

Abstract
Background
Atrial fibrillation (AF), often associated with structural, fibrotic change in cardiac tissues involving regulatory signaling mediators, becomes increasingly common with age. In the present study, we explored the role of mitogen‐activated protein kinase kinase 4 (Mkk4), a critical component of the stress‐activated mitogen‐activated protein kinase family, in age‐associated AF.
Methods and Results
We developed a novel mouse model with a selective inactivation of atrial cardiomyocyte Mkk4 (Mkk4ACKO). We characterized and compared electrophysiological, histological, and molecular features of young (3‐ to 4‐month), adult (6‐month), and old (1‐year) Mkk4ACKO mice with age‐matched control littermates (Mkk4F/F). Aging Mkk4ACKO mice were more susceptible to atrial tachyarrhythmias than the corresponding Mkk4F/F mice, showing characteristic slow and dispersed atrial conduction, for which modeling studies demonstrated potential arrhythmic effects. These differences paralleled increased interstitial fibrosis, upregulated transforming growth factor beta 1 (TGF‐β1) signaling and dysregulation of matrix metalloproteinases in Mkk4ACKO, compared to Mkk4F/F, atria. Mkk4 inactivation increased the sensitivity of cultured cardiomyocytes to angiotensin II–induced activation of TGF‐β1 signaling. This, in turn, enhanced expression of profibrotic molecules in cultured cardiac fibroblasts, suggesting cross‐talk between these two cell types in profibrotic signaling. Finally, human atrial tissues in AF showed a Mkk4 downregulation associated with increased production of profibrotic molecules, compared to findings in tissue from control subjects in sinus rhythm.
Conclusions
These findings together demonstrate, for the first time, that Mkk4 is a negative regulator of the TGF‐β1 signaling associated with atrial remodeling and arrhythmogenesis with age, establishing Mkk4 as a new potential therapeutic target for treating AF.
Keywords: aging, atrial fibrillation, cardiac remodeling
Introduction
Atrial fibrillation (AF) is the most common chronic cardiac arrhythmia. It contributes significantly to population morbidity and mortality with a prevalence increasing with age, affecting ≈5% of the population over 65 years. It is widely recognized that development of atrial fibrosis leading to remodeling contributes to persistence of AF in human subjects. The genes and signaling pathways modulating this development are thus potential novel therapeutic targets, but identification of their relevant key signaling mediators would be critical to this end.
Mitogen‐activated protein kinases (MAPKs) are a group of serine/threonine kinases that play a central role in cellular responses to diverse stimuli, including environmental stress, proinflammatory cytokines, and developmental cues. Their cardiac roles have become a focus of recent research through their involvement in the pathogenesis of cardiac hypertrophy and cardiac failure.1 They have also been implicated in the pathogenesis of AF, through their mediation of angiotensin II (Ang II) action.2–3 Ang II type 1 receptor activation is known to induce a cascade of phosphorylations that activate MAPKs, which, in turn, stimulates fibroblast proliferation, cellular hypertrophy, and apoptosis.4–6 Tachypacing in canine hearts increases atrial Ang II production and expression of extracellular signal‐regulated kinase (ERK), c‐Jun N‐terminal kinase (JNK), and p38‐kinase. This was accompanied by substantial changes in their phosphorylated forms. Such findings implicate MAPK activation in arrhythmogenic atrial structural remodeling. However, Ventura et al.7 recently demonstrated that JNK regulated transforming growth factor beta 1 (TGF‐β1)‐mediated biological responses by suppressing TGF‐β1 gene expression, indicating a negative regulatory role in TGF‐β1 signaling. Moreover, attenuation of cardiac p38 activity produced a progressive growth response and myopathy in the heart that correlated with the degree of enzymatic inhibition. Furthermore, dominant‐negative p38α, mitogen‐activated protein kinase kinase (Mkk)3, and Mkk6 transgenic mice all showed enhanced cardiac hypertrophy after aortic banding, Ang II infusion, isoproterenol infusion, or phenylephrine infusion for 14 days.8 These apparent discrepancies reflect the complexity of the MAPK cascades in the heart, highlighting the need for further study of their role and the related signal transduction processes in cardiac tissues.
MKK4 is a critical member of the MAPK family. Its function is associated with JNK signaling through its action as the upstream kinase of JNK. MKK4 and its closely related family member, MKK7, preferentially phosphorylate JNK on its threonine 183 and tyrosine 185 residues, respectively.9–10 In contrast to MKK7, MKK4 was also reported additionally to activate p38 in some cell types.11–12 Targeted deletion of either the Mkk4 or Mkk7 gene leads to embryonic lethality, providing genetic evidence that Mkk4 and Mkk7 have nonredundant roles in vivo.13 These data indicate a central role for MKK4 in the JNK signal transduction pathway. Furthermore, recent studies, including ours, have provided strong evidence demonstrating the functional importance of MKK4 in the heart, where it is required for protecting the heart from maladaptive pathological hypertrophy through activation of the JNK pathway.14–15 We further demonstrated that MKK4 prevented stress‐associated ventricular arrhythmias through regulation of connexin 43 (Cx43) expression.15
Despite the biological importance of MKK4 being demonstrated in the ventricles, its role in the atrium has not been previously explored. The present study developed a conditional knockout mouse model with an atrial cardiomyocyte‐selective deletion of Mkk4 using the natriuretic peptide precursor A (Nppa) promoter‐driven Cre transgene16 by using the Cre‐LoxP system. It then investigated the effects of Mkk4 deletion on atrial electrophysiological and structural properties with age.
Methods
Animal Models
Generation of Mkk4ACKO mice
Mkk4 was specifically deleted from the atria of the heart using the Cre‐LoxP system. Mkk4 flox/flox (Mkk4F/F) mice14 were crossed with Nppa‐Cre4 mice, which express CRE only in atrial cardiomyocytes,16 to generate atrial‐specific Mkk4 knockout mice (Mkk4ACKO). The Nppa‐Cre4 line (kindly provided by Dr. V.M. Christoffels) is a well‐established model that provides efficient atrial‐specific Cre recombinase activity, without causing any abnormality in cardiac morphology or function. Young adult mice in this study were 3 months of age, whereas old mice were 12 months of age. All mice used in this study were maintained in a pathogen‐free facility at the University of Manchester (Manchester, UK). Animal studies were performed in accord with the UK Home Office and institutional guidelines. The procedures followed were in accord with institutional guidelines.
Neonatal rat cardiomyocytes (NRCMs) were isolated from 1‐ to 2‐day‐old Sprague‐Dawley rats using the standard enzymatic method described previously.17 Using the same protocol as for NRCMs, neonatal rat cardiac fibroblasts (NRCFs) were prepared from hearts of 1‐ to 2‐day‐old Sprague‐Dawley rats. NRCMs were infected with adenovirus encoding green fluorescent protein (Ad‐GFP), as a control or dominant negative MKK4 adenovirus (Ad‐dnMKK4; Seven Hills Bioreagents, Cincinnati, OH) at 25 multiplicities of infection in serum‐free medium, 48 hours after plating of the cells. Twenty‐four hours after infection, the virus was removed. After adenovirus infection and removal, NRCMs were treated with Ang II (500 nmol/L) for 24 hours and RNA was extracted using the Qiagen RNeasy Minikit (Qiagen, Tübingen, Germany), following the manufacturer's guidelines. To assess the interplay between MKK4‐inactivated cardiomyocytes and fibroblasts, NRCM‐conditioned media were then transferred to plated cardiac fibroblasts that were then incubated for 24 hours before RNA was extracted. Real‐time polymerase chain reaction (PCR) was carried out on target genes from NRCMs and fibroblasts, as described below.
Human Tissue Samples
Right atrial appendages were dissected from 15 control patients and 15 chronic AF patients (patients' clinical information is shown in Table 1). The study was approved by an institutional review committee and subjects gave informed consent. Thus, experimental protocols were approved by the ethics committee of the Dresden University of Technology (EK790799; Dresden, Germany). Each patient gave written informed consent. After excision, atrial appendages were flash‐frozen in liquid nitrogen for subsequent biochemical studies.
Table 1.
Clinical Features of Patients Studied
| SR | AF | |
|---|---|---|
| Patient number, n | 15 | 15 |
| Gender, male/female | 12/3 | 11/4 |
| Age, y | 69.5±1.9 | 69.3±2.0 |
| Height, cm | 169±1 | 172±2 |
| Weight, kg | 80.9±2.6 | 88.1±4.1 |
| Body mass index, kg/m2 | 28.3±0.8 | 29.8±1.2 |
| CAD, n | 14 | 5* |
| MVD/AVD, n | 10 | 14 |
| CAD+MVD/AVD, n | 10 | 4 |
| Pulmonary hypertension, n | 4 | 9 |
| Hypertension, n | 14 | 15 |
| Diabetes, n | 8 | 8 |
| Hyperlipidemia, n | 11 | 6 |
| LVEF, % | 52.0±2.8 | 47.3±3.9 |
| LVEDD, mm | 49.8±2.2 | 50.2±3.0 |
| LVSTD, mm | 13.5±0.7 | 13.3±0.5 |
| LVWTD, mm | 12.3±0.7 | 12.3±0.4 |
| LADD, mm | 42.2±1.6 | 51.6±1.2* |
| Digitalis, n | 0 | 3 |
| ACE inhibitors, n | 10 | 9 |
| AT1 blockers, n | 2 | 3 |
| β‐Blockers, n | 14 | 13 |
| Ca2+‐antagonists, n | 3 | 3 |
| Diuretics, n | 9 | 12 |
| Nitrates, n | 0 | 1 |
| Lipid‐lowering drugs, n | 13 | 7* |
ACE indicates angiotensin‐converting enzyme; AF, atrial fibrillation; AT, angiotensin receptor; AVD, aortic valve disease requiring valve replacement; CAD, coronary artery disease; LADD, left atrial diastolic diameter; LVEDD, left ventricular end diastolic diameter; LVEF, left ventricular ejection fraction; LVSTD, left ventricular septum thickness at diastole; LVWTD, left ventricular wall thickness at diastole; MVD, mitral valve disease requiring valve replacement; SR, sinus rhythm.
P<0.05 values from unpaired Student t test for continuous variables and from chi‐square test for categorical variables.
Electrocardiography
Cardiac rhythms were assessed by in vivo ECG analysis on isoflurane‐anesthetized mice (2.5%) that measured RR intervals, P wave durations, PR intervals, and QRS, JT, and QT durations.
