

Abstract
Background
Diabetes mellitus (DM) has multifactorial detrimental effects on myocardial tissue. Recently, carbonic anhydrases (CAs) have been shown to play a major role in diabetic microangiopathy but their role in the diabetic cardiomyopathy is still unknown.
Methods and Results
We obtained left ventricular samples from patients with DM type 2 (DM‐T2) and nondiabetic (NDM) patients with postinfarct heart failure who were undergoing surgical coronary revascularization. Myocardial levels of CA‐I and CA‐II were 6‐ and 11‐fold higher, respectively, in DM‐T2 versus NDM patients. Elevated CA‐I expression was mainly localized in the cardiac interstitium and endothelial cells. CA‐I induced by high glucose levels hampers endothelial cell permeability and determines endothelial cell apoptosis in vitro. Accordingly, capillary density was significantly lower in the DM‐T2 myocardial samples (mean±SE=2152±146 versus 4545±211/mm2). On the other hand, CA‐II was mainly upregulated in cardiomyocytes. The latter was associated with sodium‐hydrogen exchanger‐1 hyperphosphorylation, exaggerated myocyte hypertrophy (cross‐sectional area 565±34 versus 412±27 μm2), and apoptotic death (830±54 versus 470±34 per 106 myocytes) in DM‐T2 versus NDM patients. CA‐II is activated by high glucose levels and directly induces cardiomyocyte hypertrophy and death in vitro, which are prevented by sodium‐hydrogen exchanger‐1 inhibition. CA‐II was shown to be a direct target for repression by microRNA‐23b, which was downregulated in myocardial samples from DM‐T2 patients. MicroRNA‐23b is regulated by p38 mitogen‐activated protein kinase, and it modulates high‐glucose CA‐II–dependent effects on cardiomyocyte survival in vitro.
Conclusions
Myocardial CA activation is significantly elevated in human diabetic ischemic cardiomyopathy. These data may open new avenues for targeted treatment of diabetic heart failure.
Keywords: apoptosis, carbonic anhydrase, diabetes mellitus, hypertrophy, microRNA
Introduction
The global prevalence of diabetes mellitus (DM) is forecast to reach 300 million by 2025, and more than three‐fourths of deaths among this population are expected to result from cardiovascular disease.1 Individuals with DM are indeed at a significantly greater risk of developing both microvascular and macrovascular disease, and they have an excess morbidity and mortality for coronary artery disease (CAD) and post–myocardial infarction (MI) heart failure.2–3
Increased reactive oxygen species (ROS) production in the diabetic heart is a contributing factor in the development and progression of diabetic cardiomyopathy.4–6 Cumulative superoxide‐mediated damage or cellular dysfunction results when ROS generation supersedes ROS‐degrading pathways. Ischemic injury in DM is typically accompanied by a deeper grade of myocardial tissue hypoxia, generating increased ROS production, which in turn activates maladaptive signaling pathways, leading to myocardial cell death and maladaptive cardiac remodelling.7 Carbonic anhydrases (CAs) are a family of ubiquitous metalloenzymes that are responsible for the rapid conversion of carbon dioxide to bicarbonate and protons and thus are involved in a variety of physiological and pathological processes that involve pH regulation, CO2 and HCO3− transport, ion transport, and biosynthetic reactions.8 The CA inhibitor ethoxyzolamide prevents rat cardiomyocyte hypertrophy in vitro and reverses it once it is established.9 Elevated CA‐II expression has been detected in rats with spontaneous hypertension and heart failure.10 Mice that develop angiotensin II–induced cardiac hypertrophy and age‐dependent dilated cardiomyopathy show increased expression of CA‐II and other CAs.11 Overexpression of a catalytically inactive dominant negative CA‐II suppresses the response to hypertrophic stimuli in rat cardiomyocytes in vitro.12 Furthermore, CA‐II–deficient (Car2) mice exhibit physiological cardiac hypertrophy without any decrease in cardiac function, and isolated cardiomyocytes from Car2 mice do not respond to prohypertrophic stimulation.12 Concurrently, there was a recent report that CA‐II and CA‐IV mRNA levels are significantly increased in hypertrophied and failing human hearts of ischemic and nonischemic origin, proposing CA‐II as biomarker for the early detection of myocyte hypertrophy and heart failure.13 CAs work with the Anion Exchange 3 Cl−/HCO3− exchanger and Na+/H+ exchanger 1 (NHE‐1) to promote cardiomyocyte hypertrophy.14 Indeed, cytosolic CA‐II activates NHE‐1,15 which is a cardiac‐specific integral membrane glycoprotein of the NHE family.14 Different forms of myocardial stress, including ischemia, lead to ROS generation and NHE‐1 hyperactivity, which results in further ROS generation and Ca2+ overload, myocardial dysfunction, hypertrophy, apoptosis, and failure.14,16 Recently, CA‐I increased concentration, and activity has been shown to be detrimental in diabetic retinopathy.17 However, the expression and activity status of CAs in ischemic myocardium of patients with DM are currently unknown.
In the present study, we assessed CA‐I and CA‐II expression in human cardiac samples from post‐MI patients with or without DM type 2 (DM‐T2). Here, we determined whether CA‐I myocardial expression correlates with capillary density and endothelial cell death in DM. Also, we evaluated NHE‐1 activation in human diabetic ischemic cardiomyopathy and its dependence from CA‐II activity in cardiomyocytes. Finally, we endeavored to uncover the specific molecular mechanisms underlying CA‐II modulation in DM.
Methods
Patient Selection and Cardiac Sample Collection
Left ventricular cardiac biopsy samples were derived from patients affected by post‐MI cardiomyopathy undergoing surgical coronary revascularization as described previously.18 For each patient, 6 biopsy samples were harvested: 3 from the infarct border area (peri‐infarct zone) and 3 from the nonischemic, remote myocardium (remote zone). Samples were either immediately snap‐frozen in liquid nitrogen and stored at −80°C until processed for RNA or protein extraction or formalin‐fixed for immunohistochemistry analysis. Patients with DM‐T2 (n=20) and without diabetes (NDM, n=20) were included in the study and did not differ significantly in any clinical parameter other than the presence of DM‐T2 (Table). All diabetic patients were treated with oral hypoglycemic agents and had an acceptable glycemic control (HbA1c <8%), and for 72 hours after surgery they received insulin therapy.
Table 1.
Characteristics of the Patients Enrolled in the Study
| Diabetic Patients, n=20 | Nondiabetic Patients, n=20 | |
|---|---|---|
| Sex, M/F | 10/10 | 10/10 |
| Age, y | 64.3±2.1 | 62.6±2.3 |
| Duration of diabetes, y | 9.2±1.1 | — |
| BMI, kg/m2 | 28.9±0.9 | 27.2±0.7 |
| HbA1c, % | 7.2±0.1 | 5.3±0.1* |
| FBG, mg/dL | 151.3±9.5 | 81.5±3.9* |
| LDL cholesterol, mg/dL | 132.3±7.1 | 128±5.1 |
| HDL cholesterol, mg/dL | 43.3±1.6 | 48.2±1.8* |
| TG, mg/dL | 167.5±10.3 | 126±8.5* |
| SBP, mm Hg | 133.9±3.7 | 127.3±3.2 |
| DBP, mm Hg | 76.3±2.1 | 74.1±2.4 |
| Three‐vessel CHD, n | 18 | 18 |
| Two‐vessel CHD, n | 2 | 2 |
| Therapy, n | ||
| Statins | 16 | 16 |
| Aspirin | 20 | 20 |
| β‐Blockers | 8 | 9 |
| ACEIs/ARBs | 15 | 14 |
| Nitrates | 16 | 15 |
Quantitative data are expressed as mean±SE. Binary data are reported by counts. *P<0.01 vs diabetic patients. Comparisons of the quantitative data have been made through use of Student's t test for independent samples. The χ2 test was used to compare binary data. BMI indicates body mass index; FBG, fasting blood glucose; LDL, low‐density lipoprotein; HDL, high‐density lipoprotein; TG, triglycerides; SDP, systolic blood pressure; DBP, diastolic blood pressure; CHD, coronary heart disease; ACEI, angiotensin‐converting enzyme‐inhibitor; ARB, angiotensin II receptor blocker.
Bioptic specimens were taken after informed consent disclosing future use for research. The investigation conformed to the principles outlined in the Helsinki Declaration and to Italian laws and guidelines and was authorized by the Ethical Committee of the Second University of Naples, Italy.
Histology and Immunohistochemical Analysis
Bioptic samples were washed with PBS and fixed in 10% formalin, and paraffin‐embedded. 5‐μm sections were prepared on a microtome (Leika) and mounted onto microscope slides.19–20
To identify and localize CA‐I, CA‐II, and NHE‐1, human cardiac sections were stained with antibodies against CA‐I, CA‐II, and NHE‐1 (anti‐human CA‐I antibody, Abcam; anti‐human CA‐II antibody, R&D Systems; rabbit polyclonal anti–NHE‐1, Santa Cruz Biotechnology). Cardiac myocytes were identified with antibodies against α–sarcomeric actin (Sigma), cardiac troponin I (Santa Cruz Biotechnology), or slow (cardiac) myosin heavy chain (Sigma).19–20 Endothelial cells and capillaries were detected by staining for von Wildebrand factor (vWF) (rabbit polyconal, Dako). Images were acquired using a confocal microscopy (Zeiss 710 LSM); 3 slides per sample were assessed.
