

Abstract
Here, we describe the isolation of two nickel-induced genes in Paramecium caudatum, NCI16 and PcGST1, by subtractive hybridization. NCI16 encoded a predicted four-transmembrane domain protein (∼16 kDa) of unknown function, and PcGST1 encoded glutathione S-transferase (GST; ∼25 kDa) with GST and glutathione peroxidase (GPx) activities. Exposing cells to cobalt chloride also caused the moderate upregulation of NCI16 and PcGST1 mRNAs. Both nickel sulfate and cobalt chloride dose dependently induced NCI16 and PcGST1 mRNAs, but with different profiles. Nickel treatment caused a continuous increase in PcGST1 and NCI16 mRNA levels for up to 3 and 6 days, respectively, and a notable increase in H2O2 concentrations in P. caudatum. NCI16 expression was significantly enhanced by incubating cells with H2O2, implying that NCI16 induction in the presence of nickel ions is caused by reactive oxygen species (ROS). On the other hand, PcGST1 was highly induced by the antioxidant tert-butylhydroquinone (tBHQ) but not by H2O2, suggesting that different mechanisms mediate the induction of NCI16 and PcGST1. We introduced a luciferase reporter vector with an ∼0.42-kb putative PcGST1 promoter into cells and then exposed the transformants to nickel sulfate. This resulted in significant luciferase upregulation, indicating that the putative PcGST1 promoter contains a nickel-responsive element. Our nickel-inducible system also may be applicable to the efficient expression of proteins that are toxic to host cells or require temporal control.
INTRODUCTION
Nickel is used extensively for electroplating metals, alloys such as cupronickel, and rechargeable batteries. Occupational exposure to nickel occurs in industrial workers, in particular those involved in mining, smelting, and refining, the production of steel and other metals, and electronic devices (1). Nickel compounds are released into the environment from power plants that burn oil, trash incinerators, wastewater from nickel mines, and industries that manufacture nickel products for industrial and consumer use. Nickel has toxic and carcinogenic effects on most microorganisms and animals and is considered to impose an industrial health hazard (2, 3). Nickel compounds such as nickel subsulfide (Ni3S2) are potent carcinogens, but soluble nickel salts such as nickel chloride (NiCl2) exert weaker effects. The molecular mechanisms involved in the cytotoxicity and carcinogenicity of nickel compounds are not fully understood, but nickel might be associated with the intracellular production of reactive oxygen species (ROS), including superoxide, H2O2, singlet oxygen, and hydroxyl radicals (4–8). Nickel also increases lipid peroxide (LPO) levels, resulting in the generation of peroxyl radicals, lipid hydroperoxides, and alkoxyl radicals (9–11). Carcinogenesis related to nickel is explained by several types of DNA damage, such as cleavage, depurination, cross-linking, and DNA base damage caused by ROS (12). Nickel inhibits processes in the DNA repair system, such as DNA ligation and DNA polymerization, that are involved in rejoining DNA breaks (6). Nickel also modifies the antioxidant system; for example, nickel chloride alters hepatic reduced glutathione levels (13) as well as renal glutathione S-transferase (GST) (14) and hepatic glutathione peroxidase (GPx) activities (4) in rodents.
Nickel ions inhibit the ciliary beat of the unicellular protozoan Paramecium. Transferring Paramecium caudatum into a solution containing nickel ions causes a gradual decrease in the frequency and the amplitude of the ciliary beat without influencing the orientation of the cilia (15). The effects of nickel ions on ciliary beat also were examined in the ATP-Mg2+ reactivated cilia of detergent-extracted Paramecium models (16). Nickel ions have detrimental effects on microtubule translocation mediated by 14S dynein, which might be one of the factors directly affected during the nickel-induced paralysis of axonemal beats (17). The effects of nickel ions on the cellular functions of paramecia other than ciliary movement have not been documented in detail. Paramecium and other ciliated protozoa were used in bioassays designed to measure the cytotoxic and carcinogenic effects of soluble and particulate nickel compounds (18, 19) or of waste treatment plants (20). Although the cytotoxic effects of nickel ions on Paramecium are documented, genes for which the expression levels are altered by nickel ions remain unidentified.
We performed subtractive cDNA hybridization to identify nickel-induced genes to elucidate mechanisms mediating nickel toxicity and associated detoxifying systems in Paramecium. Two genes, NCI16 and P. caudatum GST1 (PcGST1), obviously were upregulated in P. caudatum that had been treated with NiSO4 but not in control cells. NCI16 encoded a predicted four-transmembrane domain protein of unknown function, and PcGST1 encoded a GST protein that exhibits the enzymatic activities of both GST and GPx. A region of the putative PcGST1 promoter (∼0.42 kb) was isolated and cloned into the pBsc-tel3 vector (21) with a luciferase gene to generate a reporter construct. This putative PcGST1 promoter drove a significant increase in nickel-dependent luciferase activity, implying that PcGST1 upregulation is mediated by cis-acting sequences in the promoter region. Our nickel-inducible system also may be applicable to the efficient expression of proteins that are toxic to host cells or require temporal control.
MATERIALS AND METHODS
Strains and culture methods.
The Paramecium caudatum BW6-1 strain (syngen 12, odd mating type) was used for cDNA subtraction, and the NH2 strain (syngen 3, even mating type) was transformed with the pGT1-MpLuc1H vector. Cells were cultured either in 1.25% (wt/vol) fresh lettuce juice diluted with K-DS (Dryl's solution modified by the substitution of KH2PO4 for NaH2PO4), pH 7.0 (22), or in wheat grass powder (WGP) (23) medium. Both culture media were inoculated with Klebsiella pneumoniae 1 day before use.
cDNA subtraction of nickel-induced genes.
BW6-1 cultures were grown until early log phase (approximately 300 cells/ml) in 200 ml of WGP medium in 500-ml glass flasks. Half (100 ml) of each such culture was transferred into identical 500-ml glass flasks. A stock solution of NiSO4 (0.1 M) was added to one culture at a final concentration of 10 μM, and the other served as a control culture without additives. Both cultures were incubated for 3 days without additional medium supply. Cells were collected onto a 5-μm membrane filter (Millipore). Total RNA and mRNA were isolated using Isogen (Nippon Gene) followed by Oligotex-dT30 (TaKaRa Bio), and then cDNA was synthesized from 300 ng of mRNA using the Smart rapid amplification of cDNA ends (RACE) cDNA synthesis kit (TaKaRa Bio). cDNAs that were specifically expressed in nickel-treated cells were enriched using the DsDD cDNA subtraction kit (Wako Pure Chemical) according to the manufacturer's instructions. The subtracted cDNAs were cloned in pGEM-T Easy vector (Promega) and introduced into DH5α; transformed cells were spread and grown on LB agar containing carbenicillin. Plasmid vectors (n = 39) containing a ≥0.85-kb insert identified by colony PCR were purified using Wizard plus SV minipreps (Promega) and sequenced. The nickel-induced expression of candidate cDNAs in Paramecium was assessed by agarose gel electrophoresis of the reverse transcription-PCR (RT-PCR) products.