Programmed Electrical Stimulation
Propensity to atrial arrhythmias in Langendorff‐perfused hearts from 3‐ to 4‐ and 12‐month‐old mice was assessed by programmed electrical stimulation (PES). Pacing trains of eight stimuli (S1) delivered at a basic cycle length of 100 ms were followed by a single (S2) premature extrastimuli introduced at S1S2 intervals progressively shortened with successive stimulus cycles repeated until arrhythmia was induced or the atrial refractory period was reached. A burst‐pacing protocol applied atrial pacing in trains of 20 S1 stimuli at progressively shorter intervals from an initial cycle length of 100 ms until an arrhythmia was induced or CL of 30 ms was reached. Arrhythmia was defined as three or more consecutive premature atrial waveforms. Tachycardia showing regular waveforms was defined as AT, whereas irregular fibrillating waveforms was considered to represent AF.
Epicardial Activation Mapping
Left and right atrial epicardial activation mapping in isolated Langendorff‐perfused hearts used a custom‐made multielectrode array containing 32 electrodes in a 4×8 grid, with interelectrode distances of 0.20 mm along the longitudinal and 0.35 mm along the transverse axis (0.3 mm), paced at a basic cycle length of 100 ms. Activation times were determined as the point of maximal negative slope and displayed in a grid representing the layout of the original recording array. All activation times were related to the timing of the first detected waveform. Isochrones were drawn manually around areas activated in increments of 1 ms.
Mathematical Model of Mouse Atria
A computational model for the action potential (AP) of a single mouse atrial cardiomyocyte was developed in this study. This modified the previously published mouse ventricular model18 incorporating experimental data on ionic channel properties of murine atrial cardiomyocytes. Equations for L‐type Ca2+ currents (ICaL) used experimental data of Lomax et al.19 Equations for the time‐ and voltage‐dependent transient outward (IKtof), ultrarapidly delayed rectifier (IKur) and noninactivating steady‐state K+ currents (IKss) used experimental data from Lomax et al.,19 Bou‐Abboud et al.,20 and Nakamura et al.21 Equations for the inward rectifier K+ current (IK1) used experimental data from Panama et al.22 Equations for remaining ion channel currents were the same as those used in the mouse ventricular model18 because of the lack of atrial‐specific experimental data.
The model successfully reproduced the murine atrial AP with its detailed characteristics of resting potential, AP duration (APD) and amplitude, and upstroke velocity matching experimental data and their rate dependence.19–22 The single‐cell model was then incorporated into a two‐dimensional (2D) tissue model with dimensions of 15×20 mm, which was discretized into a lattice of 150×200 nodes with a spatial resolution of 0.1 mm. The membrane voltage (Vm) for a cardiomyocyte in this tissue matrix was given by the following equation:
where D(x, y) simulates the gap junctional coupling, Iion is the membrane current, and Istim is the stimulus current.
In order to investigate the effects of cardiomyocyte‐fibroblast coupling on the conduction velocity (CV) of atrial excitation waves and assess its possible roles in causing of re‐entry, a fibroblast model23 was incorporated into the tissue model. This defined a coupling between cardiomyocyte‐fibroblast by writing the following equation:
C is the membrane capacitance of a given cardiomyocyte (Cm) or a fibroblast (Cf), Iion is the corresponding membrane current (Im or If), n is the number of coupled neighbors (either cardiomyocytes or fibroblasts), and ![]() is the gap junctional conductance between a cell (either a cardiomyocyte or a fibroblast) and its neighbor. Cm and Cf were set to 153.4 pF and 6.3 pF, respectively. Ggap was set to 3 nS.
is the gap junctional conductance between a cell (either a cardiomyocyte or a fibroblast) and its neighbor. Cm and Cf were set to 153.4 pF and 6.3 pF, respectively. Ggap was set to 3 nS.
In the 2D tissue model, the number of fibroblasts (ranging from 1 to 5) was randomly distributed among cardiomyocytes, with the overall fibroblast density scaled by the ratios of the measured population of fibrosis from Mkk4ACKO and Mkk4F/F and their related aging groups. They were either attached to one cardiomyocyte (one‐sided coupling) or interconnected neighboring cells (end‐to‐end coupling; Figure 8A). The value for D(x, y) was also chosen to simulate the measured conduction velocity from Mkk4ACKO and Mkk4F/F and their related aging groups in the control conditions.
Figure 8.
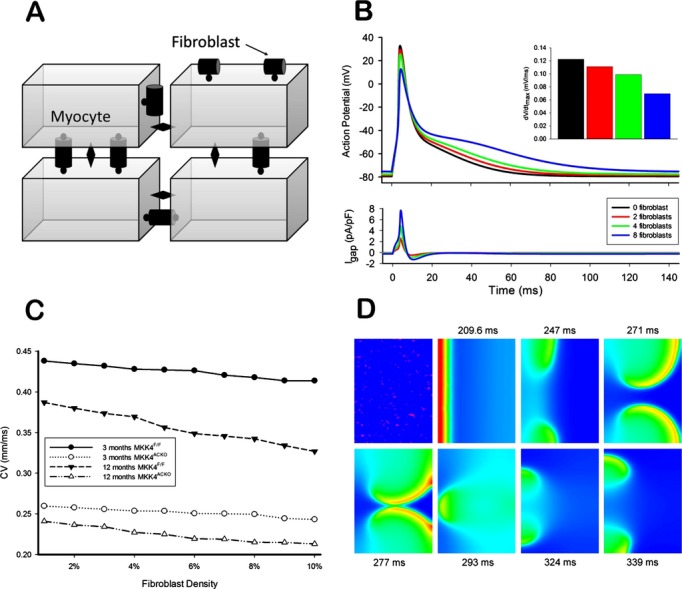
Computer simulation of the effects of fibroblast‐cardiomyocyte coupling. A, Schematic plot of a 2D tissue model with random fibroblast insertion. The diamond shape represents the gap junction between myocytes; the round shape represents the gap junction between fibroblasts and cardiomyocytes. B, Effects of fibroblast insertion on the membrane potential for the coupled myocyte. The inset shows the schematic plot of interconnected coupling. I, no fibroblast coupled; II, three fibroblasts coupling; III, four fibroblasts coupling If fibroblasts are present, gap junctions between myocytes are replaced by a fibroblast‐myocyte gap junction. C, Computer simulation of the effects of fibroblast‐cardiomyocyte coupling on conduction velocity (CV). D, Voltage snapshots showing re‐entry induced by the premature beat. The first plot shows the pattern of random fibroblast distribution, where the red spot represents fibroblast‐cardiomyocyte coupling.
Immunohistochemical Analyses
Atrial and ventricular tissue was frozen in optimal cutting temperature and 10‐μm sections were collected using a cryostat. Sections were stained for fibrosis using the picrosirius red stain and bright field images were taken at 10× magnification for quantification. For illustration purposes, 40× magnification images of representative areas were taken. Fibrotic areas were quantified using ImageJ software (National Institutes of Health, Bethesda, MD).
Quantitative Real‐Time PCR
Total RNA was extracted from frozen tissue using an RNeasy Mini Kit (Qiagen), and cDNA was synthesized using the Superscript™ III First Strand Synthesis System (Invitrogen, Carlsbad, CA) using oligo dT primers. Real‐time PCR was performed using SYBR green (Applied Biosystems, Foster City, CA), and primers were obtained from Qiagen. PCR products were detected in the ABI‐PRISM 7700 sequence detection system (Applied Biosystems), and the results were analyzed using the 2−ΔΔCT method. The mRNA levels of each gene were normalized to hypoxanthine phosphoribosyltransferase (HPRT) levels.
Immunoblot Analysis
Protein extracts (50 μg) were subjected to immunoblot analysis with antibodies (Abs) against Mkk4 (BD Pharmingen, San Diego, CA), ERK1/2 (Cell Signaling Technology, Danvers, MA), JNK (Santa Cruz Biotechnology, Santa Cruz, CA), p38 (Santa Cruz Biotechnology), and tubulin (Sigma‐Aldrich, St. Louis, MO). Immune complexes were detected by enhanced chemiluminescence with anti‐mouse, ‐rabbit, or ‐goat immunoglobulin G coupled to horseradish peroxidise as the secondary Ab (Amersham‐Pharmacia Biotech Inc., Piscataway, NJ).
Luciferase Reporter Assay
After adenovirus infection and removal, NRCMs were treated with Ang II (500 nmol/L) for 24 hours. NRCM‐conditioned media were then transferred to plated TMLC cells and left for 24 hours. TMLC cells are a TGF‐β1 reporter cell line, with a PAI‐1 promoter luciferase construct stably transfected.24 TMLC cells were harvested and luciferase activity measured, using a luciferase assay kit (Promega, Madison, WI) and following the manufacturer's guidelines.
Data Analysis
Data are presented as means±SEM, unless indicated otherwise. Statistical analysis used SPSS Statistics 19 software (SPSS, Inc., Chicago, IL) with P values <0.05 considered statistically significant. Before parametric statistical analysis, the Shapiro‐Wilk test was used to assess the null hypothesis that data sets derived from a normally distributed population where there was a probability P>0.05 would not result in rejection of the hypothesis that the data came from a normally distributed population. Student t tests were then used in comparisons where there were two groups, with the paired‐sample t test used for testing differences between paired groups. Two‐way ANOVA was used in comparisons among multiple groups. These were then followed by post‐hoc, Bonferonni‐corrected t tests. Other, chi‐squared tests, as well as extraction of correlation regression coefficients, were performed when applicable.
Results
Characterization and Electrophysiological Analyses of Atrial Cardiomyocyte‐Specific Mkk4‐Deficient Mice
To study the functional importance of Mkk4 in the atria, we developed atrial cardiomyocyte‐specific Mkk4‐deficient mice by crossing mice carrying an Nppa promoter‐driven Cre transgene16 with mice with floxed Mkk4 alleles (Mkk4F/F). Figure 1A and 1B summarizes the strategy and procedures for developing the mouse model with atrial tissue‐specific Mkk4 deletion (Mkk4ACKO). Mkk4ACKO mice that were born showed the expected Mendelian ratios developing to term and appeared normal. Immunoblot analysis of tissue lysates from adult mice demonstrated an efficient reduction of Mkk4 expression in both left and right atrial (Figure 1C, top panel), but not ventricular, tissue of Mkk4ACKO mice, compared to corresponding Mkk4F/F controls (Figure 1C). Comparisons of control Mkk4F/F with Mkk4ACKO samples confirmed that Mkk4 deletion did not result in compensatory changes in expression of other MAPK components, including JNK, p38, or ERK1/2 (Figure 1D). Mkk4ACKO mice are therefore atrial specific and suitable for studying functional roles of Mkk4 in the atrium. To determine whether loss of Mkk4 affects activation of JNK or p38, atrial tissues from two groups were collected 30 minutes after Ang II treatment (8 mg/kg). Immunoblot analysis showed that JNK activity increased in controls 30 minutes after treatment, whereas mutant atria barely showed any detectable activation (Figure 1E). In contrast, p38 activation in both groups escalated to similar levels at 30 minutes (Figure 1E). Hence, loss of Mkk4 specifically blunted atrial activation of JNK, but not p38, activity.
