

Abstract
Disruption of KSR2 in humans and mice decreases metabolic rate and induces obesity, coincident with dysregulation of glucose homeostasis. Relative to wild‐type mice, ksr2−/− mice are small prior to weaning with normal glucose tolerance at 6 weeks of age, but demonstrate excess adiposity by 9 weeks and glucose intolerance by 12–14 weeks. Defects in AICAR tolerance, a measure of whole‐body AMPK activation, are detectable only when ksr2−/− mice are obese. Food restriction prevents the obesity of adult ksr2−/− mice and normalizes glucose and AICAR sensitivity. Obesity and glucose intolerance return when ad lib feeding is restored to the diet‐restricted mice, indicating that glucose dysregulation is secondary to obesity in ksr2−/− mice. The phenotype of C57BL/6 ksr2−/− mice, including obesity and obesity‐related dysregulation of glucose homeostasis, recapitulates that of humans with KSR2 mutations, demonstrating the applicability of the C57BL/6 ksr2−/− mouse model to the study of the pathogenesis of human disease. These data implicate KSR2 as a physiological regulator of glucose metabolism during development affecting energy sensing, insulin signaling, and lipid storage, and demonstrate the value of the C57BL/6 ksr2−/− mouse model as a unique and relevant model system in which to develop and test therapeutic targets for the prevention and treatment of obesity, type 2 diabetes, and obesity‐related metabolic disorders.
Keywords: AMPK, glucose metabolism, insulin resistance, KSR2, obesity
A fraction of individuals with obesity‐induced insulin resistance and diabetes respond to diet with improved glucose metabolism. The phenotype of C57BL/6 ksr2−/− mice, including obesity and obesity‐related dysregulation of glucose homeostasis, recapitulates that of humans with KSR2 mutations. These data show that the glucose intolerance and AICAR insensitivity that accompanies KSR2 disruption in mice is preventable or reversible by diet, suggesting that dietary intervention in humans with KSR2 mutations should have similar effects.
Introduction
Obesity has become a prevalent and destructive health disorder in our society (Flier 2004) linked to some of the leading causes of preventable death including heart disease, stroke, certain types of cancer, and type 2 diabetes (NIH 1998). The prevalence of obesity and diabetes increases the need for in vivo models that reveal molecular pathways driving the pathogenesis of obesity and obesity‐related disorders. Three major physiological mechanisms produce obesity in rodents: hyperphagia, defective nonshivering thermogenesis, and the preferential deposition of calories into adipose tissue (Chua 1997). There are numerous examples (Yen et al. 1994; Zhang et al. 1994; Tecott et al. 1995; Chen et al. 1996; Lee et al. 1996; Ollmann et al. 1997; Balthasar et al. 2005; Fellmann et al. 2013) of rodent models that have been genetically engineered or developed from spontaneous mutations to investigate the contribution of a variety of signaling pathways to the regulation of these three mechanisms. For example, ob/ob and db/db mouse models were critical to identifying leptin and its receptor (Hummel et al. 1966; Zhang et al. 1994; Halaas et al. 1995; Maffei et al. 1995; Tartaglia et al. 1995; Lee et al. 1996), and identifying the JAK/STAT signaling pathway as the major mechanism through which leptin communicates satiety to the hypothalamus (Ghilardi et al. 1996; Bates et al. 2003). Thermogenic defects in ob/ob and db/db mice played an integral part in identifying the mechanisms through which leptin regulates nonshivering thermogenesis to affect organismal energetic homeostasis (Chua 1997). Subsequent identification that leptin and its receptor were mutated in rare forms of monogenic human obesity validated the importance of these models (Montague et al. 1997; Farooqi et al. 2007).
Mice with disrupted alleles of Kinase Suppressor of Ras 2 (KSR2) appear molecularly distinct from other genetic models as they become obese due to a lower metabolic rate without a decrease in locomotor activity (Costanzo‐Garvey et al. 2009). Mice lacking KSR2 are markedly insulin resistant in liver, muscle, and adipose depots. KSR2 is also mutated in a subpopulation of humans with early onset obesity (Pearce et al. 2013). Individuals carrying KSR2 mutations exhibit childhood hyperphagia, reduced basal metabolic rate, and severe insulin resistance. These observations reveal ksr2−/− mice as a useful model for understanding physiological pathways contributing to human obesity and for revealing novel biochemical mechanisms that link obesity to insulin resistance.
KSR2 is a scaffold protein in the Raf/MEK/ERK signaling cascade, where it functions along with its paralog, KSR1, to coordinate the interaction of these molecules to facilitate signal transduction and regulate the intensity and duration of ERK signaling (Dougherty et al. 2009). KSR2 also interacts with and promotes activation of the primary regulator of cellular energy homeostasis, AMPK (Costanzo‐Garvey et al. 2009; Fernandez et al. 2012). In ksr2−/− mice, decreased AMPK activation impairs the oxidation of fatty acids and increases their storage as triglycerides, promoting obesity and insulin resistance (Costanzo‐Garvey et al. 2009). Some KSR2 mutations in individuals with early onset obesity disrupt ERK activation or impair interaction of the scaffold with AMPK (Pearce et al. 2013). Together, these data implicate KSR2 as a potential sensor of cellular energy status and a key effector in whole‐body energy regulation in mice and humans.
KSR2 mutations in humans (Pearce et al. 2013) recapitulate the obesity and severe insulin resistance observed in C57BL/6 ksr2−/− mice (Costanzo‐Garvey et al. 2009), revealing them as a disease‐relevant model system, allowing investigation into mechanisms through which KSR2‐dependent signaling may contribute to the onset and progression of obesity and diabetes in humans. Here, we demonstrate that C57BL/6 ksr2−/− mice are not born with defects in glucose regulation, but that these defects arise as a result of obesity that develops later in the life of ksr2−/− adults. We show that defects in glucose homeostasis can be prevented through postweaning food restriction of young ksr2−/− mice, and reversed by restricting the diet of obese ksr2−/− adults already suffering from glucose intolerance and insulin resistance. Given the similarities in metabolic characteristics between the C57BL/6 ksr2−/− model and humans bearing KSR2 mutations, we propose the broad utility of ksr2−/− mice to the study, treatment, and prevention of human obesity and obesity‐related disorders.
Research Design and Methods
Generation and housing of mice
DBA1/LacJ ksr2−/− mice were as described previously (Costanzo‐Garvey et al. 2009). Mice were backcrossed 10 generations to generate C57BL/6 ksr2−/− mice.
First‐generation offspring (F1) from breeding of heterozygous parents were used in these experiments. From this breeding 131 KSR2+/+, 181 KSR2+/−, and 77 KSR2−/− mice (0.34:0.47:0.20) were generated, approximating the expected Medelian ratio of 0.25:0.50:0.25. The Institutional Animal Care and Use Committee (University of Nebraska Medical Center, Omaha, NE) approved all studies. Animals were maintained on a 12‐h light/dark schedule and had free access to laboratory chow (Harlan Teklad LM 485, Harlan Laboratories, Inc, Indianapolis, IN) and water except as described below.
