

Abstract
Malaria kills more than 1 million people per year worldwide, with severe malaria anemia accounting for the majority of the deaths. Malaria anemia is multifactorial in etiology, including infected erythrocyte destruction and decrease in erythrocyte production, as well as destruction or clearance of noninfected erythrocytes. We identified a panspecies Plasmodium hemolysin type III related to bacterial hemolysins. The identification of a hemolysin III homologue in Plasmodium suggests a potential role in host erythrocyte lysis. Here, we report the first characterization of Plasmodium falciparum hemolysin III, showing that the soluble recombinant P. falciparum hemolysin III is a pore-forming protein capable of lysing human erythrocytes in a dose-, time-, and temperature-dependent fashion. The recombinant P. falciparum hemolysin III-induced hemolysis was partially inhibited by glibenclamide, a known channel antagonist. Studies with polyethylene glycol molecules of different molecular weights indicated a pore size of approximately 3.2 nm. Heterologous expression of recombinant P. falciparum hemolysin III in Xenopus oocytes demonstrated early hypotonic lysis similar to that of the pore-forming aquaporin control. Live fluorescence microscopy localized transfected recombinant green fluorescent protein (GFP)-tagged P. falciparum hemolysin III to the essential digestive vacuole of the P. falciparum parasite. These transfected trophozoites also possessed a swollen digestive vacuole phenotype. Native Plasmodium hemolysin III in the digestive vacuole may contribute to lysis of the parasitophorous vacuole membrane derived from the host erythrocyte. After merozoite egress from infected erythrocytes, remnant P. falciparum hemolysin III released from digestive vacuoles could potentially contribute to lysis of uninfected erythrocytes to contribute to severe life-threatening anemia.
INTRODUCTION
An estimated 1,238,000 people died of malaria worldwide in 2010 (1). Severe malaria anemia contributes to a significant portion of malaria-related deaths. A study done in Western Kenya showed that severe malaria anemia accounted for 53% of malaria-related mortality (2). The etiology of malaria anemia is multifactorial and not fully understood (3, 4). Bone marrow suppression and increased destruction of red blood cells are the main mechanisms contributing to severe malaria anemia. Besides the obvious destruction of infected erythrocytes (parasitemias can reach 1 to 10%), increased clearance of uninfected erythrocytes is a major contributor to anemia. Studies estimate that 8 to 10 uninfected erythrocytes are destroyed for each infected erythrocyte (5, 6). Both extravascular and intravascular hemolysis of uninfected erythrocytes play important roles in severe malaria anemia pathogenesis (7).
Using the Plasmodium database, we identified a putative hemolysin in Plasmodium falciparum (gene ID, PF3D7_1455400 or PF14_0528) belonging to the hemolysin III superfamily (8). P. falciparum hemolysin III (PfHly III) is located on chromosome 14. Homologues have been identified in all Plasmodium genomes sequenced, including P. vivax, P. yoelii, P. berghei, and P. knowlesi, as well as Toxoplasma and Babesia genomes. The PfHly III gene has one intron, and the coding sequence consists of 849 bp with a GC content of 24%. The cDNA encodes a polypeptide of 282 amino acids with a predicted molecular mass of 33 kDa. PfHly III gene transcripts were detected in the erythrocytic stages, in gametocytes, and in patient samples from pregnant women and also children (9, 10). Mass spectrometry detected PfHly III in gametocytes (11). From Bacillus cereus and Vibrio vulnificus Hly III studies, Hly III was shown to be a pore-forming protein, 3 to 3.5 nm in diameter, with optimal hemolysis at 37°C (12–14). Over evolutionary time, hemolysins have adapted essential transport roles in Plasmodium, contrasting with their ancient bacterial pore-forming cytotoxic effect. For example, export protein 2 (EXP2) is a pore-forming protein that is homologous to hemolysin E. EXP2 is part of an essential protein transporter complex (PTEX) in Plasmodium, responsible for the translocation of exported proteins. PTEX was shown to function in exporting parasite proteins into the cytosol of the host erythrocyte (15, 16). The contribution of the Plasmodium hemolysin III homologue to the destruction of host erythrocytes that occurs during malaria infection is not known.
Here, we report the initial characterization of the PfHly III homologue. Recombinant PfHly III (recPfHly III) lysed human erythrocytes by a pore-forming mechanism, ruptured Xenopus laevis oocytes, and localized to the unique, essential digestive vacuole.
MATERIALS AND METHODS
Construction of pUC18-PfHly III expression vector.
The codon-optimized PfHly III gene (GenScript, Piscataway, NJ; see Fig. S1 in the supplemental material) was cloned into the pET22b plasmid (Novagen-Merck Millipore). PfHly III was amplified from pET22b-PfHly III using PCR with a 5′ primer (5′-GGATCCCATCACCACCATCATCATGAATTCATGGAATTTTACAAAAAC-3′) and 3′ primer (5′-TCTAGATCAGTGGTGGTGGTGGTGGTG-3′) to generate the BamHI and XbaI sites compatible with the pUC18 plasmid (Agilent/Stratagene, Santa Clara, CA). The DNA insert was confirmed by sequencing.
Bacterial expression of recombinant PfHly III.
The ampicillin-resistant pUC18-PfHly III expression vector was transformed into Escherichia coli HB101 competent cells and grown to an optical density at 600 nm (OD600) of 0.4, followed by a 16-h 37°C protein induction with 1 mM isopropyl-β-d-thiogalactoside. The bacterial pellet was sonicated in 2 ml of phosphate-buffered saline (PBS) followed by microcentrifugation at 12,000 × g at 4°C. recPfHly III was purified from the soluble supernatant under native conditions by addition of precharged Ni2+-nitrilotriacetic acid (NTA) resin and incubation overnight at 4°C under gentle mixing by end-over-end rotation, followed by centrifugal washes with PBS containing 5 mM imidazole and finally elution with 100 mM EDTA. The elutions were dialyzed overnight at 4°C against PBS (pH 7.5) using Slide-A-Lyzer (Thermo Scientific) to remove EDTA and imidazole. pUC18-only plasmid was transformed, induced, and purified under the same conditions and used as a negative control.
Western blot analysis of recPfHly III.
Soluble recPfHly III was separated by SDS-PAGE and transferred to a nitrocellulose membrane (Bio-Rad). The membrane was blocked with Qiagen blocking buffer at 37°C for 1 h, washed 3 times with PBS (0.05% Tween 20), and then incubated with a 1:1,000 dilution of anti-His–horseradish peroxidase (HRP) conjugate in blocking buffer at 4°C overnight. Protein was visualized with enhanced chemiluminescence.
Hemolytic activity assay.
Human erythrocytes were washed with PBS (pH 7. 5) three times and adjusted to a final concentration of 1% (vol/vol). The erythrocyte suspension (0.1 ml) was incubated with 0.1 ml of recPfHly III diluted in PBS. The mixture was incubated for a total of 60 min at 37°C. The reaction mixtures were then centrifuged at 1,000 × g for 5 min. Hemoglobin released from recPfHly III-induced hemolysis was monitored by the absorbance of the supernatant at 550 nm. One hemolytic unit (HU) was defined as the dose of recPfHly III that elicited 50% hemolysis. One hundred percent hemolysis was the amount of hemoglobin released after treating human erythrocytes (1%) with 0.1% Triton X-100. The hemolytic activity assay of recPfHly III was studied at indicated temperatures.
Osmotic protection experiments.
For osmotic protection experiments, 0.1 ml of recPfHly III (2 HU) was incubated with 0.1 ml of human erythrocytes (final concentration, 1%) suspended in PBS (pH 7.5) containing an osmotic protectant at a final concentration of 30 mM (17). Incubation was at 37°C for 60 min, and the mixture was immediately subjected to the hemolytic activity assay. Osmotic protectants included glucose, polyethylene glycol 600 (PEG 600), PEG 1500, PEG 2000, PEG 3350, PEG 4600, PEG 6000, and PEG 8000 (Sigma). The control used recPfHly III without any osmotic protectants.
Drug inhibition assay.
