

Abstract
Mitochondrial DNA (mtDNA) is highly compacted into DNA-protein structures termed mitochondrial nucleoids (mt-nucleoids). The key mt-nucleoid components responsible for mtDNA condensation are HMG box-containing proteins such as mammalian mitochondrial transcription factor A (TFAM) and Abf2p of the yeast Saccharomyces cerevisiae. To gain insight into the function and organization of mt-nucleoids in strictly aerobic organisms, we initiated studies of these DNA-protein structures in Yarrowia lipolytica. We identified a principal component of mt-nucleoids in this yeast and termed it YlMhb1p (Y. lipolytica mitochondrial HMG box-containing protein 1). YlMhb1p contains two putative HMG boxes contributing both to DNA binding and to its ability to compact mtDNA in vitro. Phenotypic analysis of a Δmhb1 strain lacking YlMhb1p resulted in three interesting findings. First, although the mutant exhibits clear differences in mt-nucleoids accompanied by a large decrease in the mtDNA copy number and the number of mtDNA-derived transcripts, its respiratory characteristics and growth under most of the conditions tested are indistinguishable from those of the wild-type strain. Second, our results indicate that a potential imbalance between subunits of the respiratory chain encoded separately by nuclear DNA and mtDNA is prevented at a (post)translational level. Third, we found that mtDNA in the Δmhb1 strain is more prone to mutations, indicating that mtHMG box-containing proteins protect the mitochondrial genome against mutagenic events.
INTRODUCTION
Individual eukaryotic cells contain a population of mitochondrial DNA (mtDNA) molecules, the number of which may reach several thousand copies. For example, aerobically grown diploid cells of the yeast Saccharomyces cerevisiae contain, on average, 100 molecules of 85-kbp mtDNA. With a distance of 0.34 nm between base pairs in DNA, the total length of mtDNA reaches almost 3 mm per cell, while the diameter of the cell does not exceed 3 to 4 μm. Analogously to its nuclear counterpart, mtDNA must be packaged into condensed nucleoprotein structures termed mitochondrial nucleoids (mt-nucleoids) (1–6). The size of mt-nucleoids in S. cerevisiae ranges from 0.2 to 0.9 μm, meaning that mtDNA in yeasts undergoes compaction of roughly 3 orders of magnitude. Although it is known that the size and shape (oval versus globular) of mt-nucleoids, the number of mt-nucleoids (ranging from 50 to 70) per diploid cell (2), and the number of copies (up to 9) of mtDNA per mt-nucleoid (3) in S. cerevisiae depend on physiological conditions, the molecular mechanisms mediating nucleoid maintenance, dynamics, and roles in mtDNA distribution/segregation during cell division are largely not understood. This is also true for mammalian mt-nucleoids, which, in contrast to their yeast counterparts, contain a relatively small number of mtDNA molecules and are thus more solitary in nature (7, 8). Description of these intra- and interspecific differences in the organization of mt-nucleoids would greatly facilitate our understanding of the mechanisms participating in the segregation behavior of mtDNA and ultimately the molecular causes of several mitochondrial diseases (9, 10).
The composition of mt-nucleoids has been examined in several eukaryotic species, including human cells (11–14). These experiments revealed a surprising diversity of proteins associated with mt-nucleoids either directly (as DNA-binding proteins) or indirectly, via protein-protein interactions. Interestingly, in addition to the proteins involved in mtDNA replication, transcription, and recombination (e.g., DNA and RNA polymerases, topoisomerases, DNA helicases) and translation (e.g., ribosomal protein Mnp1, RNA helicases), mt-nucleoids contain a substantial number of proteins involved in seemingly unrelated cellular processes, such as metabolism, membrane transport, cytoskeletal dynamics, and protein quality control (for a recent review, see reference 11). Although the roles of individual components in the maintenance of mt-nucleoids are in most cases unknown, the wide repertoire of nucleoid-associated proteins underlines an intimate connection of DNA transactions with other biochemical activities taking place in mitochondria.
Comparative analysis of the composition of mt-nucleoids from distantly related eukaryotic species revealed that although many nucleoid-associated proteins are specific for a given group of organisms, all mt-nucleoids examined contain one principal group of DNA-packaging proteins containing 1 to 2 HMG (high-mobility-group) box domains. The first representative of this large and heterogeneous family of proteins, Abf2p, was identified by biochemical means (15, 16), and its gene was cloned by Diffley and Stillman (17). Abf2p contains two HMG box domains mediating nonspecific binding of the protein to DNA, accompanied by its bending and compaction (18, 19). Chromatin immunoprecipitation (ChIP) analysis of Abf2p binding to mtDNA in vivo revealed its preference for GC-rich sequences (20). Analyses of mutants lacking the ABF2 gene indicated that the protein plays an essential role in the stabilization of mtDNA on media with fermentable carbon sources (17), regulates the organization of mt-nucleoids (21, 22), and influences recombination, the copy number of mtDNA (23, 24), the segregation of mtDNA into daughter cells (25), and the susceptibility of mtDNA to DNA damage (26). Interestingly, in spite of all these functions, S. cerevisiae is able to maintain mtDNA without Abf2p when grown on nonfermentable carbon sources (12, 27). This is possible due to the presence of a backup system of mtDNA maintenance dependent on apparently multifunctional proteins such as aconitase (12, 13).
The fact that HMG box-containing proteins fulfill the roles of DNA-packaging factors in mitochondria was further demonstrated by the discovery of the TFAM (mitochondrial transcription factor A) protein, the major DNA-packaging protein in mammalian mitochondria (28, 29). Like Abf2p, TFAM contains two HMG boxes, and its expression in S. cerevisiae Δabf2 cells suppresses the loss of mtDNA on media with fermentable carbon sources (30, 31). However, in contrast to Abf2p, TFAM exhibits high affinity toward promoter sequences on mtDNA and, in addition to other sequence dissimilarities, contains a charged C terminus involved in the stimulation of transcription (32). Crystallographic analyses of TFAM bound to the promoter revealed that it promotes DNA bending, reversing the direction of the DNA helix and forming loops that allow the initiation of transcription (33, 34). Importantly, whereas heterozygous TFAM+/− mice have ∼35 to 40% reductions in mtDNA copy numbers, homozygous knockouts (TFAM−/−) die in midgestation due to lack of mtDNA and severe respiratory chain deficiency (35). Furthermore, manipulation of the levels of TFAM caused changes in the mtDNA copy number (36, 37), and removal of TFAM using a conditional knockout system (cre-loxP) resulted both in depletion of mtDNA and mitochondrial transcripts and in severe respiratory chain deficiency (35, 38–42). All these results demonstrate that TFAM is essential for mtDNA maintenance in vivo.
Comparison of the biochemical properties as well as the in vivo roles of S. cerevisiae Abf2p (ScAbf2p) and mammalian TFAM indicates that whereas some functions of mitochondrial HMG box-containing proteins (mtHMG proteins) are shared, many seem to be species specific. Indeed, studies on mtHMG proteins have revealed an exceptionally high rate of evolutionary divergence of their amino acid sequences (43–49). Based on the number of putative HMG boxes (1 or 2) and the presence or absence of a coiled-coil domain, they have been categorized into 3 classes and several subclasses (49). Importantly, only a small fraction of different types of mtHMG proteins was subjected to detailed biochemical and/or genetic analysis: S. cerevisiae Abf2p (see above), Candida albicans (49) and Candida parapsilosis (46) Gcf1 proteins, Podospora anserina mthmg1p (43), and Physarum polycephalum protein Glom (48, 50). In addition, for various reasons, a detailed comparison of the role(s) of TFAM and its fungal counterparts is lacking. For example, in contrast to human cells, S. cerevisiae is a facultative anaerobe that can readily grow without mtDNA on fermentable carbon sources such as glucose. On the other hand, the molecular genetic analysis of mt-nucleoids of strictly aerobic fungal species is hampered either by a lack of suitable tools or by their diploid and highly heterozygous nature, or both (51).
In this study, we decided to investigate mt-nucleoids in the yeast Yarrowia lipolytica, a species belonging to basal lineages of hemiascomycetes, making it a very useful organism for comparative analyses (52). Importantly, in contrast to S. cerevisiae, Y. lipolytica is a strictly aerobic and petite mutant-negative species (53) that is totally dependent on a functional mitochondrial genome. Furthermore, it can be stably propagated in a haploid state, which makes it suitable for phenotypic analysis of the consequences of a gene deletion. We purified mt-nucleoids of Y. lipolytica and identified their principal protein component, YlMhb1p (Mitochondrial HMG box-containing protein 1). The combination of biochemical assays with molecular and genetic techniques available for Y. lipolytica enabled us to perform a detailed analysis of YlMhb1p and to gain insight into general and species-specific properties of mtHMG proteins and their role in mtDNA maintenance.
MATERIALS AND METHODS
Microbial strains and growth conditions.
Yarrowia lipolytica Po1h (MATA ura3-302 xpr2-322 axp1-2) provided by C. Madzak (INRA, Thiverval-Grignon, France) was used as the parental strain for the construction of Y. lipolytica Δmhb1 (MATA Δmhb1::URA3 ura3-302 xpr2-322 axp1-2). Y. lipolytica strains E122 (MATA ura3-302 leu2-270 lys11-23) and E129 (MATA ura3-302 leu2-270 lys11-23 xpr2-322), provided by C. Gaillardin (AgroParisTech, Micalis, Jouy-en-Josas, France), were used as the sources of mt-nucleoids and the genomic DNA for the amplification of the YlMHB1 gene. Yeast cultures were grown at 28°C in a complex medium (1% [wt/vol] yeast extract [Difco], 2% [wt/vol] Bacto peptone [Difco]) supplemented either with 2% (wt/vol) glucose (YPD medium) or with 3% (vol/vol) glycerol (YPG medium). Alternatively, a synthetic medium (0.17% [wt/vol] yeast nitrogen base without amino acids or ammonium sulfate [Difco], 0.5% [wt/vol] ammonium sulfate [Lachema]) supplemented with appropriate amino acids and bases and with a corresponding carbon source (2% [wt/vol] glucose [SD medium] or 3% [vol/vol] glycerol [SG medium]) was used. Where indicated, hygromycin B (HygB; 500 μg/ml; Roche) or ethidium bromide (EtBr; 5 μM; Sigma-Aldrich) was added. Unless otherwise indicated, Y. lipolytica cells were inoculated into a complex medium to a final concentration of 5 × 104/ml and were cultivated for 16 h at 28°C to mid-exponential phase (5 × 107 cells/ml). Escherichia coli DH5α [F− ϕ80dlacZΔM15 Δ(lacZYA-argF)U169 deoR recA1 endA1 hsdR17(rK− mK+) λ thi-1 gyrA96 relA1 glnV44 nupG] (Life Technologies) was used for the amplification of plasmid constructs. E. coli BL21 Star(DE3) [F− ompT hsdSB(rB− mB−) gal dcm rne131] (Life Technologies) was used for the production of the recombinant YlMhb1–glutathione S-transferase (GST) protein. Bacterial cultures were grown in LB medium (1% [wt/vol] Bacto peptone [Difco], 0.5% [wt/vol] yeast extract [Difco], 1% [wt/vol] NaCl [pH 7.5]) containing 100 μg/ml ampicillin. Y. lipolytica was transformed as described by Chen et al. (54) or Wang et al. (55).
DNA manipulations.
Enzymatic manipulations with DNA, cloning procedures, and DNA labeling were performed according to the instructions provided by the suppliers. Oligonucleotides (see Table S1 in the supplemental material) were synthesized by Metabion or Microsynth. Genomic DNA was isolated as described by Linda Breeden (http://labs.fhcrc.org/breeden/Methods/). PCRs were performed in a 10- to 50-μl volume using 1 U Taq DNA polymerase (Life Technologies), DreamTaq DNA polymerase (Thermo Scientific), or Phusion Hot Start II high-fidelity DNA polymerase (Thermo Scientific). PCR mixtures contained all four deoxynucleoside triphosphates (dNTPs) (final concentration, 200 μM each), the corresponding primers (final concentration, 1 μM) (see Table S1), and either 100 ng of genomic DNA or 10 ng of plasmid DNA. PCR fragments were purified from agarose gels using a QIAquick gel extraction kit (Qiagen) or a Zymoclean gel DNA recovery kit (Zymo Research).
Purification and analysis of mt-nucleoids.
