

Abstract
The infectious agent of the disease anthrax is the spore of Bacillus anthracis. Bacterial spores are extremely resistant to environmental stresses, which greatly hinders spore decontamination efforts. The spore cortex, a thick layer of modified peptidoglycan, contributes to spore dormancy and resistance by maintaining the low water content of the spore core. The cortex is degraded by germination-specific lytic enzymes (GSLEs) during spore germination, rendering the cells vulnerable to common disinfection techniques. This study investigates the relationship between SleB, a GSLE in B. anthracis, and YpeB, a protein necessary for SleB stability and function. The results indicate that ΔsleB and ΔypeB spores exhibit similar germination phenotypes and that the two proteins have a strict codependency for their incorporation into the dormant spore. In the absence of its partner protein, SleB or YpeB is proteolytically degraded soon after expression during sporulation, rather than escaping the developing spore. The three PepSY domains of YpeB were examined for their roles in the interaction with SleB. YpeB truncation mutants illustrate the necessity of a region beyond the first PepSY domain for SleB stability. Furthermore, site-directed mutagenesis of highly conserved residues within the PepSY domains resulted in germination defects corresponding to reduced levels of both SleB and YpeB in the mutant spores. These results identify residues involved in the stability of both proteins and reiterate their codependent relationship. It is hoped that the study of GSLEs and interacting proteins will lead to the use of GSLEs as targets for efficient activation of spore germination and facilitation of spore cleanup.
INTRODUCTION
Bacterial spores from the Bacillus and Clostridium genera are metabolically dormant and are known for their extreme resistance to heat, desiccation, UV radiation, chemicals, and other insults (1–3). These resistance properties allow spores to survive in the environment for extended periods and have made eradication from contaminated sites incredibly difficult (4). Spore dormancy and wet heat resistance are largely dependent on spore core dehydration, which is maintained by a thick layer of modified peptidoglycan (PG) known as the cortex (2, 5). While vegetative cell wall PG consists of alternating N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM) sugars, approximately 50% of NAM residues in the cortex are converted to muramic-δ-lactam, while an additional portion of the NAM side chains is generally cleaved to a single l-alanine (6–11).
The spore form of Bacillus anthracis is the etiological agent for all types of anthrax infections: inhalational, gastrointestinal, and cutaneous anthrax, as well as the newest form described, injectional anthrax (12, 13). When the spore senses the availability of nutrients, such as when it enters a suitable host, germination is triggered, causing a chain of events that ultimately result in a vegetative cell capable of producing deadly toxins (1, 12). After germinant contact with receptors at the spore inner membrane, the spore releases its large pool of Ca2+-dipicolinate (Ca2+-DPA) stored in the core, which becomes partially rehydrated through an influx of water (1). This is followed by cortex depolymerization, allowing the spore core to rehydrate to levels necessary for metabolism to resume (14). Spore germination concomitantly results in the loss of resistance properties; thus, triggering this process at a high efficiency is a potentially attractive approach to spore decontamination.
Cortex degradation during spore germination is accomplished by germination-specific lytic enzymes (GSLEs) already present within the dormant spore (15, 16). These enzymes exhibit specificity for PG containing the muramic-δ-lactam modification, ensuring that only the cortex PG is broken down (17–21). Bacillus anthracis contains the four GSLEs SleB, CwlJ1, CwlJ2, and SleL; SleB and CwlJ1 are partially redundant enzymes responsible for the majority of cortex hydrolysis (22–25). SleB or CwlJ1 alone is sufficient for cortex hydrolysis, and the absence of both proteins results in spores that are unable to degrade the cortex and complete germination (23–25). Identical roles for SleB and CwlJ have been demonstrated in Bacillus subtilis (26–28) and Bacillus megaterium (29). CwlJ2, a homolog of CwlJ1, appears to play a minor role at best during B. anthracis spore germination, while SleL further breaks down PG fragments first generated by SleB and/or CwlJ1 (20, 22–25).
The arrangement of sleB upstream of ypeB in an operon is highly conserved across Bacillus species and in a few Clostridiales species possessing sleB (16, 23, 30–32). In the Bacillus species examined, SleB and YpeB are expressed from the forespore under the control of σG during sporulation and are then translocated across the inner membrane by way of their N-terminal signal sequences (28, 30, 33, 34). Unlike the YpeB signal sequence, which is not predicted to be cleaved, the SleB signal sequence is removed during sporulation, and SleB is present in its mature form within the dormant spore (28, 30, 33, 35). The uncleaved signal sequence likely anchors YpeB to the inner membrane, an idea supported by a study with B. subtilis that found both SleB and YpeB associated with the inner membrane of the spore (26). The same study, as well as work with Bacillus cereus, placed SleB and YpeB at a second location near the outer region of the cortex (26, 30). Not only do SleB and YpeB appear to localize within the same regions of the spore, but it has been demonstrated in B. subtilis that YpeB is required for SleB activity and for stable incorporation of SleB into the spore (26, 34). Additionally, the relationship appears to be mutual, such that SleB is also needed to stabilize YpeB (36).