Figure 1.
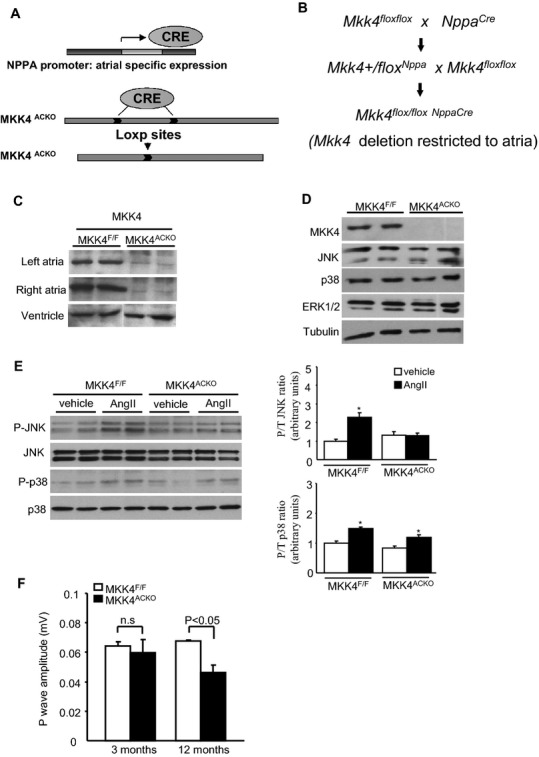
Generation of Mkk4ACKO mice and their electrophysiological characterization. A and B, Schematic diagram illustrating the use of the Cre‐LoxP system in generating atrial‐specific deletion of Mkk4. C, Immunoblot analysis of Mkk4 expression from atrial and ventricular extracts shows efficient deletion of Mkk4 from atrial tissue in Mkk4ACKO, but not control, Mkk4F/F, mice. D, Deletion of Mkk4 does not lead to compensatory changes in the MAPK proteins, JNK, p38, or ERK1/2. Tubulin illustrates equal loading. E, Ang II–induced activation of JNK or p38 was examined by immunoblot analysis, indicating that the deficiency of Mkk4 exclusively blocked JNK activation in atria. Immunoblot signals were quantified using ImageJ software (National Institutes of Health, Bethesda, MD) and the ratios of p‐JNK to total JNK expression, and p‐p38 to total p38 expression were calculated and presented as means±SEMs in the bar graphs (n=5 or 6 per group). Following Shapiro‐Wilk tests for normality, data were compared using the paired Student t test, with a value of P<0.05 vs. corresponding vehicle group considered to indicate statistical significance. F, Comparison of P wave amplitudes between young and old Mkk4F/F and Mkk4ACKO. ECG recordings were made over 5 minutes from each mouse. ECG traces from a 1‐minute record showing a stable baseline were obtained from within this recording period. Results were averaged over such ECG traces from 10 independent mice for each group. The mean and SEM from these was then obtained. Data are presented as means±SEMs (n=10 per group). Ang II indicates angiotensin‐II; CRE, CRE Recombinase; ERK, extracellular signal‐regulated kinase; JNK, c‐Jun N‐terminal kinase; MAPK, mitogen‐activated protein kinase; Mkk4ACKO, atrial cardiomyocyte specific mitogen‐activated protein kinase kinase 4 knockout mice; Mkk4F/F, Mkk4 flox/flox; NPPA, natriuretic peptide precursor A.
Effects of Mkk4 deficiency on atrial electrophysiological properties were then studied through comparisons between Mkk4F/F and Mkk4ACKO mice. Because incidence of atrial arrhythmia increases with age, ECG properties were examined at regular intervals between 3 and 12 months of age. Under baseline conditions, there were no significant differences in heart rate, P wave duration, PR, QRS, or QT intervals in surface ECGs between Mkk4ACKO and Mkk4F/F mice in all age groups (Table 2). However, P wave amplitude became significantly reduced in Mkk4ACKO, relative to that of age‐matched Mkk4F/F mice, between 5 and 6 to 12 months of age (Figure 1F), suggesting altered atrial electrical activity in Mkk4ACKO mice. Furthermore, by age 5 to 6 months, the occasional Mkk4ACKO (3 of 12) mouse, but none of the Mkk4F/F mice (0 of 11) showed spontaneous episodes of atrial tachycardia or polymorphic atrial ectopic beats (Figure 2A).
Table 2.
Role of Mkk4 in Atrial Function in Aging: In Vivo ECGs
| 3 months | 12 months | |||
|---|---|---|---|---|
| Mkk4F/F (n=6) | Mkk4ACKO (n=6) | Mkk4F/F (n=4) | Mkk4ACKO (n=6) | |
| RR interval, ms | 131.87±4.99 | 134.32±4.57 | 146.01±5.36 | 142.59±3.89 |
| Heart rate, bpm | 460.85±17.30 | 452.12±16.03 | 416.00±15.45 | 424.55±12.66 |
| PR interval, ms | 34.95±1.79 | 32.35±1.31 | 39.63±1.76 | 37.99±1.99* |
| P duration, ms | 15.81±1.33 | 14.88±2.30 | 12.63±0.57* | 12.13±1.07 |
| QRS interval, ms | 9.00±0.47 | 9.54±0.37 | 9.25±0.34 | 9.13±0.39 |
| QT interval, ms | 15.13±0.48 | 14.84±0.41 | 20.96±2.37 | 18.57±2.05 |
| QTc, ms | 41.32±1.36 | 40.18±1.16 | 54.95±6.21 | 49.16±5.00 |
| JT interval, ms | 5.93±0.36 | 5.59±0.26 | 11.71±6.21 | 9.57±1.78 |
| Q amplitude, mV | −0.14±0.04 | −0.13±0.03 | −0.03±0.01* | −0.05±0.02* |
| R amplitude, mV | 0.86±0.16 | 0.97±0.11 | 0.87±0.10 | 0.95±0.08 |
| S amplitude, mV | −0.71±0.08 | −0.59±0.10 | −0.34±0.10* | −0.19±0.07** |
| ST height, mV | 0.007±0.013 | −0.007±0.02 | 0.07±0.03* | 0.04±0.02* |
| T amplitude, mV | 0.20±0.05 | 0.20±0.04 | 0.16±0.03 | 0.16±0.03 |
Surface, in vivo ECGs in Mkk4F/F and Mkk4ACKO mice at 3 and 12 months of age reveal comparable ECG parameters. Following a Shapiro‐Wilk test for normality, data were compared using paired Student t tests or two‐way ANOVA followed by post‐hoc multiple comparison tests, where appropriate, assuming P<0.05 indicating statistically significance. Mkk4ACKO indicates atrial cardiomyocyte specific mitogen‐activated protein kinase kinase 4 knockout mice; Mkk4F/F, Mkk4 flox/flox.
P<0.05, compared to Mkk4F/F at the same age group.
P<0.05, compared to 3 month old at the same genotype group.
Figure 2.
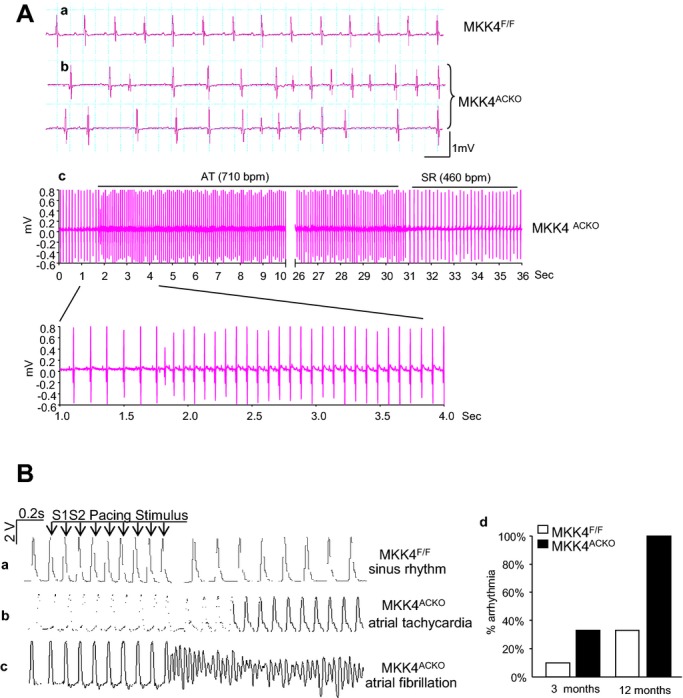
In vivo and ex vivo electrophysiological characterizations. A, Representative ECG recordings showing normal rhythm in a Mkk4F/F mouse (a) and polymorphic atrial ectopic beats and spontaneous atrial tachycardic episodes recorded from Mkk4ACKO mice (b and c). B, Representative MAP traces recorded from ex vivo hearts subjected to the PES S1S2 protocol recorded from atrial epicardium in Mkk4F/F (a) and Mkk4ACKO (b and c) hearts. Atrial arrhythmias (AT and AF) were frequently observed in Mkk4ACKO after PES. Bar graph shows summary of the occurrence of atrial arrhythmic events in Mkk4F/F and Mkk4ACKO hearts (young Mkk4F/F, n=10; young Mkk4ACKO, n=9; old Mkk4F/F, n=10; old Mkk4ACKO, n=9). AF indicates atrial fibrillation; AT, atrial tachycardial MAP, mitogen‐activated protein; Mkk4ACKO, atrial cardiomyocyte specific mitogen‐activated protein kinase kinase 4 knockout mice; Mkk4F/F, Mkk4 flox/flox; PES, programmed electrical stimulation.
