure was recorded for at least four 12/12 light‐dark cycles using the Dataquest A.R.T system (Data Sciences International).
Isothermal Titration Calorimetry Analysis
Thermodynamic parameters of fatty acids (FAs) binding to lipocalin‐2 were determined by isothermal titration microcalorimetry utilizing an iTC200 microcalorimeter (MicroCal, Northampton, MA). FAs (300 μmol/L) were prepared in a buffer containing 20 mmol/L of Tris·HCl (pH 8.0) and 50% glycerol. FAs were titrated (2.0 μL/injection) into a solution of protein (20 μmol/L in 20 mmol/L of Tris·HCl [pH 8.0]/50% glycerol) at 25°C, and the amount of heat released or absorbed was measured. Three sets of controls were performed for each experiment: buffer injected into buffer; FAs injected into buffer; and buffer injected into protein. Data were analyzed using the MicroCal Origin v7.0383 software supplied with the iTC200 microcalorimeter.
Molecular Docking
The program AutoDock Version 4.0 (Scripps Research Institute, La Jolla, CA) was used for docking flexible ligands into hlipocalin‐2 binding sites. For each ligand, the number of genetic algorithm runs was 80, the population size was set to 150, and the maximal number of energy evaluations was 2 500 000 per run. The grid box was centered on the coordinates 51.294, 100.067, and 35.44 with a spacing of 0.581. All other parameters were kept at their default settings. The protein structure was 3CMP from the Protein Data Bank. Bacterial siderophore was used as a reference. Chemical structures of all ligands were downloaded from Pubchem and modified by Accelrys Discovery Studio Visualizer 2.5 software.
Statistical Analysis
All Western blotting and histological analysis were repeated at least 3 times using different sets of experimental animals. For comparing endothelial functions, statistical calculations were performed by one‐way ANOVAs followed by Tukey's multiple comparisons using Prism version 5 (GraphPad Software Inc., San Diego, CA). Statistical calculations for blood pressure differences between WT and age‐matched Lcn2‐KO mice were compared by Student t test. Mann‐Whitney's nonparametric U test was used for evaluating the differences of blood pressure in mice subjected to acute treatment with or without recombinant lipocalin‐2 and/or LA, lipocalin‐2 levels in mice under standard chow and HFD, and interactions between lipocalin‐2 and NEFAs. All values are presented as means±SEM. For all statistical comparisons, a P value <0.05 was accepted to indicate significant differences.
Results
Lipocalin‐2 Protein Is Accumulated in Aortae of Obese Mice With Elevated Blood Pressure
Circulating lipocalin‐2 level is significantly elevated in obese humans and genetically obese animals.5 The present study demonstrated that obesity induced by long‐term consumption of HFD in WT mice also caused a significant elevation of circulating lipocalin‐2, as revealed by both ELISA and Western blotting analysis (Figure 1A and 1B). In line with the deterioration of endothelial vasodilator functions,7 lipocalin‐2 protein progressively accumulated in aortic tissues of WT mice receiving a HFD (Figure 1C). Positive immunostaining signals of lipocalin‐2 were mainly detected in the intima layer of the aortic wall (Figure 1D). The mRNA of lipocalin‐2 was undetectable in aortae of obese WT mice, suggesting that accumulation of this protein was not the result of local gene expression. The aortic expression of lipocalin‐2 receptors and serum adiponectin levels were not significantly different between WT and Lcn2‐KO mice (Figure S1, upper panel).
Figure 1.
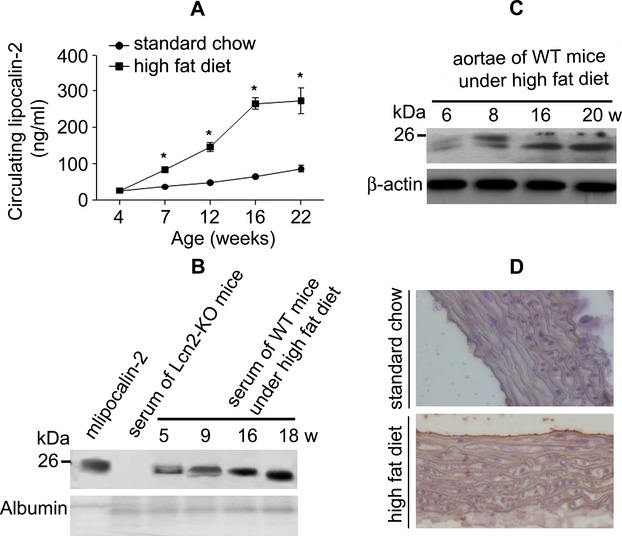
Accumulation of lipocalin‐2 in arteries of obese mice. A, Serum concentrations of lipocalin‐2 in WT mice given high fat diet (HFD) for different periods were measured by an in‐house ELISA. *P<0.05 versus standard chow group; n=3 to 5 in all groups. B, Western blotting analysis of sera collected from WT mice after different periods of HFD. Recombinant mlipocalin‐2 was used as the standard and serum from Lcn2‐KO mice was included for comparison. C, Aortic accumulation of lipocalin‐2 was monitored by Western blotting. The same amount (50 μg) of proteins was loaded for each sample. β‐actin was probed as the loading control. D, Immunohistochemistry was performed in aortae sections collected from WT mice under standard chow or HFD. Magnification, ×400. ELISA indicates enzyme‐linked immunosorbent assay; Lcn2‐KO, lipocalin‐2 knockout; WT, wild type.
Radiotelemetry measurement demonstrated that obese mice deficient with lipocalin‐2 exhibited significantly lower mean arterial pressure (MAP), systolic blood pressure (SBP), and diastolic blood pressure (DBP) than those of obese WT mice (Table 2). Moreover, after 8 weeks of HFD, the diurnal rhythms in blood pressure were blunted in WT mice, but maintained in Lcn2‐KO mice. HFD‐induced arterial inflammation was significantly attenuated by lipocalin‐2 deficiency (Figure S1, lower panel). These findings suggest that accumulation of lipocalin‐2 in arteries of obese mice contributes to the development of dietary obesity‐induced endothelial dysfunction and hypertension.
Table 2.
Blood Pressures in Conscious Mice (12 Weeks Old, High‐Fat Diet) Implanted With Telemetry Devices
| Mean Arterial Pressure (mm Hg) | Systolic Blood Pressure (mm Hg) | Diastolic Blood Pressure (mm Hg) | ||||
|---|---|---|---|---|---|---|
| WT | Lcn2‐KO | WT | Lcn2‐KO | WT | Lcn2‐KO | |
| 24‐hour average | 133.1±15.5 | 115.5±16.6# | 142.8±16.3 | 122.4±17.1# | 123.4±15.6 | 108.7±16.2# |
| Light cycle average (7:00 am to 7:00 pm) | 129.5±15.5 | 107.3±13.8# | 138.3±16.2 | 113.7±14.2# | 120.6±15.3 | 100.9±13.4# |
| Dark cycle average (7:00 pm to 7:00 am) | 136.8±14.7 | 123.7±14.5* | 147.3±15.1 | 131.1±14.9* | 126.2±15.3 | 116.4±14.3* |
Lcn2‐KO indicates lipocalin‐2 knockout; WT, wild type.
*P<0.05 and #P<0.01 versus the corresponding WT mice controls; n=4 in both groups.