Cells were cultivated aerobically at 30°C to stationary phase in YPD medium. Spheroplasts were formed according to the method of Kerscher et al. (56). Cells (about 40 g [wet weight]) were resuspended in 120 ml of 0.1 M Tris-HCl (pH 9.3)–10 mM dithiothreitol (DTT) and were incubated for 10 min at 30°C. After a wash with buffer D (2 M sorbitol, 1 mM MgCl2, 1 mM EDTA, 25 mM potassium phosphate [pH 7.5]), cells were treated with Zymolyase 20T (Seikagaku Kogyo, Japan) for 2 h in 300 ml of buffer D at 30°C. After another wash with buffer D, spheroplasts were homogenized with a Waring blender in NES1 buffer (0.3 M sucrose, 20 mM Tris-HCl [pH 7.6], 1 mM EDTA, 0.4 mM spermidine, 7 mM 2-mercaptoethanol, 0.4 mM phenylmethylsulfonyl fluoride [PMSF]). The mt-nucleoids were isolated as described previously (3, 46). The protein composition of mt-nucleoids was analyzed by SDS-PAGE (57). DNA-binding proteins in mitochondrial fractions were detected using SDS-DNA-polyacrylamide gel electrophoresis as described by Miyakawa et al. (46). For determination of the N-terminal amino acid sequence of the 30-kDa band, proteins were separated by SDS-PAGE and were transferred to a PVDF membrane, and the 30-kDa protein was sequenced using a PPSQ-21 protein sequencer (Shimazu, Japan).
Construction of plasmid vectors.
The YlMHB1 open reading frame (ORF) was amplified from Y. lipolytica strain E129 genomic DNA by using primers YlMHB1_UP and YlMHB1_DWN, and the PCR fragment was inserted into the pDrive vector by using a PCR cloning kit (Qiagen). Plasmid pGEX-6P-2-YlMHB1, expressing the recombinant version of the YlMhb1 protein in fusion with glutathione S-transferase, was constructed by amplification of the YlMHB1 ORF from pDrive-YlMHB1 by using YlMHB1_UP and YlMHB1_DWN, followed by insertion of the PCR fragment into pGEX-6P-2 (GE Healthcare) linearized with SmaI. Alternatively, the YlMHB1 ORF lacking the first 14 amino acids, which represent a cleavable mitochondrial import sequence, was amplified by PCR from Y. lipolytica strain E122 genomic DNA by using primers YlMHB1_WT_UP and YlMHB1_WT_DWN. The sequence encoding the N-terminal region of YlMhb1p (YlMhb1p-N-term; amino acids 15 to 156) (see Fig. 2A) was amplified by PCR using primers YlMHB1_NT_UP and YlMHB1_NT_DWN. The sequence encoding the C-terminal region of YlMhb1p (YlMhb1p-C-term; amino acids 157 to 238) (see Fig. 2A) was amplified using primers YlMHB1_CT_UP and YlMHB1_CT_DWN. The PCR products containing BamHI and EcoRI flanking sites were inserted into vector pGEX-6P-1 (GE Healthcare) digested with BamHI and EcoRI. For complementation of the Δmhb1 deletion, we constructed plasmid pUB4-YlMHB1 (pUB4 was provided by Ulrich Brandt, Institut für Biochemie II, Goethe Universität, Frankfurt am Main, Germany). A DNA fragment carrying the YlMHB1 ORF, as well as upstream (1,141 bp) and downstream (423 bp) sequences necessary for the initiation and termination of transcription, respectively, was obtained by PCR amplification from Y. lipolytica Po1h genomic DNA using primers YlMHB1_UP1141 and YlMHB1_DN423, each containing a SalI restriction site at its 5′ end. The PCR fragment treated with SalI was cloned into the pUB4 vector (58) linearized with SalI. All plasmid constructs were verified by restriction enzyme mapping and DNA sequencing of inserted fragments.
FIG 2.

Recombinant YlMhb1p binds DNA in vitro. (A) Schematic representation of four versions of YlMhb1p produced in E. coli and used for in vitro experiments. YlMhb1p+MTS, full-length protein containing a mitochondrial targeting sequence (MTS); YlMhb1p-WT, wild-type protein lacking MTS; YlMhb1p-N-term and YlMhb1p-C-term, truncation mutants containing amino acids 15 to 156 and amino acids 157 to 238, respectively. (B) Analysis by EMSA indicates that increasing concentrations of YlMhb1p bind to DNA fragments in vitro. For comparison, purified mitochondrial HMG box-containing proteins from S. cerevisiae (ScAbf2p) and C. albicans (CaGcf1p) were subjected to the same assay. Purified glutathione S-transferase (GST) was used as a negative control. (C) EMSAs of various forms of YlMhb1p bound to HindIII fragments of bacteriophage λ DNA. Purified proteins were added at the same molar concentrations to compensate for the differences in molecular weight. (D) mtDNA was compacted in vitro as described in Materials and Methods. The reaction mixtures contained 50 ng of purified mtDNA and either 0.2 μg of YlMhb1p-WT, 0.16 μg of YlMhb1p-N-term, or 0.1 μg of YlMhb1p-C-term.
Purification of recombinant YlMhb1p from E. coli.
An overnight bacterial culture (20 ml) was inoculated into 500 ml of LB medium containing 1% (wt/vol) glucose and 100 μg/ml ampicillin and was grown at 37°C to a final optical density at 600 nm (OD600) of 0.7. The culture was then cooled to 25°C, supplemented with 0.8 mM isopropyl-β-d-thiogalactopyranoside (IPTG) (Thermo Scientific), and cultivated for an additional 3 h at 25°C. Cells were washed with ice-cold Milli-Q water, and the pellet was frozen at −20°C. After thawing on ice, the cells were resuspended in 15 ml of buffer A (20 mM Tris-HCl [pH 7.5], 100 mM NaCl, 0.1 mM EDTA-NaOH [pH 7.5]) containing 1 mg/ml lysozyme (U.S. Biochemicals), 1× Complete (EDTA-free) protease inhibitors (Roche), 10 mM MgCl2, 100 U of DNase I (Promega), and 200 μg of PureLink RNase A (Life Technologies) and were incubated for 50 min on ice with occasional mixing. The cells were broken by sonicating six times (with a UP50H sonicator), for 20 s each time, and each sonication cycle was followed by 20 s of incubation on ice. Triton X-100 was added to a final concentration of 0.1% (vol/vol), and the suspension was incubated for an additional 10 min on ice and was centrifuged at 16,000 × g for 30 min at 4°C to remove insoluble material. The supernatant was mixed with 0.45 ml of glutathione-agarose (Sigma-Aldrich) prewashed with buffer A, and the mixture was incubated for 60 min at 4°C with continual mixing. Beads were loaded onto a 10-ml column (Bio-Rad) and were washed with 50 ml of buffer A and 15 ml of buffer B (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1 mM EDTA-NaOH [pH 7.5], 1 mM DTT). The washed beads were resuspended in 0.6 ml of buffer B supplemented with 15 μg of PreScission protease (GE Healthcare) and were incubated for 3 h at 4°C with continual mixing. The cleaved YlMhb1p was eluted six times in 0.6 ml of buffer B and was stored at −80°C. Fractions were assayed for purity by SDS-PAGE (57) and were stained with Coomassie brilliant blue R-250. ScAbf2p and C. albicans Gcf1p (CaGcf1p) were purified essentially as described for YlMhb1p. YlMhb1p was further purified with a low-pressure liquid chromatography system (BioLogic LP; Bio-Rad) on columns with heparin Sepharose using buffer B with an NaCl gradient ranging from 0.15 to 1.5 M. GST, used as a negative control in electrophoretic mobility shift assays (EMSAs), was purified by glutathione-agarose as described for YlMhb1-GST except that it was eluted from the beads by 10 mM glutathione in buffer A.
For the purification of various forms of recombinant YlMhb1p, E. coli BL21(DE3) cells were transformed with the recombinant plasmids and were cultivated in 100 ml of 2× YTA (1.6% [wt/vol] tryptone, 1% [wt/vol] yeast extract, 0.5% [wt/vol] NaCl, 50 μg/ml ampicillin) to an OD600 of 0.6; then IPTG was added to a final concentration of 0.1 mM, and expression was induced for 6 h at 30°C. Cells were harvested by centrifugation, resuspended in 5 ml of phosphate-buffered saline (PBS) (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 10 mM KH2PO4 [pH 7.3]), and sonicated with a UP50H sonicator (amplitude, 90%; cycle 0.5; 10 times). Triton X-100 and DTT were added to the cell homogenate to 1% (vol/vol) and 5.0 mM, respectively, and the mixture was then incubated for 30 min on ice. After centrifugation, 10 mM MgCl2 and 100 U DNase I–0.4 mM PMSF were added to the supernatant, and the mixture was incubated for 15 min at 25°C. The supernatant was mixed with 200 μl of a 50% (vol/vol) slurry of glutathione-Sepharose 4B and was gently mixed overnight. The beads were washed five times with PBS and were then resuspended in 1 ml of PreScission cleavage buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1 mM EDTA, 1 mM DTT); 40 μl of PreScission protease (GE Healthcare) was added, and the mixture was incubated for 2 h at 5°C. After protease digestion, the suspension was transferred to a minicolumn. The proteins were eluted twice with 100 μl of PreScission cleavage buffer.
EMSA.
Purified recombinant YlMhb1p, ScAbf2p, and CaGcf1p (0 to 400 ng) were mixed with 160 ng of plasmid pUC19 (New England BioLabs) digested with PvuI and were incubated for 1 h at room temperature in 20 mM Tris-HCl (pH 7.5)–5 mM EDTA-NaOH (pH 7.5). Alternatively, bacteriophage λ DNA (75 ng) digested with HindIII was incubated with various forms of YlMhb1p for 15 min at 25°C in 10 mM Tris-HCl (pH 7.5), 1 mM EDTA-NaOH (pH 7.5). Where indicated, purified proteins were added at the same molar concentrations to compensate for the difference in molecular weight. Samples were electrophoretically separated in a 0.6-to-0.8% (wt/vol) agarose gel in 0.5× TBE buffer (45 mM Tris-borate, 1 mM EDTA-NaOH [pH 8.0]), and DNA was visualized by staining the gel with EtBr (0.5 μg/ml).
Compaction of mtDNA in vitro.
The reaction mixture contained 50 ng of mtDNA, 0.25 M sucrose, 10 mM Tris-HCl (pH 7.5), 0.25 mM EDTA, 0.4 mM spermidine, and the amounts of proteins indicated in Fig. 2D. The mixtures were incubated for 10 min at 25°C. For the observation of mtDNA compaction, 3 μl of the sample was mixed with 3 μl of a 4′,6-diamidino-2-phenylindole (DAPI) solution (2 μg/ml DAPI, 0.25 M sucrose, 20 mM Tris-HCl [pH 7.5], 1 mM EDTA, 1 mM MgCl2, 0.1 mM ZnSO4, 0.1 mM CaCl2, 0.8 mM PMSF, 0.05% [vol/vol] 2-mercaptoethanol). All observations were made with an epifluorescence microscope (BHS-RFK; Olympus Optical Co.).
Fluorescence microscopy.
Y. lipolytica wild-type and Δmhb1 mutant cells cultivated in complex glucose medium were fixed with 70% (vol/vol) ethanol at room temperature for 10 min. Samples were centrifuged, washed with 10 mM Tris-HCl (pH 7.5), and stained at room temperature for 30 min with DAPI to a final concentration of 0.5 μg/ml. For the observation of the mitochondrial network, Y. lipolytica cells were stained with the fluorescent dye DiOC6 (Molecular Probes) added directly to a cultivation medium (YPD) to a final concentration of 437.5 nM and were observed immediately. All observations were made with an Olympus BX50 microscope equipped with a DP70 camera (Olympus Optical Co.) and an appropriate filter set, or on an Olympus BHS-RFK microscope.