The presence or otherwise, as well as the frequency, of arrhythmic tendency in both Mkk4F/F and Mkk4ACKO in atria in isolated Langendorff‐perfused hearts from mice between 3 and 12 months of age were next compared during PES, in which extrasystolic S2 stimuli followed successive trains of eight pacing S1 stimuli delivered at 8 Hz at S1S2 intervals progressively decremented by 1 ms with each successive pacing cycle. Protocols were terminated when hearts either became refractory or showed arrhythmia. Mkk4ACKO hearts showed higher atrial arrhythmic incidences than the corresponding control Mkk4F/F. At 3 months, 3 of 9 (33%) Mkk4ACKO hearts displayed single or multiple episodes of atrial tachycardia (AT) or nonsustained AF, whereas only 1 of 10 (10%) of Mkk4F/F showed a short AT (Table 3). Both Mkk4F/F and Mkk4ACKO hearts showed increased atrial arrhythmic susceptibility with age (Figure 2B), but to significantly greater extents in Mkk4ACKO hearts. At 12 months, all (10 of 10) Mkk4ACKO, but only 3 of 9 (33%) Mkk4F/F, hearts exhibited multiple episodes of nonsustained or sustained AT/AF. Applications of Fisher's exact test to such incidences stratified successively by age and genotype indicated no difference in arrhythmic incidences between Mkk4ACKO (n=9) and Mkk4F/F (n=10) at 3 months, but significantly increased arrhythmic incidences in Mkk4ACKO (n=9), but not Mkk4F/F (n=10), at 12 months. Yet, all age groups of Mkk4ACKO and Mkk4F/F mice showed statistically similar monophasic action potential (MAP) durations (MAPD90) and atrial effective refractory periods (AERP), whether in their left or right atria (Table 3).
Table 3.
Ex Vivo Measurements
| 3 months old | 12 months old | |||
|---|---|---|---|---|
| Mkk4F/F (n=10) | Mkk4ACKO (n=9) | Mkk4F/F (n=9) | Mkk4ACKO (n=10) | |
| HR, bpm | 415±18 | 495±24** | 382±28* | 372±22* |
| LA APD90, ms | 52.7±5.6 | 59.3±9.9 | 75.5±10.5 | 84.5±12.6 |
| LA ERP, ms | 49.21±2.8 | 38.2±7.9 | 51.5±11.5 | 45.8±7.4 |
| RA APD90, ms | 56.0±4.5 | 51.5±8.3 | 66.0±7.7* | 77.14±9.4* |
| RA ERP, ms | 41.93±3.6 | 40.3±5.2 | 49.78±4.5 | 48.28±6.5 |
| % arrhythmia | 10% | 33% | 33% | 100% |
Ex vivo parameters measured by monophasic action potential recordings, from left atria (LA), right atria (RA), in young (3 month) and old (12 month), Mkk4F/F, and Mkk4ACKO mice. Heart rate (HR), action potential duration 90 (APD90), and effective refractory period (ERP) were calculated and arrhythmic events, including periods of atrial tachycardia (AT) or atrial fibrillation (AF), were recorded. Following Shapiro‐Wilk tests for normality, data were compared using the paired Student t tests or two‐way ANOVA followed by post‐hoc multiple comparison tests, where appropriate, assuming P<0.05 indicating statistical significance. Mkk4ACKO indicates atrial cardiomyocyte specific mitogen‐activated protein kinase kinase 4 knockout mice; Mkk4F/F, Mkk4 flox/flox.
P<0.05, compared to Mkk4F/F at the same age group.
P<0.05, compared to 3 month old at the same genotype group.
Epicardial multielectrode array (MEA) mapping of both left and right atrial conduction properties were performed during either sinus rhythm or PES, in isolated Langendorff‐perfused hearts. Figure 3A exemplifies activation maps through five successive cardiac cycles from young (a and b) and old (c and d), Mkk4F/F (a and c), and Mkk4ACKO hearts (b and d). Most activation maps from Mkk4F/F hearts showed a sequential activation pattern, beginning with a localized activation of one region of the matrix followed by its successive spread to the remaining area from which recordings were made. This suggests an orderly spread of excitation within the plane of the epicardium (a and c). However, the old Mkk4ACKO hearts often showed a more disordered excitation sequence.
Figure 3.
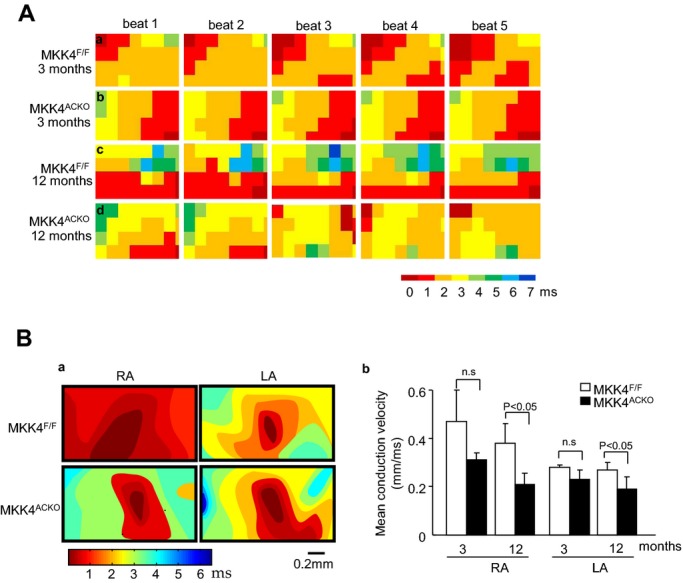
Epicardial electrical mapping with a multielectrode array (MEA). A, Representative activation maps of five successive cardiac cycles under sinus rhythm in Mkk4F/F (a and c) and Mkk4ACKO hearts (b and d) grouped by age in 3‐ (a and b) and 12 month‐old hearts (c and d). B, Representative pacing‐induced activation maps generated by pacing in the center of array on the epicardium of the right (RA) and left atria (LA) of Mkk4F/F and Mkk4ACKO mice (a). Velocities in the left (LA) and right atrium (RA) of young and old Mkk4F/F and Mkk4ACKO mice are shown as bar graphs (means±SEMs). Following Shapiro‐Wilk tests for normality, data were compared between groups using the paired Student t test, with a value of P<0.05 considered to indicate statistical significance (b). (n=5 per group for both young and old Mkk4F/F and Mkk4ACKO). Mkk4ACKO indicates atrial cardiomyocyte specific mitogen‐activated protein kinase kinase 4 knockout mice; Mkk4F/F, Mkk4 flox/flox.
It was also possible to determine conduction velocities within the right and left atria by using MEA recordings to follow the pattern of spread from stimuli delivered through a point in the center of, rather than adjoining, the array and color coding the delays in arrival times as the resulting AP spreads from the site of stimulation. The resulting velocities were comparable in young (3‐month) Mkk4F/F and Mkk4ACKO hearts (Figure 3Ba, b). However, old Mkk4ACKO atria showed slower conduction than old Mkk4F/F hearts. The isochronal maps thus illustrated a pattern consistent with a slower conduction in both left and right Mkk4ACKO than in Mkk4F/F atria from 12‐month‐old hearts (Figure 3Ba, b). Mkk4 deletion thus both alters conduction properties and increases atrial arrhythmic susceptibility, particularly in old Mkk4ACKO mice.
Mkk4 Deficiency Leads to Increased Fibrosis and Altered TGF‐β1 Signaling in the Atrium With Age
Three series of experiments then explored the structural and molecular basis of the slowed conduction and increased arrhythmogenecity in aging Mkk4ACKO atria. First, levels of fibrosis were examined by picrosirius red staining of histological sections from young (3‐ to 4‐month), and old (12‐month) Mkk4F/F and Mkk4ACKO atrial tissue. Mkk4F/F and Mkk4ACKO atria showed comparable levels of fibrotic tissue at 3 months (1.7±0.4% Mkk4F/F vs. 1.8±0.3% Mkk4ACKO; Figure 4A, B). However, aging increased fibrosis in both Mkk4F/F and Mkk4ACKO mice, with significantly greater effects in Mkk4ACKO (6.1±0.4% at 12 months) than Mkk4F/F mice (2.8±0.2% at 12 months).
Figure 4.
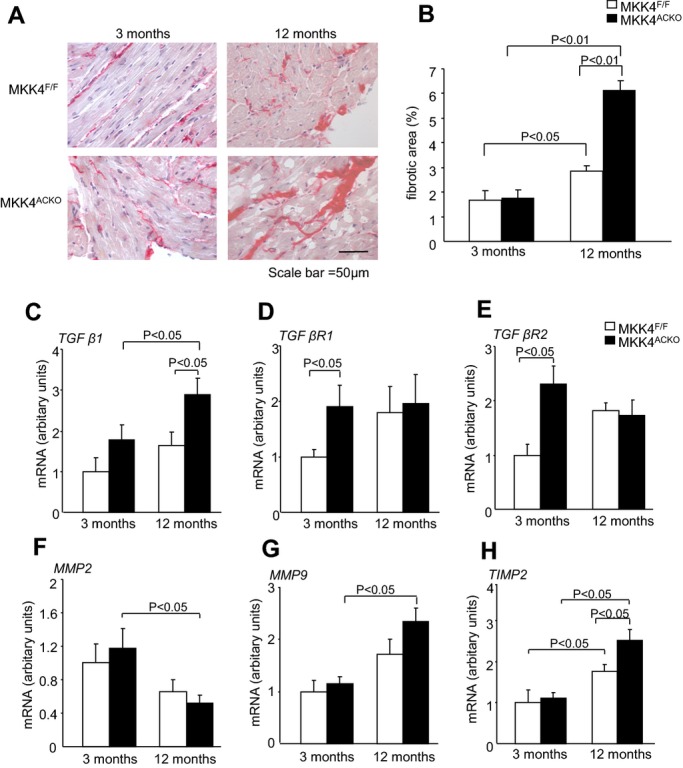
Structural and molecular characterizations. A, Representative images of picrosirius red stained tissue, with fibrotic areas stained dark red, taken at 40× magnification in Mkk4F/F and Mkk4ACKO mice at ages of 3 (n=5) and 12 months (n=7). B, Quantification of the percentage area of fibrosis in young (left) and old (right), Mkk4F/F (white), and Mkk4ACKO (black) mice by picrosirius red staining. C through H, Real‐time PCR analysis of TGF‐β1 (C), TGF‐β receptor 1 (TGF‐β R1; D), TGF‐β receptor 2 (TGF‐β R2; E), MMP2 (F), MMP9 (G), and TIMP2 mRNA expression (H), from young and old Mkk4F/F and Mkk4ACKO left atria. The derived data are normalized to the housekeeping gene (HPRT) content, and the relative expression was calculated using the 2−ΔΔCT method. A Shapiro‐Wilk test was used to check whether data came from a normal distribution before data analysis using two‐way ANOVA followed by Bonferonni‐corrected post‐hoc t tests, with a value of P<0.05 considered to indicate statistical significance. Data are presented as means±SEMs (n=4 per group for both young and old Mkk4F/F and Mkk4ACKO). HPRT, hypoxanthine phosphoribosyltransferase; Mkk4ACKO indicates atrial cardiomyocyte specific mitogen‐activated protein kinase kinase 4 knockout mice; Mkk4F/F, Mkk4 flox/flox; MMP, matrix metalloproteinase; PCR, polymerase chain reaction; TGF‐β, transforming growth factor‐β; TIMP, tissue inhibitors metalloproteinase.