SDS-PAGE and Western blotting.
Cells were fixed in medium containing 10% trichloroacetic acid for 10 min at 4°C, pelleted, washed with distilled water, and lysed in an appropriate volume of 5 M urea, 2 M thiourea, 2% 3-[(3-cholamidopropyl)-dimethylammonio]-1-propanesulfonate (CHAPS), 65 mM dithiothreitol. Cell lysates (2 to 10 μg) were resolved by electrophoresis on 13.5% SDS-PAGE gels, and proteins were stained using Coomassie brilliant blue (CBB) or the silver stain MS kit (Wako). Proteins were blotted onto polyvinylidene difluoride (PVDF) membrane. The membrane was blocked for 1 h at room temperature in 1% Western blocking reagent (Roche Applied Science), incubated with anti-His6 (2) antibody (Roche Applied Science) diluted (1:2,500) in 0.5% Western blocking reagent, and then developed with horseradish peroxidase (HRP)-conjugated sheep anti-mouse IgG antibody and ECL prime (GE Healthcare). Chemiluminescent signals were captured on Hyperfilm ECL (GE Healthcare).
Phylogenetic analyses.
The deduced amino acid sequences of NCI16 and PcGST1 were aligned using MUSCLE with the default parameters in the MEGA5 program (24). Phylogenetic trees were generated using maximum-likelihood (ML) analysis.
qPCR.
NCI16 and PcGST1 mRNA expression in control and nickel-treated cells were measured using Thunderbird SYBR quantitative real-time PCR (qPCR) mix (Toyobo) and an ABI Prism 7900HT sequence detection system (Life Technologies). The primer sequences were the following: for NCI16 cDNA, Ni46UP1 (5′-AATTAACTCTCCTCGGCACTGCTTTTG-3′) and Ni46LP1 (5′-TCCAGCCCATAGAGTGAGTTTATTTTT-3′); for PcGST1 cDNA, Nif66UP3 (5′-CTTAACAAGAATGGGAAGAAGACTAT-3′) and Nif66LP3 (5′-AAGAAATCGGTAGAAATCCTCAAGCA-3′); for P. caudatum α-tubulin cDNA, a-Tub RT-UP1 (5′-GCAACAATCAAGACAAAGAGAACC-3′) and a-Tub RT-LP1 (5′-ACAAAGGCTCTCTTGGCATACATA-3′). The primer sequences for quantitative RT-PCR (qRT-PCR) analyses of four PcGST1 homologs are listed in Table S2 in the supplemental material. All primers were designed using Oligo 7 primer analysis software.
Expression and purification of recombinant proteins.
Each coding sequence of NCI16 and PcGST1 was synthesized de novo (Eurofins Genomics) to optimize codon usage for protein expression in Escherichia coli and subcloned into pET-16b and pET-20b vectors (Novagen), respectively. The pET16b-NCI16 construct carried a full-length NCI16 open reading frame (ORF) with an N-terminal 10× His tag; the pET20b-PcGST1 construct carried a full-length PcGST1 ORF with a C-terminal 6× His tag. The recombinant proteins were expressed in E. coli BL21(DE3) cells (Stratagene) cultivated in 10 ml of ZYM5052 medium (25) at 30°C for 24 h with vigorous shaking. Pelleted cells expressing PcGST1–6× His-tagged protein were resuspended in 2 ml of 20 mM Tris-HCl, pH 8.0, 10 mM MgCl2, 0.5 M NaCl, and 5 mM imidazole and sonicated on ice. PcGST1–6× His was bound to Talon Superflow (GE Healthcare) and eluted in 1 ml of 20 mM Tris-HCl, pH 8.0, 10 mM MgCl2, 0.5 M NaCl, and 500 mM imidazole (Im500).
GST and GPx assays.
GST and GPx assays with purified PcGST1–6× His were performed using a glutathione S-transferase assay kit (Cayman Chemical) and glutathione peroxidase activity colorimetric assay kit (BioVision), respectively, according to the manufacturers' instructions.
Measurement of H2O2.
H2O2 formation was measured using an ROS-Glo H2O2 assay kit (Promega). Cells were treated with 10 μM NiSO4 or 10 μM tert-butylhydroquinone (tBHQ) for 18 h. Cell cultures (80 μl) were transferred to each well of a 96-well plate, mixed with 20 μl of H2O2 substrate, and incubated for an additional 6 h (24 h of total treatment). Resulting luminescent signals were measured using a Varioskan Flash microplate reader (Thermo Scientific).
Cloning of the PcGST1 promoter and construction of inducible expression vector.
Genomic DNA (1 μg) isolated from BW6-1 cells was digested with DraI at 37°C for 16 h, extracted with phenol, and precipitated with isopropanol. Digested genomic DNA was self-ligated and served as a template to amplify the putative PcGST1 promoter region by PCR using the primers Ni66UP309, 5′-AGTGCTTGAGGATTTCTACCGATTTCTT-3′, and Ni66LP265, 5′-CTAACAATTCTGGTTTCTTAGCCTTATGAG-3′. The resultant PCR product (∼1.1 kb) was cloned into pGEM-T Easy and sequenced. A smaller region of the putative PcGST1 promoter (∼0.42 kb) was amplified from the plasmid DNA using the primers Ni66ProSpeIUP1, 5′-ACTAGTCCAGAAGAAATATATAATCAACAA-3′, and Ni66ProEcoRILP1, 5′-GAATTCTTAATTATCTTCCTAAAATTCCAT-3′. The PCR fragment was subcloned into an SpeI-EcoRI-digested pBsc-tel3 vector (21) to generate a potentially nickel-inducible expression vector, designated pGT1-MCS. Sequences encoding a secreted luciferase from the marine copepod Metridia pacifica, MpLuc1 (AB195233), were amplified from plasmid DNA harboring a cloned luciferase cDNA (26); the luciferase sequence was subcloned into EcoRI- and SacI-digested pGT1-MCS to generate the plasmid pGT1-MpLuc1H. Plasmid DNA was isolated using a Qiagen-tip 500 (Qiagen), linearized by BamHI digestion, purified by phenol-chloroform extraction, precipitated with isopropanol, and resuspended in sterile distilled water at a final concentration of 1 μg/μl.