To assess cardiomyocyte size, sections were stained with hematoxylin and eosin (H&E), according to standard procedures.20 Myocyte diameter was measured across the nucleus in 3 transverse H&E sections per sample, on a light microscope (Nikon E1000M) using Lucia G software. A total of 300 myocytes per section were analyzed for each sample.
The density of capillaries in the infarct region was evaluated by staining with an antibody against vWF (Dako).20 The secondary antibody used was a donkey anti‐rabbit, conjugated with horseradish peroxidase (Santa Cruz). Endogenous peroxidase in the section was blocked with 3% hydrogen peroxide in PBS for 15 minutes at room temperature. The chromogen 3,3′‐diaminobenzidine (DAB, Sigma) was used to visualize the blood vessels. The slides were counterstained with hematoxylin for identification of nuclei before being examined by using light microscopy. The number of capillaries (defined as 1 or 2 endothelial cells spanning the vWF‐positive vessel circumference) was determined by counting 10 fields per section at ×40 magnification. A total of 3 slides per sample were assessed. The amount of capillaries was expressed per mm2.
To detect myocyte cell apoptosis, sections were stained with rabbit anti‐human activated caspase‐3 primary antibody (R&D Systems).20 The chromogen DAB (Sigma) were used to visualize the apoptotic cardiomyocytes. Sections were then counterstained with hematoxylin and permanently mounted before being examined with light microscopy. Further, parallel staining using fluorescence detection was instituted to measure activated caspase‐3–positive apoptotic myocytes with confocal microscopy. The number of caspase‐3–positive myocytes was determined by counting 20 random fields per section at ×40 magnification. A total of 3 slides per sample were assessed. The amount of caspase‐3–positive myocytes was expressed as percentage relative to the total number of myocytes counted.
Endothelial cell apoptosis was detected by use of the terminal deoxynucleotidyl transferase (TdT) assay kit (Roche) or caspase 3 staining.19–20 Apoptotic endothelial cells were counterstained with a vWF antibody (Dako) and quantified using confocal microscopy.20
Immunoblots and Immunoprecipitation
For immunoblotting, cardiac samples (or cells) were homogenized and proteins were extracted as previously described.18 After PAGE, proteins were transferred and exposed to rabbit polyclonal CA‐II antibody (Abcam), mouse monoclonal CA‐I (Abcam), ERK1/2 (Cell Signaling) antibodies, and rabbit polyclonal p‐38, phospho‐p‐38, JNK, phospho‐JNK, and phospho‐ERK1/2 (Cell Signaling) antibodies. Blots were incubated with the primary antibody, at a concentration suggested by the manufacturers, overnight at 4°C in Tris‐buffered saline with Tween (TBS‐T), 5% nonfat dried milk. After extensive washing in TBS‐T, blots were incubated with the secondary antibody (horseradish peroxidase–conjugated anti‐mouse or anti‐rabbit immunoglobulins, Santa Cruz Biotechnology) at a dilution of 1:1000 for 45 minutes at room temperature. Specific proteins were detected by enhanced chemiluminescence (Amersham) and evaluated by using densitometry.
For immunoprecipitation of NHE‐1 and immunoblotting for phospho‐serine, myocardial protein extracts were incubated overnight at 4°C with mouse monoclonal anti–NHE‐1 (Santa Cruz Biotechnology). Subsequently, protein A–agarose was added. Immunoprecipitated proteins were separated on 8% SDS‐PAGE, transferred onto nitrocellulose filters, and exposed to rabbit polyclonal anti–phospho‐serine antibodies (Cell Signaling).19,21
Quantitative Real‐Time RT‐PCR
RNA was extracted from human cardiac samples or cultured cells using Qiagen RNeasy columns and was reverse transcribed using first‐strand cDNA synthesis with random or oligo‐dT primers (Applied Biosystems).18 Quantitative RT‐PCR (qRT‐PCR) was performed using SYBR Green (Bio‐Rad) on a MyIQ thermocycler (Bio‐Rad). The PCR included 2 μL of template cDNA and 300 nmol/L forward and reverse primers. PCR efficiency was evaluated by using a standard curve of 5 serial dilution points. Data were analyzed using Bio‐Rad IQ software, and mRNA was normalized to the housekeeping gene GAPDH. All reactions were carried out in triplicate. Primers were designed using the Primer 3 software, and the specific sequences are as follows (forward, followed by reverse primer in each case): rat GAPDH (5′‐CGTCTCATAGACAAGAT‐3′ and 5′‐TGATGGCAACAATGTCCACT‐3′), rat ANF (5′‐GGGGGTAGGATTGACAGGAT‐3′ and 5′‐GGATCTTTTGCGATCTGCTC‐3′), and rat CA‐II (5′‐ACCAGAGAACTGGCACAAGG‐3′ and 5′‐ATGAGCAGAGGCTGTAGGGA‐3′), human CA‐II (5′‐CAATGGTCATGCTTTCAACG‐3′ and 5′‐CCCCATATTTGGTGTTCCAG‐3′), human Actin (5′‐CCACGAAACTACCTTCAACTCC‐3′ and 5′‐TCATACTCCTGCTGCTTGCTGATCC‐3′), rat BNP (5′‐GACGGGCTGAGGTTGTTTTA‐3′ and 5′‐ACTGTGGCAAGTTTGTGCTG‐3′), and rat beta‐MHC (5′‐CCTCGCAATATCAAGGGAAA‐3′ and 5′‐TACAGGTGCATCAGCTCCAG‐3′).
Cell Culture and Reagents
Primary cultures of neonatal cardiomyocytes (nCMs) or adult ventricular cardiomyocytes (arCMs) were prepared from ventricles of 1‐ to 2‐day‐old or adult (≈9 weeks old) Wistar rats, respectively, as previously described.20,22 Freshly isolated cardiomyocytes were plated onto laminin (20 mg/mL)‐coated chamber slides at a density of 2×104 cells/cm2 and were cultured in DMEM, containing 10% FBS and 0.1% penicillin‐streptomycin, at 37°C, in an atmosphere containing 5% CO2. Human coronary endothelial cells (hCECs) were obtained from Cell Applications.
6‐Ethoxyzolamide (ETZ), [5‐(2‐methyl‐5‐fluorophenyl)furan‐2‐ylcarbonyl]guanidine (KR‐32568, an NHE‐1 inhibitor), and human recombinant CA‐I were obtained from Sigma.
Human CA‐II and rat CA‐IIshRNA plasmids were obtained from Origene, and short hairpin RNA (shRNA) construct to silence CA‐I (CA‐IshRNA) was purchased from Sigma–Aldrich. Efficiency of plasmid construct transfection was verified by green fluorescence protein (GFP) fluorescence of nCM or arCMs transfected with a plasmid encoding the GFP. An empty plasmid vector was used as additional control.
Endothelial Cell Permeability and Apoptotic Death
Human recombinant CA‐I protein (Sigma) was added at 100 μmol/L to hCEC culture in vitro. Permeability across endothelial cell monolayers was measured using gelatin‐coated Transwell units (Boyden chambers) (6.5‐mm diameter, 0.4‐μm‐pore polycarbonate filter; Corning Costar), as previously described.23 Briefly, hCECs were plated at a density of 0.6×105 cells (or 1×105 in the case of transduced cells) per well (upper chamber) and were cultured for 3 days until a tight monolayer formed. Endothelial permeability was determined by measuring the passage of FITC‐labeled dextran (1 mg/mL of fluorescein isothiocyanate‐dextran, molecular mass 40 kDa; Sigma–Aldrich) through the hCEC monolayer. Then, NG or HG (NG: low/normal, 5.5 mmol/L glucose; HG: high, 33 mmol/L glucose) was added to the upper chamber in the presence of 1 mg/mL FITC‐labeled dextran. After 30 minutes of treatment, 100 μL was collected from the lower compartment and fluorescence was evaluated using a Fluoroskan Ascent Microplate Fluorometer following the manufacturer's protocol (Thermo Scientific).
hCEC apoptosis were detected using the terminal deoxynucleotide transferase–mediated dUTP nick‐end labeling assay20 48 hours after NG or HG stimuli. Quantitative data are expressed as mean±SE from 4 independent experiments.
Cardiomyocyte Hypertrophy and Apoptosis In Vitro
nCMs were exposed to NG or HG in the presence or absence of ETZ (100 μmol/L) or the NHE‐1 inhibitor (10 μmol/L) for 48 hours. Hypertrophy was assayed by measurement of the cell surface area of cells. Cell surface area was measured before and after intervention with drugs. Images of cultured cardiomyocytes were collected with a QICAM fast cooled 12‐bit color camera (QImaging Corporation). Images were analyzed with Image‐Pro Plus software (Media Cybernetics) to measure cell surface area. In each group, surface areas were measured for 200 cells. Cell surface area (percent relative to control)=surface area (after treatment)/surface area (before treatment)×100. Additionally, nCMs were incubated in the same media with 1.0 μCi/mL [3H]leucine for 12 hours, and then processed for determination of incorporated radioactivity after precipitation with 10% TCA as described previously.24
Apoptotic arCMs were detected using the terminal deoxynucleotide transferase–mediated dUTP nick‐end labeling assay 20 48 hours after NG or HG stimuli. The number of positively stained cells was counted from 20 fields per slide.