Endogenous Lipocalin‐2 Is Polyaminated
Western blotting analysis revealed that in the serum of WT mice fed with standard chow, a single slow‐migrating band of lipocalin‐2 was detected in SDS‐PAGE gel. In serum of WT mice fed with HFD, lipocalin‐2 was not only upregulated, but also the protein migrated as a high‐ (HMW) and a low‐molecular‐weight (LWM) species (Figure 2A, left panel). Lipocalin‐2 mainly accumulated as the LMW species in both blood and aortae of WT mice after prolonged HFD (Figure 1B and 1C). In adipose tissues of WT mice, both HMW and LWM species of lipocalin‐2 were detectable, whereas only HMW species was present in liver samples (Figure 2A, right panel). In Lcn2‐KO mice, after intraperitoneal administration of recombinant mlipocalin‐2 (left panel) or hlipocalin‐2 (right panel), a species equivalent to the injected protein was present in the serum of animals fed either standard chow or HFD (Figure 2B). In addition, an immunoreactive protein band with a lower molecular weight was revealed by Western blotting in serum derived from HFD‐fed Lcn2‐KO mice.
Figure 2.
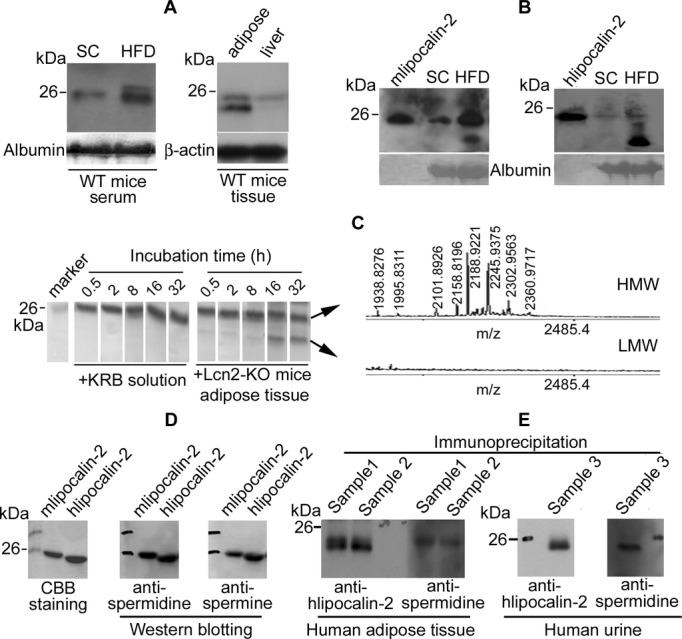
Lipocalin‐2 is modified by polyamination. A, Western blotting was performed for detection of lipoclain‐2 in serum (1.5 μL/lane) derived from WT mice fed with standard chow (SC) or high fat diet HFD (left panel), and in tissue lysates (epididymal adipose fat and liver, 50 μg/lane) derived from WT mice fed with HF (right panel). B, Lipocalin‐2 knockout (Lcn2‐KO) mice fed with SC or HF were injected intraperitoneally with murine lipocalin‐2 (mlipocalin‐2, left panel) or human lipocalin‐2 (hlipocalin‐2, right panel). Six hours after treatment, sera were collected and subjected to Western blotting. Purified recombinant protein was included in each gel for comparison. C, Recombinant hlipocalin‐2 protein was incubated in KRB solution with or without the adipose tissues derived from obese Lcn2‐KO mice for different time points. Protein in solution was then analyzed by SDS‐PAGE and Coomassie Brilliant Blue (CBB) staining (left panel). Protein bands were cut for in‐gel trypsin digestion and mass spectrometric analysis by MALDI‐TOF.22 Mass finger‐printing spectra containing the polyaminated peptide peaks are shown (right panel). D, Purified recombinant mlipocalin‐2 and hlipocalin‐2 were separated by SDS‐PAGE and visualized by CBB staining or Western blotting using antibodies recognizing spermidine or spermine. E, Coimmunoprecipitation was performed to precipitate lipocalin‐2 protein from adipose tissue lysates (500 μg, left panel) and concentrated urine sample (right panel) from human subjects. Immune complexes were separated by SDS‐PAGE and Western blotting was performed using antibodies recognizing hlipocalin‐2 or spermidine. HMW indicates high molecular weight; KRB, Krebs‐Ringer bicarbonate; LMW, low molecular weight; MALDI‐TOF, matrix‐assisted laser desorption/ionization time‐of‐flight; WT, wild type.
The differences between HMW and LMW species of lipocalin‐2 were further investigated. After incubation in KRB buffer for different periods of time, recombinant hlipocalin‐2 remained as one species, as revealed by Coomassie Brilliant Blue staining of the SDS‐PAGE gels (Figure 2C, left panel). After prolonged incubation with adipose tissue derived from obese Lcn2‐KO mice, which contained no endogenous lipocalin‐2, a LMW protein species appeared (Figure 2C, left panel). In‐gel digestion and MS analysis confirmed the identities of both HMW and LMW protein bands as hlipocalin‐2. Moreover, the peptides corresponding to NH2‐ and COOH‐terminus of hlipocalin‐2 were present in both spectra (Figure S2), suggesting that the LMW species was not a cleaved fragment of the HMW species. On the other hand, a cluster of masses differing by 57, 87, 144, and 201 Da (1938.8276 versus 1995.8311, 2101.8926 versus 2188.9211, 2101.8926 versus 2145.9375, and 2101.8926 versus 2302.9563) was observed in the spectrum of the HMW species, but was absent in that of LMW hlipocalin‐2 (Figure 2C, right panel), suggesting that this protein is modified by polyamination and that the polyamine groups are removed in the presence of adipose tissue from obese mice. The cluster of the modified peaks also existed in the HMW species of recombinant mlipocalin‐2 (Figure S3). Western blotting confirmed that mlipocalin‐2 and hlipocalin‐2 were both cross‐reactive to Abs recognizing the 2 widely distributed aliphatic polyamines,23 spermidine and spermine (Figure 2D). Immunoprecipitation was performed using adipose tissues and urine collected from human subjects. Western blotting confirmed that the precipitated hlipocalin‐2 was polyaminated in both adipose tissue and urine (Figure 2E). Because mlipocalin‐2 and hlipocalin‐2 showed similar properties, unless specified otherwise, subsequent studies used recombinant hlipocalin‐2 and were performed in Lcn2‐KO mice to avoid the influence from endogenous lipocalin‐2.
LA Promotes Lipocalin‐2 Deamidation
The above data demonstrate that human and murine lipocalin‐2 are polyaminated. Deamidation (removal of polyamine groups) of lipocalin‐2 is facilitated under conditions such as diet‐induced obesity. HFD increases circulating levels of NEFAs.24–25 Therefore, the effects of individual NEFAs on circulating hlipocalin‐2 were evaluated. Recombinant hlipocalin‐2 protein was given intraperitoneally to Lcn2‐KO mice maintained on standard chow, with or without combined administration of saturated PA, monounsaturated OA, long‐chain polyunsaturated LA, DHA, or EPA. Two hours after the injection, two strongly immunoreactive protein bands were detected by Western blotting in the serum collected from mice treated with hlipocalin‐2 plus LA (Figure 3A, upper panel). ELISA results revealed that combined administration of hlipocalin‐2 and LA significantly elevated the circulating amount of hlipocalin‐2 more than 8‐fold, when compared to animals injected with hlipocalin‐2 alone (Figure 3A, bottom panel). Protein of hlipocalin‐2 remained low or undetectable in sera samples collected from those with combined treatments of other NEFAs. Time‐course evaluation by Western blotting demonstrated that combined LA administration prevented the clearance of circulating hlipocalin‐2 from Lcn2‐KO mice receiving standard chow. Without LA, hlipocalin‐2 was undetectable in serum 4 hours after injection. By contrast, deamidated hlipocalin‐2 could still be readily detected in serum of mice 12 hours after injection of the protein in combination with LA (Figure 3B).