Food restriction of young, lean ksr2−/− mice
For food restriction studies, female ksr2−/− and age‐matched WT control mice were placed into one of four groups: (1) WT ad libitum (WT ad lib), (2) WT food restricted (WT FR), (3) ksr2−/− ad libitum (ksr2−/− ad lib), or (4) ksr2−/− food restricted (ksr2−/− FR). At 5 weeks of age, daily ad libitum food intake was determined for 1 week for each mouse by subtracting the weight of remaining chow from the weight of total chow provided. ksr2−/− FR and WT FR mice received 70% of their daily ad libitum food intake once daily from 6 to 12 weeks of age. Following 6 weeks of food restriction, animals were subjected to glucose homeostasis tests (see glucose, insulin and 5‐aminoimidazole‐4‐carboxamide‐1‐β‐d‐ribofuranoside [AICAR] tolerance tests described below) during which time food restriction continued, totaling 8 weeks of food restriction. Following tests of glucose homeostasis, previously FR mice were allowed ad libitum access to food at 14 weeks of age. Daily food intake was again determined for these ksr2−/− FR‐ad lib and WT FR‐ad lib mice as well as their ad lib fed controls. Animals were refed for 13 weeks. Glucose homeostasis testing was repeated on these 6‐month‐old FR‐ad lib and ad lib control animals.
Food restriction of adult, obese ksr2−/− mice
Female ksr2−/− and age‐matched WT control mice were placed into one of four groups: (1) WT ad lib, (2) WT FR, (3) ksr2−/− ad lib, or (4) ksr2−/− FR. All mice were fed ad libitum from weaning until 16 weeks of age. Daily ad libitum food intake was determined for each mouse for 1 week starting at 15 weeks of age. At 16 weeks of age, WT FR and ksr2−/− FR mice received 70% of their daily ad libitum food intake once daily from 16 to 22 weeks of age. WT ad lib and ksr2−/− ad lib mice were fed ad libitum throughout the study.
Dual Energy X‐ray Absorptiometry (DEXA)
Mice were weighed weekly on a digital scale. Quantification of lean mass, fat mass, and % body fat was performed every 2 weeks using dual energy X‐ray absorptiometry (DEXA). Mice were anesthetized using a mixture of inhaled isoflurane and oxygen (anesthetization using 3% isoflurane and 1 L/min oxygen; maintenance using 1–2% isoflurane and 1 L/min oxygen) and placed prone on the imaging positioning tray. Mice were scanned using a Lunar PIXImus™ densitometer (GE Medical‐Lunar, Madison, WI).
Glucose, insulin, and 5‐aminoimidazole‐4‐carboxamide‐1‐β‐d‐ribofuranoside (AICAR) tolerance tests
To determine the role KSR2 plays in glucose homeostasis, glucose (GTT), insulin (ITT), and AICAR (ATT) tolerance tests were performed in ksr2−/− and WT mice. The AICAR tolerance test measures whole‐body response to the activation of AMPK by the AMPK agonist AICAR (Viollet et al. 2003). GTT were performed after a 10‐h fast. ITT and ATT were performed following a 4‐h fast. Mice were injected intraperitoneally (IP) with D‐glucose (20% solution, 2 g/kg of body weight) for GTT, human insulin (1 unit/kg) for ITT, or AICAR (0.25 g/kg) for ATT. Blood glucose levels were determined following injection at the indicated times.
Metabolite assays
Blood was collected by tail bleeds of live animals or via cardiac puncture of euthanized animals. Animals were fasted overnight for 10–12 h prior to collection for blood glucose and serum insulin measurement. Blood glucose was measured with an Ascensia Glucometer Elite (Bayer Corp., Elkhart, IN). For serum analysis, blood was allowed to clot at 4°C for 8–24 h, and the serum was separated by centrifugation for 10 min at 100 g. Serum was transferred to a new tube and stored at −20°C until assay. Serum insulin was measured with the Mouse Insulin ELISA Kit (ChrystalChem, Downers Grove, IL) using mouse standards. Serum leptin was measured with a Mouse Leptin ELISA kit (Millipore, Billerica, MA).
Leptin responsiveness
Responsiveness to leptin was assessed by IP injecting 4‐ to 5‐month‐old ksr2−/− and WT mice with 100 μL phosphate‐buffered saline (PBS; as a control) followed 2 days later by an injection of 5 mg/kg leptin in 100 μL and quantifying total food intake during the 24‐h period following each injection. Thus, each animal received two injections and served as its own control.
Lipid quantification
Serum triglyceride and free fatty acid (FFA) levels were quantified from ad lib and FR WT and ksr2−/− mice using Triglyceride Reagent (T2449; Sigma‐Aldrich, St. Louis, MO) and a Free Fatty Acids Half Micro Test (11383175001; Roche, Indianapolis, IN). Lipid accumulation in ad lib and FR WT and ksr2−/− mice was visualized by hematoxylin and eosin staining of white adipose tissue (WAT) and brown adipose tissue (BAT) sections.
Statistical analysis
GraphPad Prism version 5.04 for Windows (GraphPad Software, San Diego, CA) was used for graphics design and statistical analyses. To determine the extent to which disruption of KSR2 affected the indicated response variable at a single time point, we used a Student's t‐test, applying a Bonferroni adjustment when multiple pairwise comparisons were made (Sokal and Rohlf 1995). To determine the effect of KSR2 disruption on the indicated response variable over time or under various treatments, we applied a two‐way analysis of variance (ANOVA) with genotype and time, age, or treatment as dependent factors (Sokal and Rohlf 1995). When multiple data points were drawn sequentially from the same animal, pseudoreplication was avoided by performing a repeated measures two‐way ANOVA. When results from the two‐way ANOVA indicated that genotype had a significant effect, we performed an additional series of Bonferroni‐adjusted Student's t‐tests to identify the time point at which or treatment under which the effect of KSR2 disruption became significant. Data are shown as the mean ± standard deviation (SD). Significance was accepted at P < 0.05. Unless indicated otherwise for clarity, significant comparisons are represented as follows: *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Results
C57BL/6 ksr2−/− mice are small and lean at weaning, but become obese as adults
We reported previously that DBA1/LacJ ksr2−/− mice are obese (Costanzo‐Garvey et al. 2009). We generated and characterized congenic C57BL/6 ksr2−/− mice. The C57BL/6 strain is commonly used to study the genetics and physiology of obesity and diabetes. At birth, ksr2−/− mice are not distinguishable from WT littermates by size. However, at 2.5 weeks of age and while still nursing, ksr2−/− neonates weigh 25% less than their WT littermates (Fig. 1A). After weaning, ksr2−/− mice demonstrate accelerated growth rates that allow them to surpass their WT littermates in body weight by 9 weeks of age (Fig. 1B). This increase in weight is due primarily to an increase in fat mass (Fig. 1C and D). By 13 weeks of age ksr2−/− mice have three times more fat mass than a WT mouse and a 10% increase in lean mass (Fig. 1D). All adipose depots in ksr2−/− mice are increased relative to the same depots in WT mice (Fig. 1E).
Figure 1.