When a plasmid construct was transfected, cell size, [3H]leucine, or apoptosis rate was measured 96 hours after initial transfection (48 hours transfection+48 hours NG or HG). Quantitative data are expressed as mean±SE from 4 independent experiments.
MicroRNA 23b Detection and Biological Function
Micro RNA (miRNA)/mRNA were extracted from cell culture or human cardiac samples as previously described.25–26 Specifically, for miRNA/mRNA extraction from isolated cells, the mirVana miRNA Isolation Kit (Ambion, Inc) was used while Trizol was used for miRNA/mRNA extraction from solid tissues. Specific cDNA was obtained using the high‐capacity cDNA Reverse transcription kit (Applied Biosystems). TaqMan MicroRNA Assays and TaqMan Gene Expression Assays (Applied Biosystem) were used to quantify miRNAs (miR‐23b and miR‐1) levels by qRT‐PCR (see earlier).
Precursor miRNA expression clone for rno‐mir‐23b (a pEZX‐MR04 vector expressing the specific precursor miR‐23b under the CMV promoter with an eGFP reporter gene) and miRNA inhibitor against rno‐miR‐23b (a pEZX‐AM04 vector expressing the specific miR‐23b inhibitor under the U6 promoter and a mCherry reporter gene) were purchased from Tebu‐Bio. A precursor miRNA scrambled control for pEZX‐MR04 and an miRNA inhibitor scrambled control clone for pEZX‐AM04 were also obtained from Tebu‐Bio. In addition, pre–miR‐23b, scrambled pre‐miR, anti–miR‐23b, or scrambled anti‐miR was introduced into nCMs or arVMS via reverse transfection using Lipofectamine 2000 (Invitrogen) as previously described.25–26 The miR or anti‐miR transfection was confirmed via eGFP or mCherry fluorescence and quantified by using qRT‐PCR.
Luciferase reporter and activity assay was performed as previously described.25 Briefly, wild‐type and mutant CA‐II 3′‐UTR sequences were cloned into luciferase‐reporter constructs and then separately cotransfected with scrambled miR precursor, miR‐23b precursor, scrambled anti‐miR, or anti–miR‐21b into C2C12 myoblasts using Lipofectamine 2000. Forty‐eight hours post‐transfection, luciferase activity was measured and normalized to β‐galactosidase activity.
All the in vitro experiments were performed in quadruplicate, and the data shown as mean±SE.
Statistics
Statistical analysis was performed with GraphPad Prism version 6.00 for Macintosh (GraphPad Software). Quantitative data are reported as mean±SE and binary data by counts. Significance between 2 groups was determined by Student's t test or paired t test as appropriate. For comparison between multiple groups, ANOVA was used. A P value <0.05 was considered significant. Bonferroni post‐hoc method was used to locate the differences. In these cases, the Type 1 error (α=0.05) was corrected by the number of statistical comparisons performed.
For the in vitro cell and molecular biology experiments with an n=4 sample size, giving the low number of the sample, the Kruskal–Wallis test (for multiple‐group comparison), and the Mann–Whitney U test (for comparison between 2 groups) were performed.
Results
CA‐I and CA‐II Are Increased in Diabetic Ischemic Cardiomyopathy
We first compared protein expression of CA‐I and CA‐II in the cardiac samples from NDM and DM‐T2 patients with ischemic cardiomyopathy by using Western blot analysis. CA‐I and CA‐II increased in the peri‐infarct zone compared with the remote area in both NDM and DM‐T2 (Figure 1A and 1B). More relevant, CA‐I and CA‐II were increased in DM‐T2 compared with NDM patients in both the remote zone and the peri‐infarct area (Figure 1A and 1B). Specifically, CA‐I and CA‐II increased by 6‐ and 11‐fold, respectively, in DM‐T2 versus NDM.
Figure 1.
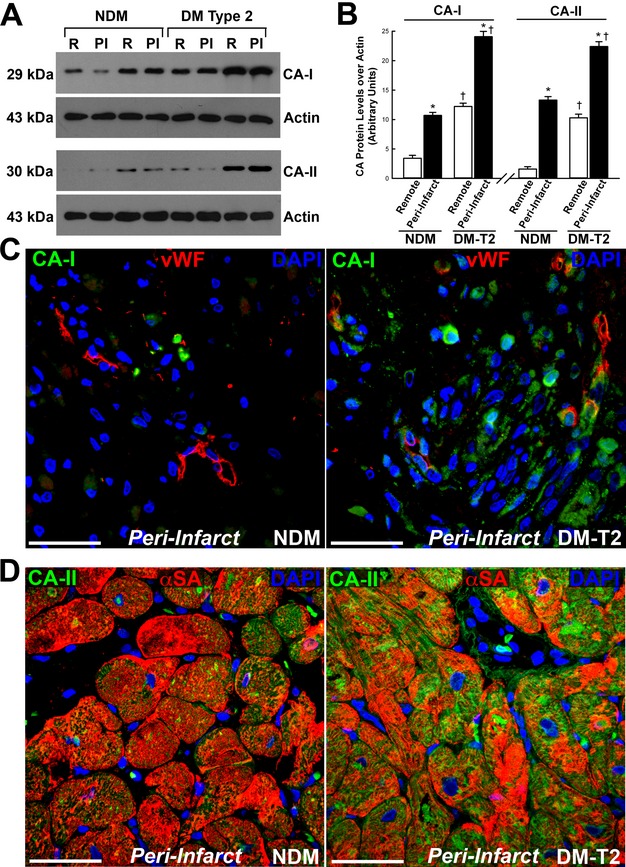
Carbonic anhydrase (CA)‐I and ‐II are increased in diabetic ischemic cardiomyopathy. A, Representative Western blot showing CA‐I and ‐II levels in the remote (R) and peri‐infarct (PI) zone of diabetes mellitus type 2 (DM‐T2) and non–diabetes mellitus (NDM) cardiac samples. B, Optical density (O.D.) analysis of CA‐I and ‐II protein levels in DM‐T2 compared with NDM patients; *P<0.0001 vs remote, †P<0.0001 vs NDM, n=20 per group. C, Representative confocal microscopy image showing CA‐I localization in capillary endothelial cells in the peri‐infarct area of cardiac samples from NDM and DM‐T2 patients (vWF, red fluorescence; CA‐I, green fluorescence; DAPI (4′,6‐diamidino‐2‐phenylindole), nuclei blue fluorescence); scale bar 50 μm. D, CA‐II was upregulated in cardiomyocytes from DM‐T2 compared with NDM (α‐SA, red fluorescence; CA‐II, green fluorescence; DAPI, nuclei blue fluorescence); scale bar 50 μm. Quantitative data are expressed as mean±SE. CA indicates carbonic anhydrases; vWF, von Willebrand factor.
CA‐I and CA‐II are prevalently cytosolic CA isoforms.8 We assessed CA‐I and CA‐II localization by using immunohistochemistry and confocal microscopy. An increased myocardial expression of CA‐I in DM‐T2 versus NDM patients was evident whereby CA‐I was prevalently detectable in the interstitial space (Figure 1C). However, CA‐I was also increased in capillary endothelial cells in the peri‐infarct area of cardiac samples from DM‐T2 compared with NDM patients (Figure 1C). On the other hand, CA‐II was mostly upregulated in cardiomyocytes from DM‐T2 compared with NDM patients (Figure 1D).
Finally, for a complete illustration of the data, all the blots related to CA‐I and CA‐II expression from each patient cardiac sample are accordingly presented with the relative semiquantitative analysis as box‐plot graphs (Figure 2A and 2B).
Figure 2.

Carbonic anhydrase (CA)‐I and‐II expression in all diabetic and nondiabetic patients included in the study. A, Western blot showing CA‐I and ‐II expression in the remote and peri‐infarct (Peri‐MI) zone of all diabetes mellitus type 2 (DM‐T2) and non–diabetes mellitus (NDM) cardiac samples included in the study. B, Box‐plot graphs showing optical density analysis of CA‐I and ‐II protein levels in DM‐T2 compared with NDM patients; *P<0.0001 vs remote, #P<0.0001 vs NDM, n=20 per group. MI indicates myocardial infarction.