Far more is known about the structure and function of SleB than about those of YpeB. At the N terminus of the mature protein, SleB has a PG-binding domain (pfam01471), while the C terminus contains a catalytic domain (pfam07486) (21, 28, 33, 37). As anticipated, the N-terminal domain plays the dominant role in PG binding; however, the C-terminal domain appears to be responsible both for the lytic transglycosylase activity of SleB on cortex PG and for its specificity for muramic-δ-lactam (21, 37, 38). Mutational analysis and crystal structure determination of the SleB C-terminal domain in B. anthracis (39) and B. cereus (38) have revealed a conserved catalytic glutamate residue and a unique substrate-binding cleft thought to mediate specificity for cortex PG. Apart from its signal sequence, the only recognizable features of YpeB are the three putative PepSY domains positioned in the C-terminal 60% of the protein (40). These domains are best characterized in the M4 family of metallopeptidases, where the PepSY domains serve as intramolecular inhibitors of protease activity until the protease is secreted from the cell (40). Like a large number of other proteins containing PepSY domains, YpeB does not show homology with proteases; thus, the function of the PepSY domains in YpeB and the other proteins constituting this group is unknown (40).
The current study investigates the relationship between SleB and YpeB in B. anthracis and demonstrates the contribution of the PepSY domains to this interaction. The data solidify the codependent nature of this relationship and reveal that SleB and YpeB are each rapidly degraded following expression in the absence of the other protein. The C terminus of YpeB containing the PepSY domains is critical for the stability and function of SleB, and certain highly conserved residues within the PepSY domains are important for the stability of both proteins. By understanding GSLEs and regulators of their activity, it may be possible to trigger germination at the stage of cortex hydrolysis, resulting in cells that are readily killed.
MATERIALS AND METHODS
Bacterial strains and general growth conditions.
The strains and plasmids used in this work are listed in Table 1. All B. anthracis strains used in this study are derived from the Sterne strain 34F2 and were grown at 37°C in brain heart infusion (BHI; Difco) with 5 μg/ml erythromycin or 10 μg/ml tetracycline where appropriate. B. anthracis strains maintaining pBKJ236-derived plasmids extrachromosomally were incubated at 25°C. Following plasmid integration into the chromosome (at 42°C in most instances), these strains were grown at 37°C. Escherichia coli strains were grown in LB at 37°C with antibiotics (500 μg/ml erythromycin or 100 μg/ml ampicillin for strains involved in plasmid propagation; 30 μg/ml chloramphenicol and 50 μg/ml ampicillin for strains used in protein overexpression).
TABLE 1.
Bacterial strains and plasmids
| Strain or plasmid | Relevant genotype/phenotypea | Constructionb | Source or reference |
|---|---|---|---|
| Strains | |||
| E. coli | |||
| DPVE13 | BL21 λ(DE3) pLysS (Cmr) | Novagen | |
| DPVE440 | pDPV426 (His6-MBP-YpeB21–446 Ampr) Cmr | pDPV426 → DPVE13 | This study |
| B. anthracis | |||
| Sterne 34F2 | pXO1+ pXO2− | P. Hanna | |
| DPBa38 | ΔsleB | 23 | |
| DPBa57 | ΔsleB pDPV346 (sleB+ Err) | 23 | |
| DPBa89 | ΔypeB | pDPV392 → 34F2 | This study |
| DPBa113 | ΔypeB::pDPV416 (ypeB+ Err) | pDPV416 → DPBa89 | This study |
| DPBa124 | ΔypeB::pDPV421 (YpeB1–208-His6 Err) | pDPV421 → DPBa89 | This study |
| DPBa125 | ΔypeB::pDPV422 (YpeB1–283-His6 Err) | pDPV422 → DPBa89 | This study |
| DPBa126 | ΔypeB::pDPV423 (YpeB1–368-His6 Err) | pDPV423 → DPBa89 | This study |