Second, RNA was isolated from left atrial tissue from 3‐ and 12‐month‐old Mkk4F/F and Mkk4ACKO mice for real‐time PCR analysis of gene expression of profibrotic signaling components. Of these, the TGF‐β1 pathway is one of the major known profibrotic pathways; JNK is known to repress TGF‐β1 transcription.7 Mkk4ACKO atrial tissue showed significantly greater TGF‐β1 mRNA expression, compared to Mkk4F/F controls, in 12‐, but not 3‐month‐old, mice (Figure 4C). These positive findings concerning TGF‐β1 mRNA expression in aged Mkk4F/F and Mkk4ACKO mice was complemented by similarly positive differences in TGF‐β1 protein expression (Figure S1). It also showed significantly upregulated levels of both TGF‐β receptor I and II, compared to Mkk4F/F, at 3 months of age, but these expression levels were equal by 12 months (Figure 4D, E). These findings are consistent with deletion of atrial Mkk4 increasing expression of TGF‐β signaling components, thereby providing a potential key mechanism for increased atrial tissue fibrosis in 12‐month‐old Mkk4ACKO, although consistent with an initially increased TGF‐β receptor expression followed by its downregulation by a subsequently increased TGF‐β expression.
A further important mechanism regulating fibrosis arises from activity of matrix metalloproteinases (MMPs) and their tissue inhibitors, tissue inhibitors of metalloproteinases (TIMPs). The JNK pathway has been reported to regulate expression of many MMPs through activator protein 1 (AP1) transcriptional activity.25 Real‐time PCR analysis of two of the major cardiac MMPs, MMP2 and MMP9, revealed significant changes with age only in Mkk4ACKO tissue. However, whereas MMP2 expression was decreased, MMP9 expression was significantly increased at 12 months (Figure 4F, G). Nevertheless, of the major cardiac TIMPs, Mkk4F/F and Mkk4ACKO tissue showed similar TIMP2 expression levels at 3 months and both were increased at 12 months, but with significantly higher levels in the Mkk4ACKO than the Mkk4F/F. Thus, aging and Mkk4 deletion appear to exert additive effects on TIMP2 expression (Figure 4H).
Third, we examined expression of other candidate proteins in atrial tissue from both young and old Mkk4ACKO and Mkk4F/F mice that could contribute to slow conduction and atrial arrhythmogenesis at either mRNA or protein levels. These included the Na+ channel protein, Nav1.5, the calcium handling proteins, ryanodine receptor 2 (RyR2), Na+/Ca2+ exchanger (NCX), and inositol 1,4,5‐trisphosphate receptor (IP3R), the transient receptor potential (TRP) channel proteins, TRPC1, TRPC3, and TRPC6, and the gap junction proteins, Cx40 and Cx43 (Figures 5 through 7). There were no significant differences in mRNA or protein expression between Mkk4F/F and Mkk4ACKO mice.
Figure 6.
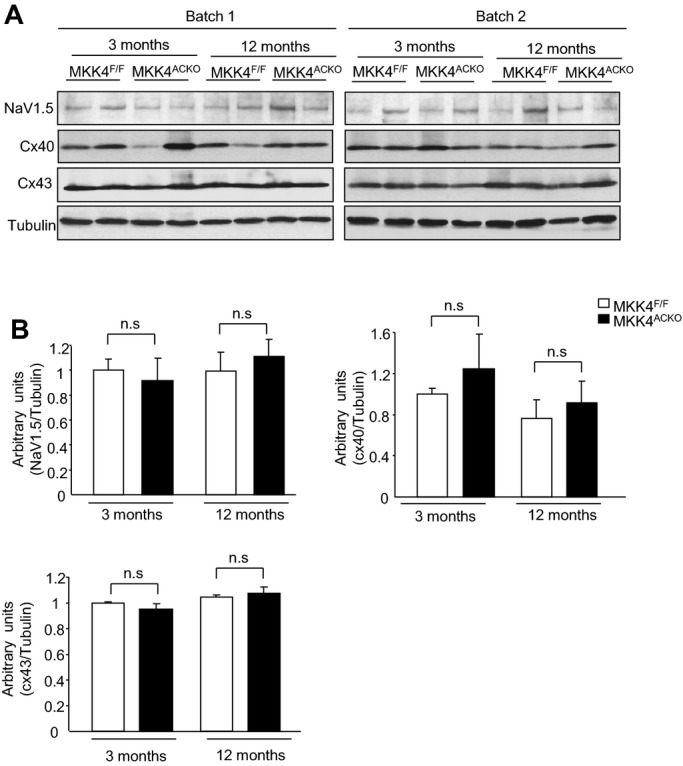
Expression of Nav1.5, connexin 40 (Cx40), and connexin 43 (Cx43) in the atria from young and old Mkk4F/F and Mkk4ACKO mice. A, Immunoblot analysis on two batches of samples. Tubulin expression is the protein loading control. B, Immunoblot signals were quantified using ImageJ software (National Institutes of Health) and the ratios of Nav1.5/tubulin, connexin 40/tubulin, and connexin 43/tubulin are represented by the bar graphs. Following a Shapiro‐Wilk test confirming normality, data were analyzed using the paired Student t test, with a value of P<0.05 considered to indicate statistical significance. Data are expressed as means±SEMs (n=4 per group for both young and old Mkk4F/F and Mkk4ACKO). Mkk4ACKO indicates atrial cardiomyocyte‐specific mitogen‐activated protein kinase kinase 4 knockout mice; Mkk4F/F, Mkk4 flox/flox.
Figure 7.
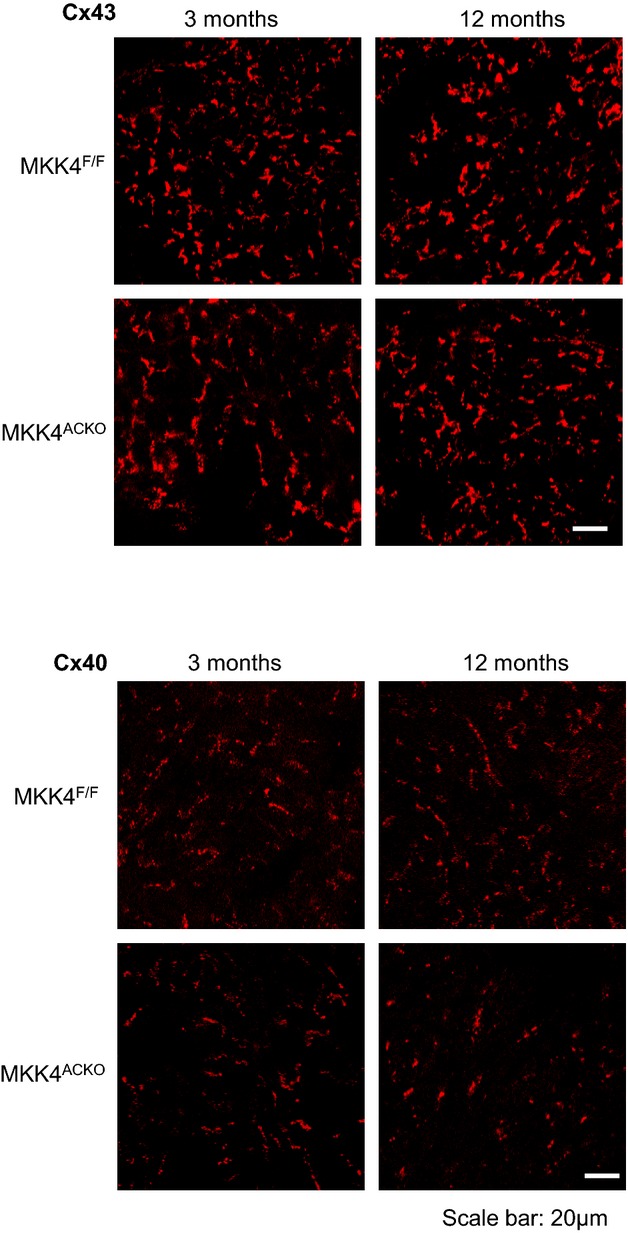
Immunohistochemical staining of connexin 40 (Cx40) and connexin 43 (Cx43) in the atria shows similar distribution between Mkk4F/F and Mkk4ACKO mice in young (3 months) and old age (12 months). Scale bar, 20 μm (n=4 per group for both young and old Mkk4F/F and Mkk4ACKO). Mkk4ACKO indicates atrial cardiomyocyte‐specific mitogen‐activated protein kinase kinase 4 knockout mice; Mkk4F/F, Mkk4 flox/flox.
Figure 5.
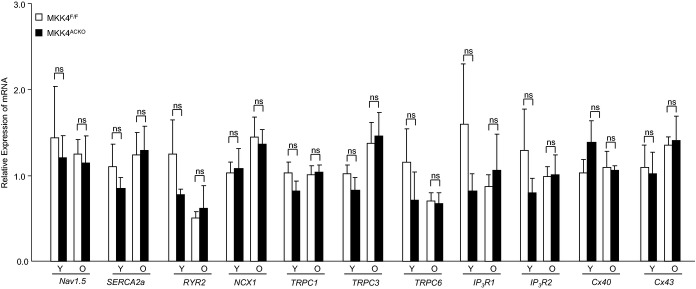
Transcriptional expression of Ca2+ handling proteins RyR2, SERCA2a, and NCX1, the TRPC channels, TRPC1, 3, and 6, the IP3 receptors, IP3R1 and IP3R2, and major connexins (Cx) Cx40 and Cx43. Total RNA was extracted from atria of young (3‐month) and old (12‐month) Mkk4F/F and Mkk4ACKO mice. This was followed by cDNA synthesis and qPCR analysis. The derived data are normalized to the housekeeping gene (HPRT) content, and the relative expression was calculated using the 2−ΔΔCT method. Following a Shapiro‐Wilk test for normality, data were analyzed using the paired Student t test or two‐way ANOVA followed by Bonferroni‐corrected post‐hoc multiple comparison tests, where appropriate, with a value of P<0.05 considered to indicate statistical significance. Y: young (3 month old); O: old (12 month old); ns: not significant; results are presented as means±SEM (n=4 per group for both young and old Mkk4F/F and Mkk4ACKO). Mkk4ACKO indicates atrial cardiomyocyte‐specific mitogen‐activated protein kinase kinase 4 knockout mice; Mkk4F/F, Mkk4 flox/flox; NCX, sodium calcium exchanger; PCR, polymerase chain reaction; RyR, Ryanodine receptor; SERCA, sarcoendoplasmic reticulum Ca2+ATPase; TRPC, transient receptor potential cation channel.