CA I Mediates Endothelial Cell Death
Altered vascular permeability and endothelial dysfunction are hallmarks of diabetic microangiopathy.4 CA‐I is a key player in mediating vascular leakage diabetic retinopathy,17 while CA‐IV has also been linked to endothelial cell apoptosis in retinitis pigmentosa.27 We then measured the number of capillaries in the peri‐infarct zone of DM‐T2 and NDM patients. Consistent with the severe dysfunction of the microcirculation in ischemic heart disease with diabetes,4 we detected a significant decrease in capillary density in the peri‐infarct region in DM‐T2 compared with NDM patients (Figure 3A through 3C). Also, an increased rate of endothelial cell apoptosis in DM‐T2 versus NDM patients was detected (Figure 3D and 3E). Thus, we tested whether CA‐I could directly affect endothelial cell survival in vitro. hCECs were challenged with CA‐I in vitro, and this treatment significantly increased hCEC death at levels comparable to those for HG (Figure 3F). Concurrently, CA‐IshRNA significantly reduced HG‐induced hCEC apoptosis in vitro (Figure 3F). Also, we ascertained whether CA‐I induced an increase in endothelial cell permeability. hCECs cultivated as tight monolayers in Boyden chambers were challenged with CA‐I for 90 minutes, and cell permeability was determined by measuring the passage of FITC‐labeled dextran across the endothelial cell monolayer. HG and CA‐I increased hCEC permeability and CA‐IshRNA prevented the HG‐induced effect (Figure 3G). Thus, these data suggest that the increased myocardial expression of CA‐I in ischemic myocardium of diabetic patients alters endothelial permeability and increases endothelial cell death with a net reduction in capillary density. Additionally, it appears that microvascular dysfunction and leakage in DM sustain CA‐I accumulation in the cardiac interstitium (Figure 1C) through an increased ischemic hemorrhage and consequent erythrocyte lysis.28
Figure 3.
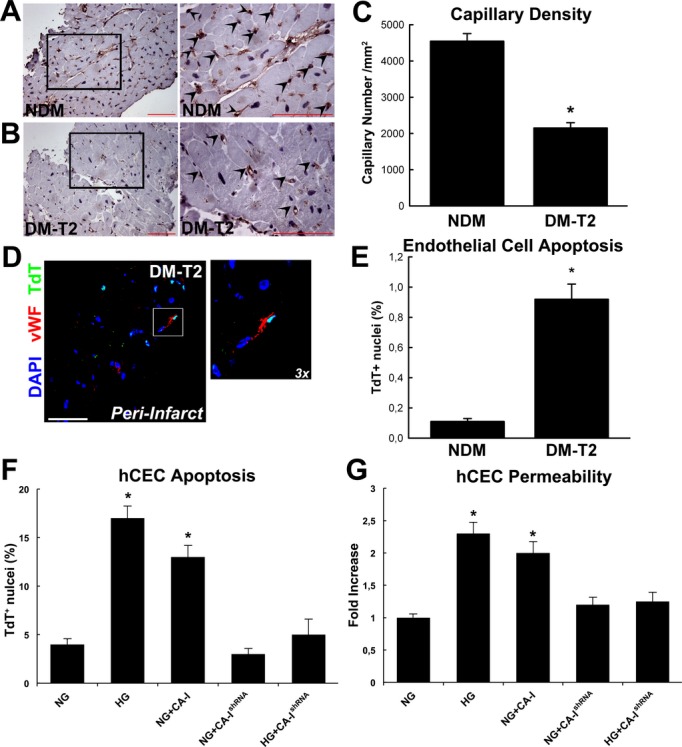
Carbonic anhydrase (CA)‐I and decreased capillary density in diabetes mellitus type 2 (DM‐T2). A, Representative DAB staining (vWF, brown) in the peri‐infarct showing capillary density in non–diabetes mellitus (NDM); right panel shows higher magnification of the inset from left panel. B, Representative DAB staining (vWF, brown) showing capillary density in DM‐T2; right panel shows higher magnification of the inset from left panel. Scale bar 50 μm. C, Myocardial capillary density was decreased in DM‐T2 compared with NDM patients. *P<0.0001 vs NDM, n=20 per group. D, Representative confocal microscopy image of TdT‐positive apoptotic endothelial cells in DM‐T2 peri‐infarct zone (vWF, white fluorescence; TdT, green fluorescence; DAPI, nuclei blue fluorescence). Scale bar 50 μm. E, Bar graph showing increased endothelial cell apoptosis in DM‐T2 vs NDM patients; *P<0.0001 vs NDM, n=20 per group. F, CA‐I treatment and CA‐IshRNA transfection on hCEC death as assessed by TdT staining; *P<0.0001 vs normal glucose (NG), NG+CA‐IshRNA, and high glucose+CA‐IshRNA (HG+CA‐IshRNA), n=4 per group. G, CA‐I increased hCEC permeability in the Boyden chamber in vitro *P<0.001 vs normal glucose (NG), NG+CA‐IshRNA, and high glucose+CA‐IshRNA (HG+CA‐IshRNA), n=4 per group. Quantitative data are expressed as mean±SE. hCEC indicates human coronary endothelial cells; DAB, 3,3′‐diaminobenzidine; Tdt, terminal deoxynucleotidyl transferase; vWF, von Willebrand factor.
Increased Myocyte CA‐II Hyperphosphorylates the NHE‐1, Worsening Myocyte Hypertrophy and Death in Diabetic Ischemic Cardiomyopathy
One of the main targets of CA‐II activity in cardiomyocytes is the NHE‐1.14 Thus, we measured, through immunoprecipitation and immunoblotting, the serine‐phosphorylation status of NHE‐1 in diabetic versus nondiabetic myocardial samples. NHE‐1 protein expression was increased in both remote and peri‐infarct areas of DM‐T2 samples compared with NDM samples (Figure 4A through 4D). More importantly, NHE‐1 was hyperphosphorylated in T2‐DM versus NDM (Figure 4A through 4D). The increased phosphorylation was confirmed when normalizing for NHE‐1 total protein levels (Figure 4A through 4D). Immunofluorescence and confocal microscopy detected NHE‐1 expression specifically within myocytes that was increased in DM‐T2 versus NDM samples (Figure 4E and 4F).
Figure 4.

Myocyte NHE‐1 is hyper‐phosphorylated in diabetic ischemic cardiomyopathy. A, Representative WB analysis showing NHE‐1 protein expression and phosphorylation (immunoprecipitation, IP, with NHE‐1 antibody and immunoblot, IB, with phospho‐serine, p‐ser, antibody) in both remote and peri‐infarct areas of DM‐T2 samples compared with NDM. B, Bar graph showing optical density analysis of NHE‐1 protein expression n DM‐T2 and NDM samples; *P<0.0001 vs remote, #P<0.0001 vs NDM, n=20 per group. C, Bar graph showing optical density analysis of NHE‐1 phosphorylation levels in DM‐T2 and NDM samples; *P<0.04 vs remote NDM, **P<0.0001 vs remote DM‐T2, #P<0.0001 vs NDM, n=20 per group. D, Bar graph showing optical density analysis of phosphorylated NHE‐1 to total NHE‐1 ratio DM‐T2 and NDM samples; *P<0.0001 vs remote, #P<0.0001 vs NDM, n=20 per group. E and F, Representative confocal microscopy images showing NHE‐1 localization and increased expression within myocytes in DM‐T2 vs NDM samples. Scale bar 20 μm. G and H, Bar graphs showing cardiomyocyte size and apoptosis in peri‐infarct areas of DM‐T2 samples compared with NDM; *P<0.01 vs NDM, n=20 per group. (I through L) Representative confocal microscopy and DAB‐staining light microscopy images showing an activated caspase‐3 (Act‐Casp‐3)–positive apoptotic myocyte in the peri‐infarct zone of a DM‐T2 sample compared with NDM. Quantitative data are expressed as mean±SE. DAB indicates 3,3′‐diaminobenzidine; DM‐T2, diabetes mellitus type 2; NDM, non–diabetes mellitus; NHE‐1, sodium‐hydrogen exchanger‐1; WB, Western blot.
Because NHE‐1 overexpression and hyperactivity mediate maladaptive myocardial hypertrophy and death,14,16 we then measured myocyte size and apoptosis in the samples from patients included in the study. Myocyte hypertrophy and apoptosis, evaluated by cell size and activated caspase‐3 staining, respectively, were increased in peri‐infarct areas of DM‐T2 samples compared with NDM samples (Figure 4G through 4L).
Rat neonatal or adult cardiac myocytes (nCMs or arCMs, respectively) were then isolated, transfected with a CA‐II or empty plasmid (Figure 5A), and exposed to NG or HG concentration in the presence or absence of a specific NHE‐1 inhibitor, KR‐32568, in vitro. CA‐II induced CM hypertrophic growth assessed by cell size measurement and [3H]leucine incorporation at levels similar to HG treatment (Figure 5B and 5C). CA‐II inhibition by ETZ administration reduced nCM hypertrophy induced by HG, while NHE‐1 inhibition (KR‐32568, named as NHE‐1‐i) prevented nCM hypertrophy induced by CA‐II overexpression (Figure 5B and 5C). ETZ and NHE‐1‐i, respectively reduced HG‐ and CA‐II–induced molecular hypertrophic response (Figure 5B and 5C). ANF, BNP, and β‐MHC mRNAs, markers of cardiac hypertrophy, were upregulated in nCMs treated with HG or CA‐II overexpression (Figure 5D). Furthermore, HG and CA‐II overexpression increased adult rat ventricular myocyte (arCM) apoptosis as measured by TdT staining (Figure 5E). The latter was attenuated by ETZ or NHE‐1 inhibition, respectively (Figure 5E). Finally, a specific shRNA silencing CA‐II expression (CA‐IIshRNA) was transfected in CMs and it blocked NHE‐1 phosphorylation by HG (Figure 5F).
Figure 5.