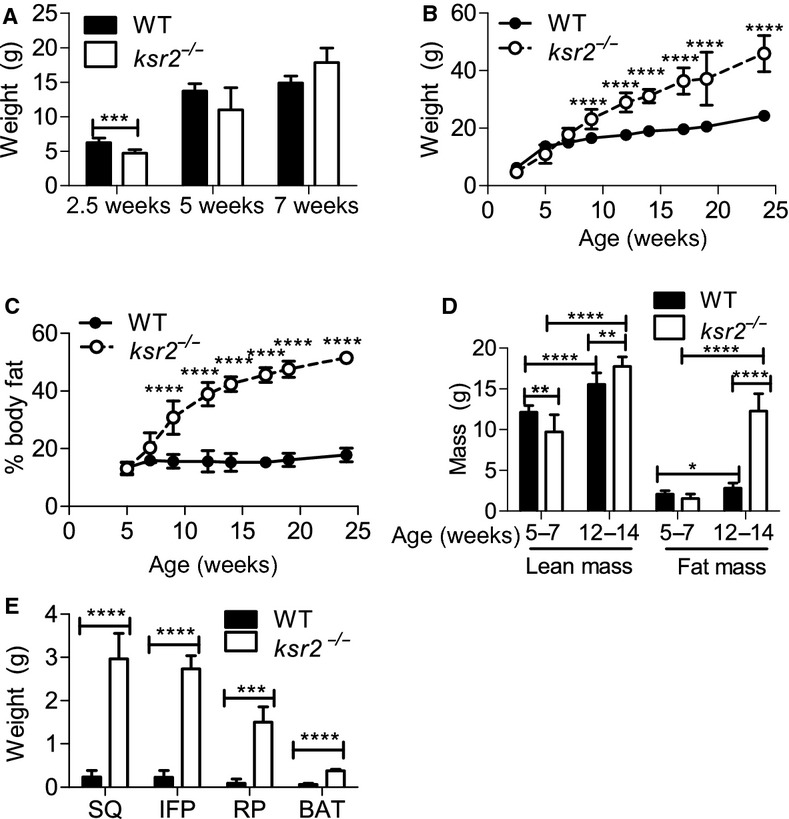
C57BL/6 ksr2−/− mice are small and lean at weaning, but become obese as adults. (A) Body weights of WT and ksr2−/− mice compared at 2.5 (n = 8–9), 5 (n = 3–5), and 7 (n = 3–5 per genotype) weeks of age. Body weights (B) and percentage body fat (C) determined by DEXA of WT and ksr2−/− mice compared from 2.5 to 24 weeks of age. 2.5 weeks, n = 8–9; 5–7 weeks, n = 3–5; 9–14 weeks, n = 4–9; and 17–24 weeks, n = 2–4 per genotype. (D) Lean and fat mass determined by DEXA of WT and ksr2−/− mice at 5–7 weeks of age (n = 8) and 12–14 weeks of age (n = 8–11 per genotype). (E) The weight of adipose distinct depots was determined in WT and ksr2−/− mice. SQ, subcutaneous; IFP, inguinal fat pad; RP, retroperitoneal; BAT, brown adipose tissue. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Defects in glucose homeostasis in ksr2−/− mice are coincident with the development of obesity
ksr2−/− mice display many obesity‐related changes in glucose regulation including glucose and AICAR intolerance and high fasting blood glucose and insulin levels (Fig. 2A–G; Costanzo‐Garvey et al. 2009). To determine the extent to which glucose homeostasis was deregulated prior to the onset of obesity, 6‐ and 7‐week‐old ksr2−/− mice were challenged with GTT and ATT prior to becoming obese. Six‐week‐old ksr2−/− mice responded similar to WT mice when injected with glucose (Fig. 2A). In contrast, 5‐month‐old ksr2−/− mice are glucose intolerant (Fig. 2B). In addition, fasting glucose and insulin levels are normal in 5‐ to 6‐week‐old ksr2−/− mice, but elevated in 5‐ to 6‐month‐old ksr2−/− mice (Fig. 2C and D). HOMA‐IR was calculated from the fasting level of glucose and insulin in 6‐month‐old WT and ksr2−/− mice, revealing significant insulin resistance in the knockout mice (Fig. 2E). While 5‐month‐old ksr2−/− mice respond to AICAR by inappropriately increasing their blood glucose (Fig. 2G), 7‐week‐old ksr2−/− mice do not display a defect in AICAR tolerance when compared to WT mice of the same age (Fig. 2F). These data indicate that the dysregulation of glucose homeostasis is not present in younger ksr2−/− mice but coincides with, or occurs subsequent to, rapid weight gain.
Figure 2.
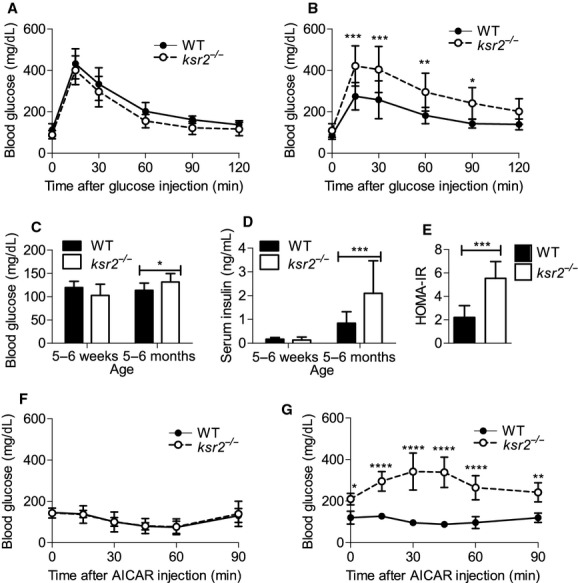
Defects in glucose homeostasis in ksr2−/− mice are coincident with the development of obesity. GTT of (A) 6‐week‐old (WT n = 6; ksr2−/− n = 4) and (B) 5‐month‐old (WT n = 8; ksr2−/− n = 8) WT and ksr2−/− mice. (C) Blood glucose (5–6 weeks old, n = 10; 5–6 months old, n = 10–13 per genotype) and (D) serum insulin levels (5–6 weeks, n = 8–9; 5–6 months old, n = 14–16 per genotype) of 5‐ to 6‐week‐old and 5‐ to 6‐month‐old fasted WT and ksr2−/− mice. (E) HOMA‐IR, calculated from fasting glucose and insulin levels in 6‐month‐old WT and KSR2−/− mice (n = 6–8 per genotype), revealed significant insulin resistance in the knockout mice. (F) ATT of 7‐week‐old (n = 5 per genotype) and (G) 5‐month‐old (n = 4 per genotype) WT and ksr2−/− mice. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Food restriction of young ksr2−/− mice prevents obesity
Contrary to DBA1/LacJ ksr2−/− mice, which do not eat more than WT mice (Costanzo‐Garvey et al. 2009), C57BL/6 ksr2−/− mice are hyperphagic (Fig. 3A). At 5 weeks of age C57BL/6 ksr2−/− mice eat the same amount of food as WT mice, but by 7 weeks of age ksr2−/− mice eat significantly more food per day than WT mice (Fig. 3A). Whereas lean 5‐week‐old ksr2−/− mice have normal serum leptin levels, obese 5‐month‐old ksr2−/− mice display high serum leptin levels in comparison to WT controls (Fig. 3B; Costanzo‐Garvey et al. 2009). DBA1/LacJ ksr2−/− mice, which are normophagic, retained sensitivity to leptin administration (Costanzo‐Garvey et al. 2009). To determine if the hyperphagia present in C57BL/6 ksr2−/− mice is due to leptin resistance, mice were injected with leptin and food intake monitored. Acute leptin administration suppressed food intake by 26% in ksr2−/− mice and by 37% in WT mice (Fig. 3C). These data indicate that C57BL/6 ksr2−/− mice, like DBA1/LacJ ksr2−/− mice, remain responsive to leptin.