Pharmacological agents clotrimazole, dantrolene, furosemide, glibenclamide, and phlorizin were selected from the Johns Hopkins Clinical Compound Library (18). Each 10 mM drug was diluted in PBS (pH 7.5) to a final concentration of 100 μM. Each drug was incubated with recPfHly III (0.5 HU) and 1% erythrocytes at 37°C for 60 min and immediately subjected to the hemolytic activity assay. The control used recPfHly III without any pharmacological agent. All samples had an equivalent amount of dimethyl sulfoxide (DMSO).
Immunofluorescence assay (IFA).
recPfHly III (5 HU) was incubated with 1% human erythrocytes and 30 mM PEG 4600 at 37°C for 60 min. The erythrocytes were pelleted and washed with PBS containing 30 mM PEG 4600, a total of 3 times. The cells were then spotted on polylysine-coated slides and fixed in 4% formalin for 30 min. The cells were washed with PBS and permeabilized with PBS containing 0.1% Triton X-100 for 5 min. Cells were again washed and incubated with blocking buffer at 4°C overnight. Cells were washed and incubated with penta-His Alexa Fluor 488 conjugate (Qiagen). Cells were washed with PBS 3 times and sealed under a coverslip, and images were captured on a Nikon Eclipse 90i fluorescence microscope.
Oocyte expression.
The plasmid pGS21a_PfHly III-flag with an NheI site was constructed using the QuikChange Lightning site-directed mutagenesis kit (Stratagene), digested with EcoRI and NheI, and ligated into pXβG-ev1-myc to produce pXβG-ev1-myc-PfHly III-flag. We received pXβG-ev1-myc and pXβG-ev1-hAQP1 cDNA as a generous gift from Peter Agre. Capped cRNA was produced using in vitro transcription from either the pXβG-ev1-myc-PfHly III-flag or pXβG-ev1-hAQP1 plasmid template and linearized with XbaI, using T3 RNA polymerase and the RNeasy minikit (Qiagen). X. laevis oocytes generously donated by Craig Montell were defolliculated and injected with 25 to 44 ng of cRNA or 50 nl of diethyl pyrocarbonate (DEPC)-treated water, followed by incubation at 16°C for 3 to 5 days in oocyte recipe 3 medium. Oocytes were collected 72, 96, and 120 h postinjection for Western blot analysis and swelling assays, described below.
For Western blot analysis, 10 oocytes per treatment were pooled into 200 μl of ice-cold lysis buffer (20 mM Tris-HCl, pH 7.5, 140 mM NaCl, 2% Triton X-100, 1× protease inhibitor [Sigma fast protease inhibitor cocktail tablet, EDTA free]) and incubated on ice for 30 min. Oocytes were homogenized by pipetting samples repeatedly and centrifuged at 4,500 × g for 15 min at 4°C to remove yolk and cellular debris. The supernatant was transferred to a new tube and incubated on ice for 30 min, with occasional vortexing. The sample was spun at 15,000 × g for 15 min at 4°C to remove insoluble materials, and the supernatant was stored in SDS loading buffer at −20°C. Samples were thawed on ice (not heated) and then run on an SDS-PAGE gel. Proteins were then transferred to a nitrocellulose membrane and probed with anti-Flag M2 (Sigma), anti-myc-HRP (Invitrogen), anti-aquaporin 1 (anti-AQP1) B-11 (Santa Cruz), or anti-beta-actin antibodies (AbCam). Swelling assays were modified from previous protocols (19). Briefly, 5 to 6 oocytes per group were transferred to a small petri dish of water (hypotonic) and monitored with videomicroscopy at room temperature for swelling and rupture over a time course of 0 to 60 min. Still pictures were taken at 0, 1, 5, 10, 15, 30, 45, and 60 min, and the number of intact oocytes was determined based on the number of oocytes which did not rupture in water after each time point.
Parasite culture, vector construction, and transfection.
P. falciparum Dd2attB strain parasites and transfection plasmids (pLN and pINT) were kindly provided by Sean Prigge at Johns Hopkins Bloomberg School of Public Health. Parasites were cultured in RPMI 1640 medium (Cellgro) supplemented with 10% human serum at 2% hematocrit under 5% O2, 5% CO2, and 90% N2 atmosphere at 37°C. PfHly III was PCR amplified from P. falciparum 3D7 cDNA using a 5′ primer (5′-CCTAGGATGGAATTTTATAAGAACTTTTTTTCAC-3′) and a 3′ primer (5′CGTACGTTTGTCATCGTCATCTTTGTAATCTGTTCTTTTAATAACACTACAATTTAG-3′). The PCR product was then ligated into the AvrII and BsiWI sites of the pLN plasmid (20). The DNA insert was confirmed by DNA sequencing. The pLN had a mitochondrial ribosomal protein L2 (mRPL2) promoter (21). Parasites were transfected as described previously (22). Briefly, uninfected red blood cells were loaded into 0.2-cm cuvettes and electroporated (Bio-Rad Gene Pulser; 310 V, 950 μF, infinite resistance) with 75 μg of each plasmid, pLN-PfHly III and pINT. Dd2attB strain parasites were added to transfected erythrocytes. Transgenic parasites were selected with 2.5 μg/ml blasticidin (Invitrogen). Integration into the genomic attB site was verified by PCR as described previously (22).
Fluorescence microscopy, nucleic acid, and Western blot analyses of transgenic parasites.
The localization of recPfHly III-green fluorescent protein (GFP) fusion protein within the transgenic parasite was determined using live fluorescence microscopy (23). Mature parasites (trophozoites and schizonts) were purified using a 62% Percoll gradient and then washed 3 times with RPMI medium. Transgenic parasites were streaked on a slide, and images were captured on a Nikon Eclipse 90i fluorescence microscope and deconvoluted with Volocity software (Improvision). Genomic DNA was isolated from transgenic parasites using DNAzol reagent (Invitrogen). To verify integration of our construct in Dd2attB, the following PCR primers were used to detect integration both before and after the fusion protein (22): 5′1, forward primer located in Cg6 5′ untranslated region (UTR) (5′-GCGCAATTAACCCTCACTAAAGGG-3′); 5′2, reverse primer located in backbone of pLN vector about 200 bp downstream of attP sites (5′-GCACAGATGCGTAAGGAGAAAATACC-3′); 3′1, forward primer located in pLN vector in hrp2 3′ UTR (5′-GATAGCGATTTTTTTTACTGTCTG-3′); and 3′2, reverse primer located in Cg6 3′ UTR between attB/attR sites and dihydrofolate reductase (DHFR) gene (5′-TGTATAAAAGATGAACATGGTGAATTC-3′).
PCR primers p1 and p2 were used to detect the presence of GFP DNA in transgenic parasites, and PCR primers p2 and p3 were used to detect the presence of PfHly-GFP fusion DNA: p1 primer, 5′-CGTACGAGTAAAGGAGAAGAACTTTTCACTGG-3′; p2 primer, 5′-CTTAAGTTATTTGTATAGTTCATCCATGCC-3′; p3 primer, 5′-GTCTAGGAAGTGCTGTATGCACATTTGTCC-3′.
Mature parasites were pelleted, washed 3 times in PBS, and resuspended in SDS-PAGE sample loading buffer. A Western blot assay was performed as described above using 1:1,000 anti-GFP antibody (Invitrogen).
Genomic and cDNA amplification.
Total RNA was isolated from P. falciparum 3D7 cells, treated with RNase-free DNase I (Roche) in 100 mM sodium acetate (pH 5.0), repurified, and mixed with Superscript II reverse transcriptase (RT) and poly(T) to synthesize cDNA. Full-length primers for P. falciparum PfHly III were as follows: forward, 5′-CTGCAGAATTCCATGGAATTTTATAAGAACTTTTTTTCAC-3′, and reverse, 5′-CTGCAGAATTCCATGGTGTTCTTTTAATAACACTACAATTTAGG-3′. Analysis was performed on a 1% agarose gel with genomic DNA at 200 ng, cDNA, and no reverse transcript RNA for PCR amplicons.