Computer Simulations
The results above suggested that fibrotic processes might lead to the electrophysiological phenotypes and atrial arrhythmogenesis in Mkk4ACKO mice. Our computer modeling studies then explored biophysical mechanisms through which such increases in fibroblast population might alter atrial electrical excitation and its propagation to result in these proarrhythmic effects. First, we investigated effects of an electrical coupling between fibroblasts and cardiomyocytes (Figure 8A) on atrial APs at the single‐cell level. Coupling to fibroblasts decreased the overshoots and maximal upstroke velocities of atrial APs, prolonged their APDs, and made resting potentials more positive (Figure 8B) to extents that increased with the number of coupled fibroblasts. Figure 8B (inset) exemplifies this for the maximal AP upstroke velocity. A coupling of more than 10 fibroblasts resulted in the membrane potential of the atrial cell model failing to attain the activation threshold of INa to generate an AP. These atrial AP modulations reflected the dual action of coupled fibroblasts as both a passive load and a driver to a cardiomyocyte over the AP time course. Thus, the more depolarized fibroblast resting potential would drive in a depolarizing current into the coupled cardiomyocyte when this was at its resting potential and the late repolarization phase of its AP. This would elevate the cardiomyocyte resting potential and prolong its APD. However, it would drive a repolarizing current during the AP upstroke and phase 1 and 2 repolarization. This would decrease the overshoot and the maximal upstroke velocity of the atrial AP (Figure 8B, bottom panel).
Use of a 2D model demonstrated that fibroblast‐cardiomyocyte couplings modified the CV of atrial excitation waves to extents that varied with fibroblast density (Figure 8C), thereby replicating observed alterations in control and aged Mkk4ACKO or Mkk4F/F. Measured CVs decreased monotonically with increased fibroblast density. The simulations that incorporated Mkk4ACKO and aging effects adopted diffusion coefficients chosen to reproduce experimentally measured CVs in the respective young and old groups of Mkk4ACKO and Mkk4F/F. This then yielded computed CVs with a greater negative dependence on fibroblast density in the old than the young group. Finally, the presence of fibroblast‐cardiomyocyte coupling disturbed the wavefronts of atrial excitation waves. Figure 8D exemplifies results from a standard S1 to S2 stimulation protocol26 (basic cycle length=150 ms) applied at the left edge of the 2D model containing randomly distributed fibroblast populations, resulting in a range from one to five fibroblasts coupled to any given cardiomyocyte. The snapshots of the excitation waves evoked by the S2 stimulus, applied at a S1S2 coupling interval of 60 ms, demonstrated that the wavefront of atrial excitation wave became broken. This led to formation of re‐entrant excitation waves (Figure 8D).
TGF‐β Signaling in Mkk4‐Inactivated Cardiomyocytes and Cardiomyocyte‐Fibroblast Crosstalk
Experiments on cultured NRCMs infected with either wild‐type (WT) Ad‐GFP or Ad‐dnMKK4 directly demonstrated that MKK4 inactivation altered profibrotic signaling whether in the absence or presence of Ang II stimulation.
First, as shown in Figure S2, the transcriptional levels of TGF‐β1, collagen, type 1, alpha 2 (Col1a2), MMP2, and TIMP were compared in the NRCMs infected with or without Ad‐GFP followed by 24 hours of stimulation of Ang II. There was no difference in these transcripts between these two groups, indicating that Ad‐GFP does not affect the transcriptional levels of TGF‐β1, Col1a2, MMP2, and TIMP in NRCMs under the Ang II stress condition. MKK4‐inactivated NRCMs, but not WT NRCMs showed increased TGF‐β1 mRNA expression (Figure 9A) under basal conditions. In contrast, Col1a1 and MMP2 expression was not significantly different between the two groups. After 24 hours of treatment with Ang II (500 nmol/L), MKK4‐deficient NRCMs showed significantly higher levels of TGF‐β1 and TIMP transcripts than did the corresponding WT NRCMs. MKK4 deficiency thus enhances Ang II–induced upregulation of TGF‐β1 signaling and TIMP activity (Figure 9A). This effect likely involves alterations in the crosstalk between cardiomyocytes and fibroblasts. This was suggested in experiments in which WT NRCMs and MKK4‐inactivated NRCMs were first treated with Ang II (500 nmol/L) for 24 hours. NRCM‐conditioned medium was then transferred to plated NRCFs. These were then further incubated for 24 hours before RNA was extracted. Cultured NRCFs that received conditioned medium from MKK4‐deficient NRCMs yielded significantly higher Col1a1 mRNA levels than did NRCFs that received conditioned medium from WT NRCMs (Figure 9B). Thus, MKK4 inactivation in NRCMs modified their profibrotic paracrine signaling to NRCFs, likely, in part, by increasing transcriptional activity of Col1a1 in NRCFs. The importance of TGF‐β1 activity in MKK4‐mediated regulation of fibrosis was demonstrated in experiments in which cell culture media from NRCMs, treated as described above, was transferred to a TGF‐β reporter cell line (TMLC cells). Their TGF‐β1 activity was then measured using luciferase reporter assays. Media from NRCM cells infected with dominant negative MKK4, resulted in a significant increase in TGF‐β1 activity in the TMLC cell line (Figure 9C). Thus, MKK4 may regulate the amount of active TGF‐β1 produced by cardiomyocytes and can directly affect TGF‐β1 transcriptional activity in colocalized cells. Together, these findings suggest mechanisms through which deletion of MKK4 in atrial cardiomyocytes could lead to increased profibrotic signaling in fibroblasts.
Figure 9.

TGF‐β signaling in NRCMs and NRCFs. A, NRCMs were infected with either adenovirus encoding GFP (Ad‐GFP, as control) or dominant negative MKK4 adenovirus (Ad‐d/nMKK4). Transcriptional levels of TGF‐β1, Colalpha1, MMP2, and TIMP2 in these NRCMs were examined by qPCR in response to Ang II stimulation at 500 nmol/L for 24 hours. B, Conditional media from NRCMs treated by Ang II were transferred to NRCFs for 24 hours. Transcriptional levels of TGF‐β1, Colalpha1, MMP2, and TIMP2 in NRCFs were measured by qPCR. C, Conditional media from NRCMs treated by Ang II was transferred to a TGF‐β reporter cell line (TMLC cells) in which the TGF‐β activity was indicated by a luciferase reporter. The TGF‐β1 activity in the media was then measured using luciferase reporter assays. Data were derived from three independent batches of cells obtained on different dates studied in triplicate and presented as means±SEMs. Following a Shapiro‐Wilk test to confirm normality, data were analyzed using paired Student t tests or two‐way ANOVA followed by post‐hoc multiple comparison tests, where appropriate, with a value of P<0.05 considered to be statistically significant (n=3 per group). Ang II indicates angiotensin II; GFP, green fluorescent protein; MKK4, mitogen‐activated protein kinase kinase 4; MMP, matrix metalloproteinase; NRCMs, neonatal rat cardiomyocytes; NRCFs, neonatal rat cardiac fibroblasts; PCR, polymerase chain reaction; TGF, transforming growth factor; TIMP, tissue inhibitors metalloproteinase.
MKK4 Expression Is Reduced in Atrial Tissue in Patients With AF
The translational significance of the results described above was explored through assessment of expression levels at the mRNA and/or protein levels of key MAPK components and profibrotic signaling molecules in atrial tissues from age‐matched cardiac surgery patients in sinus rhythm (SR) and AF. AF patients showed increased left atrial diastolic diameters (LADD; Table 1), suggesting the dilated left atria characteristic of impaired left ventricular function, a risk factor for AF. Quantitative PCR (qPCR) analysis demonstrated significant downregulation in MKK4, but not MKK7, P38, and JNK, transcription in AF patients, relative to control patients in SR (Figure 10A). Western blot analysis further confirmed decreased MKK4 protein levels in AF patients (Figure 10B). qPCR analysis of profibrotic gene expression revealed significant increases in levels of collagens, including Col1α1 and Col3α1, upregulation of the profibrotic connective tissue growth factor (CTGF) and downregulation of MMP2 in AF patients, suggesting an increased profibrotic signaling in their atria (Figure 10A). Finally, because AF is strongly associated with aging, we further divided these patients into three age groups (<65, 65 to 75, and >75 years old) to analyze the correlation between MKK4 expression with aging. In both SR and AF patients, MKK4 mRNA levels were significantly decreased in those >75 years, compared to the younger age groups (<65 and 65 to 75; Figure 10C).
Figure 10.