Figure 3.
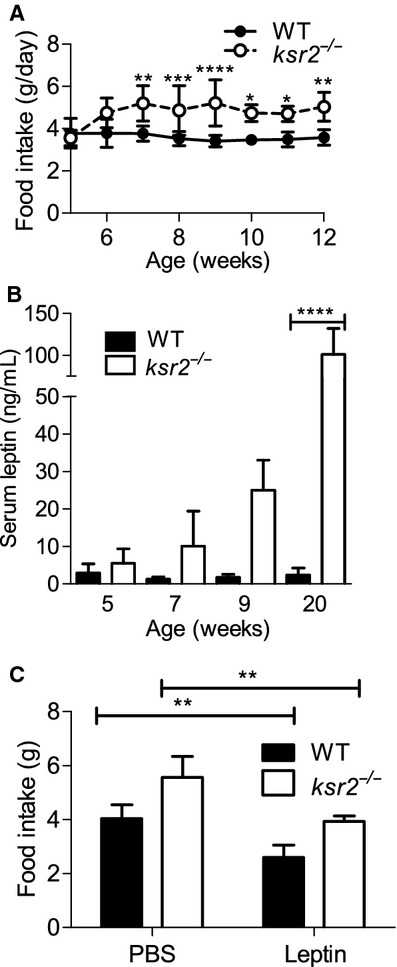
C57BL/6 ksr2−/− mice are hyperphagic, but responsive to exogenous leptin. (A) Food intake of WT and ksr2−/− mice at 5–12 weeks of age. Five to seven weeks, n = 3–5; 8–12 weeks, n = 5–9 per genotype. (B) Serum leptin levels of WT (n = 3–6) and ksr2−/− (n = 4–5) mice at 5, 7, 9, and 20 weeks of age. (C) 24‐h food intake following IP injection of 100 μL PBS or 100 μL of 5 mg/kg leptin in 4‐ to 5‐month‐old WT and ksr2−/− mice (n = 4 per genotype). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Since weight gain and fat accumulation were coincident with hyperphagia in adult C57BL/6 ksr2−/− mice, we sought to determine the extent to which food restriction in ksr2−/− young mice would prevent or forestall obesity when they were adults. WT and ksr2−/− mice were food restricted (FR) for 6 weeks. Food intake was measured at 5 weeks of age (3.95 g/day ± 0.21 for WT; 3.70 g/day ± 0.21 for ksr2−/−). Each FR mouse was fed 70% of their daily ad libitum amount from 6 to 12 weeks of age. Food restriction prevented obesity in ksr2−/− mice (Fig. 4A and B). Although not statistically significant, ksr2−/− FR mice tended to weigh less and be leaner than WT FR mice at 12 weeks of age (Fig. 4A and B), an age when ksr2−/− mice fed an ad lib diet are significantly heavier than WT mice (Fig. 4A). In addition, food restriction in young ksr2−/− mice normalized serum triglyceride levels in adult ksr2−/− FR mice (Fig. 4C) and led to a significant reduction in FFA in the serum of adult ksr2−/− FR mice compared to WT FR mice (Fig. 4D). Food restriction also led to a reduction in adipocyte cell size in WAT and BAT as well as to the reduction of lipid storage in BAT of ksr2−/− mice compared to ad lib fed controls (Fig. 4E).
Figure 4.
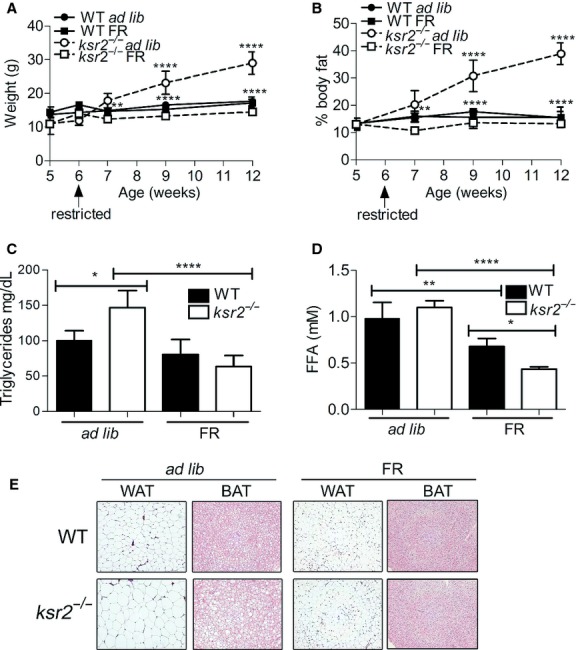
Food restriction of young ksr2−/− mice prevents obesity. Body weight (A) and percentage body fat (B) of WT ad lib, WT FR, ksr2−/− ad lib, and ksr2−/− FR mice from 5 to 12 weeks of age. Ad lib 5–7 weeks, n = 3–5 per genotype; ad lib 8–12 weeks, n = 4–9 per genotype; FR 5–12 weeks, n = 4 per genotype. The top set of asterisks identifies significant comparisons between WT ad lib and ksr2−/− ad lib mice. The bottom set of asterisks identifies significant comparisons between ksr2−/− ad lib and ksr2−/− FR mice. Serum triglyceride (C) and FFA (D) levels in 12‐ to 14‐week‐old WT ad lib, WT FR, ksr2−/− ad lib, and ksr2−/− FR mice (n = 4–5 per treatment). (E) Hematoxylin and eosin staining of WAT and BAT sections from 16‐week‐old ad lib and FR WT and ksr2−/− mice. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Food restriction of young ksr2−/− mice ameliorates defects in glucose regulation
To determine what role KSR2 plays in glucose homeostasis, GTT and ATT were performed in 12‐ to 14‐week‐old food restricted ksr2−/− and WT mice and compared to results in ad lib fed ksr2−/− and WT controls. Food restriction improved glucose tolerance and AICAR sensitivity in ksr2−/− mice (Fig. 5A–B). Food restricted ksr2−/− mice responded to exogenous glucose or AICAR to the same degree as WT FR mice (Fig. 5A–B). These data suggest that the disrupted glucose homeostasis of ksr2−/− mice is secondary to the obesity that results in these mice when fed ad libitum.
Figure 5.