Microinjection.
Linearized pGT1-MpLuc1H was microinjected as described previously (27). About 2.0 × 106 copies of plasmid DNA in a volume of ∼10 pl were injected into the macronucleus of NH2 cells. Viable recipient cells were transferred to a glass depression slide containing fresh lettuce juice medium at 25°C.
Luciferase assay.
Three transformed clonal cell lines were grown in 100 ml of WGP medium. Early-log-phase cultures (100 ml of ∼300 cells/ml) were equally divided into two flasks each. Thereafter, NiSO4 (0.1 M) was added to one flask at a final concentration of 10 μM, and nothing was added to the other. Cells were collected onto a 5-μm membrane filter at 1, 3, and 6 days after NiSO4 addition. Bacterized medium with or without NiSO4 was supplied once on day 3 immediately after harvest on the same day (see Fig. 7C). Pelleted cells from 1 ml of each culture were resuspended in 100 μl of 20 mM Tris-HCl, pH 8.0, 10 mM MgCl2 and then flash frozen at −80°C for luciferase assays. Thawed cellular lysate was vortexed and separated by centrifugation at 21,600 × g for 1 min. Supernatants (10 μl) were transferred to tubes and then placed in a MiniLumat LB 9506 luminometer (Berthold). Coelenterazine (50 μl of 1 ng/μl in 20 mM Tris-HCl, pH 8.0, 50 mM MgCl2) was added to the lysate, and then 10-s measurements were immediately started.
FIG 7.

Analysis of PcGST1 gene promoter. (A) Putative promoter sequence of the PcGST1 gene. Canonical TATA binding boxes are outlined. The bent arrow indicates the putative transcription initiation point. (B) Map of expression vector pGT1-MpLuc1H, carrying the planktonic luciferase gene (MpLuc1) downstream of the SpeI-EcoRI fragment of the PcGST1 gene promoter. Telomere sequences were designed to stabilize a linearized vector in the macronucleus of P. caudatum. (C) Schema of pGT1-MpLuc1H transformant clones incubated with (Ni+) or without (Ni−) 10 μM NiSO4. Control (Ni−) cells were cultured in regular medium at the same time as Ni+ cells. prep, preparation. (D) Relative luciferase activities were measured in pGT1-MpLuc1H transformant clones at 1, 3, and 6 days after treatment with or without 10 μM NiSO4. Values represent mean fold induction determined from triplicate measurements. The activity of untreated cells is defined as 1.0. (E) Induction kinetics of MpLuc1 mRNA in 3 transformant clones after treatment with (Ni+) or without (Ni−) 10 μM NiSO4. Values obtained for MpLuc1 mRNA were normalized to those for P. caudatum α-tubulin mRNA.
Nucleotide sequence accession numbers.
The complete sequences of genes NCI16 and PcGST1 were deposited in the DDBJ/EMBL/GenBank database under the accession numbers AB921148 and AB921149, respectively.
RESULTS
Nickel cytotoxicity in P. caudatum.
Cells were incubated with various concentrations of NiSO4, CoCl2, or CdCl2 for 2 days to compare the cytotoxic effects of nickel ions on P. caudatum to those of other metal ions. Of the three metal ions, NiSO4 was moderately toxic (Fig. 1), since at ≤15 μM, NiSO4 did not significantly decrease cell viability determined as a ratio (i.e., a percentage of untreated cells). Exposure to 5 μM NiSO4 (see Fig. S1 in the supplemental material) and 10 to 20 μM CoCl2 (data not shown) for 2 days slightly increased cell density, whereas CdCl2 was significantly lethal at all tested concentrations (P < 0.01). The 50% lethal doses (LC50) for exposure to NiSO4, CdCl2, and CoCl2 for 2 days were 18.6, 2.10, and 36.5 μM, respectively.
FIG 1.
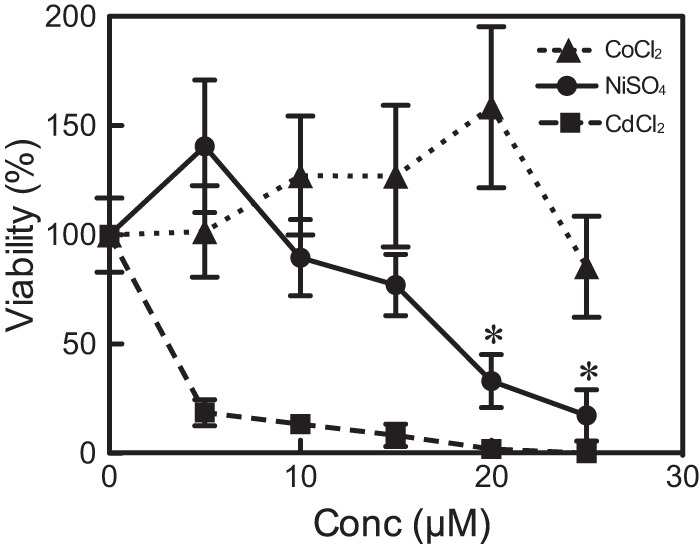
Cytotoxicity of NiSO4, CoCl2, and CdCl2 in P. caudatum. Cells were incubated with NiSO4, CoCl2, or CdCl2 at various concentrations (Conc) between 0 and 25 μM for 2 days. Each point indicates the number of treated cells as a percentage ± standard deviations (SD) of untreated cells; moreover, each point represents cell counts from 3 replicate cultures of cells with and without exposure to metal ions. *, P < 0.01 by Student's t test (only for NiSO4).
Nickel ions induce NCI16 and PcGST1 genes in P. caudatum.