PFGE.
mtDNA was analyzed by pulsed-field gel electrophoresis (PFGE) essentially as described previously (59). Briefly, whole-cell DNA samples were prepared in agarose plugs containing Zymolyase 20T (final concentration, 0.5 mg/ml; Seikagaku Corp.) and were incubated overnight in solution I (10 mM Tris-HCl, 0.45 M EDTA-NaOH [pH 8.8], 7.5% [vol/vol] 2-mercaptoethanol) at 37°C. The agarose plugs were transferred to solution II (0.45 M EDTA-NaOH [pH 8.8], 1% [wt/vol] N-lauroylsarcosine) and were incubated with proteinase K (1 mg/ml) overnight at 55°C. For DNA digestion, the agarose plugs were washed three times with 50 ml of TE buffer (10 mM Tris-HCl [pH 7.5], 10 mM EDTA-NaOH [pH 8.0]) for 30 min at room temperature, twice with 1 ml of TE buffer supplemented with 0.04 mg/ml PMSF for 1 h at 50°C, twice with 50 ml of TE buffer for 15 min at room temperature, and once with 1 ml of 2× restriction enzyme buffer with bovine serum albumin (BSA) for 1 h at room temperature. Finally, the agarose plugs were immersed in 0.5 ml of 1× restriction enzyme buffer with BSA, and after the addition of 50 U of ApaI, the plugs were incubated for 1 h at 4°C and subsequently overnight at 25°C for DNA digestion. Before electrophoresis, the agarose plugs were washed with 1 ml of 0.45 M EDTA-NaOH (pH 8.8) at room temperature. Electrophoretic separation was performed using a 1% (wt/vol) agarose gel in 0.5× TBE buffer in a Pulsaphor apparatus (LKB) in a contour-clamped homogeneous electric field (CHEF) configuration with the pulse switching from 5 to 50 s (linear interpolation) for 24 h at 150 V and 9°C throughout (60). DNA bands were stained with EtBr (0.5 μg/ml) and were visualized under UV light. The gel was then incubated for 30 min in 0.125 M HCl for depurination, 40 min in denaturation buffer (1.5 M NaCl, 0.5 M NaOH), 30 min in neutralization buffer (0.5 M Tris-HCl [pH 7.4], 1.5 M NaCl), and finally 30 min in 20× SSC buffer (pH 7.0) (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate). DNA was subsequently transferred to a Hybond N+ membrane (GE Healthcare) with a VacuGene XL vacuum blotting system (Amersham) in 20× SSC. For the detection of Y. lipolytica mtDNA, the YlATP6-specific probe was prepared as follows. The 760-bp fragment of the YlATP6 gene was amplified by primers YlATP6_F and YlATP6_R. The PCR product was purified from the agarose gel (with a QIAquick gel extraction kit; Qiagen) and was labeled with [α-32P]dCTP using a Prime-a-Gene labeling kit (Promega). Finally, the membrane was prehybridized for 1 h at 65°C and was then hybridized overnight at 65°C in Church's buffer (1% [wt/vol] BSA, 1 mM EDTA, 0.5 M sodium phosphate, 7% [wt/vol] SDS [pH 7.2]) containing the labeled YlATP6 probe. The membrane was rinsed once with washing buffer I (2× SSC, 0.1% [wt/vol] SDS) for 20 min at room temperature and then once with washing buffer II (0.1× SSC, 0.1% [wt/vol] SDS) for 60 min at 50°C. Membranes were exposed to storage phosphor screens (Kodak) for 24 to 72 h, and signal detection of screens was performed with a phosphorimager (Personal Molecular Imager FX; Bio-Rad).
Quantification of mtDNA by qPCR.
For the isolation of total cellular DNA, a modified protocol described by Philippsen et al. (61) was used. Y. lipolytica cells were grown in 50 ml of YPD medium to mid-exponential phase (5 × 107 cells/ml), washed once with Milli-Q water, resuspended in 0.3 ml of 0.9 M sorbitol–80 mM EDTA with 0.4 mg/ml Zymolyase 20T, and incubated for 1 h at 37°C with occasional shaking. Protoplasts were resuspended in 0.275 ml of 50 mM Tris-HCl (pH 7.4)–20 mM EDTA-NaOH (pH 7.5)–1% (wt/vol) SDS and were incubated for 30 min at 65°C. After the addition of potassium acetate to a final concentration of 4.5 M, samples were incubated for 1 h on ice. Samples were centrifuged, and the supernatant was extracted with phenol-chloroform (1:1). Nucleic acids were precipitated with an equal volume of ice-cold 2-propanol, sedimented by centrifugation, washed with 70% (vol/vol) ethanol, and dried in a vacuum. The pellet was dissolved in 150 μl of TE buffer, and after the addition of RNase A to a final concentration of 0.1 mg/ml, samples were incubated for 30 min at 37°C. Nucleic acids were precipitated with 0.1 volume of 3 M sodium acetate and 1 volume of 2-propanol on ice. After centrifugation, the pellet was dried in a vacuum, resuspended in 50 μl of TE buffer, and used for quantitative real-time PCR (qPCR). Four to 20 ng of total cellular DNA was analyzed by qPCR using the StepOne real-time PCR system (Applied Biosystems) with the following program: 40 cycles of 95°C for 15 s, 65°C for 30 s, and 72°C for 30 s. Maxima SYBR green qPCR master mix (Thermo Scientific) was used to amplify a 133-bp portion of the YlACT1 gene, a 187-bp portion of the YlATP9 gene containing an upstream sequence, and a 145-bp portion of the YlGPD1 gene using the qPCR primers listed in Table S1 in the supplemental material. Experiments were performed on five biological replicates for each strain and were analyzed in duplicate. The relative mtDNA copy number was calculated according to the relative standard curve method (62), and the results were evaluated using a nonparametric statistical test.
RNA isolation and Northern blot analysis.
Total RNA was purified from Y. lipolytica strains according to a modified protocol described by Cross and Tinkelenberg (63). Cell cultures were grown in 100 ml of YPD medium to mid-exponential phase (5 × 107 cells/ml), harvested by centrifugation, washed twice with ice-cold diethyl pyrocarbonate (DEPC)-treated deionized water, and resuspended in 350 μl ice-cold TES buffer (10 mM Tris-HCl [pH 7.5], 10 mM EDTA-NaOH [pH 8.0], 0.5% [wt/vol] SDS) and 350 μl of acidic phenol (pH 5.0). Glass beads (200 μl) were added, and samples were vortexed at maximum speed six times, for 30 s each time, with 30-s pauses on ice. Samples were centrifuged at 13,000 × g for 5 min at 4°C, and the supernatant was extracted with phenol-chloroform (1:1). RNA was precipitated by the addition of 0.3 M sodium acetate and 2.5 volume of 96% (vol/vol) ethanol for 1 h at −80°C. RNA was sedimented by centrifugation, and the pellet was dried in a vacuum and was dissolved in DEPC-treated deionized water. The integrity of RNA samples was determined using an RNA nanochip on an Agilent 2100 Bioanalyzer. For transcriptome-sequencing (RNA-seq) analysis, total-RNA samples (100 μg) were treated with 20 U of DNase I (New England BioLabs), extracted with phenol-chloroform (1:1), and cleaned up with a GeneJET RNA purification kit (Thermo Scientific). Northern blot analysis of total RNA samples was performed essentially as described by Kroczek and Siebert (64). RNA was electrophoretically separated in a 1.2% (wt/vol) agarose gel in 1× morpholinepropanesulfonic acid (MOPS) buffer (20 mM MOPS, 5 mM sodium acetate, 1 mM EDTA [pH 7.0]) for 1.5 h at 10 V/cm. RNA was visualized under UV light and was vacuum blotted (VacuGene XL; Amersham) to a Hybond N+ membrane (GE Healthcare) using 20× SSC. The membrane was rinsed with 5× SSC, and RNA was stained with methylene blue (0.02% [wt/vol] methylene blue, 0.3 M sodium acetate). The membrane was then destained with 0.2× SSC–1% (wt/vol) SDS, rinsed with 5× SSC, and fixed for 2 h at 80°C. DNA probes labeled with [α-32P]dCTP (MHB1, 23SrRNA, ACT1, ATP6, NUBM, NUCM, NESM) were prepared as described above using the primers listed in Table S1 in the supplemental material. The membrane was prehybridized for 1 h at 42°C in a hybridization buffer (50% [vol/vol] deionized formamide, 5× SSC, 50 mM phosphate buffer [pH 7.0], 5× Denhardt‘s buffer [0.1% {wt/vol} Ficoll-400, 0.1% {wt/vol} polyvinylpyrrolidone, 0.1% {wt/vol} BSA fraction V], 100 μg/ml denatured salmon sperm DNA) and was then hybridized overnight at 42°C in a hybridization buffer containing a corresponding probe. The membrane was rinsed once with washing buffer I for 20 min at room temperature and then once with washing buffer II for 30 min at 50°C. Signal detection was performed as described above.
In silico analyses.
The mitochondrial import sequence was predicted using MitoProt II (http://ihg.gsf.de/ihg/mitoprot.html) (65). The HMG boxes were predicted using Phyre (http://www.sbg.bio.ic.ac.uk/phyre2/html/page.cgi?id=index) (66). Sequence alignments and comparisons were carried out using ClustalW2 (http://www.ebi.ac.uk/Tools/msa/clustalw2/) (67).
Immunoblot analysis.
Yeast cultures were grown in YPD or YPG medium to mid-exponential phase (5 × 107 cells/ml), harvested by centrifugation (5 min, 3,000 × g), and washed with ice-cold Milli-Q water. Whole-cell protein extracts were prepared by using the NaOH lysis/trichloroacetic acid (TCA) precipitation method according to the work of Knop et al. (68). Proteins were separated by SDS-PAGE (57) and were electrotransferred to a nitrocellulose membrane (Hybond-ECL; GE Healthcare), and immunodetection was performed as described by Tomaska et al. (69). For the detection of Atp6p, membranes were incubated with a polyclonal antibody raised against ScAtp6p (dilution, 1:10,000; provided by Jean-Paul di Rago, Institut de Biochemie et Génetique Cellulaires, Université Bordeaux, Bordeaux, France). Cox2p was visualized by a rabbit polyclonal anti-Cox2p antibody (dilution, 1:1,000) provided by Alexander Tzagoloff (Columbia University, New York, NY, USA). Antigen-antibody complexes were visualized using peroxidase-conjugated (dilution, 1:15,000) or alkaline phosphatase-conjugated (dilution, 1:10,000) (both from Sigma-Aldrich) anti-rabbit antibodies. For the detection of subunits of respiratory complex I, undiluted cell culture supernatants of mouse hybridoma cell lines recognizing NUCM (49-kDa subunit), NUBM (51-kDa subunit), and NESM (provided by Volker Zickermann, Institut für Biochemie II, Goethe Universität, Frankfurt am Main, Germany) (70) were used. Antigen-antibody complexes were visualized using an alkaline phosphatase-conjugated anti-mouse antibody (diluted 1:10,000; Sigma-Aldrich).
MnCl2-induced mutagenesis of mtDNA.
Mutagenesis was performed as described previously (71). Yeast cells were pregrown to stationary phase in liquid YPG medium and were stored overnight at 4°C. Cells were inoculated into 10 ml of “mutagenic” medium (YPD supplemented with 8 mM MnCl2, with the pH adjusted to 6.0) or 10 ml of YPD medium to a final concentration of 5 × 105/ml and were incubated at 28°C for 8 h at 200 rpm. Cells were centrifuged (5 min, 3,000 × g) and were resuspended in the same medium to a concentration of 8 × 106/ml, and 0.1 ml was plated onto YPG plates containing oligomycin (10 μg/ml; Sigma-Aldrich). Samples of 10−4 dilutions were plated onto YPG plates for the determination of CFU. Oligomycin-resistant (OliR) mutant colonies were counted after 8 days of incubation at 28°C.
RESULTS
Purified mt-nucleoids of Y. lipolytica contain a protein with two HMG boxes.