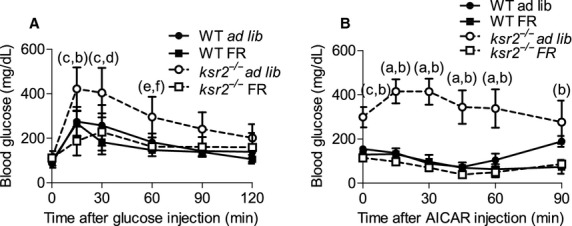
Food restriction of young ksr2−/− mice ameliorates defects in glucose regulation. (A) GTT (ad lib, n = 8; FR, n = 4 per genotype) and (B) ATT (ad lib, n = 4; FR, n = 3–4 per genotype) of 12‐ to 14‐week‐old WT ad lib, WT FR, ksr2−/− ad lib, and ksr2−/− FR mice. a = P < 10−5; c = P < 10−4; e = P < 0.05 for comparisons between WT ad lib and ksr2−/− ad lib mice. b = P < 10−5; d = P < 10−4; f = P < 0.05 for comparisons between ksr2−/− ad lib and ksr2−/− FR mice.
Food restriction of young ksr2−/− mice does not permanently alter glucose metabolism
To determine if food restriction resulted in permanent changes in glucose metabolism, mice were food restricted from 6 to 14 weeks of age, then allowed free access to food (FR‐ad lib). Immediately upon gaining free access to food, both WT and ksr2−/− mice significantly increased their food intake (data not shown). However, after 1 week of free access to food, food intake in both WT and ksr2−/− mice returned to levels observed in mice of the same age (14 weeks) that had not been restricted (Fig. 6A). While FR‐ad lib ksr2−/− mice became once again hyperphagic compared to FR‐ad lib WT mice after 1 week of refeeding, there was no difference in food intake between FR‐ad lib ksr2−/− mice and age‐matched ad lib ksr2−/− controls (Fig. 6A). This result indicates that food restriction from weaning did not reprogram the set point of food intake in either ksr2−/− or WT mice.
Figure 6.
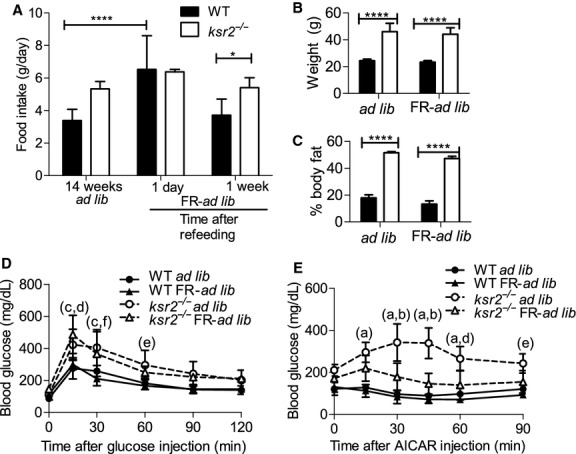
Food restriction of young ksr2−/− mice does not permanently alter glucose metabolism. (A) WT (black bars, A–C) and ksr2−/− (white bars, A–C) mice (fed ad lib, food restricted [FR], or food restricted then refed [FR‐ad lib]) at the indicated ages. Refeeding was performed in 14‐week‐old FR mice for 1 week. Fourteen‐week‐old ad lib (n = 8), 14‐week‐old FR‐ad lib 1 day after refeeding (n = 4), and 15‐week‐old FR‐ad lib (n = 4). Body weight (B) and percentage body fat (C) of 6‐month‐old WT and ksr2−/− ad lib and FR‐ad lib fed mice (n = 4 per treatment). *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. (D) GTT of 6‐month‐old WT ad lib (n = 8), WT FR‐ad lib (n = 4), ksr2−/− ad lib (n = 8), and ksr2−/− FR‐ad lib (n = 4) mice. a = P < 10−5; c = P < 10−4; e = P < 0.05 for comparisons between WT ad lib and ksr2−/− ad lib mice. b = P < 10−5; d = P < 10−3; f = P < 0.05 for comparisons between WT FR‐ad lib and ksr2−/− FR‐ad lib mice. (E) ATT of 6‐month‐old WT ad lib (n = 4), WT FR‐ad lib (n = 3), ksr2−/− ad lib (n = 4), and ksr2−/− FR‐ad lib (n = 4) mice. a = P < 10−5; c = P < 10−4; e = P < 0.05 for comparisons between WT ad lib and ksr2−/− ad lib mice. b = P < 10−5; d = P < 10−3; f = P < 0.05 for comparisons between ksr2−/− ad lib and ksr2−/− FR‐ad lib mice.
After 13 weeks of refeeding, 6‐month‐old FR‐ad lib ksr2−/− and WT mice had similar weights compared to their respective ad lib fed controls (Fig. 6B). FR‐ad lib ksr2−/− mice became obese and had similar body composition to ksr2−/− ad lib fed controls (Fig. 6C). Although food restriction delayed obesity in ksr2−/− mice, upon ad lib refeeding they regained weight quickly, most likely due to their persistent hyperphagia on the C57BL/6 background (Figs. 3A and 6A; Revelli et al. 2011) and decreased metabolic rate (Costanzo‐Garvey et al. 2009; Pearce et al. 2013).
We next tested the effect of food restriction followed by free access to food on glucose tolerance and AICAR sensitivity. FR‐ad lib WT mice had glucose tolerance indistinguishable from ad lib WT mice (Fig. 6D). Importantly, after FR, ad lib feeding restored glucose intolerance in ksr2−/− mice (Fig. 6D). Although obesity for FR‐ad lib ksr2−/− mice was delayed, the same defects in glucose handling observed in ad lib ksr2−/− mice returned to FR‐ad lib ksr2−/− mice once they became obese.
AICAR tolerance was not altered between FR‐ad lib and ad lib groups in WT mice (Fig. 6E). In ksr2−/− mice, food restriction followed by free access to food did significantly improve AICAR tolerance, although FR‐ad lib ksr2−/− mice remained somewhat AICAR insensitive compared to WT controls (Fig. 6E). However, the degree of insensitivity was less than what was detected in ad lib ksr2−/− mice (Fig. 6E). This result may indicate that AICAR tolerance in adult ksr2−/− mice is affected by food restriction, or it may reflect the fact that ksr2−/− mice need to be obese for a longer length of time before AICAR insensitivity develops fully.
Food restriction in adult ksr2−/− mice reduces obesity
To determine whether adult obesity and dysregulation of glucose homeostasis is reversible in adult ksr2−/− mice, the diet of 16‐week‐old ksr2−/− and WT mice was restricted to 70% of their daily food intake for 6 weeks. Adult WT and ksr2−/− mice lost weight under food restriction (Fig. 7A). Weight loss in food restricted adult ksr2−/− mice, but not WT mice, was visible as a significant reduction in adipocyte size in WAT and decreased lipid accumulation in BAT when compared to ad lib controls (Fig. 7B).
Figure 7.