Figure 3.

Deamidation of lipocalin‐2. A, Recombinant hlipocalin‐2 was injected intraperitoneally into Lcn2‐KO mice under standard chow (SC). Immediately before protein treatment, mice were administered with or without palmitic acid, oleic acid, linoleic acid (LA), DHA, and EPA, respectively. Sera were collected 2 hours after treatment for measuring circulating lipocalin‐2 levels by Western blotting (upper panel). ELISA results were shown for sera collected at 6 hours after injection (bottom panel). Murine IgG was probed as a loading control. *P<0.05 versus hlipocalin‐2‐treated samples; n=12 to 15 in all groups. B, Lcn2‐KO mice under SC were treated with hlipocalin‐2, combined with or without LA as in (A). At different time points after injection, serum was collected from tail vein and subjected to Western blotting analysis of circulating hlipocalin‐2. Albumin, as shown in membrane stained with Ponceau S, was used as the loading control. C, Recombinant hlipocalin‐2 (5 μg) was incubated in 100 μL of KRB solution containing serum (5%) or 100 mg of tissues (epididymal fat, liver, or kidney) collected from Lcn2‐KO mice (8 weeks of age) under SC. The solution was collected at 1, 2, and 4 hours of incubation and subjected to Western blotting analysis using antibody recognizing hlipocalin‐2. D, Recombinant hlipocalin‐2 (5 μg) was incubated with serum or tissues as in (C) in the presence of LA (1 mg/well) and analyzed by Western blotting. E, Recombinant hlipocalin‐2 (5 μg) was incubated in KRB solution containing serum, adipose tissue, liver, or kidney collected from Lcn2‐KO mice (8 weeks of age) under HFD. Western blotting was performed as in (C and D). F, Recombinant hlipocalin‐2 (5 μg) was incubated with serum or tissues collected from Lcn2‐KO mice (under SC, treated with LA [3 μg/mouse] for 1 hour) as in (C) and analyzed by Western blotting. DHA indicates docosahexaenoic acid; ELISA, enzyme‐linked immunosorbent assay; EPA, eicosapentaenoic acid; HF, high fat diet; IgG, immunoglobulin G; KRB, Krebs‐Ringer bicarbonate; Lcn2‐KO, lipocalin‐2 knockout.
Next, in vitro incubation of recombinant protein with sera or tissues derived from Lcn2‐KO mice was performed to test the effects of LA on hlipocalin‐2 deamidation. After coincubation with serum, adipose tissue, liver, or kidney derived from mice fed standard chow, hlipocalin‐2 remained as one species (Figure 3C). A small amount of LMW species of hlipocalin‐2 appeared when LA was present during coincubation with serum, adipose tissue, and kidney (Figure 3D). By contrast, without the addition of LA, a large amount of deamidated hlipocalin‐2 was readily formed after incubation with adipose tissue or kidney derived from Lcn2‐KO mice fed HFD (Figure 3E). Moreover, coincubation with adipose tissue or kidney collected from Lcn2‐KO mice treated with LA (intraperitoneal injection) promoted hlipocalin‐2 deamidation (Figure 3F). Thus, adipose tissue represents a major site for deamidation of hlipocalin‐2, which can be facilitated by LA or dietary obesity.
Reduced Interactions Between Deamidated Lipocalin‐2 and MMP9
Isothermal titration calorimetry was applied to investigate the molecular interactions between lipocalin‐2 and NEFAs. Thermodynamic parameters were measured to indicate binding strength and interaction force. The dissociation constants for the lipocalin‐2/NEFA complex at pH 7.4 were 0.487, 2744.343, and 1467.45 μmol/L for LA, PA, and OA, respectively (Figure 4), suggesting that lipocalin‐2 could bind to LA with a stronger affinity than the other two NEFAs. In fact, molecular docking simulation suggested that among all tested FAs, lipocalin‐2 possessed the highest binding affinity with LA (Table 3).
Figure 4.
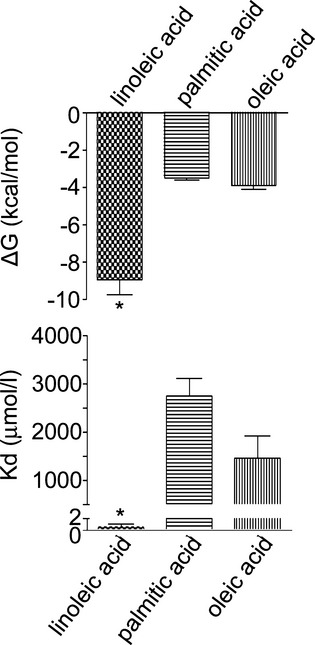
Lipocalin‐2 binds with linoleic acid with high affinity. Thermodynamic parameters of nonesterified free fatty acids (NEFAs) binding to hlipocalin‐2 were determined by isothermal titration microcalorimetry. Changes in Gibbs free energy (ΔG) and equilibrium dissociation constant (Kd) are presented as means±SEM. *P<0.01 versus other conditions; n=5 in all groups.
Table 3.
Calculated Binding Affinities Between Lipocalin‐2 and Various Fatty Acids by Molecular Docking
| Ligand | ΔG (kcal/mol) | Kd (nmol/L) |
|---|---|---|
| Arachidonic acid | −6.1 | 35.8 |
| 20‐Hydroxyeicosatetraenoic acid | −6.2 | 30.2 |
| 12‐Hydroxyeicosatetraenoic acid | −4.0 | 1026.7 |
| 5‐Hydroxyeicosatetraenoic acid | −4.4 | 620.4 |
| Prostaglandin F2 | −6.2 | 30.2 |
| Prostaglandin H2 | −5.9 | 50.1 |
| Prostaglandin D2 | −4.6 | 443.4 |
| Linoleic acid | −7.3 | 4.7 |
| Oleic acid | −4.2 | 867.9 |
| Palmitic acid | −3.2 | 4649.3 |
| Arachidic acid | −3.1 | 5499.1 |
| Stearic acid | −3.2 | 4649.3 |
| Enterochelin* | −8.7 | 0.5 |
A ligand of lipocalin‐2 (http://www.rcsb.org/pdb/explore.do?structureId=3CMP) was included as the reference control.
MMP9 and lipocalin‐2 form heterodimers.26 The cysteine 87 (C87) residue of human lipocalin‐2 may play a role in covalent binding with MMP9.27 SDS‐PAGE and Western blotting revealed that the hlipocalin‐2 mutant, C87A (C87 replaced by alanine), contained a significantly lesser amount of polyamines, when compared to that of WT hlipocalin‐2 (Figure 5A, left panel). Analysis with nonreducing SDS‐PAGE demonstrated that the ratio of homodimer/monomer was below 1:10 for WT hlipocalin‐2, but over 10:1 for C87A (Figure 5A, right panel). In addition, C87A formed even higher molecular weight species that contained no polyamine signals. In Lcn2‐KO mice, interaction between C87A and MMP9 was significantly decreased, compared to that of WT hlipocalin‐2 (Figure 5B, left panel). Combined treatment with LA also abolished interactions between MMP9 and hlipocalin‐2 (Figure 5B, right panel). In serum of obese humans, interactions between MMP9 and lipocalin‐2 were reduced (Figure S4).
Figure 5.