Although the complete sequence of mtDNA in Y. lipolytica was determined more than 10 years ago (72), virtually nothing was known about its organization into higher-order nucleoprotein structures. To initiate a systematic analysis, we purified mt-nucleoids from wild-type Y. lipolytica cells and analyzed their major constituents. Whereas the most abundant protein present in the mitochondrial matrix (the major mitochondrial nuclease, Nuc1p) was completely absent in a fraction containing mt-nucleoids, the fraction was enriched for a 30-kDa protein that was also detectable by EtBr staining, suggesting that it possesses DNA-binding activity (Fig. 1A and B). The 30-kDa band was transferred to a PVDF membrane, excised, and subjected to N-terminal sequencing. The sequence of the peptide (KEAATKTKASKAKAP) was used as a query for a BLAST search of the Y. lipolytica genome. As a result, we identified a DNA sequence localized at chromosome E, 186 bp upstream of the ATG codon of the annotated ORF YALI0E07623g. Since this region did not contain any in-frame ATG codon, we hypothesized that the ORF may start with GTG (a codon for valine). This phenomenon has been experimentally confirmed for a number of ORFs, such as ScGRS1 (encoding glycyl-tRNA synthetase) (73) and several mammalian genes (74). Indeed, when GTG was used as a start codon in a conceptual translation, the resulting 238-amino-acid protein contained not only the sequence of the peptide identified but also 14 amino acids representing a putative mitochondrial targeting sequence (MTS) (MitoProt score, 0.9783) (Fig. 1C). To test whether this sequence is able to direct a protein into mitochondria, we constructed a plasmid encoding the predicted protein in fusion with green fluorescent protein (GFP) at its C terminus. When expressed in S. cerevisiae, the fusion protein was targeted into mitochondria and colocalized with mt-nucleoids stained with DAPI, while the GFP without the protein at the N terminus was localized in the cytoplasm (see Fig. S1A in the supplemental material). In addition to the mitochondrial targeting sequence, the protein contains two putative HMG boxes, as predicted by the Phyre server (66). Based on its mitochondrial localization and the fact that an HMG box is a common feature of mtDNA-packaging proteins (e.g., TFAM, ScAbf2p, CaGcf1p, Glom), we named the protein YlMhb1p (Y. lipolytica mitochondrial HMG box-containing protein 1). The fact that YlMhb1p is a putative functional orthologue of the prototypic yeast HMG box-containing protein ScAbf2 prompted us to investigate its ability to restore defects exhibited by the Δabf2 mutant, namely, its rapid loss of mtDNA on a fermentable carbon source such as glucose. Indeed, expression of YlMHB1 from an inducible GAL1 promoter in an Δabf2 mutant almost completely prevented the formation of petite colonies composed of respiration-deficient cells (see Table S2 and Fig. S1B in the supplemental material). Interestingly, as observed by Zelenaya-Troitskaya et al. (24), overexpression of ScABF2 in wild-type as well as Δabf2 cells grown on galactose (inducing conditions for GAL1-driven transcription) interfered with mtDNA maintenance, resulting in increased formation of respiration-deficient cells. Although a mitochondrial HMG box-containing protein (DhmtHMG1) from Debaryomyces hansenii (49), a yeast species more closely related to S. cerevisiae, exhibited the same property as ScAbf2p, YlMhb1p prevented the formation of petite colonies under both derepressed and inducing conditions (see Table S2 and Fig. S1B). These results, together with the dramatically different cellular physiologies of S. cerevisiae (a facultative anaerobe) and Y. lipolytica (a strict aerobe) manifested by different demands on the stability of mtDNA (petite mutant positivity versus petite mutant negativity), resulted in our decision to pursue a detailed study of YlMhb1p.
FIG 1.

Y. lipolytica nucleoids contain a novel type of HMG box-containing protein, YlMhb1. (A) Y. lipolytica mitochondria were fractionated, and mt-nucleoids were purified as described in Materials and Methods. Proteins (5 μg per lane) were separated by 12% SDS-PAGE and were visualized by silver staining. (B) DNA-binding proteins in mitochondrial fractions were detected using SDS-DNA-polyacrylamide gel electrophoresis as described previously (46). The 38-kDa band exhibiting strong DNA-binding activity most likely corresponds to the Nuc1 nuclease as observed in C. parapsilosis (46). (C) Schematic representation of the amino acid sequence of YlMhb1p highlighting the putative N-terminal mitochondrial targeting sequence, the peptide identified by N-terminal sequencing, and the positions of two putative HMG boxes predicted by the Phyre server (66).
Recombinant YlMhb1p binds and compacts DNA in vitro.
To test whether YlMhb1p is a DNA-binding protein, we first expressed the entire ORF in E. coli in fusion with N-terminally located glutathione S-transferase (GST). The crude protein fraction was loaded onto glutathione agarose, followed by extensive washing of the beads, subsequent cleavage of YlMhb1p+MTS-GST with PreScission protease, and further purification on heparin Sepharose (see Fig. S2A in the supplemental material). The resulting protein (YlMhb1p+MTS) (Fig. 2A; see also Fig. S2A in the supplemental material) was used for in vitro experiments using linear DNA fragments as substrates in electrophoretic mobility shift assays (EMSAs). As with its full-length counterparts from S. cerevisiae (ScAbf2p) and C. albicans (CaGcf1p), the increasing concentrations of YlMhb1p yielded slower-migrating DNA-protein complexes, indicating gradual covering of DNA with protein particles (Fig. 2B).
To analyze the DNA-binding properties of YlMhb1p in more detail, we prepared three different versions of the protein. YlMhb1p-WT, like the protein identified in Y. lipolytica mitochondria, lacks the first 14 amino acids representing the mitochondrial targeting sequence. In addition, to investigate the contributions of individual HMG boxes to the DNA-binding properties of YlMhb1p, we prepared recombinant versions of YlMhb1p containing only the N-terminal HMG box (YlMhb1p-N-term) or only the C-terminal HMG box (YlMhb1p-C-term) (Fig. 2A; see also Fig. S2B in the supplemental material). We tested the properties of all three versions of YlMhb1p by three biochemical assays. First, we performed EMSAs using a mixture of linear DNA fragments as substrates. The three versions of the protein exhibited very different binding properties. In comparison with wild-type YlMhb1p, the N-terminal half of the protein apparently bound DNA with much higher affinity, whereas the C-terminal portion of YlMhb1p bound DNA only at much higher concentrations (Fig. 2C). Although it is possible that the migration of DNA is caused by aggregation of the N-terminal fragment, thus preventing the movement of DNA through the gel, we observed in the second assay that in contrast to YlMhb1p-C-term, both YlMhb1p-WT and YlMhb1p-N-term are relatively strongly retained on DNA cellulose (see Fig. S2C in the supplemental material). Together with the EMSA findings, these results indicate that the centrally located HMG box is crucial for DNA binding. In the third assay, we tested the ability of YlMhb1p to compact mtDNA in vitro. In this case, the N-terminal half of the protein was the most potent version of YlMhb1p, whereas incubation of YlMhb1p-C-term with mtDNA resulted only in subtle compaction of mtDNA (Fig. 2D). All these experiments indicate that YlMhb1p exhibits the in vitro characteristics (DNA binding, compaction of DNA) of a typical HMG box-containing protein and that the centrally located HMG box plays a pivotal role in DNA binding, while the C-terminal HMG box seems to modulate the behavior of full-length YlMhb1p.
Deletion of the YlMHB1 gene results in altered mt-nucleoids and a lower mtDNA copy number.
To investigate the role(s) of YlMhb1p in vivo, we tried to delete the YlMHB1 gene from the wild-type Po1h strain lacking a functional YlURA3 gene. By two-step PCR, we constructed a deletion cassette (75) carrying the YlURA3 gene (including promoter and terminator sequences) flanked by 610-bp (5′-end) and 603-bp (3′-end) sequences derived from the regions upstream and downstream of the YlMHB1 ORF (see Fig. S3A in the supplemental material). The cassette was then introduced into the Po1h strain of Y. lipolytica, and the Ura+ clones were screened by PCR to distinguish homologously and nonhomologously integrated cassettes (see Fig. S3A and B in the supplemental material). We identified one clone exhibiting the correct patterns of DNA fragments by a battery of four verification PCRs (see Fig. S3A and B). Lack of the YlMHB1 ORF was also demonstrated by both Southern and Northern blot analyses employing the entire YlMHB1 coding sequence as a probe (see Fig. S3C to E). Furthermore, we also addressed the question of whether the Y. lipolytica genome contains a putative paralogue of YlMHB1 that might substitute for its function in the Δmhb1 mutant. However, neither bioinformatic analysis nor Southern blot hybridization under less stringent conditions (see Fig. S3D) revealed the presence of a sequence exhibiting reasonable similarity to that of YlMHB1. Finally, the absence of YlMHB1-derived transcripts was confirmed by RNA-seq analysis of the transcriptome from the deletion mutant (data not shown). All these results confirm that we have constructed a Δmhb1 deletion mutant lacking a functional YlMHB1 allele, representing the first example of a viable null mutant of a mitochondrial HMG box-containing protein in a strictly aerobic organism.
The viability of the mutant enabled us to compare mt-nucleoids in wild-type and Δmhb1 Y. lipolytica cells by DAPI staining. We observed that the number of mt-nucleoids per cell decreases substantially in the mutant cells, while the morphology of mitochondria visualized by DiOC6 is indistinguishable from that of the wild-type (Fig. 3A). To confirm that the difference observed in mt-nucleoids is due to the absence of YlMhb1p, the YlMHB1 gene, including 5′ and 3′ regulatory sequences, was cloned into a pUB4 plasmid and was introduced into both wild-type and Δmhb1 strains. As shown in Fig. 3A, the Δmhb1 mutant harboring the pUB4-YlMHB1 plasmid exhibits a similar number of mt-nucleoids as the wild-type strain, demonstrating that the phenotype observed can be ascribed to the lack of YlMhb1p in the mutant cells.
FIG 3.

The absence of YlMhb1p leads to a decreased number of mt-nucleoids as well as a lower mtDNA copy number per cell. (A) Wild-type Po1h (WT) (top) and the Δmhb1 mutant (bottom) were stained with DAPI or DiOC6 and were visualized by fluorescence microscopy. Cells transformed with an empty pUB4 vector or with pUB4 carrying the wild-type YlMHB1 gene were grown in YPD medium supplemented with hygromycin B (HygB). (B) PFGE analysis of mtDNA from the wild-type Po1h strain and the Δmhb1 strain. Whole-cell DNA was released from the cells in agarose blocks and was subjected to electrophoresis either without (undigested) or with prior treatment with the restriction endonuclease ApaI. Mitochondrial DNA was visualized by Southern blotting using a radioactively labeled probe derived from the ATP6 gene. EtBr, ethidium bromide-stained gel; SB, Southern blot. (C) The amount of mtDNA in the Δmhb1 mutant relative to that in the WT strain Po1h was measured by qPCR. The data presented are means of results from five independent experiments.
The decreased number of mt-nucleoids in the Δmhb1 mutant indicates that the mutant may have less mtDNA per cell than the wild type strain. To address this question, we performed two types of experiments. First, whole-cell DNA from the wild-type and Δmhb1 strains was separated by PFGE and was analyzed by Southern blot hybridization using an mtDNA-derived probe. The results showed that the mutant cells lacking YlMhb1p have a substantially smaller amount of mtDNA (Fig. 3B, left). Since others have shown previously that ScAbf2p affects mtDNA recombination in S. cerevisiae mitochondria (23), we hypothesized that the smaller amount of mtDNA released from the Δmhb1 cells could be due to the formation of recombination intermediates unable to enter the gel. We therefore treated the DNA in agarose blocks with ApaI, which has a single recognition site within the mtDNA of Y. lipolytica (72). Although the DNA was efficiently digested, the difference in the amount of mtDNA between the wild-type and mutant strains remained the same (Fig. 3B, right). To compare the amounts of mtDNA quantitatively, we performed qPCR using a set of primers derived from mtDNA and nuclear DNA. This analysis revealed that the mutant cells contain about 40% of the number of mtDNA copies present in the wild-type cells (Fig. 3C). All these data confirm that the Δmhb1 mutant contains a smaller amount of mtDNA, as well as a lower number of mt-nucleoids, than the wild-type strain, supporting the conclusion that YlMhb1p plays a role in mtDNA packaging and maintenance in Y. lipolytica.
The Δmhb1 mutant exhibits normal growth and respiratory capacity due to avoidance of mitonuclear imbalance resulting from decreased mtDNA transcription.
The observation that the Δmhb1 mutant has a smaller amount of mtDNA is perhaps not surprising, since both ScAbf2p and mammalian TFAM have been shown to regulate the mtDNA copy number (24, 36). On the other hand, the unexpected finding was that the dramatic decrease in the amount of mtDNA and the number of mt-nucleoids did not result in any apparent growth defects. We tested a number of growth conditions, but with only one exception (see below), we observed no difference in growth between the strains (see Table S3 and Fig. S4 in the supplemental material). We also investigated whether the smaller amount of mtDNA in mutant cells leads to lower respiratory capacity. However, respiration in the mutant cells seemed not to be affected (see Fig. S5A in the supplemental material). Furthermore, we wanted to know what effect the absence of YlMhb1p had on the protein composition of mt-nucleoids in the mutant cells. We observed that due to the absence of YlMhb1p, there was a decreased level of protein(s) comigrating as a 30-kDa band. Other than that, there were no obvious changes in other proteins copurifying with mt-nucleoids (see Fig. S5B), although a more detailed analysis would be needed to identify subtler differences.