GST and MBP fusion protein expression and purification, anti-PfHly III antibody production, Western blot assays, and GSTF80AA competition.
The first 80 amino acids of PfHly III were expressed as a glutathione S-transferase (GST) fusion protein (GSTF80AA) by cloning a 240-bp insert encoding the codon-optimized N-terminal 80 amino acids into the pGEXT vector. Primers for expression from optimized genomic template were as follows: forward, 5′-GAATTCAATGGAATTTTACAAAAACTTC-3′, and reverse, 5′-GAATTCCTAAGCTTGCCGCGAAACAGGGTTTTG-3′. Expression of the GST fusion protein was conducted in BL21*RIL cells in LB broth plus ampicillin and chloramphenicol induced with 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) at an optical density at 600 nm (OD600) of 0.6 and incubated for 10 h at 20°C. The GSTF80AA fusion protein was purified after high-pressure cell homogenization (EmulsiFlex C5 cell disruptor; Avestin; 100 MPa), centrifugation, and incubation of supernatant with glutathione-Sepharose 4B resin (GE Healthcare), followed by elution with 10 mM glutathione. Purified GSTF80AA was used to immunize a rabbit at Cocalico Biologicals, and preimmune serum, test bleeds, and the final bleed were received and tested by Western blotting. Separately, a maltose binding protein (MBP) construct was also made using the same PCR insert cloned into the MBP-tev pRSF vector, and the MBPF80AA fusion protein was expressed in LB plus kanamycin and 0.2% glucose, induced as described above for the GST fusion protein, and purified using amylose resin (NEB catalog no. E8021L), eluted with 10 mM maltose. The MBPF80AA protein was used for testing of anti-PfHly III antibodies as well as for affinity purification involving coupling the MBPF80AA fusion protein to an N-hydroxy-succinimide (NHS)-activated HiTrap column (GE Healthcare), running the antiserum over the column for 1 h, washing with binding buffer (0.05 M NaH2PO4, 0.15 NaCl, 0.01 M EDTA), and eluting with 0.1 M glycine, 0.15 M NaCl, pH 2.6. Antibody responses were determined by Western blot analysis as described above with antiserum concentrations at 1:10,000 and affinity-purified test bleed 2 (APTB2) at 1:1,000. The GSTF80AA fusion protein was used in competition for native or recombinant PfHly III antigen by preincubating APTB2 anti-PfHly III antiserum (20 μl, 0.34 mg/ml with majority of protein present as bovine serum albumin [BSA] from purification) with 100 μl of GSTF80AA fusion protein (0.35 mg/ml), at an approximately 1:50 ratio in 1 ml of blocking buffer (5% milk in PBS-Tween 0.1%) for 1 h, followed by dilution to 20 ml for Western blot analysis.
Statistical methods.
Each experimental group consisted of three replicates. The data obtained from the study were expressed as mean ± standard deviation. Student's t test was used to determine statistical significance. A P value of <0. 05 was considered statistically significant.
RESULTS
Bacterial expression of recombinant PfHly III.
In this study, we were able to express and enrich recPfHly III. Previous studies experienced difficulty purifying the Hly III protein. This was mainly due to the remarkable toxicity of the protein (12). We also had similar toxicity issues while trying to express recPfHly III using highly efficient expression plasmids such as pGS-21a (GenScript). Previous Hly III-related studies used the weak-expression pUC vector to avoid toxicity problems (12–14). To enrich recPfHly III, we developed an expression construct that had 6×His tags at both ends of the Hly III insert, ligated into a pUC18 plasmid (Fig. 1A). We were able to demonstrate the presence of soluble expressed recPfHly III by SDS-PAGE and Western blot analysis (Fig. 1B and C). An immunofluorescence assay (IFA) was performed using anti-His antibody conjugated to Alexa Fluor 488 on recPfHly III-treated erythrocytes. PEG 4600 was added to the assay to prevent hemolysis but not binding of Hly III to the erythrocyte membrane. Immunofluorescence microscopy showed recPfHly III coating the erythrocyte membrane (Fig. 1C and D).
FIG 1.
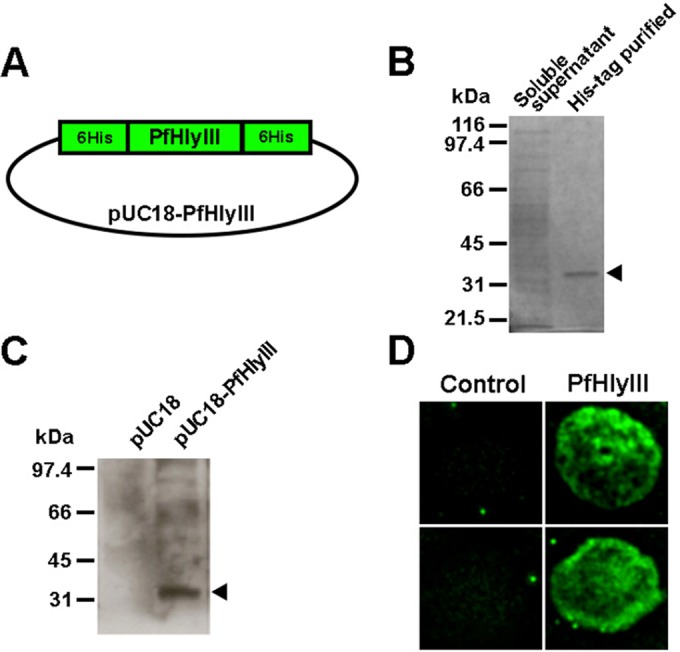
Characterization of PfHly III. (A) Schematic of pUC-PfHly III expression vector showing recPfHly III flanked by His6 tags inserted in the pUC18 plasmid. (B) SDS-PAGE gel of purified His-tagged recPfHly III compared to soluble supernatant. (C) Western blot assay with anti-His antibody confirming expression of recPfHly III. (D) IFA with anti-His antibody conjugated with Alexa 488 showing Hly III coating the erythrocyte membrane.
recPfHly III demonstrated hemolytic activity.
Washed human erythrocytes were incubated at various temperatures with recPfHly III (2 HU), and hemolysis was determined by absorbance at 550 nm, an isosbestic point of hemoglobin. The amount of 1 hemolytic unit was roughly equivalent to 0.25 μg ml−1, which is approximately 7 nM. Lysis of erythrocytes by recPfHly III was optimal at 37°C (Fig. 2A). No significant hemolysis was seen at 4°C and with the negative controls. Binding of Hly III to erythrocytes has been previously reported to be temperature dependent (13). Hemolytic activity was completely lost when recPfHly III was heated at 65°C for 15 min. recPfHly III-induced hemolysis was the same at pH 5.0.
FIG 2.