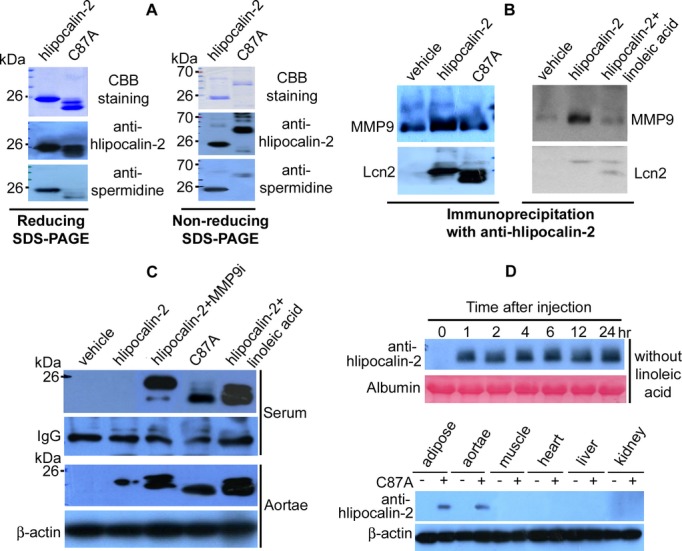
Reduced polyamination of human lipocalin‐2 mutant C87A. A, Recombinant hlipocalin‐2 and C87A mutant (3 μg) were separated by reducing (left panel) or nonreducing (right panel) SDS‐PAGE and staining with Coomassie Brilliant Blue (CBB). After transferring to PVDF membrane, Western blotting was performed to detect spermidine. B, Coimmunoprecipitation was performed using 30 μL of serum from Lcn2‐KO mice treated with vehicle, hlipocalin‐2, C87A, or hlipocalin‐2 plus linoleic acid (LA). Immune complexes were separated by SDS‐PAGE and subjected to Western blotting using antibodies recognizing MMP9 or hlipocalin (Lcn2). C, Wild‐type hlipocalin‐2 or C87A was given to Lcn2‐KO mice under standard chow. Before administration of hlipocalin‐2, mice were treated with or without MMP9 Inhibitor I (MMP9i; 2 μg/mouse, tail vein injection) or LA (3 mg/mouse, intraperitoneal injection). Six hours after treatment, sera and aortae were collected for Western blotting to evaluate hlipocalin‐2 levels. Nonspecific serum IgG and β‐actin was probed as the loading controls, respectively. D, Lcn2‐KO mice under standard chow were treated with C87A, without combined administration of LA. At different time points after injection, serum was collected from tail vein and subjected to Western blotting analysis of circulating C87A using hlipocalin‐2 antibody (upper panel). At the end of treatment, tissues were also collected for analyzing the accumulated C87A protein (lower panel). IgG indicates immunoglobulin G; Lcn2‐KO, lipocalin‐2 knockout; MMP9, matrix metalloproteinase 9.
Binding with LA may contribute to deamidation of hlipocalin‐2 and subsequent dissociation from MMP9, or vice versa. The latter possibility was tested in Lcn2‐KO mice treated with MMP9 Inhibitor I (MMP9i; 2 μg/mouse) before administration of hlipocalin‐2. Although the total amount of hlipocalin‐2 in the circulation was significantly augmented, MMP9i did not facilitate deamidation of this protein as efficiently as that of LA (Figure 5C, upper panel). In Lcn2‐KO mice, the amount of C87A in the circulation was still detectable by Western blotting at 6 hours after treatment. In SDS‐PAGE, C87A migrated to the same position as the deamidated lipocalin‐2 species in samples from mice subjected to combined treatment with LA and hlipocalin‐2 (Figure 5C, upper panel). Analysis of aortae derived from these mice revealed a large amount of accumulated C87A, similar to those treated with WT hlipocalin‐2 plus LA (Figure 5C, lower panel). On the other hand, hlipocalin‐2 was barely detectable in aortae of mice given the protein alone. Aortae of mice treated with MMP9i contained 2 species of hlipocalin‐2, both of which migrated at a higher molecular weight position than C87A (Figure 5C, lower panel). Time‐course studies revealed that after treatment, C87A remained stable in the circulation of Lcn2‐KO mice throughout the 24‐hour observation period (Figure 5D, upper panel). C87A protein mainly accumulated in adipose tissue and aortae (Figure 5D, lower panel).
The above results suggest that polyaminated lipocalin‐2 binds to MMP9 and is rapidly cleared from the circulation, whereas deamidated lipocalin‐2 accumulates in the circulation and tissues, such as the aortae.
Deamidated Lipocalin‐2 Causes Endothelial Dysfunction and Hypertension
Next, arterial function in mice treated with polyaminated (WT hlipocalin‐2) and deamidated (C87A) lipocalin‐2 was evaluated. After in vivo administration to Lcn2‐KO mice with recombinant hlipocalin‐2 alone, only a small amount of polyaminated lipocalin‐2 was detected in aortae (Figure 5C), and no significant effects were observed on expression of endothelial NOS (eNOS), cyclooxygenase‐2 (COX‐2), and cytochrome P450 2C (Cyp2C) (Figure 6A). Combined treatment with hlipocalin‐2 plus LA or administration of C87A triggered accumulation of a large amount of deamidated lipocalin‐2 in aortae of Lcn2‐KO mice (Figure 5C), accompanied by eNOS uncoupling as well as significant induction of COX‐2 and Cyp2C (Figure 6A). Quantitative PCR (QPCR) analysis revealed that C87A elicited similar proinflammatory effects to combined administration of lipocalin‐2 and LA. Expressions of inflammatory genes, including E‐selectin, P‐selectin, tumor necrosis factor alpha (TNF‐α), and intercellular adhesion molecule‐1 (ICAM‐1), were augmented significantly by both treatments (Figure 6B).
Figure 6.
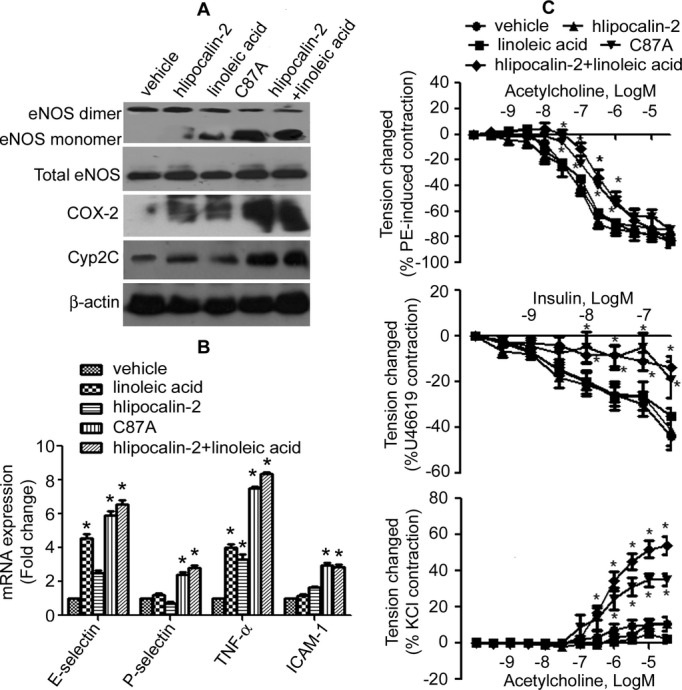
Deamidated lipocalin‐2 causes vascular inflammation and endothelial dysfunction in Lcn2‐KO mice. A, Lcn2‐KO mice (12 weeks old, under standard chow [SC]), were treated with vehicle, hlipocalin‐2, linoleic acid (LA), C87A, or hlipocali‐2 plus LA. Six hours after treatment, aortae were collected and analyzed by Western blotting using specific antibodies as indicated. β‐actin was monitored as the loading control. B, QPCR was performed to evaluate the inflammatory gene expressions in aortae collected from mice treated as in (A). C, Aortae were collected from mice treated as in (A) to evaluate relaxations to acetylcholine (ACh) (upper panel) or insulin (middle panel). Contractions to ACh (in the presence of 10−4 mol/L of l‐NAME) were obtained in quiescent carotid arteries (lower panel) of these mice. *P<0.05 versus vehicle control group; n=8 to 10 in all groups. COX‐2 indicates cyclooxygenase 2; Cyp2C, cytochrome P450 2C; eNOS, endothelial nitric oxide synthase; ICAM‐1, intercellular adhesion molecule‐1; Lcn2‐KO, lipocalin‐2 knockout; l‐NAME, N‐nitro‐l‐arginine methyl ester; PE, phenylephrine; QPCR, quantitative reverse‐transcription polymerase chain reaction; TNF‐α, tumor necrosis factor alpha.