The only assay where we observed clear physiological differences between the wild type and the mutant was the assay of growth in the presence of EtBr. This intercalating agent, used to induce deletions in the mtDNA of petite mutant-positive yeast species, strongly inhibited the growth of Δmhb1 cells on both glucose (Fig. 4) and glycerol (data not shown) media. Importantly, reintroduction of YlMHB1 restored the growth of the Δmhb1 mutant in the presence of EtBr, demonstrating that the EtBr sensitivity is due to the lack of functional YlMhb1p (Fig. 4). The sensitivity of the Δmhb1 mutant to EtBr is reminiscent of the situation in S. cerevisiae Δabf2 cells, which are hypersensitive to EtBr on a nonfermentable carbon source (13). Since DNA accessibility to EtBr is informative with regard to the organization of DNA-protein complexes (76), this phenotype of Δmhb1 cells further supports our conclusion that mt-nucleoids in the mutant differ not only in number but also in architecture from wild-type mt-nucleoids. One possible explanation of the pronounced effect of EtBr on mutant cells may be that mtDNA is less compacted and thus more accessible to the drug. We reasoned that this could also lead to increased transcription of mtDNA, thus compensating for the smaller amount of template mtDNA. However, the levels of mitochondrial 23S rRNA and of atp6 RNA transcripts from mtDNA in the Δmhb1 mutant were decreased to a similar extent as mtDNA (Fig. 5). On the other hand, the levels of mRNA for mitochondrial proteins encoded by nuclear DNA, including subunits of complex I of the respiratory chain, were similar in the two strains (Fig. 5). In agreement, comparison of transcriptomes of the wild-type and Δmhb1 strains by RNA-seq analysis has not revealed reproducible differences in the levels of nuclear mRNAs (data not shown). This indicates that the decrease in the mtDNA copy number associated with lower levels of mitochondrial transcripts is probably not reflected in dramatic changes in the transcription of nuclear genes. The fact that mutant cells exhibit no apparent growth defect indicates that the potential imbalance between organellar proteins encoded by the nuclear and mitochondrial genomes is solved at the protein level. We therefore analyzed mitochondrial proteins by immunoblotting. For subunits of the respiratory chain encoded by the nuclear genome, we used antibodies against three protein components of complex I (70), and we found that the wild-type and mutant strains did not differ in the amount of either the NUCM (49-kDa), the NUBM (51-kDa), or the NESM subunit (Fig. 5). Because no antibodies against Y. lipolytica mtDNA-encoded proteins are available, and because, due to its resistance to cycloheximide (77), Y. lipolytica is not suitable for pulse-chase labeling of mtDNA-encoded proteins, we tried several antibodies raised against their S. cerevisiae counterparts. Of these, only anti-Atp6p and anti-Cox2p recognized their corresponding antigens in Y. lipolytica protein extracts. Importantly, there was no apparent difference in the amounts of these two mtDNA-encoded protein subunits of mitochondrial F1Fo-ATPase and cytochrome oxidase, respectively (Fig. 5), indicating that the deficit in mtDNA-derived transcripts is compensated for at the level of translation and/or assembly of the respiratory protein complexes. Importantly, the S. cerevisiae Δabf2 mutant, which was previously shown to contain a lower number of mtDNA molecules (24), did not exhibit changes in the levels of Atp6, Atp9, and Cox2 proteins encoded by mtDNA (data not shown), indicating that, as in the Y. lipolytica Δmhb1 mutant, the mitochondrial translation system in S. cerevisiae Δabf2 cells is able to provide a sufficient amount of proteins for the assembly of respiratory complexes in spite of decreased mtDNA levels.
FIG 4.
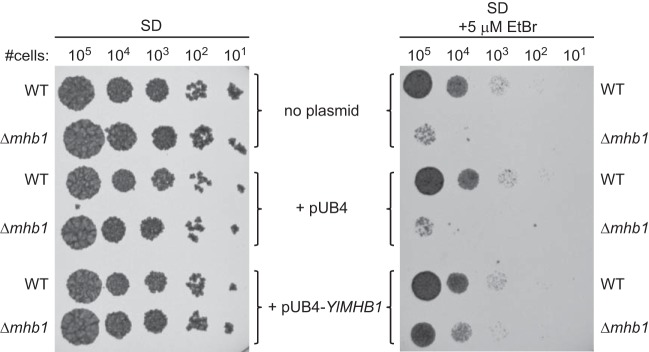
The absence of YlMhb1p results in sensitivity to ethidium bromide. Cells of the wild-type strain Po1h (WT) and the Δmhb1 strain either carrying no plasmid or transformed with the pUB4 vector without or with the YlMHB1 gene were first grown overnight in YPD medium alone or supplemented with HygB and were then spotted onto solid SD medium without (left) or with (right) EtBr (5 μM). Plates were photographed after 1 day (left) or 3 days (right) of growth at 28°C.
FIG 5.
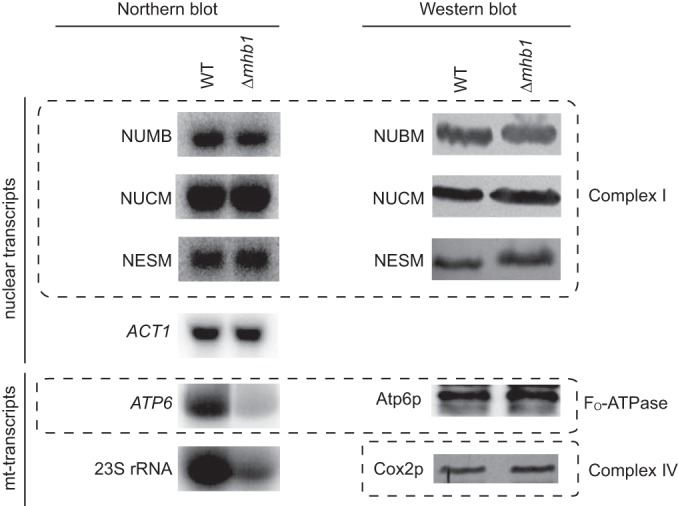
Comparison of the levels of RNA and proteins encoded by nuclear DNA and mtDNA. (Left) Northern blot analysis of RNA resulting from transcription of nuclear DNA (ACT1, NUBM, NUCM, NESM) and mtDNA (23S rRNA, ATP6). (Right) Western blot analysis of the levels of mitochondrial proteins encoded by nuclear DNA (NUCM [49-kDa], NUBM [51-kDa], and NESM subunits of complex I) and mtDNA (Atp6p and Cox2p). The WT (strain Po1h) and Δmhb1 protein extracts were prepared from the same numbers of cells. Note that whereas anti-NUCM, anti-NUMB, and anti-NESM antibodies were raised against Y. lipolytica proteins, anti-Atp6p and anti-Cox2p antibodies were raised against S. cerevisiae Atp6p and Cox2p, respectively.
Lack of YlMhb1p leads to increased frequencies of spontaneous and MnCl2-induced mutants resistant to oligomycin.
Although our results show that YlMhb1p is the major mtDNA-packaging protein in Y. lipolytica, its loss does not lead to any observable growth defect. The question is why the cells maintain the gene in spite of its apparent dispensability. The sensitivity of the mutant to EtBr indicates that the less-condensed mtDNA in the mutant cells might be more prone to genotoxic agents, leading to an accelerated mtDNA mutation rate. To address this possibility, we compared the frequencies of spontaneous and induced mtDNA mutations in the wild-type and Δmhb1 strains. Due to the petite mutant negativity of Y. lipolytica, we could not measure the frequency of mitochondrial respiration-deficient mutants. However, we found that Y. lipolytica is sensitive to oligomycin, a specific inhibitor of the Fo subunit of F1Fo-ATP synthase (78). Resistance to this antibiotic in yeast is mediated by point mutations in the atp6 (oli2), and atp9 (oli1, oli3) genes located on mtDNA (79–81). We therefore investigated whether the absence of YlMhb1p results in an increased frequency of oligomycin-resistant (OliR) mutants in Y. lipolytica. Indeed, we observed that the Δmhb1 strain exhibited a frequency of spontaneous generation of OliR mutants nearly 16-fold higher than that for the wild-type strain and that this frequency was further increased when the cells were pretreated with MnCl2, which is known to induce mutations in mtDNA (71) (Fig. 6). In contrast, the frequency of mutations in nuclear DNA leading to resistance to 5-fluoroorotic acid was the same for both strains (data not shown), demonstrating that the effect was specific for mtDNA. These results indicate that mtDNA in the Δmhb1 strain is much more prone to spontaneous mutagenesis, emphasizing the importance of YlMhb1p in protecting the mitochondrial genome against mutagenic events.
FIG 6.
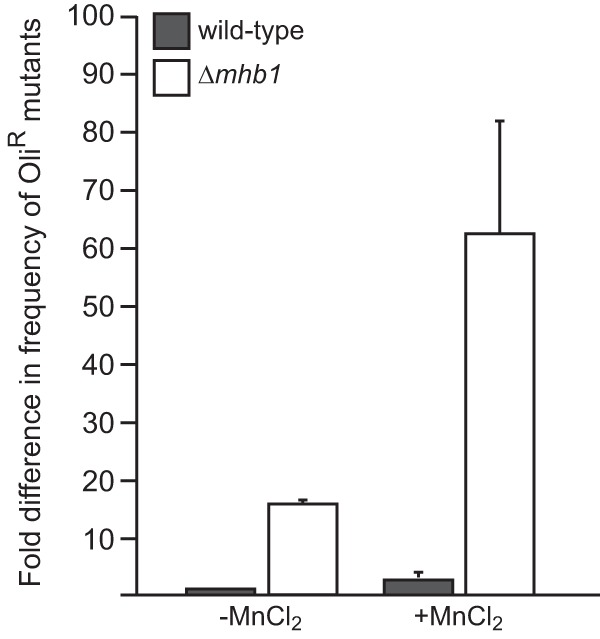
Measurement of frequencies of spontaneous and MnCl2-induced OliR mutations. MnCl2-induced mutagenesis of mtDNA was performed as described previously (71), and the frequency of OliR mutants was calculated based on the number of colonies appearing after 8 days of cultivation on plates containing 10 μg/ml oligomycin A. Wild-type, strain Po1h.
DISCUSSION
The first yeast-like organisms appeared on Earth almost a billion years ago (82). Therefore, it is not surprising that Ascomycota represent a highly heterogeneous group of microorganisms differing in various aspects of their cellular anatomy, metabolism, or genome composition. This biodiversity, together with the availability of molecular tools for many nonconventional species, makes yeasts an ideal group of organisms for comparative studies.
The goal of the present study was to explore a nonconventional yeast, Y. lipolytica, as a model for the study of mt-nucleoids. The main motivation stemmed from the fact that although studies of mt-nucleoids in S. cerevisiae have been highly informative (13), the petite mutant positivity of this yeast and its ability to grow without a functional respiratory chain are characteristics very different from those exhibited by mammalian cells. We therefore initiated studies of mt-nucleoids in the strictly aerobic species C. parapsilosis (46) and C. albicans (49). In the latter case, we constructed a conditional knockdown mutant of the gene coding for an mtHMG protein (CaGcf1p) and investigated the impact of CaGcf1p depletion on mtDNA maintenance (49). However, due to its nature, the gcf1− mutant may have contained residual CaGcf1 protein, which, together with the highly heterozygous genome of C. albicans (51), makes interpretation of the observed phenotypes difficult. Y. lipolytica combines the features of many types of mammalian cells whose proper functioning is dependent on functional mtDNA; it can reproduce in the haploid state; and molecular tools enabling genetic manipulations are available. In addition, Y. lipolytica belongs to the basal phylogenetic lineages of Ascomycota (52, 83). These characteristics make Y. lipolytica a suitable model for comparative analysis of mt-nucleoids, with possible implications for higher eukaryotes.
To identify a putative mtHMG box-containing protein, we first tried a bioinformatic approach. However, a BLAST search of the Y. lipolytica genome yielded no suitable candidates. This negative result is probably due to the fact that besides the HMG box, other regions of mtHMG proteins undergo very rapid diversification and thus do not exhibit significant similarity even among relatively closely related species (47). In fact, these proteins seem to exhibit one of the highest rates of evolutionary diversification among all proteins associated with mt-nucleoids (46). Since the bioinformatic approach failed, we pursued biochemical methods. We purified mt-nucleoids and identified YlMhb1p as their abundant protein component (Fig. 1). Biochemical analyses revealed that in most characteristics YlMhb1p resembles the prototypic mtHMG protein ScAbf2. It binds and compacts DNA in vitro (Fig. 2; see also Fig. S2 in the supplemental material); when expressed heterologously in fusion with GFP, it is targeted to the mitochondria of S. cerevisiae; and introduction of the YlMHB1 gene into S. cerevisiae prevents the loss of mtDNA in Δabf2 mutant cells grown on a fermentable carbon source (see Fig. S1 in the supplemental material). Also, the observation that the centrally located HMG box, rather than the C-terminal HMG box, seems to play a dominant role in DNA binding is consistent with the properties of both ScAbf2p (84) and human TFAM (85). The possibility that in addition to the similarities with other mtHMG proteins, YlMhb1p may also possess some unique properties will be assessed in more detail in our further studies.