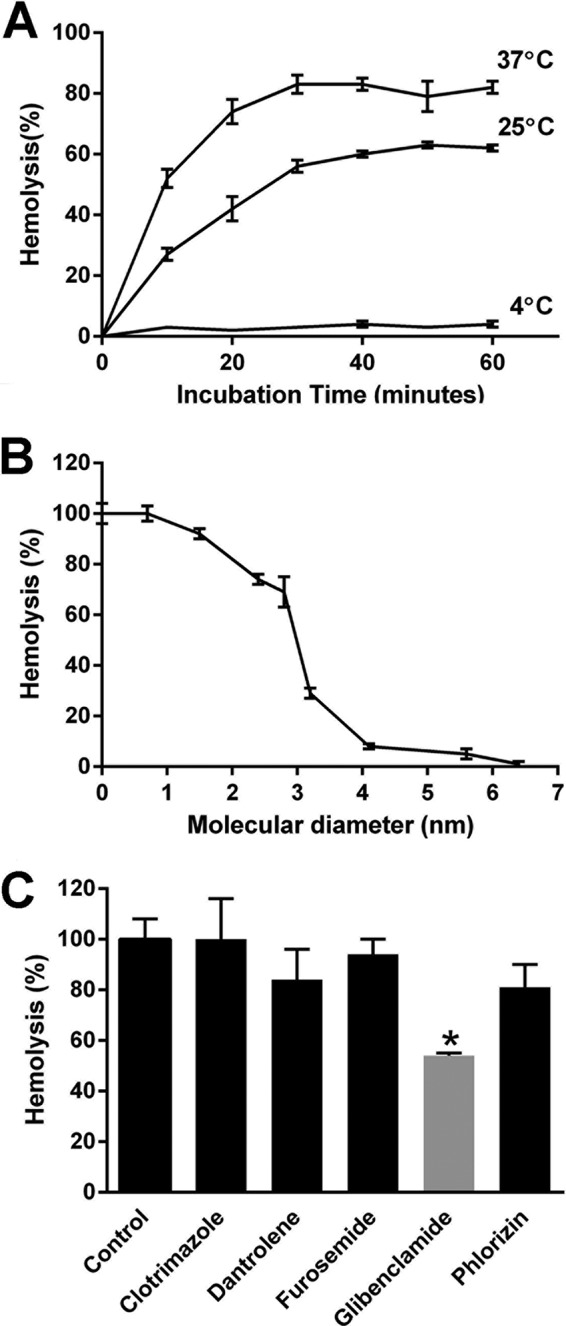
Hemolytic activity of recPfHly III. (A) Time and temperature dependence of hemolysis determined by absorbance at 550 nm with 2 hemolytic units of recPfHly III and washed human erythrocytes. (B) Increasing size of the osmotic protectant PEG at 30 mM and 2 hemolytic units of recPfHly III at 37°C for 60 min. The zero control used hemolysin without protectants. Increasing pore size was determined with glucose (0.7 nm), PEG 600 (1.5 nm), PEG 1500 (2.4 nm), PEG 2000 (2.8 nm), PEG 3350 (3.2 nm), PEG 4600 (4.1 nm), PEG 6000 (5.6 nm), and PEG 8000 (6.4 nm). (C) Partial drug inhibition by glibenclamide. Drugs at 100 μM and recPfHly III (0. 5 HU) were incubated with 1% human erythrocytes at 37°C for 60 min. *, P value < 0.05 (Student's t test). Each value is the mean ± standard deviation of three replicates from one experiment and is representative of two independent experiments.
Osmotic protectants inhibit recPfHly III-induced hemolysis.
Inhibition of hemolysis by osmotic protectants is considered a distinctive feature of pore-forming hemolysins (13). In addition, the protectants can allow estimation of the size of pores present in erythrocyte membranes created by pore-forming hemolysin. Osmotic protectants with hydrodynamic radii equal to or larger than those of the pores can block the pores effectively and thus prevent the lysis of erythrocytes (24). To assess the effect of osmotic protectants on recPfHly III-induced hemolysis, we estimated hemolytic activity in the presence of glucose and PEG solutes (final concentration, 30 mM). There was a significant inhibition of hemolysis (>50% protection from hemolysis) by PEG 3350, with a hydrodynamic diameter of 3.2 nm, and almost complete inhibition by PEG 4600 and above (Fig. 2B). The inhibitory effects were similar to those in a previous study that showed the pore size for Hly III to have a hydrodynamic diameter of 3. 0 to 3.5 nm (13). Washing erythrocytes and resuspending them in PBS induced hemolysis, indicating that PEG solutes had no effect on binding.
Glibenclamide partially inhibits recPfHly III-induced hemolysis.
We also wanted to determine if recPfHly III-induced hemolysis could be inhibited by pharmacological agents. Several drugs were selected from the Johns Hopkins Clinical Compound Library (18). They were selected based on having antimalarial and channel-inhibitory effects. The selected agents were clotrimazole, dantrolene, furosemide, glibenclamide, and phlorizin. Drugs (100 μm) and Hly III (0.5 HU) were incubated with 1% human erythrocytes at 37°C for 60 min. Hemolysis was quantified by measuring absorbance at 550 nm. Glibenclamide was the only drug to show a statistically significant inhibition of recPfHly III-induced hemolysis (Fig. 2C). The quinoline drugs did not demonstrate any significant inhibition of hemolysis (data not shown).
Rupture of Xenopus oocytes expressing PfHly III.
We wanted to investigate recPfHly III lytic activity in a nonbacterial system. We constructed an expression plasmid with recPfHly III flanked by Myc and Flag tags, pXβG-ev1-myc-PfHly III-flag, and produced RNA with in vitro transcription (Fig. 3A and B), along with human aquaporin 1 (hAQP1) RNA for use as a positive control. Myc-PfHly III-Flag and hAQP1 RNA were injected at 25- to 44-ng concentrations into X. laevis oocytes along with diethyl pyrocarbonate (DEPC)-treated water-injected controls, and protein expression was monitored from 72 to 120 h postinjection using anti-Myc, anti-Flag, and anti-hAQP1 antibodies for Western blot assays. hAQP1 was expressed in only hAQP1 RNA-injected oocytes (20 kDa), whereas a 25-kDa product was consistently identified in Myc-PfHly III-Flag RNA-injected oocytes with both anti-Myc and anti-Flag antibodies but not in the hAQP1- or water-injected controls (Fig. 4A). A 30-kDa product was also identified with the anti-Myc antibody. While the predicted size of Myc-PfHly III-Flag is 36 kDa, the data suggest that the protein may be processed or run at a different size on the gel. In order to determine whether expression of Myc-PfHly III-Flag would result in sensitivity to hypotonic lysis similar to that of hAQP1, we conducted swelling assays 120 h postinjection and monitored swelling and rupture of H2O-, hAQP1-, and Myc-PfHly III-Flag-injected oocytes for 1 h. Myc-PfHly III-Flag-expressing oocytes showed an intermediate phenotype, rupturing at a slightly lower rate than hAQP1-expressing oocytes, compared to little to no rupturing of water-injected controls (Fig. 4B and C). A time course analysis is depicted in Fig. S2 in the supplemental material. Finally, addition of 30 mM osmotic protectant polyethylene glycol (PEG) at increasing molecular weights (1,500, 3,350, and 6,000) resulted in decreased rupture of Myc-PfHly III-Flag expressing oocytes, with PEG 6000 protecting almost 100% of oocytes from rupturing up to 1 h (Fig. 5).
FIG 3.
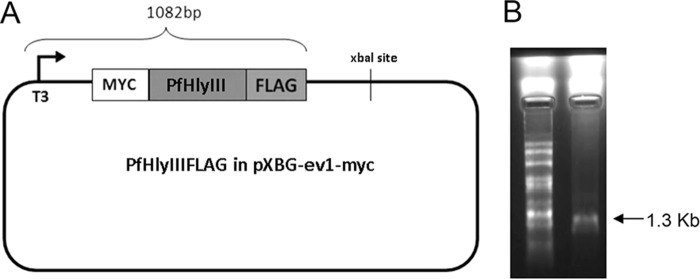
(A) pXβG-ev1-myc-PfHly III-flag construct. (B) In vitro transcription product, RNA (1,315 bp), on a 1% agarose-6.6% formaldehyde denaturing gel. Lane 1, 0.5- to 10-kb RNA ladder; lane 2, RNA sample.
FIG 4.