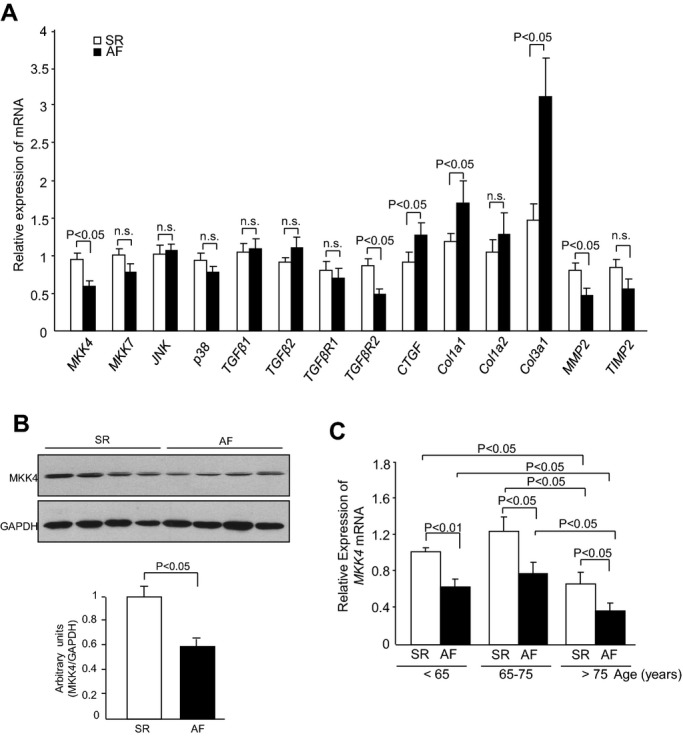
MKK4 expression is reduced in atrial fibrillation patients. A, qPCR analysis shows transcriptional levels of MAPK signaling components: MKK4, MKK7, p38, and JNK and fibrotic genes: TGF‐β1, TGF‐β2, TGF‐βR1, TGF‐βR2, CTGF, Col1α1, Col1α2, Col3α1, MMP2, and TIMP2 in the atria from sinus rhythm (SR) and atrial fibrillation (AF) patients. B, Immunoblot analysis demonstrates the protein level of MKK4 in the atria of SR and AF patients. GAPDH expression is the protein loading control. Immunoblot signals were quantified using ImageJ software (National Institutes of Health), and the ratios of MKK4/GAPDH are represented by the bar graphs. Following a Shapiro‐Wilk test to confirm normality, data were compared between groups using paired Student t tests, with a value of P<0.05 taken to indicate statistical significance. Data are expressed as means±SEMs. C, Transcription level of MKK4 in the atria from SR (n=15) and AF (n=15) patients in different age groups (<65, 65 to 75, and >65). Following Shapiro‐Wilk normality tests, data were analyzed using paired Student t tests or two‐way ANOVA followed by post‐hoc multiple comparison tests, where appropriate, with a value of P<0.05 considered statistically significant. Data are presented as means±SEM. CTGF indicates connective tissue growth factor; GADPH, glyceraldehyde 3‐phosphate dehydrogenase; JNK, c‐Jun N‐terminal kinase; MAPK, mitogen‐activated protein kinases; MKK4, mitogen‐activated protein kinase kinase 4; MMP, matrix metalloproteinase; PCR, polymerase chain reaction; TGF, transforming growth factor; TIMP, tissue inhibitors metalloproteinase.
Discussion
The principal outcome of this study is the demonstration that Mkk4 acts as a negative regulator of atrial remodeling and arrhythmogenesis with age, that it does so through a mechanism associated with TGF‐β1 signaling, and that the latter is likely mediated through JNK‐regulated signaling. A corroborating clinical study reports that AF patients show remarkably decreased MKK4 expression. Together, these findings argue, for the first time, that MKK4 is functionally important in the pathogenesis of human AF.
Mkk4 Signaling and Age‐Related Atrial Remodeling
Atrial fibrosis is a key substrate for the initiation and maintenance of AF through its associated alterations in tissue architecture, composition, and electrophysiological function.27–29 The precise signaling mechanisms underlying its development with age remain unclear. The present study implicates Mkk4 in age‐dependent atrial fibrosis and its associated electrical remodeling. Our experimental electrophysiological studies demonstrate that Langendorff‐perfused Mkk4ACKO, but not Mkk4F/F, hearts became more susceptible to atrial arrhythmias with age. Aged Mkk4ACKO atria also showed reduced P wave amplitudes, dispersed and heterogeneous activation patterns suggesting spatial nonuniformities in propagation, slower atrial tissue conduction, and increased histological evidence for interstitial fibrosis. In contrast, ex vivo Mkk4ACKO and Mkk4F/F hearts showed similar APD90 and ERP values, as well as indistinguishable connexin expression levels and distribution through all age groups. Such findings suggest an increased arrhythmogenecity attributable to fibrotic events, as opposed to altered cellular electrophysiology. Such changes are known to occur in both animal models of, and patients with, AF, the latter including the lone AF observed in the absence of other cardiovascular disease.29–31
Our computational studies correspondingly demonstrated that atrial tissue containing randomly distributed fibroblast populations, modeled in two dimensions, became more heterogeneous. Fibroblast‐cardiomyocyte coupling then slowed down atrial CV and prolonged APD more in the localized regions with greater fibroblast populations. Premature S2 stimuli at short coupling time intervals did not permit full recovery of such localized regions with high fibroblast densities from their previous excitation because of their long refractory periods. This resulted in formation of regions of functional conduction block fragmenting the atrial excitation wavefronts and leading to formation of re‐entrant excitation waves. Thus, increases in fibroblast density or alterations in fibroblast‐cardiomyocyte coupling alter atrial electrophysiological properties and result in an arrhythmogenic substrate offering sources for local micro re‐entry that might form the trigger for the AT/AF observed in Mkk4ACKO hearts and also account for the widespread, generalized process of junctional remodeling that has been observed in patients with AF.29–32 Mkk4 might then function in maintaining normal atrial electrical function during aging through limiting such structural remodeling through regulation of atrial fibrosis.
Mechanistic Links Between Mkk4 and Transduction by TGF‐β1 Signaling
The TGF‐β‐signaling pathway constitutes one of the central regulators for cardiac fibrotic alterations, which have been associated with various cardiac disease conditions that include AF.28–29 Cardiac overexpression of constitutively active TGF‐β1 is known to cause selective atrial fibrosis, conduction heterogeneity, and AF susceptibility.33 Mkk4 deficiency led to an increase in expression of TGF‐β‐signaling components, providing a possible mechanism by which atrial fibrosis may be exacerbated with age in Mkk4ACKO mice.
How does MKK4 deletion upregulate the TGF‐β‐signaling pathway? First, MKK4 is a well‐recognized upstream kinase in the JNK‐signaling pathway and regulates diverse physiological processes through activating JNKs.34 Recent studies, including ours, have suggested that MKK4 in the heart is an important regulator of activation of the JNK pathway.14–15,17 Our findings suggest that MKK4 acts as a negative regulator of TGF‐β1 signaling through JNK activity. This is consistent with recent findings that JNK‐deficient fibroblasts isolated from Jnk1−/−Jnk2−/− embryos containing the embryonically lethal Jnk1−/−Jnk2−/− genotype constitutively express TGF‐β1 (Ventura et al.7). These studies had demonstrated that JNK/c‐Jun signaling represses Tgf‐β1 gene expression, in an important crosstalk between the TGF‐β1‐ and JNK‐signaling pathways.7 Our present study further demonstrates that TGF‐β1 mRNA as well as both TGF‐β receptors I and II were significantly upregulated in Mkk4ACKO, relative to Mkk4F/F atria. Our results further demonstrated alterations in MMPs and their inhibitors, TIMPs25 in Mkk4ACKO atria, in the form of additive effects of aging and Mkk4 deletion on TIMP2 expression. However, these findings were less clear cut, in that whereas MMP2 expression was decreased, MMP9 expression was significantly increased at 12 months. These findings were reinforced by our further in vitro comparative studies in Mkk4‐inactivated and WT NRCMs.
Effects of Cardiomyocyte‐Fibroblast Crosstalk on Profibrotic Signaling
Our studies in NRCMs and NRCFs revealed that crosstalk between two cell types affected profibrotic signaling. Cardiomyocytes account for ≈45% of the atrial myocardium by volume, whereas nonmyocytic cells are thought to compose ≈70% of cardiac cells by number.30 There is a complex interplay among these two cell types, the more numerous of which are cardiac fibroblasts. The fibroblast was traditionally thought to be a passive bystander in the myocardium, but is now recognized to participate actively in shaping and responding to the cardiac milieu.30 Both fibroblasts and cardiomyocytes are important profibrotic mediators.30 Cardiac overexpression of constitutively active TGF‐β1 causes selective atrial fibrosis, conduction heterogeneity, and susceptibility to AF.33 TGF‐β1 functions as a downstream mediator of Ang II through both para‐ and autocrine mechanisms. It acts primarily through the SMAD pathway to stimulate fibroblast activation and collagen production. Knockdown of MKK4 in cultured cardiomyocytes increased their sensitivity to Ang II stimulation on TGF‐β1‐signaling activation; such a change, in turn, enhanced paracrine expression of profibrotic molecules in cultured cardiac fibroblasts. Ang II has been shown to induce extracellular matrix protein synthesis and secretion in cardiac fibroblasts.35 Cardiomyocytes are not likely to directly synthesize collagen, but instead interact with neighboring fibroblasts to stimulate collagen synthesis and secretion, thereby resulting in the structural remodeling that we observed in Mkk4ACKO mice. Author Summary Figure 11 illustrates such a prediction.
Figure 11.

Signaling mechanism that MKK4 deficiency leads to enhanced TGF‐β1 signaling activity. MKK4 deficiency that leads to enhanced TGF‐β1 signaling activity is likely the result of the reduced inhibitory effect of JNK/c‐Jun. Increased TGF‐β1 activates intracellular TGF‐β1 signaling cascades, such as phosphorylated SMADS, and leads to enhanced proliferation and synthesis of collagens of cardiac fibroblasts. AP indicates action potential; GPCR, G protein coupled receptors; JNK, c‐Jun N‐terminal kinase; MAPK, mitogen‐activated protein kinases; MKK4, mitogen‐activated protein kinase kinase 4; MMP, matrix metalloproteinase; TGF, transforming growth factor.
MKK4 Dysregulation in Atrial Tissue in Patients With AF
A key question concerns the applicability of the results obtained from our mouse models and in vitro studies to human conditions. Our study on human atrial tissue demonstrated that MKK4, but not MKK7, p38, and JNK, was significantly downregulated in AF patients at both the mRNA and protein levels. A significant increase in collagens, including Col1α1 and Col3α1, was accompanied by upregulation of CTGF and downregulation of MMP2 in AF patients, compared to control patients in SR, which suggests a correlation between AF and increased profibrotic signaling pathways in the atrium in these patients.
Conclusions and Limitations of the Study
In summary, we suggest, for the first time, a critical role for MKK4 in age‐related atrial structural remodeling associated with AF. MKK4 is therefore a potential novel therapeutic target for management of, and a potential novel biomarker for susceptibility to, AF. We also report clear‐cut evidence implicating MKK4 as a negative regulator for profibrotic signaling through TGF‐β1, likely through its regulation by JNK activity, but provide less‐clear‐cut outcomes concerning roles of profibrotic signaling pathways involving MMPs and their tissue inhibitors (TIMPs), therefore justifying further study of these (Figure 11). Further studies may also increase the population from which human biopsy results are obtained and extend our MEA and computational modeling studies to evaluate further possible mechanisms by which increased fibroblast populations alter atrial electrical excitation and its propagation to produce arrhythmic effects. Finally, a broader analysis would recognize that although atrial fibrosis may contribute to AF, AF also occurs in the absence of fibrosis, and this has implications for our understanding of the development of this condition and the role of other predisposing risk variants.