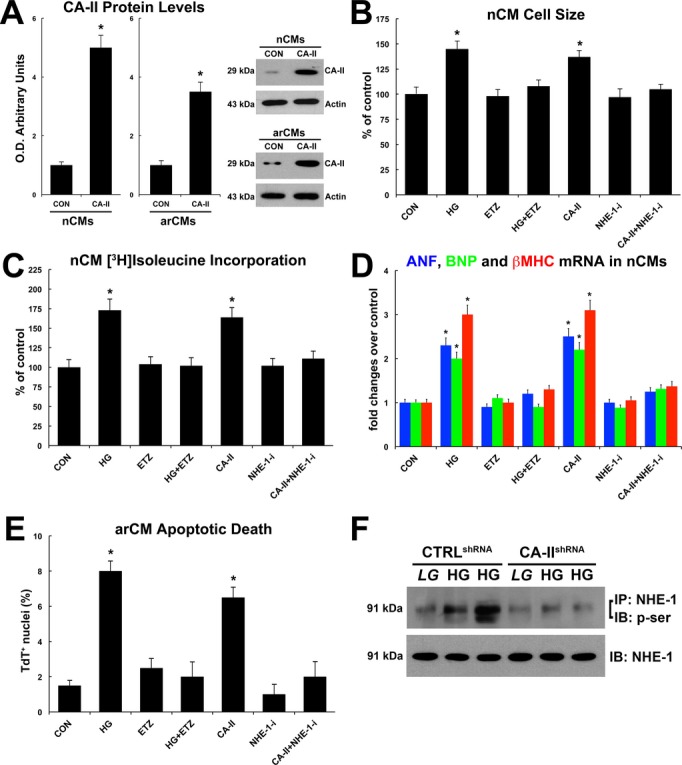
Carbonic anhydrase (CA)‐II exacerbates cardiomyocyte hypertrophy and death in vitro. A, CA‐II overexpression in neonatal and adult cardiomyocytes (nCMs and arCMs, respectively); *P<0.001 vs control empty plasmid (CON). B, Effects of normal/low glucose (CON), high glucose (HG), CA‐II overexpression, CA‐II inhibition by ETZ, and specific NHE‐1 inhibitor (NHE‐1‐i) on nCM cell size; *P<0.004 vs CON, ETZ, HG+ETZ, NHE‐1‐i and CA‐II+NHE‐1‐i. When not specified, drug treated cells were grown in normal/low glucose. C, Effects of normal/low glucose (CON), high glucose, CA‐II overexpression, CA‐II inhibition by ETZ, and specific NHE‐1 inhibitor (NHE‐1‐i) on nCM hypertrophic growth assessed by [3H]leucine incorporation; *P<0.001 vs CON, ETZ, HG+ETZ, NHE‐1‐I, and CA‐II+NHE‐1i. D, Effects of normal/low glucose (CON), high glucose, CA‐II overexpression, CA‐II inhibition by ETZ, and specific NHE‐1 inhibitor (NHE‐1‐i) on ANF, BNP and β‐MHC mRNA levels in nCMs; *P<0.001 vs CON, ETZ, HG+ETZ, NHE‐1‐i and CA‐II+NHE‐1i. E, Effects of normal/low glucose (CON), high glucose, CA‐II overexpression, CA‐II inhibition by ETZ, and specific NHE‐1 inhibitor (NHE‐1‐i) on arCM apoptosis; *P<0.001 vs CON, ETZ, HG+ETZ, NHE‐1‐i and CA‐II+NHE‐1i. F, Effects of scrambled control shRNA (CTRLshRNA) and specific CA‐II shRNA (CA‐IIshRNA) on glucose‐induced NHE‐1 phosphorylation. All quantitative data are from 4 independent experiments. Quantitative data are expressed as mean±SE. EZT indicates 6‐ethoxyzolamide; NHE‐1, sodium‐hydrogen exchanger‐1.
Thus, taken together, these data suggest that HG increases myocyte CA‐II expression, which hyperactivates NHE‐1, leading to increased maladaptive myocyte growth and apoptotic death in ischemic diabetic cardiomyopathy.
miRNA‐23b Is Downregulated in Cardiomyocytes of Diabetic Ischemic Cardiomyopathy
The upregulation of CA‐II expression within cardiac myocytes in diabetic samples raised the question on how this is regulated. MicroRNAs (miRNAs) are master regulators of gene expression in plant and mammalian cells,29 and it was logical to scan miRNA libraries for the different miRNAs potentially involved in CA‐II expression regulation. From the analysis of the CA‐II 3′‐UTR by using the TargetScan algorithm, we identified miRNA‐23b, which has recently been linked to the development of immune diseases and other disorders.30–31 miR‐23b has been proposed to be a key regulator of cell cycle, migration, apoptosis, and differentiation.30–33 Yet, little is known about miR‐23b expression and function in the adult diabetic heart.
We first evaluated miR‐23b expression in rat hearts compared with other adult tissues. miR‐23b was detectable in the heart at levels comparable with the lungs and expressed more compared with the liver, kidney, spleen, and brain (Figure 6A). In particular, miR‐23b was enriched in rodent cardiomyocytes compared with cardiac endothelial cells, smooth muscle cells, and fibroblasts (Figure 6B). However, miR‐23b myocyte levels were lower than known cardiac‐enriched miRNAs, like miR‐1 (Figure 6C). We then measured miR‐23b expression in the cardiac samples from DM‐T2, compared with NDM patients. Intriguingly, miR‐23b was downregulated in the remote zone and even more in the peri‐infarct zone of DM‐T2 compared with NDM (Figure 6D). Accordingly, 48 hours of HG treatment of both nCMs and arCMs significantly reduced miR‐23b levels while increasing CA‐II mRNA and protein levels in vitro (Figure 6E and 6F).
Figure 6.

MicroRNA‐23b (miR‐23b) is downregulated in cardiomyocytes of diabetic ischemic cardiomyopathy. A, miR‐23b expression in rat hearts as compared with other adult tissues, *P<0.0001 vs other tissues. B, miR‐23b levels in nCMs and arCMs compared with aortic endothelial cells (AoECs) and smooth muscle cells (AoSMCs). *P<0.0001 vs AoECs, AoSMCs, and nCMs. C, miR‐23b and miR‐1 levels in nCMs and arCMs; *P<0.0001 vs miR‐1, #P<0.01 vs nCMs. D, miR‐23b levels in the remote zone in the peri‐infarct zone of DM‐T2 compared with NDM; *P<0.0001 vs NDM, #P<0.0001 vs remote zone. E, The 48‐hour high glucose (HG) treatment of both NCMs and arCMs significantly reduced miR‐23b levels compared with low/normal glucose controls (CON); *P<0.001 vs CON. F, The 48‐hour HG treatment of both nCMs and arCMs significantly increased CA‐II mRNA levels in vitro compared with low/normal glucose controls (CON); *P<0.001 vs CON. All quantitative data are from 4 independent experiments. Quantitative data are expressed as mean±SE. arCM indicates adult ventricular cardiomyocyte; DM‐T2, diabetes mellitus type 2; nCM, neonatal cardiomyocyte; NDM, non–diabetes mellitus.
Thus, these data show that miR‐23b is normally expressed in healthy mammalian myocardium and that it is significantly downregulated in the stressed myocardium of diabetic ischemic cardiomyopathy. As miR‐23b putatively targets CA‐II, it may be a main player in the detrimental effect of CA‐II overexpression in diabetic cardiomyopathy.
miR‐23b Directly Targets CA‐II Expression and Modulates HG‐Induced Myocyte Hypertrophy and Death In Vitro
We first assessed the effects of miR‐23b gain and loss of function on myocyte hypertrophy in nCMs and death in arCMs in vitro. nCMs or arCMs were transfected with an miR‐23b precursor or an miR‐23b inhibitor (anti–miR‐23b) in vitro. miR‐23b precursor overexpression significantly increased the expression of miR‐23b in both nCMs and arCMs (Figure 7A). miR‐23b overexpression reduced HG‐induced nCM hypertrophic growth, while miR‐23b inhibition per se induced nCM growth in vitro (Figure 7B and 7C). The latter was counteracted by ETZ treatment (Figure 7B and 7C). Accordingly, miR‐23b overexpression reduced HG‐induced arCM apoptosis, while EZT treatment prevented anti–miR‐23b–induced arCM apoptotic death in vitro (Figure 7D). mRNA levels of CA‐II were reduced or increased when overexpressing or inhibiting miR‐23b, respectively (Figure 7E). Importantly, miR‐23b overexpression reduced HG‐induced CA‐II expression in nCMs (Figure 7E). Moreover, a luciferase reporter fused to the 3′ untranslated region (UTR) of CA‐II was repressed by miR‐23b transfection in mouse C2C12 myoblast cells, and this repression was abolished when the miR‐23b seed‐complementary sequence was mutated (Figure 7F). Finally, a CA‐II construct lacking the UTRs (CA‐II‐1–Δ3′‐UTR), and therefore not targeted by miR‐23b, completely reverted the reduced myocyte hypertrophy that we observed after miR‐23b overexpression in HG‐treated nCMs in vitro (Figure 7G).
Figure 7.