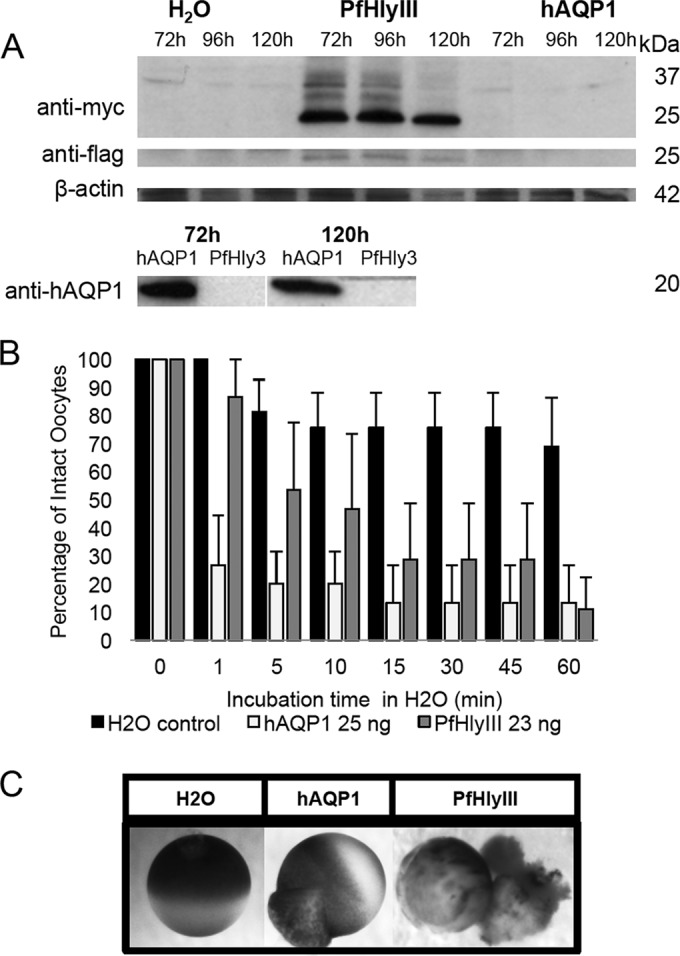
Swelling and rupture of Xenopus oocytes expressing recPfHly III. (A) X. laevis oocytes injected with pXβG-ev1-myc-recPfHly III-flag cRNA express PfHly III (25 kDa), recognized by both anti-Myc and anti-Flag antibodies, whereas pXβG-ev1-hAQP1 cRNA-injected oocytes express hAQP1 (20 kDa). (B) hAQP1- and recPfHly III-expressing oocytes swell and rupture in a time-dependent manner in hypotonic buffer (water) at a higher rate than do the water-injected controls. Three independent experiments were conducted with three to six oocytes per group, and the mean percentage of intact oocytes was calculated and reported with the standard error of the mean. (C) Still photos of water control (45 min), hAQP1 (1 min), and PfHly III (5 min).
FIG 5.
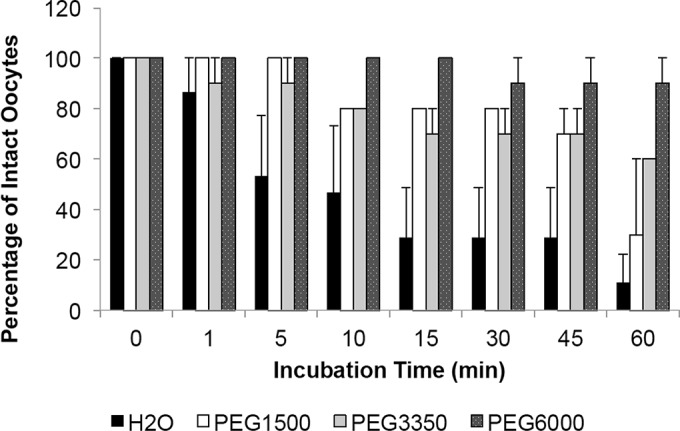
Addition of increasing-molecular-weight polyethylene glycol (PEG) decreases rupture in recPfHly III-expressing oocytes in a size-dependent manner. Mean percentage reported with standard error of the mean (3 independent experiments for each treatment).
PfHly III-GFP localizes to the parasite digestive vacuole.
We investigated the localization of PfHly III by integrating the PfHly III-GFP fusion protein into the Dd2attb genome. The integration was done by transfecting the parasite with pLN-PfHly III and pINT. PCR analysis on transfected parasites using primers (GFP for, Hly III for, and GFP rev) demonstrated the presence of pLN-PfHly III-GFP DNA (Fig. 6A to D). We confirmed that a PfHly III-GFP fusion protein of the expected size was expressed by Western blotting with anti-GFP antibody (see Fig. 9C and D). Microscopy of Giemsa-stained transfected P. falciparum with Hly III-GFP fusion protein demonstrated swollen digestive vacuole phenotypes (Fig. 7B). Live fluorescence microscopy shows localization to the digestive vacuole evident by colocalization of recPfHly III-GFP with hemozoin (Fig. 7C).
FIG 6.
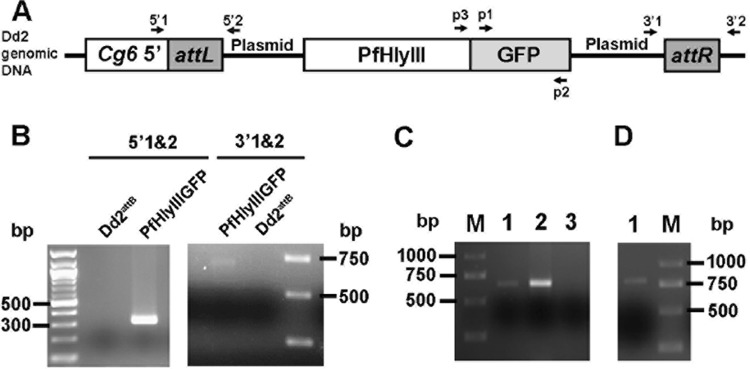
Transfection of PfHly III-GFP into P. falciparum. (A) Schematic of PfHly III-GFP integrated into Dd2 genome. 5′1 (Cg6 5′), 5′2 (pLN-PfHly III rev), 3′1 (pLN-PfHly III-GFP for), and 3′2 (attR rev) represent primer annealing sites. (B) Predicted genetic locus and PCR verification of pLN-PfHly III-GFP integration into the Dd2attB site. The 5′1 and 5′2 primers amplify a 350-bp product indicative of the attL site, and the 3′1 and 3′2 primers amplify a 680-bp product indicative of the attR site, overall indicating integration into the targeted genetic locus. (C) Lane 1, PCR 720-bp amplicon with p1 and p2 primers demonstrating the presence of GFP DNA in PfHly III-GFP-transfected parasites; lane 2, positive control; lane 3, negative control; lane M, marker. (D) PCR 780-bp amplicon with p3 and p2 primers showing presence of PfHly III-GFP DNA.
FIG 9.

Generation of rabbit polyclonal antiserum against the N terminus (first 80 amino acids) of PfHly III, affinity purification, and detection of PfHly III in asexual blood stages. (A) TMpred-generated prediction of transmembrane regions I to VII in PfHly III, with the first N-terminal 80 amino acids indicated with no transmembrane domains. (B) Test bleed antiserum recognizes recombinant MBP-tagged PfHly III N-terminal 80-amino-acid peptide (MBPF80AA, 53 kDa) compared to the preimmune serum. (C) Affinity-purified test bleed 2 (APTB2) recognizes a unique band of less than 30 kDa in three strains of P. falciparum: 3D7, Dd2attB, and Dd2attB transfected with PfHly III-GFP; anti-GFP and APTB2 both recognize a unique higher-molecular-weight product in the PfHly III-GFP-transfected strain. (D) A 25-kDa product is located in the pellet (P) and soluble (S) fractions of both 3D7 and PfHly III-GFP-transfected strains of P. falciparum for native PfHly III. The 25- and 50-kDa products in the PfHly III-GFP-expressing parasites are also found in the pellet and supernatant fractions. Approximately 10:1 amounts of protein were loaded for the pellet/supernatant ratio seen in the Western blot. (E) Comparing synchronized rings (R) and trophozoite (T) stages normalized by protein content, the native 25-kDa product was primarily observed in trophozoites, with signal from the native protein removed by competition with recombinant GSTF80AA antigen.
FIG 7.