To investigate whether nickel ions could induce the expression of specific genes in P. caudatum, cells were cultured in medium with or without NiSO4. Protein expression from these cells was resolved and compared by SDS-PAGE with silver staining. The intensity of at least two protein bands substantially increased in cells incubated with 10 or 20 μM NiSO4 (Fig. 2A, arrowheads). These two proteins were partially purified by DEAE Sepharose FF resin to determine their N-terminal amino acid sequences using conventional Edman degradation. However, no amino acid could be interpreted from chromatograms, because the peaks were quite vague (data not shown). Therefore, we used cDNA subtraction to identify genes induced by nickel ions. Thirty-nine candidate cDNAs that were ≥0.85 kb were isolated, sequenced, and evaluated by semiquantitative RT-PCR using gene-specific primers. Two genes, designated NCI16 and PcGST1, obviously were upregulated in the cells incubated with 10 μM NiSO4 (Fig. 2B). The deduced sequences of NCI16 and PcGST1 comprised 148 (16.2 kDa; pI, 10.5) and 208 (25.0 kDa; pI, 5.12) amino acids, respectively. The PcGST1 gene sequence contained a single intron, whereas the NCI16 gene had no intronic sequences. The molecular mass of NCI16 and PcGST1 proteins was evaluated by Western blotting with 10× His-tagged recombinant proteins expressed in E. coli and detected using an anti-His antibody. NCI16 protein was found exclusively in the phosphate-buffered saline (PBS)-insoluble fraction; it was detectable only on Western blots of cells that had been sonicated, denatured with SDS-PAGE sample buffer, and directly loaded onto SDS-PAGE gel. A smeared signal at a high-molecular-mass location (Fig. 2C) indicated that recombinant NCI16 tended to aggregate in E. coli. Estimating positions of nontagged protein bands by subtracting the molecular mass of the His tag and the protease recognition sequence (∼2.5 kDa), it is possible that PcGST1 corresponds to the upregulated protein band at the higher molecular mass (Fig. 2A, upper arrowhead).
FIG 2.
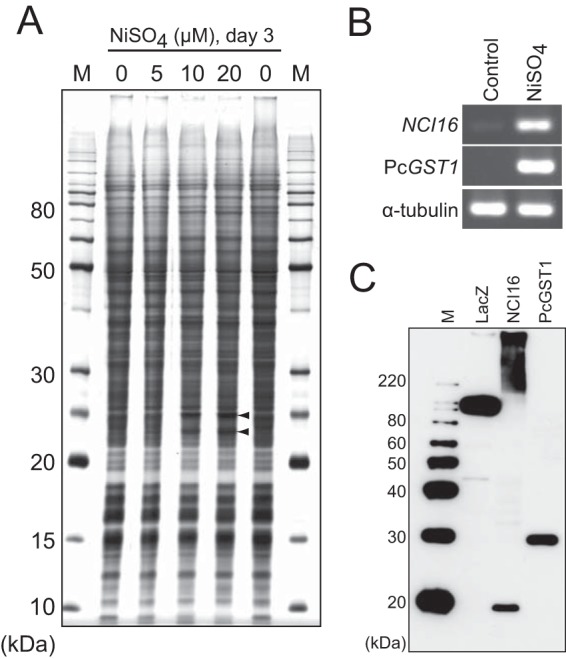
Identification of nickel ion-induced genes. (A) Identification of induced proteins in whole-cell lysates of P. caudatum incubated with various concentrations of nickel sulfate for 3 days. Cellular proteins (2 μg/lane) were separated by 12.5% SDS-PAGE and silver stained. Arrowheads indicate two major induced proteins. M, BenchMark protein molecular mass marker (Life Technologies). (B) RT-PCR analyses of two nickel-induced genes, NCI16 and PcGST1, after treatment with 10 μM NiSO4 for 3 days. (C) The molecular masses of 10× His-tagged NCI16 and PcGST1 proteins were evaluated by Western blotting probed with anti-His antibody. M, MagicMark XP protein standard (Life Technologies); LacZ, His-tagged LacZ protein as a positive control on Western blotting.
Molecular phylogeny and functions of NCI16 and PcGST1.
A BLAST homology search revealed that NCI16 was most orthologous to a protein of unknown function (XP_001432069.1) in a closely related species, Paramecium tetraurelia (75% amino acid sequence identity) (Fig. 3A). NCI16 orthologs were found in the following eukaryotic and prokaryotic microorganisms: an amoeba (Dictyostelium discoideum), cnidaria (Hydra vulgaris and Nematostella vectensis), a fungus-like oomycete (Phytophthora infestans), microalgae (Galdieria sulfuraria and Coccomyxa subellipsoidea), methylotrophs (Methylobacterium nodulans, Methylobacterium nodulans, and Hyphomicrobium denitrificans), a methanotroph (Methyloglobulus morosus), and root nodule bacteria (Rhizobium giardinii, Sinorhizobium fredii, and Sphingobium quisquiliarum) (Fig. 3A). Phylogenetic analysis of NCI16 and 16 orthologous proteins showed that NCI16 and other eukaryotic orthologs were paraphyletic, but most prokaryotic orthologs formed a monophyletic group, except for Methyloglobulus morosus, which formed a small monophyletic clade with G. sulfuraria (Fig. 3C). Both SOSUI 1.11 (http://harrier.nagahama-i-bio.ac.jp/sosui//sosui_submit.html) and TMHMM 2.0 (http://www.cbs.dtu.dk/services/TMHMM-2.0/) predicted that the amino acid sequence of NCI16 contains four-transmembrane segments (see Fig. S2 and Table S1 in the supplemental material).
FIG 3.

Phylogeny of NCI16 and PcGST1. (A) Multiple-amino-acid sequence alignment of NCI16 and 16 NCI16 orthologs. (B) Multiple-amino-acid sequence alignment of PcGST1 and 21 PcGST1 orthologs. Outlined and shaded text represents at least 90% identical amino acid residues. The ML consensus tree obtained from bootstrap analysis with 1,000 replications of NCI16 (C) and PcGST1 (D) was based on amino acid sequence alignments shown in panels A and B. Bootstrap values of >60% are given to the left of selected nodes. Accession numbers or identifiers used in ParameciumDB are indicated with species names.
A BLAST search of ParameciumDB (http://paramecium.cgm.cnrs-gif.fr/cgi/tool/blast) (28) using the amino acid sequence of PcGST1 revealed similarity to six P. caudatum, six P. multimicronucleatum, and seven P. tetraurelia GST-like proteins. However, none of these protein sequences were annotated (Fig. 3B). Among the six P. caudatum homologs found in the database, PCAUDP12020 was the most similar to PcGST1 (E value, 1e−107). We assumed that PCAUDP12020 corresponded to PcGST1 and that protein sequences differ according to strain. Aside from the Paramecium PcGST1 homologs, >50 similar proteins were identified (cutoff E value, >1e−18) by a BLAST search of the NCBI protein database, and most were designated GSTs (Fig. 3B). An ML analysis of PcGST1, five homologs, and 20 orthologs revealed that the Paramecium GST-like proteins formed a monophyletic clade (Fig. 3D) that diverged into two monophyletic clades, 1 and 2. Clade 1 contains GST-like proteins (including PcGST1) of three Paramecium species, whereas clade 2 did not contain any P. caudatum proteins. PcGST1 and a GST-like protein of P. caudatum (PCAUDP12019) formed a monophyletic clade (clade 3) with a well-supported bootstrap value (89%), suggesting that their amino acid sequences were relatively unique compared to those of the other Paramecium GST-like proteins analyzed in the present study.