Sources of Funding
The work was supported by the British Heart Foundation (PG/12/21/29473: Lei, Wang, Cartwright), the Medical Research Council (G10002647: Lei, Wang, Cartwright), a Cardiovascular Therapeutics Inc (USA) research grant (Lei), The Wellcome Trust (081809: Lei, Zhang), and a Chinese Nature Science Foundation grant (31171085: Wu, Lei). Fraser is a BBSRC David Phillips Research Fellow.
Disclosures
None.
References
- 1.Rose BA, Force T, Wang Y. Mitogen‐activated protein kinase signaling in the heart: angels versus demons in a heart‐breaking tale. Physiol Rev. 2010; 90:1507-1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sugden PH, Clerk A. Cellular mechanisms of cardiac hypertrophy. J Mol Med. 1998; 76:725-746 [DOI] [PubMed] [Google Scholar]
- 3.Li D, Shinagawa K, Pang L, Leung TK, Cardin S, Wang Z, Nattel S. Effects of angiotensin‐converting enzyme inhibition on the development of the atrial fibrillation substrate in dogs with ventricular tachypacing‐induced congestive heart failure. Circulation. 2001; 104:2608-2614 [DOI] [PubMed] [Google Scholar]
- 4.Sugden PH, Clerk A. Regulation of the ERK subgroup of MAP kinase cascades through G protein‐coupled receptors. Cell Signal. 1997; 9:337-351 [DOI] [PubMed] [Google Scholar]
- 5.Ushio‐Fukai M, Alexander RW, Akers M, Griendling KK. p38 Mitogen‐activated protein kinase is a critical component of the redox‐sensitive signaling pathways activated by angiotensin II. J Biol Chem. 1998; 273:15022-15029 [DOI] [PubMed] [Google Scholar]
- 6.Goette A, Lendeckel U, Klein HU. Signal transduction systems and atrial fibrillation. Cardiovasc Res. 2002; 54:247-258 [DOI] [PubMed] [Google Scholar]
- 7.Ventura J‐J, Kennedy NJ, Flavell RA, Davis RJ. JNK regulates autocrine expression of TGF‐β1. Mol Cell. 2004; 15:269-278 [DOI] [PubMed] [Google Scholar]
- 8.Braz JC, Bueno OF, Liang Q, Wilkins BJ, Dai YS, Parsons S, Braunwart J, Glascock BJ, Klevitsky R, Kimball TF, Hewett TE, Molkentin JD. Targeted inhibition of p38 MAPK promotes hypertrophic cardiomyopathy through upregulation of calcineurin‐NFAT signaling. J Clin Invest. 2003; 111:1475-1486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deng Y, Ren X, Yang L, Lin Y, Wu X. A JNK‐dependent pathway is required for TNFalpha‐induced apoptosis. Cell. 2003; 115:61-70 [DOI] [PubMed] [Google Scholar]
- 10.Lei K, Davis RJ. JNK phosphorylation of Bim‐related members of the Bcl2 family induces Bax‐dependent apoptosis. Proc Natl Acad Sci USA. 2003; 100:2432-2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brancho D, Tanaka N, Jaeschke A, Ventura J‐J, Kelkar N, Tanaka Y, Kyuuma M, Takeshita T, Flavell RA, Davis RJ. Mechanism of p38 MAP kinase activation in vivo. Genes Dev. 2003; 17:1969-1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang X, Nadarajah B, Robinson AC, McColl BW, Jin J‐W, Dajas‐Bailador F, Boot‐Handford RP, Tournier C. Targeted deletion of the mitogen‐activated protein kinase kinase 4 gene in the nervous system causes severe brain developmental defects and premature death. Mol Cell Biol. 2007; 27:7935-7946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lei K, Nimnual A, Zong WX, Kennedy NJ, Flavell RA, Thompson CB, Bar‐Sagi D, Davis RJ. The Bax subfamily of Bcl2‐related proteins is essential for apoptotic signal transduction by c‐Jun NH2‐terminal kinase. Mol Cell Biol. 2002; 22:4929-4942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu W, Zi M, Jin J, Prehar S, Oceandy D, Kimura TE, Lei M, Neyses L, Weston AH, Cartwright EJ, Wang X. Cardiac‐specific deletion of Mkk4 reveals its role in pathological hypertrophic remodeling but not in physiological cardiac growth. Circ Res. 2009; 104:905-914 [DOI] [PubMed] [Google Scholar]
- 15.Zi M, Kimura TE, Liu W, Jin J, Higham J, Kharche S, Hao G, Shi Y, Shen W, Prehar S, Mironov A, Neyses L, Bierhuizen MFA, Boyett MR, Zhang H, Lei M, Cartwright EJ, Wang X. Mitogen‐activated protein kinase kinase 4 deficiency in cardiomyocytes causes connexin 43 reduction and couples hypertrophic signals to ventricular arrhythmogenesis. J Biol Chem. 2011; 286:17821-17830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Lange FJ, Moorman AFM, Christoffels VM. Atrial cardiomyocyte‐specific expression of Cre recombinase driven by an Nppa gene fragment. Genesis. 2003; 37:1-4 [DOI] [PubMed] [Google Scholar]
- 17.Liu W, Min Z, Naumann R, Ke Y, Ulm S, Jin JW, Taglieri DM, Prehar S, Gui J, Xiao Y, Neyses L, Solaro RJ, Cartwright E, Lei M, Wang X. PAK1 is a novel signal transducer attenuating cardiac hypertrophy. Circulation. 2011; 124:2702-2715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bondarenko VE, Szigeti GP, Bett GCL, Kim S‐J, Rasmusson RL. Computer model of action potential of mouse ventricular myocytes. Am J Physiol Heart Circ Physiol. 2004; 287:H1378-H1403 [DOI] [PubMed] [Google Scholar]
- 19.Lomax AE, Rose RA, Giles W. Electrophysiological evidence for a gradient of G protein‐gated K+ current in adult mouse atria. Br J Pharmacol. 2003; 140:576-584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bou‐Abboud E, Nattel S. Molecular mechanisms of the reversal of imipramine‐induced sodium channel blockade by alkalinization in human cardiac myocytes. Cardiovasc Res. 1998; 38:395-404 [DOI] [PubMed] [Google Scholar]
- 21.Nakamura H, Ding W‐G, Sanada M, Maeda K, Kawai H, Maegawa H, Matsuura H. Presence and functional role of the rapidly activating delayed rectifier K+ current in left and right atria of adult mice. Eur J Pharmacol. 2010; 649:14-22 [DOI] [PubMed] [Google Scholar]
- 22.Panama BK, McLerie M, Lopatin AN. Heterogeneity of IK1 in the mouse heart. Am J Physiol Heart Circ Physiol. 2007; 293:H3558-H3567 [DOI] [PubMed] [Google Scholar]
- 23.MacCannell K, Bazzazi H, Chilton L, Shibukawa Y, Clark R, Giles W. A mathematical model of electrotonic interactions between ventricular myocytes and fibroblasts. Biophys J. 2007; 92:4121-4132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abe M, Harpel JG, Metz CN, Nunes I, Loskutoff DJ, Rifkin DB. An assay for transforming growth factor‐beta using cells transfected with a plasminogen activator inhibitor‐1 promoter‐luciferase construct. Anal Biochem. 1994; 216:276-284 [DOI] [PubMed] [Google Scholar]
- 25.Spinale FG. Myocardial matrix remodeling and the matrix metalloproteinases: influence on cardiac form and function. Physiol Rev. 2007; 87:1285-1342 [DOI] [PubMed] [Google Scholar]
- 26.Leon L, Roberge F, Vinet A. Simulation of two‐dimensional anisotropic cardiac reentry: effects of the wavelength on the reentry characteristics. Ann Biomed Eng. 1994; 22:592-609 [DOI] [PubMed] [Google Scholar]
- 27.Gramley F, Lorenzen J, Koellensperger E, Kettering K, Weiss C, Munzel T. Atrial fibrosis and atrial fibrillation: the role of the TGF‐β1 signaling pathway. Int J Cardiol. 2010; 143:405-413 [DOI] [PubMed] [Google Scholar]
- 28.Gordon KJ, Blobe GC. Role of transforming growth factor‐β superfamily signaling pathways in human disease. Biochim Biophys Acta. 2008; 1782:197-228 [DOI] [PubMed] [Google Scholar]
- 29.Nattel S, Burstein B, Dobrev D. Atrial remodeling and atrial fibrillation. Circ Arrhythm Electrophysiol. 2008; 1:62-73 [DOI] [PubMed] [Google Scholar]
- 30.Burstein B, Nattel S. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol. 2008; 51:802-809 [DOI] [PubMed] [Google Scholar]
- 31.Iwasaki Y‐K, Nishida K, Kato T, Nattel S. Atrial fibrillation pathophysiology. Circulation. 2011; 124:2264-2274 [DOI] [PubMed] [Google Scholar]
- 32.Spach MS, Dolber PC. Relating extracellular potentials and their derivatives to anisotropic propagation at a microscopic level in human cardiac muscle. Evidence for electrical uncoupling of side‐to‐side fiber connections with increasing age. Circ Res. 1986; 58:356-371 [DOI] [PubMed] [Google Scholar]
- 33.Verheule S, Sato T, Everett T, Engle SK, Otten D, Rubart‐von der Lohe M, Nakajima HO, Nakajima H, Field LJ, Olgin JE. Increased vulnerability to atrial fibrillation in transgenic mice with selective atrial fibrosis caused by overexpression of TGF‐β1. Circ Res. 2004; 94:1458-1465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang X, Destrument A, Tournier C. Physiological roles of MKK4 and MKK7: insights from animal models. Biochim Biophys Acta. 2007; 1773:1349-1357 [DOI] [PubMed] [Google Scholar]
- 35.Bouzegrhane F, Thibault GT. Is angiotensin II a proliferative factor of cardiac fibroblasts? Cardiovasc Res. 2002; 53:304-312 [DOI] [PubMed] [Google Scholar]