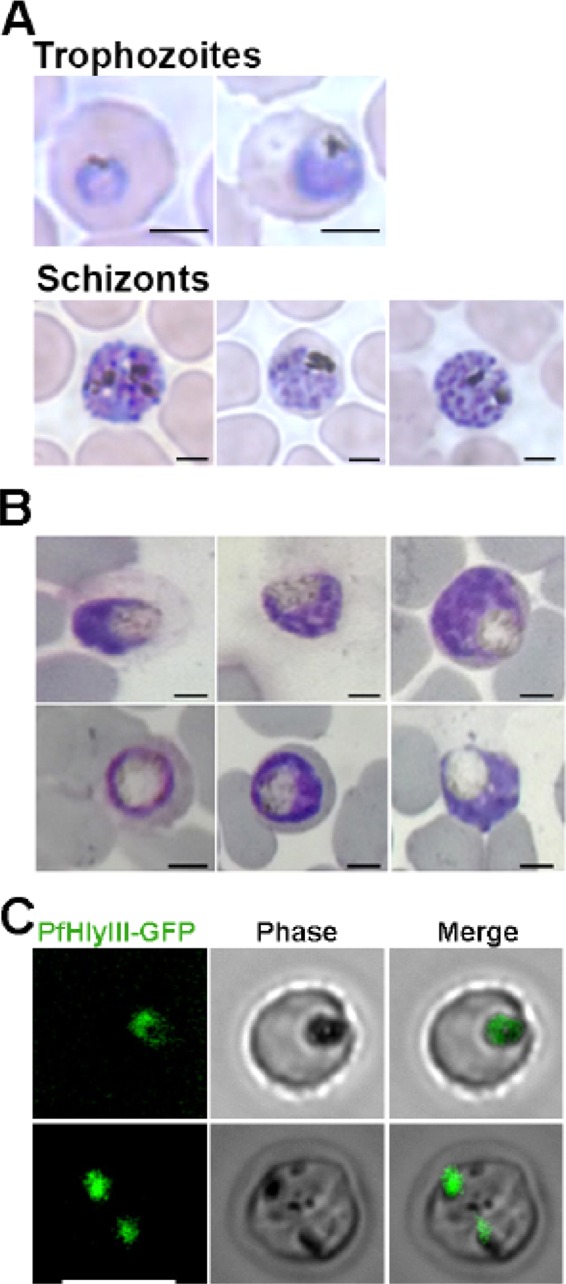
PfHly III-GFP localizes to the digestive vacuole of P. falciparum. (A) Microscopy of Giemsa-stained nontransfected Dd2attB trophozoites and schizonts. (B) Giemsa-stained transfected P. falciparum with PfHly III-GFP fusion protein. Parasites demonstrate swollen digestive vacuoles. Bar, 2 μm. (C) Live fluorescence microscopy shows colocalization of PfHly III-GFP with hemozoin. Bar, 7 μm.
PfHly III transcripts are in 3D7 erythrocytic stages, and most of the protein exists in a pelletable membrane fraction in trophozoites.
We amplified cDNA and genomic DNA, verifying that the 3′ intron was spliced out in intraerythrocytic transcripts from 3D7 parasites (Fig. 8). Rabbits were immunized with a GST–80-amino-acid soluble amino-terminal tail for PfHly III (GSTF80AA) to obtain anti-PfHly III antiserum (Fig. 9A). A maltose-binding protein fusion to the same 80-amino-acid N-terminal tail (MBPF80AA) was used to affinity purify specific antibody (Fig. 9B). In Dd2attB-transfected PfHly III-GFP parasites, the native protein was more abundant than the larger-molecular-mass PfHly III-GFP fusion of ∼50 kDa (Fig. 9C). The competition of antibody signal by GSTF80AA drastically minimized the ∼25-kDa native protein as well as PfHly III-GFP fusion at about 50 kDa. We observed some persistent higher-molecular-weight cross-reacting bands that were not competed by recombinant protein.
FIG 8.
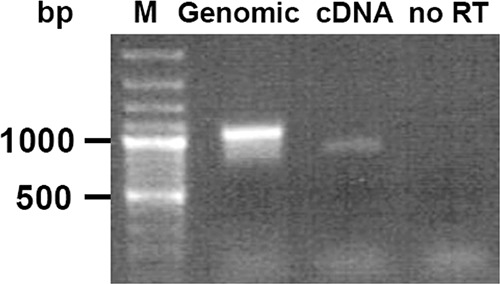
Presence of cDNA in asexual parasites for culture. Total RNA was reverse transcribed and amplified with primers for full-length PfHly III with genomic DNA in lane 2, cDNA in lane 3 with lower-base-pair amplification from intron excision, and no-reverse-transcriptase (no RT) control in lane 4. The cDNA has a predicted molecular weight with intron spliced out of 849 with a genomic DNA molecular weight of 1,063.
We investigated the relative proportions of soluble and membrane native PfHly III in asexual whole-cell lysate, which indicated that most of the native protein was in the membrane fraction rather than soluble supernatants (Fig. 9D). Finally, in a population of synchronized rings and trophozoites which were equalized to protein content, which increases the number of rings compared to the number of trophozoites, the native protein was largely absent in rings and competition with GSTF80AA ablated signal from native protein (Fig. 9E).
DISCUSSION
RecPfHly III-induced hemolysis was optimal at 37°C, with no activity at 4°C (Fig. 2A). It was previously reported that B. cereus Hly III acts via a multihit mechanism. The first step involves the binding of Hly III monomers to the erythrocyte membrane. This is followed by the assembly of monomers into an oligomeric pore-forming hemolysin. Baida and Kuzmin showed that monomeric binding and assembly were both temperature dependent and that these steps were inhibited at 4°C (13). Since recPfHly III and B. cereus Hly III share significant amino acid sequence similarity, it would be a reasonable deduction that recPfHly III acts by a temperature-dependent multihit mechanism.
Hemolysins disrupt erythrocyte membranes via channel formation (pore forming), detergent action, or lipase activity (25). recPfHly III was found to mediate hemolysis by pore formation, with a pore size of approximately 3.2 nm, in keeping with the previous B. cereus Hly III reported pore size of 3.0 to 3. 5 nm (13). Expression of recPfHly III in X. laevis oocytes resulted in sensitivity to hypotonic lysis in a time-dependent manner similar to that seen with hAQP1-expressing oocytes. Lysis of Myc-PfHly III-Flag-expressing oocytes was also reduced in the presence of an osmotic protectant of sufficient diameter, supporting a pore-forming mechanism for recPfHly III (Fig. 5). Because these proteins can disrupt the membrane bilayer, they have the ability to lyse erythrocytes and are labeled hemolysins (26). We were able to show lysis whether the recPfHly III was expressed inside an oocyte or added exogenously after soluble bacterial expression to the outside erythrocytes, thus demonstrating formation of a bidirectional pore.
Glibenclamide is a sulfonylurea that is used in the treatment of type II diabetes. Glibenclamide was shown to inhibit the transport of amino acid, sugar alcohol, an inorganic anion, and a cation across the new permeation pathways in P. falciparum-infected erythrocytes (27). In addition, glibenclamide has been shown to inhibit a wide variety of channels such as the cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channel, ATP-sensitive K+ channels, the Na+-K+ pump, and Ca2+ channels (28). This broad spectrum of channel-blocking ability might explain why glibenclamide was able to partially inhibit recPfHly III-induced hemolysis. We do not foresee the use of glibenclamide as an antimalarial, as at present it is a crude partially active inhibitor. Many proteins labeled hemolysins have other functions, such as the Bordetella adenylate cyclase toxin. In addition to pore-forming cytotoxicity, Bordetella adenylate cyclase toxin induces cell death by enzymatically increasing cyclic AMP (cAMP) production, which in turn stimulates calcium influx (29, 30). It is very possible that PfHly III has more than one function and that hemolysis is an adverse secondary effect.
Severe anemia is one of the main causes of mortality and morbidity seen with malaria infections (2, 31). Severe anemia is due in large part to the destruction of uninfected red blood cells (6, 32). The mechanism by which uninfected erythrocytes are destroyed is not fully understood. Merozoite surface protein 2 can bind uninfected erythrocytes in an aborted invasion. Knockout of this protein decreased anemia in an animal model (33). PfHly III may play a role in the destruction of uninfected erythrocytes, and the use of recPfHly III antagonists by antibodies or possibly drugs could alleviate this adverse effect, resulting in a decrease in malaria morbidity and mortality. PfHly III in the digestive vacuoles may be released into the bloodstream after merozoite rupture of the infected erythrocyte. Digestive vacuoles would then rupture in the plasma, resulting in PfHly III lysing uninfected erythrocytes. In addition to directly lysing erythrocytes, the ability of PfHly III to bind to uninfected human erythrocytes may provide another mechanism for destruction of uninfected erythrocytes. As with ring surface protein 2 (RSP2) (34), opsonization of PfHly III-bearing uninfected erythrocytes may provide a mechanism of phagocytic clearance.