Nickel ions enhance GST and GPx activities in P. caudatum.
To confirm that PcGST1 has GST activity, a 6× His-tagged PcGST1 protein was expressed in E. coli and purified on immobilized metal affinity chromatography resin charged with cobalt. Recombinant PcGST1-His protein eluted with a buffer containing Im500 migrated as a single band on SDS-PAGE (Fig. 4A). The specific GST activity of purified PcGST1-His toward the substrate, 1-chloro-2,4-dinitrobenzene (CDNB), was comparable to that of equine liver GST, which was used as a positive control (Fig. 4B). The purified PcGST1-His also exerted considerable GPx activity toward cumene hydroperoxide (Fig. 4B).
FIG 4.

GST and GPx activities of PcGST1. (A) Recombinant PcGST1 His-tagged protein was expressed in E. coli, purified by cobalt affinity chromatography, resolved by SDS-PAGE, and stained with CBB. M, protein molecular mass marker. Crude, soluble fraction of E. coli crude extract. FT, flowthrough fraction. Im20, Im50, and Im500, fractions eluted with buffers containing 20, 50, and 500 mM imidazole, respectively. (B) Specific GST and GPx activities of purified recombinant PcGST1. Values for the positive control indicate specific activities of equine liver GST and bovine erythrocyte GPx that are components of GST and GPx assay kits, respectively. Values for the empty vector indicate specific GST and GPx activities of crude extract from E. coli transformed with empty expression vector as a negative control. Specific GST (C) and GPx (D) activities of P. caudatum whole-cell lysate after treatment with 10 μM NiSO4 for 3 or 6 days. Bars show means ± SD from 5 different cultures (*, P < 0.01 by Student's t test). (E) Relative mRNA levels of PcGST1 and 4 other homologs normalized to α-tubulin expression upon nickel exposure. Cells were incubated with 10 μM NiSO4 for 3 or 6 days. Bars represent mean fold changes ± SD (n = 3 per group). The level of mRNA in untreated control cells is defined as 1.0.
Significant upregulation of PcGST1 mRNA, which encoded the protein with GST- and GPx-like activities, during exposure to nickel ions (Fig. 2B) implied an increase in the levels of intracellular GST and GPx activities. Therefore, we examined GST and GPx activities in P. caudatum lysates 3 and 6 days after exposure to 10 μM NiSO4. Both GST and GPx activities significantly increased during 6 days of exposure to nickel (P < 0.01) (Fig. 4C and D).
To estimate the contribution of PcGST1 and the five PcGST1 homologs (Fig. 3B and D) to the overall GST and/or GPx activities measured in nickel-treated P. caudatum lysate, we examined their relative expression levels by qRT-PCR analyses with gene-specific primers. PcGST1 was upregulated >3,000-fold in cells incubated with 10 μM NiSO4 compared to untreated control cells on days 3 and 6, whereas the four other PcGST1 homologs were induced only <150-fold (Fig. 4E). One homolog (PCAUDP15663) was not sufficiently amplified and was not further analyzed. Thus, it is likely that PcGST1 significantly contributes to changes in GST and/or GPx activity in cellular extract, although the enzymatic nature of the other five homologs remains undetermined. We cloned qRT-PCR products containing partial sequences of PcGST1 homologs and sequenced them to confirm the specificity of the PCR amplification.
Metal ion specificity, dose dependency, and time course of NCI16 and PcGST1 induction evaluated by quantitative RT-PCR.
Incubation with either NiSO4 or NiCl2 at 10 μM for 3 days increased the levels of NCI16 and PcGST1 mRNAs >40-fold and >6,000-fold, respectively, compared to untreated controls (Fig. 5A and B). Both genes also were significantly upregulated 11.8- and 225-fold, respectively, by CoCl2 at 10 μM. The concentration-related effects of NiSO4 on the induction of NCI16 and PcGST1 genes differed notably from those of CoCl2 (Fig. 5C and D). The mRNA levels of NCI16 and PcGST1 were not altered at NiSO4 concentrations ranging from 0 to 5 μM, whereas CoCl2 exerted dose-dependent effects on both genes. Both NCI16 and PcGST1 mRNAs were rapidly induced 10.5- and 267-fold, respectively, at 6 h after NiSO4 exposure and continued to increase up to days 6 and 3, respectively (Fig. 5E and F).
FIG 5.

Quantitative RT-PCR analyses of NCI16 or PcGST1 mRNA expression. Cells were treated with 10 μM either of the metal ions (1 μM for CdCl2 and CuSO4) for 3 days. Expression of NCI16 (A) or PcGST1 (B) was analyzed by qRT-PCR using gene-specific primers. Values obtained for both genes were normalized to those of P. caudatum α-tubulin mRNA. Concentration-dependent induction of NCI16 (C) or PcGST1 (D) mRNA. Cells were grown in medium containing various concentrations of NiSO4 or CoCl2, harvested 3 days later, and analyzed by qRT-PCR. Induction kinetic of NCI16 (E) or PcGST1 (F) mRNA after treatment with 10 μM NiSO4 also are shown. Bars and points show mean fold changes ± SD from replicate qRT-PCR assays (n = 3) of total RNA from 3 to 4 cell cultures compared to untreated control cells.
NCI16 and PcGST1 expression was enhanced by H2O2 and antioxidant tBHQ, respectively.
We investigated whether exposure to nickel causes intracellular ROS accumulation in Paramecium. Exposure to 10 μM NiSO4 caused a notable increase in the H2O2 concentration in P. caudatum (Fig. 6A). Treatment of cells with 1 mM H2O2 for 24 h significantly enhanced NCI16 expression ∼700-fold but increased PcGST1 by only ∼15-fold (Fig. 6B). The antioxidant tBHQ is capable of inducing phase II detoxification enzymes, including GSTs. When cells were incubated with 10 μM tBHQ for 24 h, PcGST1 expression was prominently induced (∼780-fold) (Fig. 6B), but tBHQ induced minimal NCI16 expression (∼2-fold) (Fig. 6B) and no intracellular H2O2 (Fig. 6A).
FIG 6.