MicroR‐23b (miR‐23b) directly targets CA‐II expression and modulate myocyte hypertrophy and death in vitro. A, miR‐23b precursor transfection increased miR‐23b levels in nCMs and arCMs in vitro compared with miR‐scrambled precursor (CON); *P<0.001 vs CON. B, Effects of normal/low glucose (CON), high glucose (HG), ETZ, miR‐23b precursor, and anti–miR‐23b on nCM hypertrophic growth assessed by [3H]leucine incorporation; *P<0.03 vs CON, ETZ, miR‐23b, HG+miR‐23b, and anti–miR23b+ETZ. When not specified, drug treated or transfected cells were grown in normal/low glucose. C, Effects of normal/low glucose (CON), high glucose (HG), ETZ, miR‐23b precursor, and anti–miR‐23b on ANF, BNP, and β‐MHC mRNA levels in nCMs in vitro; *P<0.03 vs CON, ETZ, miR‐23b, HG+miR‐23b, and anti–miR23b+ETZ. D, Effects of normal/low glucose (CON), high glucose (HG), ETZ, miR‐23b precursor, and anti–miR‐23b on arCM apoptotic death in vitro; *P<0.01 vs CON, ETZ, miR‐23b, HG+miR‐23b, and anti–miR23b+ETZ. E, mRNA levels of CA‐II in nCMs cultured in low/normal glucose (CON) or high glucose (HG) and transfected with miR‐23b precursor or anti–miR‐23b; *P<0.001 vs CON; #P<0.03 vs all. F, Luciferase activity of wild‐type (WT) and mutant (Mut) CA‐II 3′‐UTR sequences separately co‐transfected with miR‐23b precursor or anti–miR‐23b into C2C12 myoblasts. *P<0.01 vs all. G, Effects of normal/low glucose (CON), high glucose (HG), miR‐23b precursor, and a mutant CA‐II construct (lacking the 3′‐UTRs, CA‐IImut) on nCM cell size in vitro; *P<0.003 vs all, #P<0.03 vs HG. All quantitative data are from 4 independent experiments. Quantitative data are expressed as mean±SE. arCM indicates adult ventricular cardiomyocyte; EZT, 6‐ethoxyzolamide; nCM, neonatal cardiomyocyte; UTR, untranslated region.
Thus, these data show that miR‐23b directly regulates CA‐II expression and suggest that miR‐23 downregulation releases CA‐II within myocytes, leading to increased myocyte hypertrophy and death in DM‐T2.
miR‐23b Downregulation Is Dependent on p38 Mitogen‐Activated Protein Kinase Pathway
The mitogen‐activated protein kinase (MAPK) activation is regulated by miR‐23b.34 Accordingly, maladaptive myocyte hypertrophy and increased myocyte death in diabetic cardiomyopathy are regulated by p38 MAPK activity.34–36 Thus, to assess the relationship between miR‐23b and MAPK signaling pathway in cardiac myocytes and DM‐T2, the activation of p38 MAPK, ERK1/2, and JNK was evaluated by Western blotting both in the myocardial samples from DM‐T2 and NDM patients and in HG‐treated CMs in vitro. As shown in Figure 8A, the 3 MAPKs were significantly phosphorylated in the myocardial samples from DM‐T2 compared with NDM. Also, HG treatment significantly increased the phosphorylation levels of p38, ERK1/2, and JNK in nCMs and arVMs in vitro (Figure 8B and 8C). To further explore the functional relationship between miR‐23b and p38, ERK1/2, and JNK activity, we used specific inhibitors of the 3 MAPKs, SB‐203580, SP‐600125, and PD‐98059, respectively, to test whether inhibition of p38, ERK1/2, and JNK could influence the effect of miR‐23b on cardiomyocyte growth and survival. Of note, the p38 MAPK inhibitor SB203580 significantly increased miR‐23b while reducing CA‐II levels in HG‐treated nCMs and arCMs, whereas SP600125 or PD98059 (JNK and ERK1/2 inhibitors, respectively) had no such effect (Figure 8D and 8E). Concurrently, SB203580 reduced HG‐induced nCM hypertrophy and arCM apoptotic death (Figure 8F and 8G).
Figure 8.

MicroR‐23b (miR‐23b) downregulation is dependent on p38 MAPK pathway. A, p38 MAPK, ERK1/2, and JNK were significantly more phosphorylated in the myocardial samples from T2‐DM compared with NDM as shown by representative Western blots and O.D. semiquantitative analysis; *P<0.001 vs NDM, n=20 per group. B, High glucose (HG) significantly increased the phosphorylation levels of p38, ERK1/2, and JNK in nCMs in vitro. *P<0.001 vs normal glucose (NG), n=4 per group. C, HG significantly increased the phosphorylation levels of p38, ERK1/2, and JNK in arCMs in vitro. *P<0.0001 vs NG, n=4 per group. D, SB‐203580, PD‐98059, and SP‐600125 (p38, ERK1/2, and JNK specific inhibitors, respectively) effects on miR‐23b levels in high‐glucose (HG) treated nCMs or arCMs in vitro. *P<0.001 vs normal glucose (NG). E, SB‐203580, PD‐98059, and SP‐600125 effects on CA‐II mRNA levels in high‐glucose (HG)‐treated nCMs or arCMs in vitro. *P<0.03 vs normal glucose (NG), n=4 per group. F, SB‐203580 reduced HG‐induced nCM growth in vitro. *P<0.003 vs normal glucose (CON), n=4 per group. G, SB‐203580 reduced HG‐induced arCM apoptosis in vitro. *P<0.0008 vs normal glucose (CON), n=4 per group. Quantitative data are expressed as mean±SE. DM‐T2 indicates diabetes mellitus type 2; nCM, neonatal cardiomyocyte; arCM, adult ventricular cardiomyocyte; NDM, non–diabetes mellitus.
Overall, these data point to a molecular mechanism whereby HG concentration in DM triggers MAPK cascade in CMs leading to p38 activation, which downregulates miR‐23b that removes its repression on CA‐II. Increased levels of CA‐II hyperphosphorylate NHE‐1, leading to the worsened maladaptive cardiomyocyte growth and ensuing death in diabetic cardiomyopathy.
Discussion
The main findings emanating from this study are that (1) CA‐I and CA‐II are overexpressed in diabetic ischemic human myocardium; (2) CA‐I is involved in the reduced capillary density in the ischemic diabetic myocardium; (3) increased myocyte CA‐II is associated with NHE‐1 hyperphosphorylation in diabetic ischemic cardiomyopathy; (4) miRNA‐23b directly targets CA‐II for repression, and it is downregulated in cardiomyocytes of diabetic ischemic cardiomyopathy; and (5) miR‐23b regulation by hyperglycemia depends on p38 MAPK activity and modulates HG‐induced myocyte hypertrophy and death in vitro.
Burgeoning evidences point to a detrimental prohypertrophic role of CA‐II in rodent and human hearts.12–13 In the clinical setting, it is worth remembering that the membrane‐permeant CA inhibitor acetazolamide (ACTZ) has been historically used as a diuretic (before the introduction of loop diuretics) in patients with severe congestive heart failure,37 and the status of congestive heart failure in all patients receiving ACTZ at that time substantially improved.38 Recently, ACTZ has been safely used in pediatric patients with heart disease.39 Moreover, the GUARDIAN Trial 40 and the ESCAMI Trial,41 assessing the effects of NHE‐1 inhibition in ischemic cardiomyopathy, have shown promising results. Here we show that HG‐induced myocyte hypertrophy and death correlate with CA‐II expression and are prevented by NHE‐1 inhibition. In some studies, increased cardiac expression of NHE‐1 protein appears to be involved in the subsequent pathological changes.42 In human heart failure, however, enhanced NHE‐1 activity has not been correlated with increased NHE‐1 expression, suggesting a role for activation by posttranslational mechanisms.43 It is interesting to note that in the present study, we show that diabetic cardiomyopathy is characterized by an increased NHE1 protein expression along with its exaggerated posttranslation phosphorylation. Thus, the 2 mechanisms appear to have an additive detrimental effect on the maladaptive cardiomyocyte growth and ensuing death in the diabetic myocardium.
A decrease in capillary density due to an increase in endothelial cell apoptosis in the heart is implicated in cardiac ischemia in DM.28 These perturbations in vascular cell homeostasis are linked to the development of pathologies affecting large‐vessel (atherosclerosis and cardiomyopathy) and small‐vessel (retinopathy, nephropathy, and neuropathy) diabetic complications. Recently, it has been shown that increased extracellular carbonic CA‐I leads to an excessive hemorrhagic retinal vascular permeability contributing to the pathogenesis of proliferative diabetic retinopathy.17 Intriguingly, we show here that CA‐I is overly expressed in the human diabetic ischemic myocardium, and CA‐I appears to mediate HG‐induced altered endothelial permeability as well as the decreased capillary density in diabetic ischemic cardiomyopathy. Furthermore, the increase in CA‐I expression in the interstitium suggests that other cardiac cell types could be involved in CA‐I overexpression. While no strong evidence is available on a direct role of CA‐I on cardiac fibroblasts, CA‐I (as well as other CAs) is expressed by human monocytes,8 which have an established role in cardiac inflammation and fibrosis in the ischemic myocardium. Thus, it is highly tempting to speculate a potential role for CA‐I in cardiac fibrosis to be tested in future studies. Overall, these observations postulate that CA‐I inhibition, currently clinically investigated for the treatment of diabetic retinopathy, may be a potential novel therapeutic approach for diabetic cardiovascular disease.