WT hlipocalin‐2, when given alone, had no significant effect on vascular tone responsiveness. However, lipocalin‐2 plus LA treatment attenuated ACh‐ and insulin‐induced relaxations (because of activation of eNOS and release of nitric oxide [NO]7,28) in aortae and enhanced ACh‐evoked contractions (because of release of endothelium‐derived vasoconstrictor prostanoids29) in the carotid artery, whereas administration of the protein or LA alone had no significant effects (Figure 6C). Relaxation to sodium nitroprusside (10−10 to 10−5 mol/L), an endothelium‐independent dilator, was not different between groups (Figure S5). Treatment of Lcn2‐KO mice with C87A mutant alone reduced relaxations to ACh or insulin in aortae and augmented contractions to ACh in carotid arteries to a similar extent as changes observed with combined administration of hlipocalin‐2 and LA (Figure 6C).
Similar treatment was performed in WT mice fed standard chow. As shown in Figure 7A, LA promoted accumulation of endogenous deamidated lipocalin‐2 in the blood. After injection, hlipocalin‐2 appeared only transiently in the circulation, whereas combined treatment with LA significantly extended the circulating life of hlipocalin‐2, especially that of its deamidated species. In aortae, treatment with LA enhanced accumulation of endogenous mlipocalin‐2, as well as that of recombinant hlipocalin‐2 (Figure 7B, upper panel). QPCR analysis further confirmed that increased accumulation of lipocalin‐2 was associated with an augmented expression of inflammatory genes in the aortae (Figure 7B, lower panel). In line with the amount of deamidated lipocalin‐2 accumulating in the aortic wall, endothelial function was significantly reduced in mice treated with either LA or hlipocalin‐2 plus LA (Figure 7C). Moreover, this combined treatment caused a significant elevation of MAP, SBP, and DBP in WT mice (Figure 8).
Figure 7.
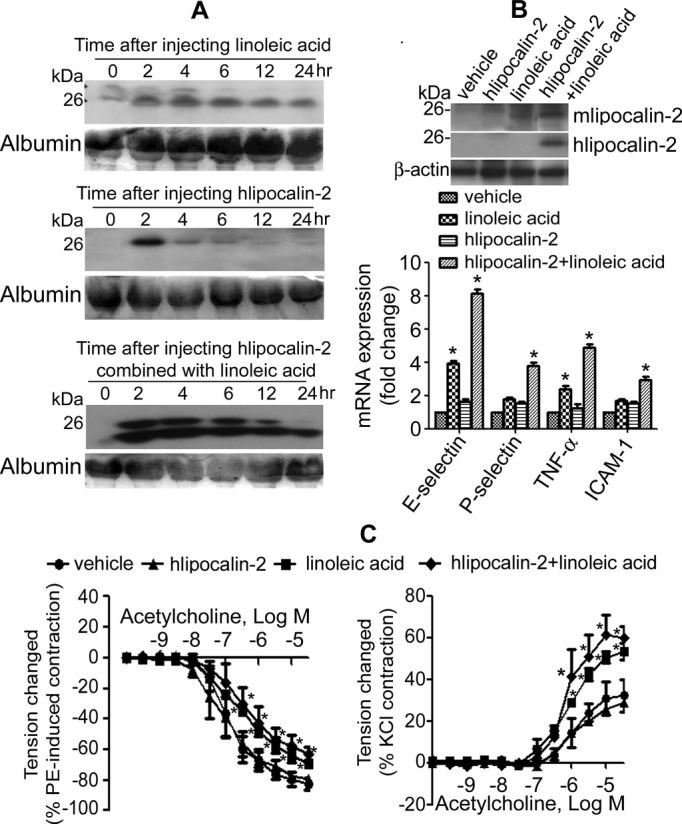
Linoleic acid (LA) promotes lipocalin‐2 deamidation and accumulation in WT mice. A, WT mice under standard chow were treated with LA, hlipocalin‐2, or hlipocalin‐2 plus LA. At different time points after injection, serum was collected from tail vein and subjected to Western blotting analysis of circulating murine (upper panel) or human lipocalin‐2 (middle and lower panels) using specific in‐house antibodies.5 Albumin stained with Ponceau S was used as the loading control. B, Aortae were collected from WT mice subjected to different treatments as above. Western blotting was performed for analyzing the amount of endogenous mlipocalin‐2 and the accumulation of exogenous hlipocalin‐2 (upper panel). β‐actin was probed as the loading control. QPCR was performed for gene expression comparison, as described in Materials and Methods (lower panel). C, Aortae were collected from the above treated mice to evaluate relaxations to acetylcholine (ACh) (left panel). Contractions to ACh (in the presence of 10−4 mol/L of l‐NAME) were obtained in quiescent carotid arteries (right panel) of these mice. *P<0.05 versus vehicle control group; n=3 to 5 in all groups. ICAM‐1 indicates intercellular adhesion molecule‐1; l‐NAME, N‐nitro‐l‐arginine methyl ester; PE, phenylephrine; QPCR, quantitative reverse‐transcription polymerase chain reaction; TNF‐α, tumor necrosis factor alpha; WT, wild type.
Figure 8.
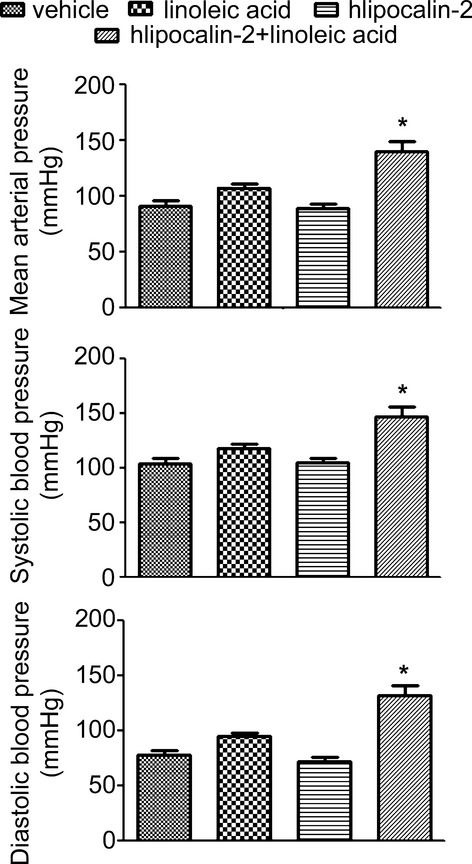
Lipocalin‐2 treatment in combination with linoleic acid (LA) promotes the elevation of blood pressure. Wild‐type (WT) mice (6 weeks old, standard chow) were implanted with the telemetry transmitter as described in Materials and Methods, and allowed 2 weeks of recovery from the surgery. Blood pressure was monitored after injection with vehicle, hlipocalin‐2, linoleic acid (LA), or hlipocalin‐2 in combination with LA. The results show that, at 5 hours after injection, systolic, diastolic, and mean arterial pressures were significantly increased in mice treated with hlipocalin‐2 in combination with LA. *P<0.05 versus other treatments; n=3 in all groups.