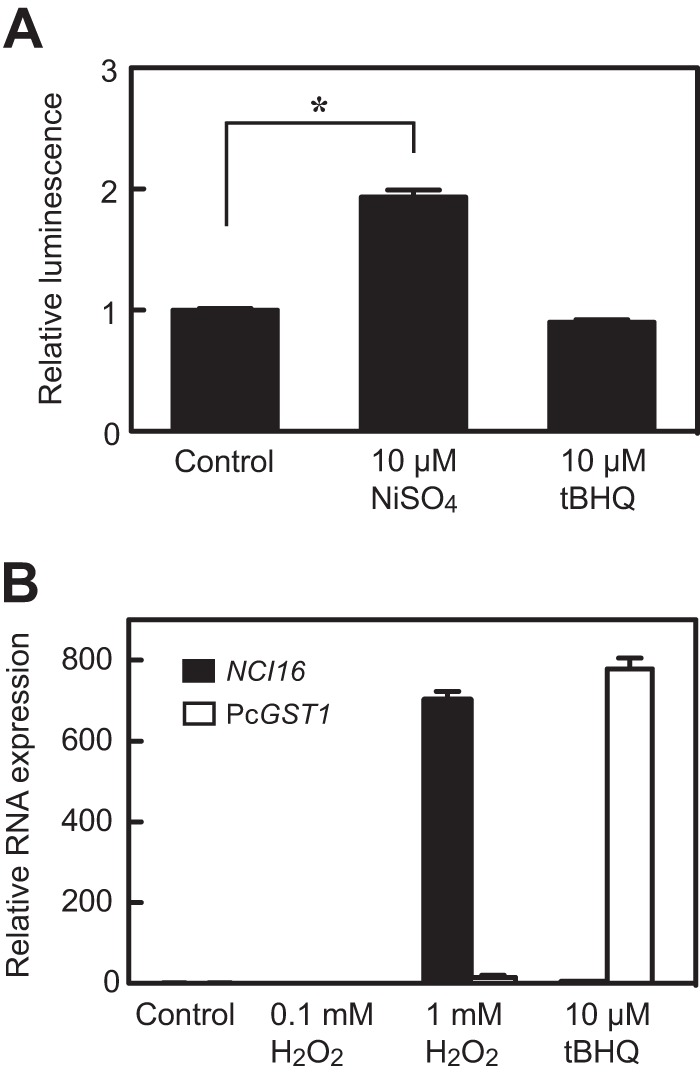
Nickel induces ROS production in P. caudatum. (A) Cells were exposed to 10 μM NiSO4 or 10 μM antioxidant tert-butylhydroquinone (tBHQ) for 24 h. Luminescent signals were obtained using an ROS-Glo H2O2 assay kit to measure H2O2 levels. Bars show mean fold changes ± SD from 4 replicate wells compared to 4 untreated control samples (*, P < 0.01, Student's t test). (B) Induction of NCI16 or PcGST1 mRNA in P. caudatum treated with either 1 mM H2O2 or 10 μM tBHQ for 24 h. Bars show mean fold changes ± SD from triplicate qRT-PCR assays compared to untreated control cells.
Putative PcGST1 promoter can induce luciferase reporter gene upon nickel exposure.
Lastly, we examined whether or not nickel-responsive expression of the PcGST1 gene is regulated by elements in a putative PcGST1 gene promoter region. Genomic sequence data for P. caudatum were not yet listed in the ParameciumDB at the time this experiment started. Therefore, we isolated PcGST genomic DNA with its 5′ and 3′ flanking regions by inverse PCR. We amplified a DNA product containing a 5′ flanking region (∼0.42 kb) immediately upstream of the PcGST1 coding sequence. This putative PcGST1 gene promoter contained two putative TATA boxes at −31 and −44 bp relative to the transcription start site for PcGST1 (Fig. 7A), but neither canonical GC boxes nor sequences similar to the consensus metal-responsive or antioxidant-responsive element were found in the mammalian metallothionein (29) or GST (30) gene promoter, respectively.
We performed a reporter gene assay to determine whether or not this putative PcGST1 gene promoter was sufficient to induce nickel-responsive transcription. We generated the expression vector pGT1-MpLuc1H that encoded the marine planktonic luciferase, MpLuc1 (26), as a reporter gene immediately downstream of the putative ∼0.42-kb PcGST1 promoter fragment (Fig. 7B). Three clonal transformant lines were established by microinjecting pGT1-MpLuc1 into the macronucleus of P. caudatum. The relative abundance of the transduced luciferase gene in the genomic DNA isolated from these three lines was determined by qPCR. When the amount of the luciferase gene in clone 1 was defined as 1.0, those of clones 2 and 3 were 0.57 and 0.14, respectively. Each clonal line was grown in medium with or without 10 μM NiSO4 (Fig. 7C). Luciferase activity was considerably enhanced by NiSO4 treatment for 3 days in clones 1 and 2 (211- and 167-fold, respectively, compared to the untreated control) and moderately enhanced in clone 3 (6.4-fold) (Fig. 7D). Luciferase activity did not seem to change in a transformant line transduced with a constitutive reporter expression vector, pTT3-MpLuc1H, that harbors an α-tubulin promoter sequence driving MpLuc1 gene transcription (Fig. 7D). Based on qRT-PCR analyses, the induction of MpLuc1 mRNA by NiSO4 began on day 1 and ended precipitously on day 3 in all inducible clone 3 lines (Fig. 7E).
DISCUSSION
The present study aimed to identify genes that are significantly induced by nickel ions. Using subtractive cDNA hybridization, we identified NCI16 and PcGST1 as genes that were upregulated in cDNA prepared from cells exposed to 10 μM NiSO4, a concentration at which cellular viability did not significantly decrease (Fig. 1). Recombinant 6× His-tagged NCI16 protein expressed in E. coli was largely insoluble, and that expressed in mammalian cultured cells could not be detected on immunoblots (data not shown). Thus, we could not use recombinant NCI16 protein for functional analysis. The hydrophobic nature of the predicted multiple membrane-spanning domains (see Fig. S2 in the supplemental material) might have rendered recombinant NCI16 insoluble. We are still in the process of expressing and purifying recombinant NCI16 to reveal its functions and roles in cellular responses to nickel and/or ROS. Recombinant PcGST1 protein was produced in and purified from E. coli at high yields (Fig. 4A). PcGST1 was predicted to have GST activity based on amino acid sequence similarity to other GSTs. Purified recombinant PcGST1 exerted GST activity in assays using CDNB as a substrate (Fig. 4B). Recombinant PcGST1 also exhibited efficient GPx activity on the substrate cumene hydroperoxide (Fig. 4B). To date, GPxs are classified into two major types (31). Proteins of the selenoprotein type contain a selenocysteine residue in the enzyme active site to act as GPxs with organic hydroperoxides and H2O2. The others do not depend on selenium for catalysis and are active only upon organic hydroperoxides. We considered that PcGST1 was the latter type of GPx due to having a GST-like amino acid sequence. This notion also was supported by the finding that PcGST1 did not react with tert-butyl hydroperoxide (data not shown), which is a suitable substrate for selenium-containing GPx. Therefore, PcGST1 is probably a GST protein with GPx-like activity. We attempted gene silencing of NCI16 and PcGST1 by feeding RNA interference (RNAi) to reveal their importance in cells exposed to nickel ions. However, we have not yet achieved sufficient knockdown of either transcript. We are currently evaluating other gene silencing methods for Paramecium.