Increasing evidence indicates that miRNAs regulate key genetic programs in cardiovascular biology, physiology, and disease.44 In particular, several miRNAs are responsible for the pathogenesis of post‐ischemic heart failure, and recently a set of miRNAs have been found to be deregulated in experimental diabetes 34 and in diabetic versus nondiabetic patients with post‐MI heart failure.45 Thus, miRNA network dysregulation is viewed as a gatekeeper underlying the specific ischemic cardiomyopathy mechanisms differentiating diabetic patients.46–47 In the present report, we show for the first time that miR‐23b is specifically downregulated in the cardiac samples from diabetic patients with ischemic cardiomyopathy. miR‐23b levels inversely correlate with CA‐II, and we demonstrated that CA‐II is a bona fide target of miR‐23b gene‐silencing activity, which is removed via HG concentration through a molecular mechanism involving p38 activation. It is also interesting that miR‐23b has a potent immune modulatory role in different diseases.30–31 Thus, it is possible that miR‐23b downregulation in the ischemic diabetic myocardium could contribute to the detrimental immune activation in DM.4 Moreover, our data raise the hypothesis that miR23b modulation in vivo could affect CA‐II regulation and post‐MI remodeling. The latter should be directly tested in future studies by myocyte‐specific miR‐23b gain and loss of function in genetically modified animals.
Finally, it is highly tempting to speculate the potential clinical relevance of the findings from the present study. Indeed, CA inhibition with existing drugs could be readily and specifically adopted for diabetic patients with post‐MI heart failure as adjuvant diuretic therapy, on one hand, and as specific treatment for the exaggerated pathological remodeling of the ischemic diabetic heart, on the other. Nevertheless, further studies are warranted to test experimentally such hypothesis before it is tested in the clinical human scenario.
Limitations
In the present study, we have obtained strong evidence that CA‐I and CA‐II are significantly induced in the diabetic human myocardium compared with nondiabetic heart tissues. However, as it was impossible for ethical and logical reasons to obtain left ventricular tissues from matched healthy donors, no direct data could be provided to actually show that ischemic heart disease itself affects CA expression by comparing ischemic tissues in diabetic or nondiabetic patients with nonischemic tissues. Despite that, the increased CA expression in the peri‐infarct versus the remote zone provides a good indication that ischemia is indeed an inducer of myocardial CA‐I and CA‐II expression. Also, we would like to point out that the recent report by Alvarez et al has already shown that failing human hearts (of ischemic and nonischemic origin) of nondiabetic patients are characterized by an increased CA‐II expression.13
We have not performed an animal study using a diabetic animal model to directly provide a causal relationship for our findings on human samples. However, previous studies have established that CA plays a major role in pathological cardiac remodeling in different animal models.9–12 Yet, the extensive in vitro gain‐ and loss‐of‐function data on endothelial cells and neonatal and adult myocytes supports a causal relationship for the observations made on human samples, which should be verified in future studies by the indisputable and additional value of in vivo animal experiments.
In the present study, we have endeavored to specifically explain how DM regulates CA‐II increase in cardiomyocytes while we have not experimentally tested the mechanisms underlying CA‐I–increased expression by DM. Our findings show that hyperglycemia through a p38/miR‐23b–dependent mechanism increases CA‐II in myocytes. In Figure 2, we show that CA‐I inhibition prevents hyperglycemia‐induced endothelial cell death and altered permeability. The latter finding suggests that hyperglycemia is a strong inducer of CA‐I in endothelial cells. It is known that hyperglycemia induces MAPK‐p38 activation in endothelial cells and that p38 inhibition prevents endothelial cell damage by hyperglycemia.48 Thus, this evidence also suggests that a mechanism similar to that underlying CA‐II increase in myocytes by DM may also explain CA‐I increase in endothelial cells by DM. Of course, this hypothesis remains highly speculative, and it remains to be properly tested in future studies.
Sources of Funding
This work was in part supported by the Italian Ministry of Education, University and Research (MIUR) FIRB‐Futuro‐in‐Ricerca (RBFR081CCS and RBFR12I3KA) and the Italian Ministry of Health (Ricerca Finalizzata GR‐2008‐1142673 and GR‐2010‐2318945).
Disclosures
None.
References
- 1.Sattar N. Revisiting the links between glycaemia, diabetes and cardiovascular disease. Diabetologia. 2013; 56:686-695 [DOI] [PubMed] [Google Scholar]
- 2.Berry C, Tardif JC, Bourassa MG. Coronary heart disease in patients with diabetes. Part I: recent advances in prevention and non‐invasive management. J Am Coll Cardiol. 2007; 49:631-642 [DOI] [PubMed] [Google Scholar]
- 3.Dries DL, Sweitzer NK, Drazner MH, Stevenson LW, Gersh BJ. Prognostic impact of diabetes mellitus in patients with heart failure according to the etiology of left ventricular systolic dysfunction. J Am Coll Cardiol. 2001; 38:421-428 [DOI] [PubMed] [Google Scholar]
- 4.Boudina S, Abel ED. Diabetic cardiomyopathy revisited. Circulation. 2007; 115:3213-3223 [DOI] [PubMed] [Google Scholar]
- 5.Khavandi K, Khavandi A, Asghar O, Greenstein A, Withers S, Heagerty AM, Malik RA. Diabetic cardiomyopathy—a distinct disease? Best Pract Res Clin Endocrinol Metab. 2009; 23:347-360 [DOI] [PubMed] [Google Scholar]
- 6.Boudina S, Bugger H, Sena S, O'Neill BT, Zaha VG, Ilkun O, Wright JJ, Mazumder PK, Palfreyman E, Tidwell TJ, Theobald H, Khalimonchuk O, Wayment B, Sheng X, Rodnick KJ, Centini R, Chen D, Litwin SE, Weimer BE, Abel ED. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation. 2009; 119:1272-1283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seddon M, Looi YH, Shah AM. Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart. 2007; 93:903-907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov. 2008; 7:168-181 [DOI] [PubMed] [Google Scholar]
- 9.Alvarez BV, Johnson DE, Sowah D, Soliman D, Light PE, Xia Y, Karmazyn M, Casey JR. Carbonic anhydrase inhibition prevents and reverts cardiomyocyte hypertrophy. J Physiol. 2007; 579:127-145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sharkey LC, McCune SA, Yuan O, Lange C, Fray J. Spontaneous pregnancy‐induced hypertension and intrauterine growth restriction in rats. Am J Hypertens. 2001; 14:1058-1066 [DOI] [PubMed] [Google Scholar]
- 11.Domenighetti AA, Wang Q, Egger M, Richards SM, Pedrazzini T, Delbridge LM. Angiotensin II‐mediated phenotypic cardiomyocyte remodeling leads to age‐dependent cardiac dysfunction and failure. Hypertension. 2005; 46:426-432 [DOI] [PubMed] [Google Scholar]
- 12.Brown B, Quon A, Casey JR. Carbonic anhydrase II promotes cardiomyocyte hypertrophy. Can J Physiol Pharmacol. 2012; 90:1599-1610 [DOI] [PubMed] [Google Scholar]
- 13.Alvarez BV, Quon AL, Mullen J, Casey JR. Quantification of carbonic anhydrase gene expression in ventricle of hypertrophic and failing human heart. BMC Cardiovasc Disord. 2013; 13:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cingolani HE, Ennis IL. Sodium‐hydrogen exchanger, cardiac overload, and myocardial hypertrophy. Circulation. 2007; 115:1090-1100 [DOI] [PubMed] [Google Scholar]
- 15.Li X, Alvarez B, Casey JR, Reithmeier RA, Fliegel L. Carbonic anhydrase II binds to and enhances activity of the Na+/H+ exchanger. J Biol Chem. 2002; 277:36085-36091 [DOI] [PubMed] [Google Scholar]
- 16.Nakamura TY, Iwata Y, Arai Y, Komamura K, Wakabayashi S. Activation of Na+/H+ exchanger 1 is sufficient to generate Ca2+ signals that induce cardiac hypertrophy and heart failure. Circ Res. 2008; 103:891-899 [DOI] [PubMed] [Google Scholar]
- 17.Gao BB, Clermont A, Rook S, Fonda SJ, Srinivasan VJ, Wojtkowski M, Fujimoto JG, Avery RL, Arrigg PG, Bursell SE, Aiello LP, Feener EP. Extracellular carbonic anhydrase mediates hemorrhagic retinal and cerebral vascular permeability through prekallikrein activation. Nat Med. 2007; 13:181-188 [DOI] [PubMed] [Google Scholar]