With the aim of investigating the role of YlMhb1p in vivo, we attempted to prepare a mutant lacking the entire ORF encoding the protein. Initially, we were quite skeptical about the feasibility of this aim; we expected that the absence of a major mt-nucleoid protein in a petite mutant-negative yeast would lead to lethality. Yet we identified one clone in which the YlURA3 marker replaced the YlMHB1 ORF, and we subjected it to a number of tests for its verification, demonstrating that the mutant lacks functional YlMhb1p (see Fig. S3 in the supplemental material). We cannot exclude the possibility that Y. lipolytica contains another HMG box-containing protein that partially replaces YlMhb1p in the mutant cells. However, our bioinformatic and Southern blot (see Fig. S3D) analyses do not indicate that there is a protein exhibiting reasonable sequence homology to YlMhb1p. Another possible explanation for the viability of the Δmhb1 mutant is that the deletion resulted in a genomic imbalance that was compensated for by secondary mutations elsewhere in a genome. In fact, it was shown recently that most gene knockout strains of S. cerevisiae acquired additional mutations selected to counteract the effect(s) of the original genetic defect (86). To explore this possibility, we tried to obtain independent clones lacking the YlMHB1 gene. To increase the frequency of homologous recombinants, we took advantage of the Ku-deficient mutant of Y. lipolytica constructed in the Barth laboratory (87). The decreased level of the nonhomologous end-joining pathway in this strain leads to an increase in the frequency (up to 85%) of homologous recombinants. In addition, the Ku-deficient strain is descended from a Y. lipolytica isolate different from the parent of the Po1h strain used in our study, enabling us to test the effect of YlMHB1 deletion in a different genetic background. We obtained three independent clones exhibiting the replacement of YlMHB1 with the deletion cassette (see Fig. S6A in the supplemental material). These homologous recombinants were visualized by fluorescence microscopy after staining with DAPI (see Fig. S6B), analyzed by PFGE to assess the content of mtDNA (see Fig. S6C), and tested for sensitivity to EtBr (see Fig. S6D). All three assays revealed that the strains tested exhibit the same phenotypes (increased sensitivity to EtBr, a decreased number of mt-nucleoids, and a decreased amount of mtDNA) as those observed for the original Δmhb1 mutant. Thus, although we still cannot exclude the possibility that the viability of the Δmhb1 mutant strain is caused by a secondary compensatory mutation, both the occurrence of the same phenotype for four independent deletion mutants constructed in two different genetic backgrounds and the restoration of wild-type mt-nucleoids by the reintroduction of the YlMHB1 gene into mutant cells indicate that the phenotypes observed are caused primarily by the lack of YlMhb1p.
The most likely reason for the viability of the Δmhb1 mutant is that mt-nucleoids are disturbed only partially and can fulfill their functions thanks to the other proteins shown to be associated with mt-nucleoids in vivo (11, 13). Although we have not investigated the whole mt-nucleoid proteome, the patterns of proteins copurifying with mt-nucleoids from wild-type and mutant cells analyzed by SDS-PAGE did not exhibit notable differences (see Fig. S5B in the supplemental material). This indicates that mt-nucleoids in the Δmhb1 mutant do not undergo dramatic changes. Rather it seems that in the wild-type cells, mt-nucleoids are packaged on two levels. The first is mediated by mt-nucleoid-associated proteins, such as aconitase and Ilv5p in S. cerevisiae (13). This level of mtDNA packaging is observed in Δmhb1 cells, although at present the nature of the corresponding proteins that would constitute an mt-nucleoid backup system in Y. lipolytica is unknown. Both aconitase and Ilv5p have paralogues in Y. lipolytica (YALI0D09361p and YALI0D03135p, respectively), but their involvement in the maintenance of mt-nucleoids remains to be tested. A systematic search for components of the mt-nucleoid backup system may be based on the screening of mutants synthetically lethal with the Δmhb1 mutation. The second level of packaging is then mediated by YlMhb1p, whose absence leads to the phenotypes observed in the mutant strain: both a decreased number of mt-nucleoids and a decreased mtDNA copy number (Fig. 3). Although the reduction in the number of mt-nucleoids is difficult to evaluate, visual assessment indicates that it might be relatively large (Fig. 3A). If so, this would raise several questions, including the mode of selection of mtDNA molecules for replication and segregation.
The absence of YlMhb1p clearly results in decreased levels of mtDNA-derived transcripts. This might be due to (i) a lower number of mtDNA templates, caused by a loss of YlMhb1p-mediated packaging, and/or to (ii) the direct or indirect involvement of YlMhb1p in the regulation of mtDNA transcription. In either case, the decrease in mitochondrial transcript levels raises an important question: how do Δmhb1 cells circumvent a situation of imbalance in the relative amounts of protein subunits of the respiratory chain encoded by nuclear DNA and mtDNA? Mitochondria employ various means to deal with unassembled protein complexes. An imbalance between nuclear and mitochondrial proteomes and/or an accumulation of unfolded mitochondrial proteins is solved by the collective action of proteases and chaperones and, at least in metazoans, by a transcriptional response program called the mitochondrial unfolded-protein stress response pathway (88–91). Since the proteins involved in these processes are encoded by nuclear DNA, it seems that the nucleus governs the response to mitonuclear imbalance. Are there any means by which mitochondria can participate actively in this process? Subunits of multiprotein complexes have recently been shown to be made in precise proportion to their stoichiometry in both E. coli and S. cerevisiae, although the molecular nature of this control is not understood (92). For mitochondria, it has been shown that translation of several mtDNA-encoded genes, such as cox1, cob, and atp6, depends on the assembly of the corresponding protein complexes (reviewed in reference 89). For instance, mutants of S. cerevisiae lacking the F1 part of ATP synthase, composed of subunits encoded by nuclear DNA, synthesize only reduced levels of Atp6 and Atp8 (93). This regulation is achieved by transcript-specific translational regulators (89) as well as by an intimate association between nucleoids and the machinery of protein synthesis in mitochondria (94). Thus, mitochondria employ feedback control loops coordinating the production of mtDNA-encoded proteins. These loops may be responsible for the compensation for a decrease in the mtDNA copy number in Δmhb1 cells, which seem to compensate for the deficit in mtDNA transcription on the level of translation and/or posttranslational assembly of the protein complexes.
If Y. lipolytica can tolerate the lack of YlMhb1p, which seems to be the major mtDNA-packaging protein, the question is why it keeps the YlMHB1 gene. The sensitivity of the Δmhb1 mutant to EtBr suggests a possible explanation. The less-compacted mtDNA in the Δmhb1 mutant may be more prone to mutations and prolonged exposure to DNA-damaging agents. Because Y. lipolytica is a petite mutant-negative yeast, we could not address this question by comparing the frequencies of respiration-deficient mutants. We therefore compared the frequencies of OliR mutants in the wild-type and Δmhb1 strains. Indeed, we observed that the frequency of spontaneous mutations leading to oligomycin resistance is about 16-fold higher in the Δmhb1 mutant. Treatment with MnCl2 increased the frequency of OliR mutants even further. Although Mn2+ cations induce nuclear mutations as well, oligomycin resistance in yeast has been shown to be mediated mostly by mutations in mtDNA (79–81). These results indicate that although the lack of YlMhb1p does not result in immediate problems associated with mitochondrial dysfunction, mtDNA in Δmhb1 cells is more susceptible to mutagenic agents, leading to an increased mutation rate that will eventually decrease the fitness of the host cell. These results may be relevant for other organisms as well. Loss of Abf2p in S. cerevisiae can be compensated for by other mt-nucleoid-associated proteins, such as aconitase (12), but in the long run, these cells may accumulate mutations in mtDNA preventing growth on nonfermentable substrates such as ethanol. Indeed, it has been shown that the loss of ScAbf2p results in increased rates of frameshift mutations and direct-repeat-mediated deletions in mtDNA (26). Similarly, less-compacted mtDNA in human cells with lower levels of TFAM may accumulate mutations at higher rates, resulting in mitochondrion-associated pathologies. Our results for YlMhb1p thus expand the repertoire of possible roles played by mitochondrial HMG box proteins in maintaining mtDNA functions and integrity.
Supplementary Material
ACKNOWLEDGMENTS
We thank Ladislav Kovac for inspiration and continuous support, Julius Subik for insightful suggestions related to phenotypic characterization of the Δmhb1 mutant, Juraj Laco, Gabriela Gerecova, Lucia Simonicova, Katarina Visacka, Slavomir Kinsky, and Jana Tomaskova for technical help, members of our laboratories for discussions, and Joachim M. Gerhold (Institute of Molecular and Cell Biology, University of Tartu, Tartu, Estonia) and Tomas Vinar and Brona Brejova (Faculty of Mathematics, Physics and Informatics, Comenius University) for technical comments. We thank Keita Miyazono (Yamaguchi University, Yamaguchi, Japan) for help with mt-nucleoid isolation, Catherine Madzak (Institut National de la Recherche Agronomique, Thiverval-Grignon, France) for strain Po1h, Claude Gaillardin (AgroParisTech, Micalis, Jouy-en-Josas, France) for strains E122 and E129, Gerold Barth (Technische Universität Dresden, Dresden, Germany) for strain H222-SW2-1, H. Y. de Steensma (Leiden University, Leiden, The Netherlands) for the S. cerevisiae strain GG595, Johannes H. Hegemann (Heinrich-Heine-Universität, Düsseldorf, Germany) for plasmid pUG35, Mathias L. Richard (Institut National de la Recherche Agronomique, Thiverval-Grignon, France) for plasmid pKSURA, Xin Jie Chen (SUNY Upstate Medical University, Syracuse, NY) for the S. cerevisiae Δabf2 strain, Volker Zickermann (Institut für Biochemie II, Goethe Universität, Frankfurt am Main, Germany) for anti-NUCM, anti-NUBM, and anti-NESM antibodies, Jean-Paul di Rago (Institut de Biochemie et Génetique Cellulaires, Université Bordeaux, Bordeaux, France) for the anti-Atp6 antibody, and Alexander Tzagoloff (Columbia University, New York, NY) for the anti-Cox2 antibody.
This work was supported in part by the Slovak grant agencies APVV (0035-11 and 0123-10) and VEGA (1/0311/12 and 1/0405/11), by National Institutes of Health grant 2R01ES013773-06A1, and by grants from the Japan Society for the Promotion of Science (22570213) and from Core Research for Evolutional Science and Technology (CREST).
Footnotes
Published ahead of print 27 June 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00092-14.