Nickel treatment caused a notable increase in the H2O2 concentration in P. caudatum (Fig. 6A), indicating the intracellular formation of various ROS, including superoxide, which is converted to H2O2 by superoxide dismutase, hydroxyl radicals, and LPO. Incubating cells with H2O2 significantly enhanced NCI16 expression ∼700-fold (Fig. 6B). Taken together, these findings suggest that NCI16 gene induction was caused by H2O2 and/or superoxide produced by nickel ions. Interestingly, the antioxidant tBHQ notably induced PcGST1 but not NCI16 (Fig. 6B). In mammals, tBHQ might have both chemoprotective and carcinogenic effects (32). The chemoprotective mechanism of tBHQ might involve the induction of phase II detoxification enzymes, such as GSTs, UDP-glucuronyltransferases, and NAD(P)H:quinone oxidoreductase. Our results indicated that P. caudatum and mammals share a mechanism by which tBHQ enhances transcription of GST and other phase II genes. To date, various mechanisms have been proposed for tBHQ-mediated phase II enzyme induction in mammals, for example, via ROS-mediated dissociation of Nrf2-Keap1, Nrf2 stabilization, mitogen-activated protein kinase pathway activation, phosphatidylinositol 3-kinase/Akt activation, or a combination of these (32). We found that incubating cells with tBHQ did not lead to intracellular H2O2 accumulation (Fig. 6A). Thus, we postulated that the PcGST1 gene was induced by oxidative intermediates derived from tBHQ, such as phenoxyl free radicals (33), rather than ROS generated during tBHQ metabolism. This hypothesis also was supported by the finding that H2O2 treatment only weakly induced PcGST1 (Fig. 6B). However, the mechanisms by which PcGST1 is induced by nickel exposure remain to be elucidated.
The 5′-flanking region of the PcGST1 gene isolated from P. caudatum genomic DNA was capable of inducing MpLuc1 luciferase transcription in transformed Paramecium organisms incubated with NiSO4. This finding indicated that the ∼0.42 kb of the PcGST1 promoter region contains a putative nickel-responsive element, although little is known about any gene regulatory element in paramecia. The kinetics of MpLuc1 mRNA expression and luciferase activity induced by nickel in transformant cells apparently differed (Fig. 7D and E). This delayed induction of luciferase activity relative to that of mRNA expression might be due to protein synthesis and proper MpLuc1 folding. The kinetics of MpLuc1 mRNA expression driven by the PcGST1 promoter (Fig. 7E) and endogenous PcGST1 expression (Fig. 5F) also differed; the former sharply declined on day 3 after nickel exposure. Differences in the expression mechanism of each gene product might explain this discrepancy. First, the reporter construct might not have the cis- and/or trans-regulatory element required to maintain long-term MpLuc1 mRNA expression. These elements might be located at a distal region extending upstream or downstream of the PcGST1 gene. Second, the reporter construct might lack epigenetic regulation, such as histone modifications, for long-term mRNA expression. Furthermore, the mRNA sequences of MpLuc1 and PcGST1 fundamentally differ, which might result in different mRNA stability and expression profiles.
Importantly, we have shown that luciferase activity as a reporter could serve for gene promoter activity in paramecia. MpLuc1, like commercially available Gaussia and Metridia longa luciferases, is a secreted luciferase isolated from marine plankton. The secretion of planktonic luciferases into the extracellular milieu might require the initial direction of these proteins to the endoplasmic reticulum by signal sequences at their N termini. Here, we found that MpLuc1 was not secreted into the culture media, whereas an intracellular fraction exhibited substantial luciferase activity (data not shown). This finding indicated that the signal sequence encoded by MpLuc1 does not function in signal-dependent secretion in Paramecium. Thus, the reporter system using MpLuc1 can serve as a highly sensitive and efficient nonsecreted luciferase assay in Paramecium. We plan to utilize this reporter system to further elucidate the mechanisms of PcGST1 and NCI16 gene regulation in nickel-treated paramecia.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to Sawako Sato and Yuka Nakano (Saitama Medical University) for excellent technical assistance.
Footnotes
Published ahead of print 7 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00112-14.
REFERENCES
- 1.Scansetti G, Maina G, Botta GC, Bambace P, Spinelli P. 1998. Exposure to cobalt and nickel in the hard-metal production industry. Int. Arch. Occup. Environ. Health 71:60–63. 10.1007/s004200050251 [DOI] [PubMed] [Google Scholar]
- 2.Sunderman FW., Jr 1989. Mechanisms of nickel carcinogenesis. Scand. J. Work Environ. Health 15:1–12 [DOI] [PubMed] [Google Scholar]
- 3.Denkhaus E, Salnikow K. 2002. Nickel essentiality, toxicity, and carcinogenicity. Crit. Rev. Oncol. Hematol. 42:35–56. 10.1016/S1040-8428(01)00214-1 [DOI] [PubMed] [Google Scholar]
- 4.Athar M, Hasan SK, Srivastava RC. 1987. Evidence for the involvement of hydroxyl radicals in nickel mediated enhancement of lipid peroxidation: implications for nickel carcinogenesis. Biochem. Biophys. Res. Commun. 147:1276–1281. 10.1016/S0006-291X(87)80208-5 [DOI] [PubMed] [Google Scholar]
- 5.Cavallo D, Ursini CL, Setini A, Chianese C, Piegari P, Perniconi B, Iavicoli S. 2003. Evaluation of oxidative damage and inhibition of DNA repair in an in vitro study of nickel exposure. Toxicol. In Vitro 17:603–607. 10.1016/S0887-2333(03)00138-3 [DOI] [PubMed] [Google Scholar]
- 6.Lynn S, Yew FH, Chen KS, Jan KY. 1997. Reactive oxygen species are involved in nickel inhibition of DNA repair. Environ. Mol. Mutagen. 29:208–216 [PubMed] [Google Scholar]