- 18.Sasso FC, Torella D, Carbonara O, Ellison GM, Torella M, Scardone M, Marra C, Nasti R, Marfella R, Cozzolino D, Indolfi C, Cotrufo M, Torella R, Salvatore T. Increased vascular endothelial growth factor expression but impaired vascular endothelial growth factor receptor signaling in the myocardium of type 2 diabetic patients with chronic coronary heart disease. J Am Coll Cardiol. 2005; 46:827-834 [DOI] [PubMed] [Google Scholar]
- 19.Urbanek K, Torella D, Sheikh F, De Angelis A, Nurzynska D, Silvestri F, Beltrami CA, Bussani R, Beltrami AP, Quaini F, Bolli R, Leri A, Kajstura J, Anversa P. Myocardial regeneration by activation of multipotent cardiac stem cells in ischemic heart failure. Proc Natl Acad Sci USA. 2005; 102:8692-8697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ellison GM, Torella D, Dellegrottaglie S, Perez‐Martinez C, Perez de Prado A, Vicinanza C, Purushothaman S, Galuppo V, Iaconetti C, Waring CD, Smith A, Torella M, Cuellas Ramon C, Gonzalo‐Orden JM, Agosti V, Indolfi C, Galiñanes M, Fernandez‐Vazquez F, Nadal‐Ginard B. Endogenous cardiac stem cell activation by insulin‐like growth factor‐1/hepatocyte growth factor intracoronary injection fosters survival and regeneration of the infarcted pig heart. J Am Coll Cardiol. 2011; 58:977-986 [DOI] [PubMed] [Google Scholar]
- 21.Torella D, Rota M, Nurzynska D, Musso E, Monsen A, Shiraishi I, Zias E, Walsh K, Rosenzweig A, Sussman MA, Urbanek K, Nadal‐Ginard B, Kajstura J, Anversa P, Leri A. Cardiac stem cell and myocyte aging, heart failure, and insulin‐like growth factor‐1 overexpression. Circ Res. 2004; 94:514-524 [DOI] [PubMed] [Google Scholar]
- 22.Ellison GM, Torella D, Karakikes I, Purushothaman S, Curcio A, Gasparri C, Indolfi C, Cable NT, Goldspink DF, Nadal‐Ginard B. Acute beta‐adrenergic overload produces myocyte damage through calcium leakage from the ryanodine receptor 2 but spares cardiac stem cells. J Biol Chem. 2007; 282:11397-11409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Simoneau B, Houle F, Huot J. Regulation of endothelial permeability and transendothelial migration of cancer cells by tropomyosin‐1 phosphorylation. Vasc Cell. 2012; 4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vinciguerra M, Santini MP, Claycomb WC, Ladurner AG, Rosenthal N. Local IGF‐1 isoform protects cardiomyocytes from hypertrophic and oxidative stresses via SirT1 activity. Aging. 2009; 2:43-62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Torella D, Iaconetti C, Catalucci D, Ellison GM, Leone A, Waring CD, Bochicchio A, Vicinanza C, Aquila I, Curcio A, Condorelli G, Indolfi C. MicroRNA‐133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ Res. 2011; 109:880-893 [DOI] [PubMed] [Google Scholar]
- 26.Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, Bauersachs J. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007; 116:258-267 [DOI] [PubMed] [Google Scholar]
- 27.Rebello G, Ramesar R, Vorster A, Roberts L, Ehrenreich L, Oppon E, Gama D, Bardien S, Greenberg J, Bonapace G, Waheed A, Shah GN, Sly WS. Apoptosis‐inducing signal sequence mutation in carbonic anhydrase IV identified in patients with the RP17 form of retinitis pigmentosa. Proc Natl Acad Sci USA. 2004; 101:6617-6622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Geraldes P, King GL. Activation of protein kinase C isoforms and its impact on diabetic complications. Circ Res. 2010; 106:1319-1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Brodersen P, Voinnet O. Revisiting the principles of microRNA target recognition and mode of action. Nat Rev Mol Cell Biol. 2009; 10:141-148 [DOI] [PubMed] [Google Scholar]
- 30.Zhang H, Hao Y, Yang J, Zhou Y, Li J, Yin S, Sun C, Ma M, Huang Y, Xi JJ. Genome‐wide functional screening of miR‐23b as a pleiotropic modulator suppressing cancer metastasis. Nat Commun. 2011; 2:554. [DOI] [PubMed] [Google Scholar]
- 31.Zhu S, Pan W, Song X, Liu Y, Shao X, Tang Y, Liang D, He D, Wang H, Liu W, Shi Y, Harley JB, Shen N, Qian Y. The microRNA miR‐23b suppresses IL‐17‐associated autoimmune inflammation by targeting TAB 2, TAB 3 and IKK‐a. Nat Med. 2012; 18:1077-1086 [DOI] [PubMed] [Google Scholar]
- 32.Zhou Q, Gallagher R, Ufret‐Vincenty R, Li X, Olson EN, Wang S. Regulation of angiogenesis and choroidal neovascularization by members of microRNA‐23‐27‐24 clusters. Proc Natl Acad Sci USA. 2011; 108:8287-8292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He J, Li Y, Yang X, He X, Zhang H, He J, Zhang L. The feedback regulation of PI3K‐miR‐19a, and MAPK‐miR‐23b/27b in endothelial cells under shear stress. Molecules. 2013; 18:1-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shen E, Diao X, Wang X, Chen R, Hu B. MicroRNAs involved in the mitogen‐activated protein kinase cascades pathway during glucose‐induced cardiomyocyte hypertrophy. Am J Pathol. 2011; 179:639-650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fiordaliso F, Leri A, Cesselli D, Limana F, Safai B, Nadal‐Ginard B, Anversa P, Kajstura J. Hyperglycemia activates p53 and p53‐regulated genes leading to myocyte cell death. Diabetes. 2001; 50:2363-2375 [DOI] [PubMed] [Google Scholar]
- 36.Frustaci A, Kajstura J, Chimenti C, Jakoniuk I, Leri A, Maseri A, Nadal‐Ginard B, Anversa P. Myocardial cell death in human diabetes. Circ Res. 2000; 87:1123-1132 [DOI] [PubMed] [Google Scholar]
- 37.Relman AS, Leaf A, Schwartz WB. Oral administration of a potent carbonic anhydrase inhibitor (Diamox). II. Its use as a diuretic in patients with severe congestive heart failure. N Engl J Med. 1954; 250:800-804 [DOI] [PubMed] [Google Scholar]
- 38.Moyer JH, Hughes WM. A comparative study of neohydrin and diamox when used alone and in combination for the treatment of severe congestive heart failure. J Chronic Dis. 1955; 2:678-686 [DOI] [PubMed] [Google Scholar]
- 39.Moffett BS, Moffett TI, Dickerson HA. Acetazolamide therapy for hypochloremic metabolic alkalosis in pediatric patients with heart disease. Am J Ther. 2007; 14:331-335 [DOI] [PubMed] [Google Scholar]
- 40.Théroux P, Chaitman BR, Danchin N, Erhardt L, Meinertz T, Schroeder JS, Tognoni G, White HD, Willerson JT, Jessel A. Inhibition of the sodium‐hydrogen exchanger with cariporide to prevent myocardial infarction in high‐risk ischemic situations. Main Results of the GUARDIAN Trial. Circulation. 2000; 102:3032-3038 [DOI] [PubMed] [Google Scholar]
- 41.Zeymer U, Suryapranata H, Monassier JP, Opolski G, Davies J, Rasmanis G, Linssen G, Tebbe U, Schröder R, Tiemann R, Machnig T, Neuhaus KLESCAMI Investigators. The Na(+)/H(+) exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction. Results of the evaluation of the safety and cardioprotective effects of eniporide in acute myocardial infarction (ESCAMI) trial. J Am Coll Cardiol. 2001; 38:1644-1650 [DOI] [PubMed] [Google Scholar]
- 42.Engelhardt S, Hein L, Keller U, Klambt K, Lohse MJ. Inhibition of Na+‐H+ exchange prevents hypertrophy, fibrosis, and heart failure in beta(1)‐adrenergic receptor transgenic mice. Circ Res. 2002; 90:814-819 [DOI] [PubMed] [Google Scholar]
- 43.Yokoyama H, Gunasegaram S, Harding SE, Avkiran M. Sarcolemmal Na+/H+ exchanger activity and expression in human ventricular myocardium. J Am Coll Cardiol. 2000; 36:534-540 [DOI] [PubMed] [Google Scholar]
- 44.Catalucci D, Gallo P, Condorelli G. MicroRNAs in cardiovascular biology and heart disease. Circ Cardiovasc Genet. 2009; 2:402-408 [DOI] [PubMed] [Google Scholar]
- 45.Greco S, Fasanaro P, Castelvecchio S, D'Alessandra Y, Arcelli D, Di Donato M, Malavazos A, Capogrossi MC, Menicanti L, Martelli F. MicroRNA dysregulation in diabetic ischemic heart failure patients. Diabetes. 2012; 61:1633-1641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shantikumar S, Caporali A, Emanueli C. Role of microRNAs in diabetes and its cardiovascular complications. Cardiovasc Res. 2012; 93:583-593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zampetaki A, Mayr M. MicroRNAs in vascular and metabolic disease. Circ Res. 2012; 110:508-522 [DOI] [PubMed] [Google Scholar]
- 48.Jansen F, Yang X, Franklin BS, Hoelscher M, Schmitz T, Bedorf J, Nickenig G, Werner N. High glucose condition increases NADPH oxidase activity in endothelial microparticles that promote vascular inflammation. Cardiovasc Res. 2013; 98:94-106 [DOI] [PubMed] [Google Scholar]