REFERENCES
- 1.Kaufman BA, Kolesar JE, Perlman PS, Butow RA. 2003. A function for the mitochondrial chaperonin Hsp60 in the structure and transmission of mitochondrial DNA nucleoids in Saccharomyces cerevisiae. J. Cell Biol. 163:457–461. 10.1083/jcb.200306132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miyakawa I, Aoi H, Sando N, Kuroiwa T. 1984. Fluorescence microscopic studies of mitochondrial nucleoids during meiosis and sporulation in the yeast, Saccharomyces cerevisiae. J. Cell Sci. 66:21–38 [DOI] [PubMed] [Google Scholar]
- 3.Miyakawa I, Sando N, Kawano S, Nakamura S, Kuroiwa T. 1987. Isolation of morphologically intact mitochondrial nucleoids from the yeast, Saccharomyces cerevisiae. J. Cell Sci. 88:431–439 [DOI] [PubMed] [Google Scholar]
- 4.Miyakawa I, Miyamoto M, Kuroiwa T, Sando N. 2004. DNA content of individual mitochondrial nucleoids varies depending on the culture conditions of the yeast Saccharomyces cerevisiae. Cytologia 69:101–107. 10.1508/cytologia.69.101 [DOI] [Google Scholar]
- 5.Williamson DH, Fennell DJ. 1975. The use of fluorescent DNA-binding agent for detecting and separating yeast mitochondrial DNA. Methods Cell Biol. 12:335–351. 10.1016/S0091-679X(08)60963-2 [DOI] [PubMed] [Google Scholar]
- 6.Williamson DH, Fennell DJ. 1979. Visualization of yeast mitochondrial DNA with the fluorescent stain “DAPI.” Methods Enzymol. 56:728–733 [DOI] [PubMed] [Google Scholar]
- 7.Brown TA, Tkachuk AN, Shtengel G, Kopek BG, Bogenhagen DF, Hess HF, Clayton DA. 2011. Superresolution fluorescence imaging of mitochondrial nucleoids reveals their spatial range, limits, and membrane interaction. Mol. Cell. Biol. 31:4994–5010. 10.1128/MCB.05694-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kukat C, Wurm CA, Spahr H, Falkenberg M, Larsson NG, Jakobs S. 2011. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc. Natl. Acad. Sci. U. S. A. 108:13534–13539. 10.1073/pnas.1109263108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Copeland WC. 2012. Defects in mitochondrial DNA replication and human disease. Crit. Rev. Biochem. Mol. Biol. 47:64–74. 10.3109/10409238.2011.632763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Friedman JR, Nunnari J. 2014. Mitochondrial form and function. Nature 505:335–343. 10.1038/nature12985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bogenhagen DF. 2011. Mitochondrial DNA nucleoid structure. Biochim. Biophys. Acta 1819:914–920. 10.1016/j.bbagrm.2011.11.005 [DOI] [PubMed] [Google Scholar]
- 12.Chen XJ, Wang X, Kaufman BA, Butow RA. 2005. Aconitase couples metabolic regulation to mitochondrial DNA maintenance. Science 307:714–717. 10.1126/science.1106391 [DOI] [PubMed] [Google Scholar]
- 13.Chen XJ, Butow RA. 2005. The organization and inheritance of the mitochondrial genome. Nat. Rev. Genet. 6:815–825. 10.1038/nrg1708 [DOI] [PubMed] [Google Scholar]
- 14.Kaufman BA, Newman SM, Hallberg RL, Slaughter CA, Perlman PS, Butow RA. 2000. In organello formaldehyde crosslinking of proteins to mtDNA: identification of bifunctional proteins. Proc. Natl. Acad. Sci. U. S. A. 97:7772–7777. 10.1073/pnas.140063197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Caron F, Jacq C, Rouviere-Yaniv J. 1979. Characterization of a histone-like protein extracted from yeast mitochondria. Proc. Natl. Acad. Sci. U. S. A. 76:4265–4269. 10.1073/pnas.76.9.4265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Certa U, Colavito-Shepanski M, Grunstein M. 1984. Yeast may not contain histone H1: the only known ‘histone H1-like' protein in Saccharomyces cerevisiae is a mitochondrial protein. Nucleic Acids Res. 12:7975–7985. 10.1093/nar/12.21.7975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diffley JF, Stillman B. 1991. A close relative of the nuclear, chromosomal high-mobility group protein HMG1 in yeast mitochondria. Proc. Natl. Acad. Sci. U. S. A. 88:7864–7868. 10.1073/pnas.88.17.7864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diffley JF, Stillman B. 1992. DNA binding properties of an HMG1-related protein from yeast mitochondria. J. Biol. Chem. 267:3368–3374 [PubMed] [Google Scholar]
- 19.Friddle RW, Klare JE, Martin SS, Corzett M, Balhorn R, Baldwin EP, Baskin RJ, Noy A. 2004. Mechanism of DNA compaction by yeast mitochondrial protein Abf2p. Biophys. J. 86:1632–1639. 10.1016/S0006-3495(04)74231-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kucej M, Kucejova B, Subramanian R, Chen XJ, Butow RA. 2008. Mitochondrial nucleoids undergo remodeling in response to metabolic cues. J. Cell Sci. 121:1861–1868. 10.1242/jcs.028605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miyakawa I, Kanayama M, Fujita Y, Sato H. 2010. Morphology and protein composition of the mitochondrial nucleoids in yeast cells lacking Abf2p, a high mobility group protein. J. Gen. Appl. Microbiol. 56:455–464. 10.2323/jgam.56.455 [DOI] [PubMed] [Google Scholar]
- 22.Newman SM, Zelenaya-Troitskaya O, Perlman PS, Butow RA. 1996. Analysis of mitochondrial DNA nucleoids in wild-type and a mutant strain of Saccharomyces cerevisiae that lacks the mitochondrial HMG box protein Abf2p. Nucleic Acids Res. 24:386–393. 10.1093/nar/24.2.386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.MacAlpine DM, Perlman PS, Butow RA. 1998. The high mobility group protein Abf2p influences the level of yeast mitochondrial DNA recombination intermediates in vivo. Proc. Natl. Acad. Sci. U. S. A. 95:6739–6743. 10.1073/pnas.95.12.6739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zelenaya-Troitskaya O, Newman SM, Okamoto K, Perlman PS, Butow RA. 1998. Functions of the high mobility group protein, Abf2p, in mitochondrial DNA segregation, recombination and copy number in Saccharomyces cerevisiae. Genetics 148:1763–1776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Okamoto K, Perlman PS, Butow RA. 1998. The sorting of mitochondrial DNA and mitochondrial proteins in zygotes: preferential transmission of mitochondrial DNA to the medial bud. J. Cell Biol. 142:613–623. 10.1083/jcb.142.3.613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sia RA, Carrol S, Kalifa L, Hochmuth C, Sia EA. 2009. Loss of the mitochondrial nucleoid protein, Abf2p, destabilizes repetitive DNA in the yeast mitochondrial genome. Genetics 181:331–334. 10.1534/genetics.108.095786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zelenaya-Troitskaya O, Perlman PS, Butow RA. 1995. An enzyme in yeast mitochondria that catalyzes a step in branched-chain amino acid biosynthesis also functions in mitochondrial DNA stability. EMBO J. 14:3268–3276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fisher RP, Clayton DA. 1985. A transcription factor required for promoter recognition by human mitochondrial RNA polymerase. Accurate initiation at the heavy- and light-strand promoters dissected and reconstituted in vitro. J. Biol. Chem. 260:11330–11338 [PubMed] [Google Scholar]
- 29.Fisher RP, Clayton DA. 1988. Purification and characterization of human mitochondrial transcription factor 1. Mol. Cell. Biol. 8:3496–3509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parisi MA, Xu B, Clayton DA. 1993. A human mitochondrial transcriptional activator can functionally replace a yeast mitochondrial HMG-box protein both in vivo and in vitro. Mol. Cell. Biol. 13:1951–1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoon YG, Koob MD, Yoo YH. 2011. Mitochondrial genome-maintaining activity of mouse mitochondrial transcription factor A and its transcript isoform in Saccharomyces cerevisiae. Gene 484:52–60. 10.1016/j.gene.2011.05.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parisi MA, Clayton DA. 1991. Similarity of human mitochondrial transcription factor 1 to high mobility group proteins. Science 252:965–969. 10.1126/science.2035027 [DOI] [PubMed] [Google Scholar]
- 33.Ngo HB, Kaiser JT, Chan DC. 2011. The mitochondrial transcription and packaging factor Tfam imposes a U-turn on mitochondrial DNA. Nat. Struct. Mol. Biol. 18:1290–1296. 10.1038/nsmb.2159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rubio-Cosials A, Sidow JF, Jimenez-Menendez N, Fernandez-Millan P, Montoya J, Jacobs HT, Coll M, Bernado P, Sola M. 2011. Human mitochondrial transcription factor A induces a U-turn structure in the light strand promoter. Nat. Struct. Mol. Biol. 18:1281–1289. 10.1038/nsmb.2160 [DOI] [PubMed] [Google Scholar]
- 35.Larsson NG, Wang J, Wilhelmsson H, Oldfors A, Rustin P, Lewandoski M, Barsh GS, Clayton DA. 1998. Mitochondrial transcription factor A is necessary for mtDNA maintenance and embryogenesis in mice. Nat. Genet. 18:231–236. 10.1038/ng0398-231 [DOI] [PubMed] [Google Scholar]
- 36.Ekstrand MI, Falkenberg M, Rantanen A, Park CB, Gaspari M, Hultenby K, Rustin P, Gustafsson CM, Larsson NG. 2004. Mitochondrial transcription factor A regulates mtDNA copy number in mammals. Hum. Mol. Genet. 13:935–944. 10.1093/hmg/ddh109 [DOI] [PubMed] [Google Scholar]
- 37.Pohjoismaki JL, Wanrooij S, Hyvarinen AK, Goffart S, Holt IJ, Spelbrink JN, Jacobs HT. 2006. Alterations to the expression level of mitochondrial transcription factor A, TFAM, modify the mode of mitochondrial DNA replication in cultured human cells. Nucleic Acids Res. 34:5815–5828. 10.1093/nar/gkl703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li H, Wang J, Wilhelmsson H, Hansson A, Thoren P, Duffy J, Rustin P, Larsson NG. 2000. Genetic modification of survival in tissue-specific knockout mice with mitochondrial cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A. 97:3467–3472. 10.1073/pnas.97.7.3467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Silva JP, Kohler M, Graff C, Oldfors A, Magnuson MA, Berggren PO, Larsson NG. 2000. Impaired insulin secretion and beta-cell loss in tissue-specific knockout mice with mitochondrial diabetes. Nat. Genet. 26:336–340. 10.1038/81649 [DOI] [PubMed] [Google Scholar]
- 40.Sorensen L, Ekstrand M, Silva JP, Lindqvist E, Xu B, Rustin P, Olson L, Larsson NG. 2001. Late-onset corticohippocampal neurodepletion attributable to catastrophic failure of oxidative phosphorylation in MILON mice. J. Neurosci. 21:8082–8090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang J, Wilhelmsson H, Graff C, Li H, Oldfors A, Rustin P, Bruning JC, Kahn CR, Clayton DA, Barsh GS, Thoren P, Larsson NG. 1999. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat. Genet. 21:133–137. 10.1038/5089 [DOI] [PubMed] [Google Scholar]
- 42.Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, Oldfors A, Westerblad H, Larsson NG. 2002. Increased mitochondrial mass in mitochondrial myopathy mice. Proc. Natl. Acad. Sci. U. S. A. 99:15066–15071. 10.1073/pnas.232591499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dequard-Chablat M, Allandt C. 2002. Two copies of mthmg1, encoding a novel mitochondrial HMG-like protein, delay accumulation of mitochondrial DNA deletions in Podospora anserina. Eukaryot. Cell 1:503–513. 10.1128/EC.1.4.503-513.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miyakawa I, Sato H, Maruyama Y, Nakaoka T. 2003. Isolation of the mitochondrial nucleoids from yeast Kluyveromyces lactis and analyses of the nucleoid proteins. J. Gen. Appl. Microbiol. 49:85–93. 10.2323/jgam.49.85 [DOI] [PubMed] [Google Scholar]
- 45.Miyakawa I, Yawata K. 2007. Purification of an Abf2p-like protein from mitochondrial nucleoids of yeast Pichia jadinii and its role in the packaging of mitochondrial DNA. Antonie Van Leeuwenhoek 91:197–207. 10.1007/s10482-006-9105-7 [DOI] [PubMed] [Google Scholar]