Treatment with difluoromethylornithine, an irreversible inhibitor of the polyamine synthase, ornithine decarboxylase, further increased the amount of deamidated lipocalin‐2 in the circulation as well as pervascular adipose tissues of WT mice receiving a HFD for 20 weeks (Figure S6). The 24‐hour average of MAP (180.3±19.1 mm Hg), SBP (185.9±17.9 mm Hg), and DBP (176.7±19.0 mm Hg) of these mice was elevated by 21%, 20%, and 23%, respectively, when compared to vehicle‐treated animals.
Taken in conjunction, the evidence suggests that increased uptake of NEFAs (in particular, LA), such as during dietary obesity, promotes accumulation of deamidated lipocalin‐2 in arteries, which subsequently causes endothelial dysfunction, vascular inflammation, and hypertension.
Discussion
Endothelial dysfunction is an early manifestation of obesity and contributes to progression of overt cardiovascular and metabolic morbidity.10,30 In obese subjects, elevated circulating NEFA levels (resulting largely from lipase‐mediated hydrolysis of lipoproteins and increased drainage from hypertrophic adipocytes) and abnormal production of adipokines cause impairment of endothelial vasomotor function, in particular, blunting endothelium‐dependent vasodilatation.17,31–32 In addition, dietary intake of saturated, monounsaturated, and polyunsaturated fatty acids has a significant effect on distribution of circulating lipids and vascular reactivity.33–35 Pharmacological interventions that decrease circulating lipid levels directly improve endothelial function and reduce vascular inflammation.36 However, the molecular mechanisms underlying impairment of vascular function by NEFAs remain largely unclear.
Adipose tissue, especially perivascular adipose tissue, is a major source of adipokines that exert paracrine, as well as endocrine, effects on vasomotor activity.37 Among them, “good” adipokines, such as adiponectin, promote vasodilatation and endothelial function, whereas “bad” adipokines aggravate obesity‐induced endothelial dysfunction and vascular inflammation.3,38 Lipocalin‐2 is such a proinflammatory adipokine upregulated in obese human subjects and animals.5 In obese mice, lipocalin‐2 treatment induces endothelial dysfunction by promoting oxidative stress and eNOS uncoupling.7 The present study reveals that lipocalin‐2 is polyaminated and that the proinflammatory and endothelial damaging effects of this adipokine are mediated by its deamidated form. Adipose tissue represents a major site for lipocalin‐2 deamidation, a process that can be facilitated by treatment with LA or HFD. Thus, it is reasonable to conclude that elevated NEFAs in dietary obesity enhance production and function of the proinflammatory form and deamidated lipocalin‐2, which negatively modulates endothelial function and vascular tone.
The composition of NEFAs, rather than their total amounts, is relevant for endothelial function. Acute treatment with long‐chain, but not medium‐chain, NEFAs attenuates endothelium‐dependent vasodilatation.16 Omega‐6 fatty acids, especially LA, cause endothelial cell dysfunction and potentiate TNF‐α‐mediated endothelial injury.39 LA is enriched in LDL cholesterol esters that promote endothelial activation by enhancing expression of adhesion molecules and decreasing eNOS activity.40–41 High intake of LA‐rich oils or fats leads to cellular oxidative stress and inflammatory responses.42 The present results from isothermal titration calorimetry analysis demonstrate that lipocalin‐2 binds to LA with a higher affinity than those of the saturated PA and monounsaturated OA. Compared to other members of the lipocalin family, lipocalin‐2 displays an unusually large ligand‐binding cavity lined with polar and positively charged amino acid residues.43 Consistent with the present findings, crystal structure analysis reveals that an FA complex is present in the ligand‐binding pocket of dimeric hlipocalin‐2, but not in that of lipocalin‐2 monomer.43 By binding to the pocket, LA may alter the protein structure of lipocalin‐2 and its interactions with other binding partners, such as MMP9, which facilitates deamidation of the adipokine. LA and polyamines may interact with lipocalin‐2 by binding to distinct cavities.44 Deamidated lipocalin‐2 has a strong tendency to form homodimers as well as HMW multimers, but shows decreased capacity to form heterodimers with MMP9. On the other hand, polyaminated lipocalin‐2 binds to MMP9 and exhibits a much shorter plasma half‐life, when compared to deamidated lipocalin‐2, indicating that formation of heterodimer with MMP9 facilitates clearance of the adipokine from the circulation. In fact, the circulating level and aortic accumulation of deamidated lipocalin‐2 is increased in mice treated with MMP9i. These results suggest that by modulating the polyamine modification of, and protein interactions with, lipocalin‐2, LA enhances the stability of this adipokine in the circulation and facilitates its accumulation in the arterial wall.
Polyamines are required for normal cellular function through interactions with cellular macromolecules.23 They are localized within secretory granules of protein‐ and peptide‐secreting cells (such as neutrophils) and are critically involved in the correct biogenesis, homeostasis, and intracellular trafficking of these vesicles.45–46 Lipocalin‐2 is produced from neutrophils, which contain a variety of granules that are exocytosed at different times to meet the demands, such as adhesion to endothelium (secretory vesicles), migration through basement membranes (gelatinase granules), and phagocytosis, killing, and digestion of microorganisms (specific granules and azurophil granules).47 Polyamination of this molecule may determine its selective packaging in specific granules and the timing of release during exocytosis of neutrophils. Spermidine and spermine also show high‐affinity binding to phospholipids and nucleic acids. Thus, in addition to protein interaction and stability, polyamines may also modulate other biological functions of lipocalin‐2, such as its cellular uptake,48–49 and its phospholipid remodeling activity.8 As a carrier, lipocalin‐2 may affect polyamine homeostasis within the vasculature. Polyamination of molecules in endothelial cells, such as RhoA, plays an important role in endothelial integrity and function.50 Because polyamines and NO are originated from the common precursor, arginine, changes of polyamine metabolites may influence the eNOS‐NO metabolic pathway. In fact, accumulation of deamidated lipocalin‐2 induces eNOS uncoupling, leading to endothelial dysfunction and vascular inflammation. On the other hand, administration of the same amount of WT hlipocalin‐2 alone does not affect vascular reactivity, largely because of the lack of deamidation triggers, including NEFAs and obesity.
Deamidated lipocalin‐2 accumulates in aortae of HFD‐fed WT mice, which exhibit deteriorated vasodilator responses, endothelial dysfunction, and elevated blood pressure.7 Lipocalin‐2 is a major gene target for mineralocorticoid receptor, agonists of which are known mediators of vascular insulin resistance and hypertension associated with obesity.51 In humans, a causal relationship between lipocalin‐2 and development of hypertension has been suggested by the association between single‐nucleotide polymorphisms in the gene encoding lipocalin‐2 (LCN2).9 Plasma concentration of lipocalin‐2 is positively correlated with indices of obesity, insulin resistance, and elevated blood pressure.5 Lifestyle factors are well known to contribute to obesity‐related hypertension. The present study demonstrates that deamidated lipocalin‐2 accumulates in arteries and causes vascular derangement, ultimately facilitating the occurrence of arterial hypertension. The findings highlight an important lipid‐adipokine interaction mechanism of excess LA intake‐mediated lipocalin‐2 activation, a pathway that links overnutrition and obesity to hypertensive conditions in human patients.