- 7.Sugiyama M. 1994. Role of cellular antioxidants in metal-induced damage. Cell Biol. Toxicol. 10:1–22 [DOI] [PubMed] [Google Scholar]
- 8.Zhong Z, Troll W, Koenig K, Frenkel K. 1990. Carcinogenic sulfide salts of nickel and cadmium induce H2O2 formation by human polymorphonuclear leukocytes. Cancer Res. 50:7564–7570 [PubMed] [Google Scholar]
- 9.Chakrabarti S, Bai C. 1999. Role of oxidative stress in nickel chloride-induced cell injury in rat renal cortical slices. Biochem. Pharmacol. 58:1501–1510. 10.1016/S0006-2952(99)00232-4 [DOI] [PubMed] [Google Scholar]
- 10.Chen CY, Wang YF, Lin YH, Yen SF. 2003. Nickel-induced oxidative stress and effect of antioxidants in human lymphocytes. Arch. Toxicol. 77:123–130. 10.1007/s00204-002-0427-6 [DOI] [PubMed] [Google Scholar]
- 11.Valko M, Morris H, Cronin MT. 2005. Metals, toxicity and oxidative stress. Curr. Med. Chem. 12:1161–1208. 10.2174/0929867053764635 [DOI] [PubMed] [Google Scholar]
- 12.Kasprzak KS. 1995. Possible role of oxidative damage in metal-induced carcinogenesis. Cancer Investig. 13:411–430. 10.3109/07357909509031921 [DOI] [PubMed] [Google Scholar]
- 13.Andersen HR, Andersen O. 1989. Effect of nickel chloride on hepatic lipid peroxidation and glutathione concentration in mice. Biol. Trace Elem. Res. 21:255–261. 10.1007/BF02917261 [DOI] [PubMed] [Google Scholar]
- 14.Cartana J, Romeu A, Arola L. 1992. Effects of copper, cadmium and nickel on liver and kidney glutathione redox cycle of rats (Rattus sp.). Comp. Biochem. Physiol. C 101:209–213 [DOI] [PubMed] [Google Scholar]
- 15.Naitoh Y. 1966. Reversal response elicited in nonbeating cilia of paramecium by membrane depolarizatin. Science 154:660–662. 10.1126/science.154.3749.660 [DOI] [PubMed] [Google Scholar]
- 16.Naitoh Y, Kaneko H. 1973. Control of ciliary activities by adenosinetriphosphate and divalent cations in triton-extracted models of Paramecium caudatum. J. Exp. Biol. 58:657–676 [DOI] [PubMed] [Google Scholar]
- 17.Larsen J, Satir P. 1991. Analysis of Ni2+-induced arrest of Paramecium axonemes. J. Cell Sci. 99(Part 1):33–40 [DOI] [PubMed] [Google Scholar]
- 18.Smith-Sonneborn J, Palizzi R, McCann E, Fisher G. 1983. Bioassay of genotoxic effects of environmental particles in a feeding ciliate. Environ. Health Perspect. 51:205–210. 10.1289/ehp.8351205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith-Sonneborn J, Leibovitz B, Donathan R, Fisher GL. 1986. Bioassay of environmental nickel dusts in a particle feeding ciliate. Environ. Mutagen. 8:621–626. 10.1002/em.2860080412 [DOI] [PubMed] [Google Scholar]
- 20.Madoni P. 2000. The acute toxicity of nickel to freshwater ciliates. Environ. Pollut. 109:53–59. 10.1016/S0269-7491(99)00226-2 [DOI] [PubMed] [Google Scholar]
- 21.Takenaka Y, Haga N, Harumoto T, Matsuura T, Mitsui Y. 2002. Transformation of Paramecium caudatum with a novel expression vector harboring codon-optimized GFP gene. Gene 284:233–240. 10.1016/S0378-1119(01)00886-1 [DOI] [PubMed] [Google Scholar]
- 22.Dryl S. 1959. Antigenic transformation in Paramecium aurelia after homologous antiserum treatment during autogamy and conjugation. J. Protozool. 6(Suppl):25 [Google Scholar]
- 23.Tokusumi Y, Takagi Y. 2000. Ectosymbiotic role of food bacteria for Paramecium: bacterial detoxification of paramecia-killing toxin contained in wheat grass powder. Zoolog. Sci. 17:341–348. 10.2108/zsj.17.341 [DOI] [PubMed] [Google Scholar]
- 24.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 28:2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Studier FW. 2005. Protein production by auto-induction in high-density shaking cultures. Protein Expr. Purif. 41:207–234. 10.1016/j.pep.2005.01.016 [DOI] [PubMed] [Google Scholar]
- 26.Takenaka Y, Masuda H, Yamaguchi A, Nishikawa S, Shigeri Y, Yoshida Y, Mizuno H. 2008. Two forms of secreted and thermostable luciferases from the marine copepod crustacean, Metridia pacifica. Gene 425:28–35. 10.1016/j.gene.2008.07.041 [DOI] [PubMed] [Google Scholar]
- 27.Haga N, Forte M, Ramanathan R, Hennessey T, Takahashi M, Kung C. 1984. Characterization and purification of a soluble protein controlling Ca-channel activity in paramecium. Cell 39:71–78. 10.1016/0092-8674(84)90192-2 [DOI] [PubMed] [Google Scholar]
- 28.Arnaiz O, Sperling L. 2011. ParameciumDB in 2011: new tools and new data for functional and comparative genomics of the model ciliate Paramecium tetraurelia. Nucleic Acids Res. 39:D632–D636. 10.1093/nar/gkq918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dalton T, Li Q, Bittel D, Liang L, Andrews G. 1996. Oxidative stress activates metal-responsive transcription factor-1 binding activity. Occupancy in vivo of metal response elements in the metallothionein-I gene promoter. J. Biol. Chem. 271:26233–26241 [DOI] [PubMed] [Google Scholar]
- 30.Wasserman WW, Fahl WE. 1997. Functional antioxidant responsive elements. Proc. Natl. Acad. Sci. U. S. A. 94:5361–5366. 10.1073/pnas.94.10.5361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mannervik B. 1985. Glutathione peroxidase. Methods Enzymol. 113:490–495. 10.1016/S0076-6879(85)13063-6 [DOI] [PubMed] [Google Scholar]
- 32.Gharavi N, Haggarty S, El-Kadi AO. 2007. Chemoprotective and carcinogenic effects of tert-butylhydroquinone and its metabolites. Curr. Drug Metab. 8:1–7 [DOI] [PubMed] [Google Scholar]
- 33.Yu R, Tan TH, Kong AN. 1997. Butylated hydroxyanisole and its metabolite tert-butylhydroquinone differentially regulate mitogen-activated protein kinases. The role of oxidative stress in the activation of mitogen-activated protein kinases by phenolic antioxidants. J. Biol. Chem. 272:28962–28970 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.