- 46.Miyakawa I, Okamuro A, Kinsky S, Visacka K, Tomaska L, Nosek J. 2009. Mitochondrial nucleoids from the yeast Candida parapsilosis: expansion of the repertoire of proteins associated with mitochondrial DNA. Microbiology 155:1558–1568. 10.1099/mic.0.027474-0 [DOI] [PubMed] [Google Scholar]
- 47.Nosek J, Tomaska L, Bolotin-Fukuhara M, Miyakawa I. 2006. Mitochondrial chromosome structure: an insight from analysis of complete yeast genomes. FEMS Yeast Res. 6:356–370. 10.1111/j.1567-1364.2005.00016.x [DOI] [PubMed] [Google Scholar]
- 48.Sasaki N, Kuroiwa H, Nishitani C, Takano H, Higashiyama T, Kobayashi T, Shirai Y, Sakai A, Kawano S, Murakami-Murofushi K, Kuroiwa T. 2003. Glom is a novel mitochondrial DNA packaging protein in Physarum polycephalum and causes intense chromatin condensation without suppressing DNA functions. Mol. Biol. Cell 14:4758–4769. 10.1091/mbc.E03-02-0099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Visacka K, Gerhold JM, Petrovicova J, Kinsky S, Joers P, Nosek J, Sedman J, Tomaska L. 2009. Novel subfamily of mitochondrial HMG box-containing proteins: functional analysis of Gcf1p from Candida albicans. Microbiology 155:1226–1240. 10.1099/mic.0.025759-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Itoh K, Izumi A, Mori T, Dohmae N, Yui R, Maeda-Sano K, Shirai Y, Kanaoka MM, Kuroiwa T, Higashiyama T, Sugita M, Murakami-Murofushi K, Kawano S, Sasaki N. 2011. DNA packaging proteins Glom and Glom2 coordinately organize the mitochondrial nucleoid of Physarum polycephalum. Mitochondrion 11:575–586. 10.1016/j.mito.2011.03.002 [DOI] [PubMed] [Google Scholar]
- 51.Graser Y, Volovsek M, Arrington J, Schonian G, Presber W, Mitchell TG, Vilgalys R. 1996. Molecular markers reveal that population structure of the human pathogen Candida albicans exhibits both clonality and recombination. Proc. Natl. Acad. Sci. U. S. A. 93:12473–12477. 10.1073/pnas.93.22.12473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Casaregola S, Neuveglise C, Lepingle A, Bon E, Feynerol C, Artiguenave F, Wincker P, Gaillardin C. 2000. Genomic exploration of the hemiascomycetous yeasts: 17. Yarrowia lipolytica. FEBS Lett. 487:95–100. 10.1016/S0014-5793(00)02288-2 [DOI] [PubMed] [Google Scholar]
- 53.Christen S, Sauer U. 2011. Intracellular characterization of aerobic glucose metabolism in seven yeast species by 13C flux analysis and metabolomics. FEMS Yeast Res. 11:263–272. 10.1111/j.1567-1364.2010.00713.x [DOI] [PubMed] [Google Scholar]
- 54.Chen DC, Beckerich JM, Gaillardin C. 1997. One-step transformation of the dimorphic yeast Yarrowia lipolytica. Appl. Microbiol. Biotechnol. 48:232–235. 10.1007/s002530051043 [DOI] [PubMed] [Google Scholar]
- 55.Wang JH, Hung W, Tsai SH. 2011. High efficiency transformation by electroporation of Yarrowia lipolytica. J. Microbiol. 49:469–472. 10.1007/s12275-011-0433-6 [DOI] [PubMed] [Google Scholar]
- 56.Kerscher SJ, Okun JG, Brandt U. 1999. A single external enzyme confers alternative NADH:ubiquinone oxidoreductase activity in Yarrowia lipolytica. J. Cell Sci. 112:2347–2354 [DOI] [PubMed] [Google Scholar]
- 57.Laemmli UK. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680–685. 10.1038/227680a0 [DOI] [PubMed] [Google Scholar]
- 58.Kerscher SJ, Eschemann A, Okun PM, Brandt U. 2001. External alternative NADH:ubiquinone oxidoreductase redirected to the internal face of the mitochondrial inner membrane rescues complex I deficiency in Yarrowia lipolytica. J. Cell Sci. 114:3915–3921 [DOI] [PubMed] [Google Scholar]
- 59.Nosek J, Fukuhara H. 1994. NADH dehydrogenase subunit genes in the mitochondrial DNA of yeasts. J. Bacteriol. 176:5622–5630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rycovska A, Valach M, Tomaska L, Bolotin-Fukuhara M, Nosek J. 2004. Linear versus circular mitochondrial genomes: intraspecies variability of mitochondrial genome architecture in Candida parapsilosis. Microbiology 150:1571–1580. 10.1099/mic.0.26988-0 [DOI] [PubMed] [Google Scholar]
- 61.Philippsen P, Stotz A, Scherf C. 1991. DNA of Saccharomyces cerevisiae. Methods Enzymol. 194:169–182. 10.1016/0076-6879(91)94014-4 [DOI] [PubMed] [Google Scholar]
- 62.Livak K. 1997. ABI Prism 7700 Sequence Detection System. User bulletin 2. PE Applied Biosystems, Foster City, CA [Google Scholar]
- 63.Cross FR, Tinkelenberg AH. 1991. A potential positive feedback loop controlling CLN1 and CLN2 gene expression at the start of the yeast cell cycle. Cell 65:875–883. 10.1016/0092-8674(91)90394-E [DOI] [PubMed] [Google Scholar]
- 64.Kroczek RA, Siebert E. 1990. Optimization of Northern analysis by vacuum-blotting, RNA-transfer visualization, and ultraviolet fixation. Anal. Biochem. 184:90–95. 10.1016/0003-2697(90)90017-4 [DOI] [PubMed] [Google Scholar]
- 65.Claros MG, Vincens P. 1996. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 241:779–786. 10.1111/j.1432-1033.1996.00779.x [DOI] [PubMed] [Google Scholar]
- 66.Kelley LA, Sternberg MJ. 2009. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protoc. 4:363–371. 10.1038/nprot.2009.2 [DOI] [PubMed] [Google Scholar]
- 67.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23:2947–2948. 10.1093/bioinformatics/btm404 [DOI] [PubMed] [Google Scholar]
- 68.Knop M, Siegers K, Pereira G, Zachariae W, Winsor B, Nasmyth K, Schiebel E. 1999. Epitope tagging of yeast genes using a PCR-based strategy: more tags and improved practical routines. Yeast 15:963–972 [DOI] [PubMed] [Google Scholar]
- 69.Tomaska L, Makhov AM, Nosek J, Kucejova B, Griffith JD. 2001. Electron microscopic analysis supports a dual role for the mitochondrial telomere-binding protein of Candida parapsilosis. J. Mol. Biol. 305:61–69. 10.1006/jmbi.2000.4254 [DOI] [PubMed] [Google Scholar]
- 70.Zickermann V, Bostina M, Hunte C, Ruiz T, Radermacher M, Brandt U. 2003. Functional implications from an unexpected position of the 49-kDa subunit of NADH:ubiquinone oxidoreductase. J. Biol. Chem. 278:29072–29078. 10.1074/jbc.M302713200 [DOI] [PubMed] [Google Scholar]
- 71.Putrament A, Branowska H, Ejchart A, Prazmo W. 1975. Manganese mutagenesis in yeast. A practical application of manganese for the induction of mitochondrial antibiotic-resistant mutations. J. Gen. Microbiol. 90:265–270 [DOI] [PubMed] [Google Scholar]
- 72.Kerscher S, Durstewitz G, Casaregola S, Gaillardin C, Brandt U. 2001. The complete mitochondrial genome of Yarrowia lipolytica. Comp. Funct. Genomics 2:80–90. 10.1002/cfg.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chang KJ, Lin G, Men LC, Wang CC. 2006. Redundancy of non-AUG initiators. A clever mechanism to enhance the efficiency of translation in yeast. J. Biol. Chem. 281:7775–7783. 10.1074/jbc.M511265200 [DOI] [PubMed] [Google Scholar]
- 74.Touriol C, Bornes S, Bonnal S, Audigier S, Prats H, Prats AC, Vagner S. 2003. Generation of protein isoform diversity by alternative initiation of translation at non-AUG codons. Biol. Cell 95:169–178. 10.1016/S0248-4900(03)00033-9 [DOI] [PubMed] [Google Scholar]
- 75.Wach A. 1996. PCR-synthesis of marker cassettes with long flanking homology regions for gene disruptions in S. cerevisiae. Yeast 12:259–265 [DOI] [PubMed] [Google Scholar]
- 76.Lawrence JJ, Daune M. 1976. Ethidium bromide as a probe of conformational heterogeneity of DNA in chromatin. The role of histone H1. Biochemistry 15:3301–3307 [DOI] [PubMed] [Google Scholar]
- 77.Barnett JA, Payne RW, Yarrow D. (ed). 2000. Yeasts: characteristics and identification. Cambridge University Press, Cambridge, United Kingdom [Google Scholar]
- 78.Lehninger AL, Nelson DL, Cox MM. (ed). 2005. Lehninger principles of biochemistry. W. H. Freeman, New York, NY [Google Scholar]
- 79.Sebald W, Wachter E, Tzagoloff A. 1979. Identification of amino acid substitutions in the dicyclohexylcarbodiimide-binding subunit of the mitochondrial ATPase complex from oligomycin-resistant mutants of Saccharomyces cerevisiae. Eur. J. Biochem. 100:599–607. 10.1111/j.1432-1033.1979.tb04207.x [DOI] [PubMed] [Google Scholar]
- 80.Ooi BG, Novitski CE, Nagley P. 1985. DNA sequence analysis of the oli1 gene reveals amino acid changes in mitochondrial ATPase subunit 9 from oligomycin-resistant mutants of Saccharomyces cerevisiae. Eur. J. Biochem. 152:709–714. 10.1111/j.1432-1033.1985.tb09251.x [DOI] [PubMed] [Google Scholar]
- 81.John UP, Nagley P. 1986. Amino acid substitutions in mitochondrial ATPase subunit 6 of Saccharomyces cerevisiae leading to oligomycin resistance. FEBS Lett. 207:79–83. 10.1016/0014-5793(86)80016-3 [DOI] [PubMed] [Google Scholar]
- 82.Hedges SB, Blair JE, Venturi ML, Shoe JL. 2004. A molecular timescale of eukaryote evolution and the rise of complex multicellular life. BMC Evol. Biol. 4:2. 10.1186/1471-2148-4-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Barth G, Gaillardin C. 1997. Physiology and genetics of the dimorphic fungus Yarrowia lipolytica. FEMS Microbiol. Rev. 19:219–237. 10.1111/j.1574-6976.1997.tb00299.x [DOI] [PubMed] [Google Scholar]
- 84.Kao LR, Megraw TL, Chae CB. 1993. Essential role of the HMG domain in the function of yeast mitochondrial histone HM: functional complementation of HM by the nuclear nonhistone protein NHP6A. Proc. Natl. Acad. Sci. U. S. A. 90:5598–5602. 10.1073/pnas.90.12.5598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gangelhoff TA, Mungalachetty PS, Nix JC, Churchill ME. 2009. Structural analysis and DNA binding of the HMG domains of the human mitochondrial transcription factor A. Nucleic Acids Res. 37:3153–3164. 10.1093/nar/gkp157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Teng X, Dayhoff-Brannigan M, Cheng WC, Gilbert CE, Sing CN, Diny NL, Wheelan SJ, Dunham MJ, Boeke JD, Pineda FJ, Hardwick JM. 2013. Genome-wide consequences of deleting any single gene. Mol. Cell 52:485–494. 10.1016/j.molcel.2013.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kretzschmar A, Otto C, Holz M, Werner S, Hübner L, Barth G. 2013. Increased homologous integration frequency in Yarrowia lipolytica strains defective in non-homologous end-joining. Curr. Genet. 59:63–72. 10.1007/s00294-013-0389-7 [DOI] [PubMed] [Google Scholar]
- 88.Baker MJ, Tatsuta T, Langer T. 2011. Quality control of mitochondrial proteostasis. Cold Spring Harb. Perspect. Biol. 3:a007559. 10.1101/cshperspect.a007559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Herrmann JM, Woellhaf MW, Bonnefoy N. 2013. Control of protein synthesis in yeast mitochondria: the concept of translational activators. Biochim. Biophys. Acta 1833:286–294. 10.1016/j.bbamcr.2012.03.007 [DOI] [PubMed] [Google Scholar]
- 90.Houtkooper RH, Mouchiroud L, Ryu D, Moullan N, Katsyuba E, Knott G, Williams RW, Auwerx J. 2013. Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497:451–457. 10.1038/nature12188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Luzikov VN. 1999. Quality control: from molecules to organelles. FEBS Lett. 448:201–205. 10.1016/S0014-5793(99)00344-0 [DOI] [PubMed] [Google Scholar]
- 92.Li GW, Burkhardt D, Gross C, Weissman JS. 2014. Quantifying absolute protein synthesis rates reveals principles underlying allocation of cellular resources. Cell 157:624–635. 10.1016/j.cell.2014.02.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rak M, Tzagoloff A. 2009. F1-dependent translation of mitochondrially encoded Atp6p and Atp8p subunits of yeast ATP synthase. Proc. Natl. Acad. Sci. U. S. A. 106:18509–18514. 10.1073/pnas.0910351106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.He J, Cooper HM, Reyes A, Di Re M, Sembongi H, Litwin TR, Gao J, Neuman KC, Fearnley IM, Spinazzola A, Walker JE, Holt IJ. 2012. Mitochondrial nucleoid interacting proteins support mitochondrial protein synthesis. Nucleic Acids Res. 40:6109–6121. 10.1093/nar/gks266 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.