Sources of Funding
This work was supported by grants from the Hong Kong Research Grant Council (General Research Fund [HKU780410M, HKU781311M, and HKU763312M]; Collaborative Research Fund [HKU4/CRF/10]); Les Laboratoires Servier (RS100503), and the State Key Laboratory of Pharmaceutical Biotechnology.
Disclosures
Félétou, Vilaine, and Villeneuve are employees of a research institution belonging to a drug company (Servier). The other authors have nothing to disclose.
References
- 1.Kjeldsen L, Bainton DF, Sengelov H, Borregaard N. Identification of neutrophil gelatinase‐associated lipocalin as a novel matrix protein of specific granules in human neutrophils. Blood. 1994; 83:799-807 [PubMed] [Google Scholar]
- 2.Flower DR, North AC, Sansom CE. The lipocalin protein family: structural and sequence overview. Biochim Biophys Acta. 2000; 1482:9-24 [DOI] [PubMed] [Google Scholar]
- 3.Wang Y. Small lipid‐binding proteins in regulating endothelial and vascular functions: focusing on adipocyte fatty acid binding protein and lipocalin‐2. Br J Pharmacol. 2012; 165:603-621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Auguet T, Quintero Y, Terra X, Martinez S, Lucas A, Pellitero S, Aguilar C, Hernandez M, del Castillo D, Richart C. Upregulation of lipocalin 2 in adipose tissues of severely obese women: positive relationship with proinflammatory cytokines. Obesity (Silver Spring). 2011; 19:2295-2300 [DOI] [PubMed] [Google Scholar]
- 5.Wang Y, Lam KS, Kraegen EW, Sweeney G, Zhang J, Tso AW, Chow WS, Wat NM, Xu JY, Hoo RL, Xu A. Lipocalin‐2 is an inflammatory marker closely associated with obesity, insulin resistance, and hyperglycemia in humans. Clin Chem. 2007; 53:34-41 [DOI] [PubMed] [Google Scholar]
- 6.Law IK, Xu A, Lam KS, Berger T, Mak TW, Vanhoutte PM, Liu JT, Sweeney G, Zhou M, Yang B, Wang Y. Lipocalin‐2 deficiency attenuates insulin resistance associated with aging and obesity. Diabetes. 2010; 59:872-882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu JT, Song E, Xu A, Berger T, Mak TW, Tse HF, Law IK, Huang B, Liang Y, Vanhoutte PM, Wang Y. Lipocalin‐2 deficiency prevents endothelial dysfunction associated with dietary obesity: role of cytochrome P450 2C inhibition. Br J Pharmacol. 2012; 165:520-531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang B, Fan P, Xu A, Lam KS, Berger T, Mak TW, Tse HF, Yue JW, Song E, Vanhoutte PM, Sweeney G, Wang Y. Improved functional recovery to I/R injury in hearts from lipocalin‐2 deficiency mice: restoration of mitochondrial function and phospholipids remodeling. Am J Transl Res. 2012; 4:60-71 [PMC free article] [PubMed] [Google Scholar]
- 9.Ong KL, Tso AW, Cherny SS, Sham PC, Lam TH, Lam KS, Cheung BM. Role of genetic variants in the gene encoding lipocalin‐2 in the development of elevated blood pressure. Clin Exp Hypertens. 2011; 33:484-491 [DOI] [PubMed] [Google Scholar]
- 10.de Jongh RT, Serne EH, IJzerman RG, de Vries G, Stehouwer CD. Impaired microvascular function in obesity: implications for obesity‐associated microangiopathy, hypertension, and insulin resistance. Circulation. 2004; 109:2529-2535 [DOI] [PubMed] [Google Scholar]
- 11.Williams IL, Chowienczyk PJ, Wheatcroft SB, Patel AG, Sherwood RA, Momin A, Shah AM, Kearney MT. Endothelial function and weight loss in obese humans. Obes Surg. 2005; 15:1055-1060 [DOI] [PubMed] [Google Scholar]
- 12.Williams IL, Wheatcroft SB, Shah AM, Kearney MT. Obesity, atherosclerosis and the vascular endothelium: mechanisms of reduced nitric oxide bioavailability in obese humans. Int J Obes Relat Metab Disord. 2002; 26:754-764 [DOI] [PubMed] [Google Scholar]
- 13.Bigornia SJ, Mott MM, Hess DT, Apovian CM, McDonnell ME, Duess MA, Kluge MA, Fiscale AJ, Vita JA, Gokce N. Long‐term successful weight loss improves vascular endothelial function in severely obese individuals. Obesity (Silver Spring). 2010; 18:754-759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ziccardi P, Nappo F, Giugliano G, Esposito K, Marfella R, Cioffi M, D'Andrea F, Molinari AM, Giugliano D. Reduction of inflammatory cytokine concentrations and improvement of endothelial functions in obese women after weight loss over one year. Circulation. 2002; 105:804-809 [DOI] [PubMed] [Google Scholar]
- 15.Imrie H, Abbas A, Kearney M. Insulin resistance, lipotoxicity and endothelial dysfunction. Biochim Biophys Acta. 2009; 1801:320-326 [DOI] [PubMed] [Google Scholar]
- 16.Steer P, Basu S, Lithell H, Vessby B, Berne C, Lind L. Acute elevations of medium‐ and long‐chain fatty acid have different impacts on endothelium‐dependent vasodilation in humans. Lipids. 2003; 38:15-19 [DOI] [PubMed] [Google Scholar]
- 17.Steinberg HO, Tarshoby M, Monestel R, Hook G, Cronin J, Johnson A, Bayazeed B, Baron AD. Elevated circulating free fatty acid levels impair endothelium‐dependent vasodilation. J Clin Invest. 1997; 100:1230-1239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sarafidis PA, Bakris GL. Non‐esterified fatty acids and blood pressure elevation: a mechanism for hypertension in subjects with obesity/insulin resistance? J Hum Hypertens. 2007; 21:12-19 [DOI] [PubMed] [Google Scholar]
- 19.Umpierrez GE, Smiley D, Robalino G, Peng L, Kitabchi AE, Khan B, Le A, Quyyumi A, Brown V, Phillips LS. Intravenous intralipid‐induced blood pressure elevation and endothelial dysfunction in obese African‐Americans with type 2 diabetes. J Clin Endocrinol Metab. 2009; 94:609-614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berger T, Togawa A, Duncan GS, Elia AJ, You‐Ten A, Wakeham A, Fong HE, Cheung CC, Mak TW. Lipocalin 2‐deficient mice exhibit increased sensitivity to Escherichia coli infection but not to ischemia‐reperfusion injury. Proc Natl Acad Sci USA. 2006; 103:1834-1839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang Y, Lam KS, Chan L, Chan KW, Lam JB, Lam MC, Hoo RC, Mak WW, Cooper GJ, Xu A. Post‐translational modifications of the four conserved lysine residues within the collagenous domain of adiponectin are required for the formation of its high molecular weight oligomeric complex. J Biol Chem. 2006; 281:16391-16400 [DOI] [PubMed] [Google Scholar]