Our results show that transfected PfHly III-GFP localizes to the parasite digestive vacuole. Due to toxicity and the lack of viable parasites, we switched from using the calmodulin promoter to the weaker mRPL2 promoter for our transfection of recPfHly III-GFP (at least a 100-fold difference [S. T. Prigge, unpublished data]). Parasites expressing recPfHly III-GFP fusion protein generated swollen vacuole phenotypes even though Western blot analysis suggested that less recombinant than native PfHly III was present in parasite lysates. The formation of swollen vacuoles in recPfHly III-transfected parasites may be a result of the following: (i) recPfHly III may be an influx channel and overexpression results in vacuolar swelling; (ii) in addition to channel activity, recPfHly III may have a regulatory effect on proteases and overexpressing recPfHly III may have inhibited these proteases. Prior reports have shown that protease inhibition can cause swollen vacuole phenotypes (35, 36). It is quite possible that overexpression of recPfHly III results in the accumulation of metabolic products. A previous study showed that cells transfected with the lysosomal membrane protein LIMPII resulted in swollen vacuoles. These swollen vacuoles had an accumulation of cholesterol (37) and more soluble PfHly III-GFP than soluble native PfHly III. We do not think that recPfHly III is being trafficked to the digestive vacuole for degradation, as we found by Western blotting only the recPfHly III-GFP fusion protein of the appropriate size with an absence of protein fragments.
We localized native protein to membrane fractions predominately in trophozoites with very little protein present in ring-stage parasites despite transcriptional evidence in ring stage in PlasmoDB. Immunolocalization will require a monoclonal antibody or a different purification strategy of the rabbit polyclonal antibody, as we still have nonspecific interaction with higher-molecular-weight proteins that were not competed in the antibody-antigen combination experiments.
The first 80 amino acids at the N terminus of recPfHly III are conserved across Plasmodium species but do not share homology with the Hly III superfamily. Accordingly, we suspected that the N-terminal region of recPfHly III may contain sequence information for trafficking. The falcipain2 lumenal domain digestive vacuole targeting motif shares significant sequence similarities with the amino acids at the N terminus of recPfHly III. They both consist of multiple lysine residues which are thought to be essential for digestive vacuole trafficking. Point mutation of the lysine residues in the falcipain2 lumenal domain resulted in mislocalization (38). The hydrophobic trafficking motif YXXφ (where φ can be F, I, L, M, or V) is the best-characterized sorting signal. YXXφ binds to adaptor molecules that traffic protein to lysosomes (38, 39). There is a YXXφ motif at amino acid positions 4 to 7 in recPfHly III. The YXXφ motif is also present in P. falciparum lysosomal membrane channels multidrug resistance protein (MDR) and chloroquine resistance transporter (CRT). We have identified a highly conserved undecameric DIIG motif in Hly III (see Fig. S3 in the supplemental material). This motif is conserved across multiple pathogens. The highly conserved nature of this motif implies that it might be important for function.
We have demonstrated that recPfHly III lyses erythrocytes via a channel formation mechanism. recPfHly III-induced hemolysis was optimal at 37°C and was inhibited by glibenclamide. We have also localized recPfHly III to the unique and essential digestive vacuole. Because of its hemolytic property, recPfHly III may play a role in the lysis of uninfected erythrocytes, resulting in severe life-threatening anemia in individuals with malaria, although it could still also function as a pore component of a protein complex in Plasmodium. This study also generates the hypothesis that neutralization of PfHly III may protect against severe malaria anemia. We plan to examine whether anti-PfHly III antibodies attenuate anemia in a mouse model.
Supplementary Material
ACKNOWLEDGMENTS
We thank the members of the Sean Prigge lab for their much-appreciated advice and provision of materials.
N.G.S. received generous support from the Emergent Biosolutions Fellowship. D.J.S. acknowledges support from the Johns Hopkins Malaria Research Institute and The Bloomberg Family Foundation.
Footnotes
Published ahead of print 22 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/EC.00088-14.
REFERENCES
- 1.Murray CJ, Rosenfeld LC, Lim SS, Andrews KG, Foreman KJ, Haring D, Fullman N, Naghavi M, Lozano R, Lopez AD. 2012. Global malaria mortality between 1980 and 2010: a systematic analysis. Lancet 379:413–431. 10.1016/S0140-6736(12)60034-8 [DOI] [PubMed] [Google Scholar]
- 2.Obonyo CO, Vulule J, Akhwale WS, Grobbee DE. 2007. In-hospital morbidity and mortality due to severe malarial anemia in western Kenya. Am. J. Trop. Med. Hyg. 77:23–28 [PubMed] [Google Scholar]
- 3.Roberts DJ, Casals-Pascual C, Weatherall DJ. 2005. The clinical and pathophysiological features of malarial anaemia. Curr. Top. Microbiol. Immunol. 295:137–168. 10.1007/3-540-29088-5_6 [DOI] [PubMed] [Google Scholar]
- 4.Weatherall DJ, Miller LH, Baruch DI, Marsh K, Doumbo OK, Casals-Pascual C, Roberts DJ. 2002. Malaria and the red cell. Hematology Am. Soc. Hematol. Educ. Program 2002:35–57. 10.1182/asheducation-2002.1.35 [DOI] [PubMed] [Google Scholar]
- 5.Kai OK, Roberts DJ. 2008. The pathophysiology of malarial anaemia: where have all the red cells gone? BMC Med. 6:24. 10.1186/1741-7015-6-24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jakeman GN, Saul A, Hogarth WL, Collins WE. 1999. Anaemia of acute malaria infections in non-immune patients primarily results from destruction of uninfected erythrocytes. Parasitology 119:127–133. 10.1017/S0031182099004564 [DOI] [PubMed] [Google Scholar]
- 7.Casals-Pascual C, Roberts DJ. 2006. Severe malarial anaemia. Curr. Mol. Med. 6:155–168. 10.2174/156652406776055159 [DOI] [PubMed] [Google Scholar]
- 8.Kissinger JC, Brunk BP, Crabtree J, Fraunholz MJ, Gajria B, Milgram AJ, Pearson DS, Schug J, Bahl A, Diskin SJ, Ginsburg H, Grant GR, Gupta D, Labo P, Li L, Mailman MD, McWeeney SK, Whetzel P, Stoeckert CJ, Roos DS. 2002. The Plasmodium genome database. Nature 419:490–492. 10.1038/419490a [DOI] [PubMed] [Google Scholar]
- 9.Vignali M, Armour CD, Chen J, Morrison R, Castle JC, Biery MC, Bouzek H, Moon W, Babak T, Fried M, Raymond CK, Duffy PE. 2011. NSR-seq transcriptional profiling enables identification of a gene signature of Plasmodium falciparum parasites infecting children. J. Clin. Invest. 121:1119–1129. 10.1172/JCI43457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lopez-Barragan MJ, Lemieux J, Quinones M, Williamson KC, Molina-Cruz A, Cui K, Barillas-Mury C, Zhao K, Su XZ. 2011. Directional gene expression and antisense transcripts in sexual and asexual stages of Plasmodium falciparum. BMC Genomics 12:587. 10.1186/1471-2164-12-587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Silvestrini F, Lasonder E, Olivieri A, Camarda G, van Schaijk B, Sanchez M, Younis Younis S, Sauerwein R, Alano P. 2010. Protein export marks the early phase of gametocytogenesis of the human malaria parasite Plasmodium falciparum. Mol. Cell. Proteomics 9:1437–1448. 10.1074/mcp.M900479-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baida GE, Kuzmin NP. 1995. Cloning and primary structure of a new hemolysin gene from Bacillus cereus. Biochim. Biophys. Acta 1264:151–154. 10.1016/0167-4781(95)00150-F [DOI] [PubMed] [Google Scholar]