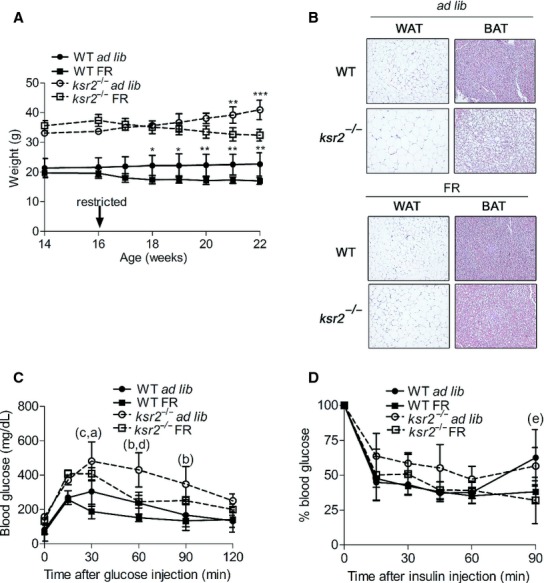
Food restriction in adult ksr2−/− mice reduces obesity and improves glucose tolerance and insulin resistance. (A) Body weight of WT ad lib (n = 3) and ksr2−/− ad lib (n = 3) mice compared to WT FR (n = 6) and ksr2−/− FR (n = 4) mice, respectively, in response to food restriction from 16 to 22 weeks of age. The top set of asterisks identifies significant comparisons between ksr2−/− ad lib and ksr2−/− FR mice. The bottom set of asterisks identifies significant comparisons between WT ad lib and WT FR mice. *P < 0.05, **P < 0.01, ***P < 0.001. (B) Hematoxylin and eosin staining of WAT and BAT sections from 6‐month‐old ad lib and FR WT and ksr2−/− mice. GTT (C) (n = 3–4 per treatment) and ITT expressed as a percentage of starting blood glucose levels (D) (n = 4–5 per treatment) of 6‐month‐old WT and ksr2−/− ad lib, and FR mice. b = P < 10−3; c = P < 0.05 for comparisons between WT ad lib and ksr2−/− ad lib mice. d = P < 10−3; e = P < 0.05 for comparisons between ksr2−/− ad lib and ksr2−/− FR mice. a = P < 10−4; f = P < 0.05 for comparisons between WT FR and ksr2−/− FR mice.
Food restriction improves glucose tolerance and insulin resistance in obese adult ksr2−/− mice
Food restriction improved glucose tolerance and insulin sensitivity in both ksr2−/− and WT adult mice. Following food restriction, ksr2−/− mice showed improved glucose tolerance relative to ad libitum fed ksr2−/− mice, although blood glucose levels of FR ksr2−/− mice remained higher than both FR and ad lib WT mice (Fig. 7C). Similarly, exogenous insulin modestly lowered blood glucose in food restricted adult ksr2−/− mice relative to ksr2−/− adults fed ad lib (Fig. 7D).
Discussion
These data demonstrate that the dysregulated glucose homeostasis of ksr2−/− mice is secondary to their obesity and can be prevented by dietary restriction prior to weaning or ameliorated by dietary restriction in obese adult knockout mice. Diet does not reverse the underlying metabolic defects caused by disruption of KSR2, however, as ad libitum feeding of diet‐restricted ksr2−/− mice restores obesity and glucose intolerance. These data likely have significance for human obesity and obesity‐dependent insulin resistance, as KSR2 mutations in humans lower resting metabolic rate and promote obesity with severe insulin resistance (Pearce et al. 2013). The phenotype exhibited by C57BL/6 ksr2−/− adult mice models a classic developmental pattern associated with an increased risk of type 2 diabetes including low neonatal body mass, accelerated rates of postnatal growth, and subsequent obesity leading to progressively worsening glucose intolerance and insulin resistance. Human infants with low birthweight or with poor postnatal nutrition have a higher risk of becoming obese as adults and developing metabolic syndrome (Barker 2005; Jimenez‐Chillaron et al. 2005; Bieswal et al. 2006; Isganaitis et al. 2009; Fernandez‐Twinn and Ozanne 2010). C57BL/6 ksr2−/− mice are small at weaning, but quickly gain weight, surpassing WT controls by 9 weeks of age to become obese as adults primarily due to an increase in fat mass and lipid accumulation. Defects in glucose regulation, including glucose intolerance and insulin resistance, arise as a consequence of morbid obesity that develops later in life in C57BL/6 ksr2−/− adults.
DBA1/LacJ ksr2−/− mice consume less food than WT mice (Costanzo‐Garvey et al. 2009), whereas C57BL/6 ksr2−/− mice are hyperphagic (Figs 3A and 6A; Revelli et al. 2011). Both DBA1/LacJ (Costanzo‐Garvey et al. 2009) and C57BL/6 (Fig. 1B–D) ksr2−/− mice become obese. Pair‐feeding ksr2−/− mice on a mixed 129SvEvBrd/C57BL/6J background reduces, but does not eliminate, obesity (Pearce et al. 2013), demonstrating that their obesity is not dependent on overeating. However, the hyperphagia of C57BL/6 ksr2−/− mice likely explains why they become more obese than do DBA1/LacJ ksr2−/− mice (Costanzo‐Garvey et al. 2009). Correspondingly, ksr2−/− mice in the C57BL/6 background suffer from more extreme glucose and AICAR intolerance as well as more severe insulin resistance than DBA1/LacJ ksr2−/− mice. Revelli et al. (2011) found ksr2−/− mice on a mixed 129SvEvBrd/C57BL/6J background to be hyperphagic and leptin resistant. We observe that ksr2−/− mice in both DBA1/LacJ and C57BL/6 backgrounds respond comparably to WT mice when treated acutely with exogenous leptin (Costanzo‐Garvey et al. 2009; Fig. 3C). The difference may be accounted for by the ability of leptin to suppress overnight feeding in this study, but not the appetite generated in ksr2−/− mice pair fed for 2 weeks (Revelli et al. 2011). The responsiveness of C57BL/6 ksr2−/− mice to leptin distinguishes them from murine models of obesity including the ob/ob mouse, db/db mouse, SHROB rat, JCR:LA‐cp rat, Zucker rat, and ZDF rat model systems (Fellmann et al. 2013). Given the rarity of cases of human obesity and obesity‐related disorders attributable to mutations in the leptin gene or receptor (Montague et al. 1997; Farooqi et al. 2002, 2007; Gibson et al. 2004; Farooqi and O'Rahilly 2006), the fact that the more common form of leptin resistance that results as a consequence of obesity is reversible in humans (Fellmann et al. 2013), and the demonstration that KSR2 mutations promote obesity in humans (Pearce et al. 2013), these data reveal C57BL/6 ksr2−/− mice as a unique model system for identifying new effectors relevant to understanding the pathogenesis of human obesity and obesity‐related disorders. Furthermore, these mice may be useful in designing novel therapies for reducing obesity in humans.