- 22.Xu A, Wang Y, Xu JY, Stejskal D, Tam S, Zhang J, Wat NM, Wong WK, Lam KS. Adipocyte fatty acid‐binding protein is a plasma biomarker closely associated with obesity and metabolic syndrome. Clin Chem. 2006; 52:405-413 [DOI] [PubMed] [Google Scholar]
- 23.Pegg AE. Recent advances in the biochemistry of polyamines in eukaryotes. Biochem J. 1986; 234:249-262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peairs AD, Rankin JW, Lee YW. Effects of acute ingestion of different fats on oxidative stress and inflammation in overweight and obese adults. Nutr J. 2011; 10:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Naderali EK, Williams G. Effects of short‐term feeding of a highly palatable diet on vascular reactivity in rats. Eur J Clin Invest. 2001; 31:1024-1028 [DOI] [PubMed] [Google Scholar]
- 26.Kjeldsen L, Johnsen AH, Sengelov H, Borregaard N. Isolation and primary structure of ngal, a novel protein associated with human neutrophil gelatinase. J Biol Chem. 1993; 268:10425-10432 [PubMed] [Google Scholar]
- 27.Kjeldsen L, Cowland JB, Borregaard N. Human neutrophil gelatinase‐associated lipocalin and homologous proteins in rat and mouse. Biochim Biophys Acta. 2000; 1482:272-283 [DOI] [PubMed] [Google Scholar]
- 28.Furchgott RF, Vanhoutte PM. Endothelium‐derived relaxing and contracting factors. Faseb J. 1989; 3:2007-2018 [PubMed] [Google Scholar]
- 29.Feletou M, Huang Y, Vanhoutte PM. Endothelium‐mediated control of vascular tone: Cox‐1 and Cox‐2 products. Br J Pharmacol. 2011; 164:894-912 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Galili O, Versari D, Sattler KJ, Olson ML, Mannheim D, McConnell JP, Chade AR, Lerman LO, Lerman A. Early experimental obesity is associated with coronary endothelial dysfunction and oxidative stress. Am J Physiol Heart Circ Physiol. 2007; 292:H904-H911 [DOI] [PubMed] [Google Scholar]
- 31.Eringa EC, Bakker W, Smulders YM, Serne EH, Yudkin JS, Stehouwer CD. Regulation of vascular function and insulin sensitivity by adipose tissue: focus on perivascular adipose tissue. Microcirculation. 2007; 14:389-402 [DOI] [PubMed] [Google Scholar]
- 32.de Jongh RT, Serne EH, Ijzerman RG, de Vries G, Stehouwer CD. Free fatty acid levels modulate microvascular function: relevance for obesity‐associated insulin resistance, hypertension, and microangiopathy. Diabetes. 2004; 53:2873-2882 [DOI] [PubMed] [Google Scholar]
- 33.Christon R, Marette A, Badeau M, Bourgoin F, Melancon S, Bachelard H. Fatty acid‐induced changes in vascular reactivity in healthy adult rats. Metabolism. 2005; 54:1600-1609 [DOI] [PubMed] [Google Scholar]
- 34.Shimabukuro M, Chinen I, Higa N, Takasu N, Yamakawa K, Ueda S. Effects of dietary composition on postprandial endothelial function and adiponectin concentrations in healthy humans: a crossover controlled study. Am J Clin Nutr. 2007; 86:923-928 [DOI] [PubMed] [Google Scholar]
- 35.Ringseis R, Eder K. Fatty acids and signalling in endothelial cells. Prostaglandins Leukot Essent Fatty Acids. 2010; 82:189-198 [DOI] [PubMed] [Google Scholar]
- 36.Stapleton PA, Goodwill AG, James ME, Brock RW, Frisbee JC. Hypercholesterolemia and microvascular dysfunction: interventional strategies. J Inflamm (Lond). 2010; 7:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Szasz T, Bomfim GF, Webb RC. The influence of perivascular adipose tissue on vascular homeostasis. Vasc Health Risk Manag. 2013; 9:105-116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu A, Vanhoutte PM. Adiponectin and adipocyte fatty acid binding protein in the pathogenesis of cardiovascular disease. Am J Physiol Heart Circ Physiol. 2012; 302:H1231-H1240 [DOI] [PubMed] [Google Scholar]
- 39.Wang L, Lim EJ, Toborek M, Hennig B. The role of fatty acids and caveolin‐1 in tumor necrosis factor alpha‐induced endothelial cell activation. Metabolism. 2008; 57:1328-1339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Caterina R, Liao JK, Libby P. Fatty acid modulation of endothelial activation. Am J Clin Nutr. 2000; 71:213S-223S [DOI] [PubMed] [Google Scholar]
- 41.Hennig B, Meerarani P, Ramadass P, Watkins BA, Toborek M. Fatty acid‐mediated activation of vascular endothelial cells. Metabolism. 2000; 49:1006-1013 [DOI] [PubMed] [Google Scholar]
- 42.Grimble RF. Dietary lipids and the inflammatory response. Proc Nutr Soc. 1998; 57:535-542 [DOI] [PubMed] [Google Scholar]
- 43.Goetz DH, Willie ST, Armen RS, Bratt T, Borregaard N, Strong RK. Ligand preference inferred from the structure of neutrophil gelatinase associated lipocalin. Biochemistry. 2000; 39:1935-1941 [DOI] [PubMed] [Google Scholar]
- 44.Goetz DH, Holmes MA, Borregaard N, Bluhm ME, Raymond KN, Strong RK. The neutrophil lipocalin ngal is a bacteriostatic agent that interferes with siderophore‐mediated iron acquisition. Mol Cell. 2002; 10:1033-1043 [DOI] [PubMed] [Google Scholar]
- 45.Hougaard DM, Larsson LI. Polyamines, molecules necessary for cell division, colocalize with peptide growth factors. Eur J Cell Biol. 1989; 48:14-18 [PubMed] [Google Scholar]
- 46.Kanerva K, Makitie LT, Back N, Andersson LC. Ornithine decarboxylase antizyme inhibitor 2 regulates intracellular vesicle trafficking. Exp Cell Res. 2010; 316:1896-1906 [DOI] [PubMed] [Google Scholar]
- 47.Borregaard N. Development of neutrophil granule diversity. Ann N Y Acad Sci. 1997; 832:62-68 [DOI] [PubMed] [Google Scholar]
- 48.Hvidberg V, Jacobsen C, Strong RK, Cowland JB, Moestrup SK, Borregaard N. The endocytic receptor megalin binds the iron transporting neutrophil‐gelatinase‐associated lipocalin with high affinity and mediates its cellular uptake. FEBS Lett. 2005; 579:773-777 [DOI] [PubMed] [Google Scholar]
- 49.Devireddy LR, Gazin C, Zhu X, Green MR. A cell‐surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell. 2005; 123:1293-1305 [DOI] [PubMed] [Google Scholar]
- 50.Lee DL, Webb RC, Jin L. Hypertension and RhoA/Rho‐kinase signaling in the vasculature: highlights from the recent literature. Hypertension. 2004; 44:796-799 [DOI] [PubMed] [Google Scholar]
- 51.Latouche C, El Moghrabi S, Messaoudi S, Cat AN, Hernandez‐Diaz I, Alvarez de la Rosa D, Perret C, Lopez Andres N, Rossignol P, Zannad F, Farman N, Jaisser F. Neutrophil gelatinase‐associated lipocalin is a novel mineralocorticoid target in the cardiovascular system. Hypertension. 2012; 59:966-972 [DOI] [PubMed] [Google Scholar]