- 13.Baida GE, Kuzmin NP. 1996. Mechanism of action of hemolysin III from Bacillus cereus. Biochim. Biophys. Acta 1284:122–124. 10.1016/S0005-2736(96)00168-X [DOI] [PubMed] [Google Scholar]
- 14.Chen YC, Chang MC, Chuang YC, Jeang CL. 2004. Characterization and virulence of hemolysin III from Vibrio vulnificus. Curr. Microbiol. 49:175–179. 10.1007/s00284-004-4288-5 [DOI] [PubMed] [Google Scholar]
- 15.de Koning-Ward TF, Gilson PR, Boddey JA, Rug M, Smith BJ, Papenfuss AT, Sanders PR, Lundie RJ, Maier AG, Cowman AF, Crabb BS. 2009. A newly discovered protein export machine in malaria parasites. Nature 459:945–949. 10.1038/nature08104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coppens I, Sullivan DJ, Prigge ST. 2010. An update on the rapid advances in malaria parasite cell biology. Trends Parasitol. 26:305–310. 10.1016/j.pt.2010.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han JH, Lee JH, Choi YH, Park JH, Choi TJ, Kong IS. 2002. Purification, characterization and molecular cloning of Vibrio fluvialis hemolysin. Biochim. Biophys. Acta 1599:106–114. 10.1016/S1570-9639(02)00407-7 [DOI] [PubMed] [Google Scholar]
- 18.Chong CR, Chen X, Shi L, Liu JO, Sullivan DJ., Jr 2006. A clinical drug library screen identifies astemizole as an antimalarial agent. Nat. Chem. Biol. 2:415–416. 10.1038/nchembio806 [DOI] [PubMed] [Google Scholar]
- 19.Preston GM, Carroll TP, Guggino WB, Agre P. 1992. Appearance of water channels in Xenopus oocytes expressing red cell CHIP28 protein. Science 256:385–387. 10.1126/science.256.5055.385 [DOI] [PubMed] [Google Scholar]
- 20.Nkrumah LJ, Muhle RA, Moura PA, Ghosh P, Hatfull GF, Jacobs WR, Jr, Fidock DA. 2006. Efficient site-specific integration in Plasmodium falciparum chromosomes mediated by mycobacteriophage Bxb1 integrase. Nat. Methods 3:615–621. 10.1038/nmeth904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Balabaskaran Nina P, Morrisey JM, Ganesan SM, Ke H, Pershing AM, Mather MW, Vaidya AB. 2011. ATP synthase complex of Plasmodium falciparum: dimeric assembly in mitochondrial membranes and resistance to genetic disruption. J. Biol. Chem. 286:41312–41322. 10.1074/jbc.M111.290973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Spalding MD, Allary M, Gallagher JR, Prigge ST. 2010. Validation of a modified method for Bxb1 mycobacteriophage integrase-mediated recombination in Plasmodium falciparum by localization of the H-protein of the glycine cleavage complex to the mitochondrion. Mol. Biochem. Parasitol. 172:156–160. 10.1016/j.molbiopara.2010.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tilley L, McFadden G, Cowman A, Klonis N. 2007. Illuminating Plasmodium falciparum-infected red blood cells. Trends Parasitol. 23:268–277. 10.1016/j.pt.2007.04.001 [DOI] [PubMed] [Google Scholar]
- 24.Bi S, Wang W, Zhao Y, Ru S, Liu Y. 2011. Studies on hemolysis of hemolysin produced by Synechocystis sp. J. Ocean Univ. China 10:362–368. 10.1007/s11802-011-1823-4 [DOI] [Google Scholar]
- 25.Alouf JE, Freer JH. (ed). 1999. The comprehensive sourcebook of bacterial protein toxins, 2nd ed. Academic Press, San Diego, CA [Google Scholar]
- 26.Hewlett E, Hughes M. 2009. Toxins, p 27–35 In Mandell GL, Bennett JE, Dolin R. (ed), Mandell, Douglas, and Bennett's principles and practice of infectious diseases, 7th ed. Churchill Livingstone Elsevier, Philadelphia, PA [Google Scholar]
- 27.Kirk K, Horner HA, Spillett DJ, Elford BC. 1993. Glibenclamide and meglitinide block the transport of low molecular weight solutes into malaria-infected erythrocytes. FEBS Lett. 323:123–128. 10.1016/0014-5793(93)81462-9 [DOI] [PubMed] [Google Scholar]
- 28.Lee SY, Lee CO. 2005. Inhibition of Na+-K+ pump and L-type Ca2+ channel by glibenclamide in guinea pig ventricular myocytes. J. Pharmacol. Exp. Ther. 312:61–68. 10.1124/jpet.104.074369 [DOI] [PubMed] [Google Scholar]
- 29.Hewlett EL, Donato GM, Gray MC. 2006. Macrophage cytotoxicity produced by adenylate cyclase toxin from Bordetella pertussis: more than just making cyclic AMP! Mol. Microbiol. 59:447–459. 10.1111/j.1365-2958.2005.04958.x [DOI] [PubMed] [Google Scholar]
- 30.Basler M, Masin J, Osicka R, Sebo P. 2006. Pore-forming and enzymatic activities of Bordetella pertussis adenylate cyclase toxin synergize in promoting lysis of monocytes. Infect. Immun. 74:2207–2214. 10.1128/IAI.74.4.2207-2214.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Murphy SC, Breman JG. 2001. Gaps in the childhood malaria burden in Africa: cerebral malaria, neurological sequelae, anemia, respiratory distress, hypoglycemia, and complications of pregnancy. Am. J. Trop. Med. Hyg. 64:57–67 [DOI] [PubMed] [Google Scholar]
- 32.Evans KJ, Hansen DS, van Rooijen N, Buckingham LA, Schofield L. 2006. Severe malarial anemia of low parasite burden in rodent models results from accelerated clearance of uninfected erythrocytes. Blood 107:1192–1199. 10.1182/blood-2005-08-3460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gomez ND, Safeukui I, Adelani AA, Tewari R, Reddy JK, Rao S, Holder A, Buffet P, Mohandas N, Haldar K. 2011. Deletion of a malaria invasion gene reduces death and anemia, in model hosts. PLoS One 6:e25477. 10.1371/journal.pone.0025477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Layez C, Nogueira P, Combes V, Costa FT, Juhan-Vague I, da Silva LH, Gysin J. 2005. Plasmodium falciparum rhoptry protein RSP2 triggers destruction of the erythroid lineage. Blood 106:3632–3638. 10.1182/blood-2005-04-1574 [DOI] [PubMed] [Google Scholar]
- 35.Moura PA, Dame JB, Fidock DA. 2009. Role of Plasmodium falciparum digestive vacuole plasmepsins in the specificity and antimalarial mode of action of cysteine and aspartic protease inhibitors. Antimicrob. Agents Chemother. 53:4968–4978. 10.1128/AAC.00882-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Armstrong CM, Goldberg DE. 2007. An FKBP destabilization domain modulates protein levels in Plasmodium falciparum. Nat. Methods 4:1007–1009. 10.1038/nmeth1132 [DOI] [PubMed] [Google Scholar]
- 37.Kuronita T, Eskelinen EL, Fujita H, Saftig P, Himeno M, Tanaka Y. 2002. A role for the lysosomal membrane protein LGP85 in the biogenesis and maintenance of endosomal and lysosomal morphology. J. Cell Sci. 115:4117–4131. 10.1242/jcs.00075 [DOI] [PubMed] [Google Scholar]
- 38.Subramanian S, Sijwali PS, Rosenthal PJ. 2007. Falcipain cysteine proteases require bipartite motifs for trafficking to the Plasmodium falciparum food vacuole. J. Biol. Chem. 282:24961–24969. 10.1074/jbc.M703316200 [DOI] [PubMed] [Google Scholar]
- 39.Robinson MS. 2004. Adaptable adaptors for coated vesicles. Trends Cell Biol. 14:167–174. 10.1016/j.tcb.2004.02.002 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.