Critical to the dissection of KSR2‐dependent signaling pathways is the identification of the tissues in which KSR2 acts to affect whole‐body energy balance. We recently identified a direct interaction between KSR2 and the AMP‐regulated kinase AMPK that is required for full activation of this cellular energy sensor (Costanzo‐Garvey et al. 2009; Fernandez et al. 2012). We proposed that defects in AMPK activation resulting from KSR2 disruption promoted adiposity by disabling acetyl CoA carboxylase regulation leading to the suppression of fatty acid metabolism and the promotion of triglyceride storage in ksr2−/− mice. However, KSR2 mRNA expression in white adipose tissue is exceedingly low (Costanzo‐Garvey et al. 2009; Guo et al. 2014), suggesting that, in vivo, noncell autonomous effects of KSR2 on lipid metabolism predominate. Data here show that defects in AICAR tolerance, which presumably reflect the activation of AMPK in vivo, are not evident prior to increased adiposity in ksr2−/− mice at 7 weeks of age (Fig. 2F). These data raise the possibility that KSR2‐mediated effects on AMPK function are regulated developmentally.
KSR2 is highly expressed in all areas of the brain, including the hypothalamus (Costanzo‐Garvey et al. 2009; Pearce et al. 2013), which serves as the main energy sensor for the body, regulating adaptive thermogenesis, food intake, and energy expenditure (Lowell and Spiegelman 2000; Lindsley and Rutter 2004; Herman and Kahn 2006). Thus, KSR2 in the hypothalamus may exert noncell autonomous effects on whole‐body energy balance. The hypothalamus contains two distinct neuron populations, those that secrete proopiomelanocortin (POMC) and cocaine‐amphetamine‐related transcript (CART) and those that secrete neuropeptide Y (NPY) and agouti‐related protein (AgRP). POMC and CART are anorexigenic and increase energy expenditure when stimulated, while NPY and AgRP are orexigenic (Cota et al. 2007). AMPK is an important signaling molecule in both neuronal types, responding to glucose levels as well as multiple hormones such as leptin, ghrelin, and adiponectin to regulate both food intake and energy expenditure. Deletion of AMPKα2 from POMC neurons results in obese mice with reduced energy expenditure and increased food intake, but which remain sensitive to leptin (Claret et al. 2007). Thus, our data suggest a role for KSR2 in the development or propagation of signals critical to the proper sensing of whole‐body energy status in the hypothalamus by potentially directing the subcellular distribution and activity of AMPK. Targeted disruption of KSR2 in the hypothalamus should reveal its potential role as a central regulator of energy sensing.
KSR2 is also detectable in skeletal muscle and liver (Guo et al. 2014). Thus, while noncell autonomous effects of KSR2 on energy balance may be mediated through the central nervous system, the potential remains for cell autonomous actions of KSR2 in muscle and liver that affect glucose homeostasis. Obese ksr2−/− mice show hepatic steatosis (Costanzo‐Garvey and Lewis, unpublished). In combination with the defective activation of AMPK of adult ksr2−/− mice, this observation is consistent with recent observations that AMPK protects against hepatic steatosis by phosphorylating SREBP‐1c, suppressing its cleavage and nuclear translocation, and repressing expression of SREBP‐1c target genes that promote lipogenesis and lipid accumulation (Li et al. 2011). Similarly, sedentary mice expressing dominant negative AMPK catalytic subunits in skeletal muscle (Mu et al. 2003) and ksr2−/− mice (Costanzo‐Garvey et al. 2009) show a similar depletion of glycogen in the gastrocnemius. Tissue‐specific deletion of KSR2 in these tissues may reveal key organs contributing to metabolic control by KSR2.
The observation that ksr2−/− mice recapitulate key metabolic characteristics identified in humans bearing mutations in KSR2 (Pearce et al. 2013) creates new opportunities for defining critical molecular mechanisms that control energy balance and for revealing their sites of action. Future studies should also reveal opportunities for early detection and prevention of obesity and its attendant metabolic disorders.
Acknowledgments
The authors thank Dr. Stephen Bonasera for the training and use of the indirect calorimeter and DEXA equipment. Members of the Lewis laboratory are thanked for their helpful comments and criticism.
Conflict of Interest
No potential conflicts of interest relevant to this article were reported.
Footnotes
Funding Information
This work is supported by National Cancer Institute grant CA90400 and US Army grant W081XWH‐10‐1‐0139 to R.E.L.
These authors contributed equally to the paper.
References
- Balthasar N., Dalgaard L. T., Lee C. E., Yu J., Funahashi H., Williams T. 2005. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell; 123:493-505. [DOI] [PubMed] [Google Scholar]
- Barker D. J. 2005. The developmental origins of insulin resistance. Horm. Res.; 64Suppl. 3:2-7. [DOI] [PubMed] [Google Scholar]
- Bates S. H., Stearns W. H., Dundon T. A., Schubert M., Tso A. W., Wang Y. 2003. STAT3 signalling is required for leptin regulation of energy balance but not reproduction. Nature; 421:856-859. [DOI] [PubMed] [Google Scholar]
- Bieswal F., Ahn M. T., Reusens B., Holvoet P., Raes M., Rees W. D. 2006. The importance of catch‐up growth after early malnutrition for the programming of obesity in male rat. Obesity; 14:1330-1343. [DOI] [PubMed] [Google Scholar]
- Chen H., Charlat O., Tartaglia L. A., Woolf E. A., Weng X., Ellis S. J. 1996. Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell; 84:491-495. [DOI] [PubMed] [Google Scholar]
- Chua S. C., Jr 1997. Monogenic models of obesity. Behav. Genet.; 27:277-284. [DOI] [PubMed] [Google Scholar]
- Claret M., Smith M. A., Batterham R. L., Selman C., Choudhury A. I., Fryer L. G. 2007. AMPK is essential for energy homeostasis regulation and glucose sensing by POMC and AgRP neurons. J. Clin. Invest.; 117:2325-2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo‐Garvey D. L., Pfluger P. T., Dougherty M. K., Stock J. L., Boehm M., Chaika O. 2009. KSR2 is an essential regulator of AMP kinase, energy expenditure, and insulin sensitivity. Cell Metab.; 10:366-378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota D., Proulx K., Seeley R. J. 2007. The role of CNS fuel sensing in energy and glucose regulation. Gastroenterology; 132:2158-2168. [DOI] [PubMed] [Google Scholar]
- Dougherty M. K., Ritt D. A., Zhou M., Specht S. I., Monson D. M., Veenstra T. D. 2009. KSR2 is a calcineurin substrate that promotes ERK cascade activation in response to calcium signals. Mol. Cell; 34:652-662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqi S., O'Rahilly S. 2006. Genetics of obesity in humans. Endocr. Rev.; 27:710-718. [DOI] [PubMed] [Google Scholar]
- Farooqi I. S., Matarese G., Lord G. M., Keogh J. M., Lawrence E., Agwu C. 2002. Beneficial effects of leptin on obesity, T cell hyporesponsiveness, and neuroendocrine/metabolic dysfunction of human congenital leptin deficiency. J. Clin. Invest.; 110:1093-1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farooqi I. S., Wangensteen T., Collins S., Kimber W., Matarese G., Keogh J. M. 2007. Clinical and molecular genetic spectrum of congenital deficiency of the leptin receptor. N. Engl. J. Med.; 356:237-247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellmann L., Nascimento A. R., Tibirica E., Bousquet P. 2013. Murine models for pharmacological studies of the metabolic syndrome. Pharmacol. Ther.; 137:331-340. [DOI] [PubMed] [Google Scholar]
- Fernandez M. R., Henry M. D., Lewis R. E. 2012. Kinase suppressor of Ras 2 (KSR2) regulates tumor cell transformation via AMPK. Mol. Cell. Biol.; 32:3718-3731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez‐Twinn D. S., Ozanne S. E. 2010. Early life nutrition and metabolic programming. Ann. N. Y. Acad. Sci.; 1212:78-96. [DOI] [PubMed] [Google Scholar]
- Flier J. S. 2004. Obesity wars: molecular progress confronts an expanding epidemic. Cell; 116:337-350. [DOI] [PubMed] [Google Scholar]
- Ghilardi N., Ziegler S., Wiestner A., Stoffel R., Heim M. H., Skoda R. C. 1996. Defective STAT signaling by the leptin receptor in diabetic mice. Proc. Natl Acad. Sci. USA; 93:6231-6235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson W. T., Farooqi I. S., Moreau M., DePaoli A. M., Lawrence E., O'Rahilly S. 2004. Congenital leptin deficiency due to homozygosity for the Delta133G mutation: report of another case and evaluation of response to four years of leptin therapy. J. Clin. Endocrinol. Metab.; 89:4821-4826. [DOI] [PubMed] [Google Scholar]
- Guo L., Volle D. J., Lewis R. E. 2014. Identification of a truncated kinase suppressor of Ras 2 mRNA in sperm. FEBS Open Bio.; 4:420-425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halaas J. L., Gajiwala K. S., Maffei M., Cohen S. L., Chait B. T., Rabinowitz D. 1995. Weight‐reducing effects of the plasma protein encoded by the obese gene. Science; 269:543-546. [DOI] [PubMed] [Google Scholar]
- Herman M. A., Kahn B. B. 2006. Glucose transport and sensing in the maintenance of glucose homeostasis and metabolic harmony. J. Clin. Invest.; 116:1767-1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummel K. P., Dickie M. M., Coleman D. L. 1966. Diabetes, a new mutation in the mouse. Science; 153:1127-1128. [DOI] [PubMed] [Google Scholar]
- Isganaitis E., Jimenez‐Chillaron J., Woo M., Chow A., DeCoste J., Vokes M. 2009. Accelerated postnatal growth increases lipogenic gene expression and adipocyte size in low‐birth weight mice. Diabetes; 58:1192-1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez‐Chillaron J. C., Hernandez‐Valencia M., Reamer C., Fisher S., Joszi A., Hirshman M. 2005. Beta‐cell secretory dysfunction in the pathogenesis of low birth weight‐associated diabetes: a murine model. Diabetes; 54:702-711. [DOI] [PubMed] [Google Scholar]
- Lee G. H., Proenca R., Montez J. M., Carroll K. M., Darvishzadeh J. G., Lee J. I. 1996. Abnormal splicing of the leptin receptor in diabetic mice. Nature; 379:632-635. [DOI] [PubMed] [Google Scholar]
- Li Y., Xu S., Mihaylova M. M., Zheng B., Hou X., Jiang B. 2011. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet‐induced insulin‐resistant mice. Cell Metab.; 13:376-388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsley J. E., Rutter J. 2004. Nutrient sensing and metabolic decisions Comp. Biochem. Physiol. B Biochem. Mol. Biol.; 139:543-559. [DOI] [PubMed] [Google Scholar]
- Lowell B. B., Spiegelman B. M. 2000. Towards a molecular understanding of adaptive thermogenesis. Nature; 404:652-660. [DOI] [PubMed] [Google Scholar]
- Maffei M., Fei H., Lee G. H., Dani C., Leroy P., Zhang Y. 1995. Increased expression in adipocytes of ob RNA in mice with lesions of the hypothalamus and with mutations at the db locus. Proc. Natl Acad. Sci. USA; 92:6957-6960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montague C. T., Farooqi I. S., Whitehead J. P., Soos M. A., Rau H., Wareham N. J. 1997. Congenital leptin deficiency is associated with severe early‐onset obesity in humans. Nature; 387:903-908. [DOI] [PubMed] [Google Scholar]
- Mu J., Barton E. R., Birnbaum M. J. 2003. Selective suppression of AMP‐activated protein kinase in skeletal muscle: update on ‘lazy mice’. Biochem. Soc. Trans.; 31:236-241. [DOI] [PubMed] [Google Scholar]
- NIH. 1998. Clinical guidelines on the identification, evaluation, and treatment of overweight and obesity in adults‐ the evidence report. Obes. Res.; 6:51S-209S. [PubMed] [Google Scholar]
- Ollmann M. M., Wilson B. D., Yang Y. K., Kerns J. A., Chen Y., Gantz I. 1997. Antagonism of central melanocortin receptors in vitro and in vivo by agouti‐related protein. Science; 278:135-138. [DOI] [PubMed] [Google Scholar]
- Pearce L. R., Atanassova N., Banton M. C., Bottomley B., van der Klaauw A. A., Revelli J. P. 2013. KSR2 mutations are associated with obesity, insulin resistance, and impaired cellular fuel oxidation. Cell; 155:765-777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Revelli J. P., Smith D., Allen J., Jeter‐Jones S., Shadoan M. K., Desai U. 2011. Profound obesity secondary to hyperphagia in mice lacking kinase suppressor of ras 2. Obesity; 19:1010-1018. [DOI] [PubMed] [Google Scholar]
- Sokal R. R., Rohlf F. J. Biometry: the principles and practice of statistics in biological research. New York: W H Freeman and Company; 1995. [Google Scholar]
- Tartaglia L. A., Dembski M., Weng X., Deng N., Culpepper J., Devos R. 1995. Identification and expression cloning of a leptin receptor, OB‐R. Cell; 83:1263-1271. [DOI] [PubMed] [Google Scholar]
- Tecott L. H., Sun L. M., Akana S. F., Strack A. M., Lowenstein D. H., Dallman M. F. 1995. Eating disorder and epilepsy in mice lacking 5‐HT2c serotonin receptors. Nature; 374:542-546. [DOI] [PubMed] [Google Scholar]
- Viollet B., Andreelli F., Jorgensen S. B., Perrin C., Geloen A., Flamez D. 2003. The AMP‐activated protein kinase alpha2 catalytic subunit controls whole‐body insulin sensitivity. J. Clin. Invest.; 111:91-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen T. T., Gill A. M., Frigeri L. G., Barsh G. S., Wolff G. L. 1994. Obesity, diabetes, and neoplasia in yellow A(vy)/‐ mice: ectopic expression of the agouti gene. FASEB J.; 8:479-488. [DOI] [PubMed] [Google Scholar]
- Zhang Y., Proenca R., Maffei M., Barone M., Leopold L., Friedman J. M. 1994. Positional cloning of the mouse obese gene and its human homologue. Nature; 372:425-432. [DOI] [PubMed] [Google Scholar]