| DPBa127 | ΔypeB::pDPV424 (YpeB1–446-His6 Err) | pDPV424 → DPBa89 | This study |
| DPBa134 | ΔsleB::pDPV346 (sleB+ Err) | DPBa57 integration | This study |
| DPBa143 | ΔypeB::pDPV435 (YpeBY410A Err) | pDPV435 → DPBa89 | This study |
| DPBa144 | ΔypeB::pDPV436 (YpeBG430A Err) | pDPV436 → DPBa89 | This study |
| DPBa148 | ΔypeB::pDPV432 (YpeBT377A Err) | pDPV432 → DPBa89 | This study |
| DPBa149 | ΔypeB::pDPV433 (YpeBY254A Err) | pDPV433 → DPBa89 | This study |
| DPBa150 | ΔypeB::pDPV434 (YpeBY329A Err) | pDPV434 → DPBa89 | This study |
| DPBa158 | ΔypeB::pDPV448 (YpeBΔ25–203-His6 Err) | pDPV448 → DPBa89 | This study |
| DPBa159 | ΔypeB::pDPV449 (YpeBΔ67–203-His6 Err) | pDPV449 → DPBa89 | This study |
| DPBa160 | ΔypeB::pDPV450 (YpeBΔ119–203-His6 Err) | pDPV450 → DPBa89 | This study |
| DPBa161 | ΔypeB::pDPV451 (YpeBΔ156–203-His6 Err) | pDPV451 → DPBa89 | This study |
| Plasmids | |||
| pBKJ236 | Err ori(Ts) | 42 | |
| pBKJ223 | Tetr Ampr Pamy-I-SceI | 42 | |
| pDEST-HisMBP-T | His6-MBP; Ampr Cmr | F. Schubot | |
| pDPV346 | sleB+ | pBKJ236::sleB | 23 |
| pDPV388 | ypeB+ | pBKJ236::ypeB | This study |
| pDPV392 | ΔypeB | pBKJ236::ΔypeB | This study |
| pDPV416 | ypeB+ | pBKJ236::ΔsleB ypeB | This study |
| pDPV421 | YpeB1–208-His6 | pBKJ236::ΔsleB ypeB1–208-His6 | This study |
| pDPV422 | YpeB1–283-His6 | pBKJ236::ΔsleB ypeB1–283-His6 | This study |
| pDPV423 | YpeB1–368-His6 | pBKJ236::ΔsleB ypeB1–368-His6 | This study |
| pDPV424 | YpeB1–446-His6 | pBKJ236::ΔsleB ypeB1–446-His6 | This study |
| pDPV426 | His6-MBP-YpeB21–446 | pDEST-HisMBP-T::ypeB21–446 | This study |
| pDPV432 | YpeBT377A | pBKJ236::ΔsleB ypeBT377A | This study |
| pDPV433 | YpeBY254A | pBKJ236::ΔsleB ypeBY254A | This study |
| pDPV434 | YpeBY329A | pBKJ236::ΔsleB ypeBY329A | This study |
| pDPV435 | YpeBY410A | pBKJ236::ΔsleB ypeBY410A | This study |
| pDPV436 | YpeBG430A | pBKJ236::ΔsleB ypeBG430A | This study |
| pDPV448 | YpeBΔ25–203-His6 | pBKJ236::ΔsleB ypeBΔ25–203-His6 | This study |
| pDPV449 | YpeBΔ67–203-His6 | pBKJ236::ΔsleB ypeBΔ67–203-His6 | This study |
| pDPV450 | YpeBΔ119–203-His6 | pBKJ236::ΔsleB ypeBΔ119–203-His6 | This study |
| pDPV451 | YpeBΔ156–203-His6 | pBKJ236::ΔsleB ypeBΔ156–203-His6 | This study |
Cmr, chloramphenicol resistance; Ampr, ampicillin resistance; Err, erythromycin resistance; ori(Ts), temperature-sensitive origin of replication; Tetr, tetracycline resistance.
Strains were constructed by conjugation or electroporation. The plasmid name precedes the arrow, while the recipient strain designation follows the arrow. Single strain designations indicate that the existing plasmid was integrated into the chromosome.
Mutant construction.
The sequences of all primers used during plasmid construction are listed in Table S1 in the supplemental material. All plasmids created were verified by DNA sequencing. To create a ΔypeB strain, ypeB and approximately 500 bp flanking each side of the gene were PCR amplified from the B. anthracis chromosome. The PCR product was inserted into vector pBKJ236 (41) by digestion with the restriction enzymes SacII and NotI and ligation of the DNA to create pDPV388. Inverse PCR of the plasmid using primers with BglI restriction sites at the 3′ ends resulted in a linear PCR product with the majority of ypeB deleted, leaving only three codons from each end of the gene. Subsequent BglI digestion and ligation of the PCR product formed pDPV392. This plasmid, containing the ypeB deletion, was introduced into B. anthracis by using the markerless gene replacement strategy as described previously (41). Gene deletion was verified by PCR amplification and sequencing.
The sleB complementation strain published previously (DPBa57) contains an extrachromosomally maintained pBKJ236 derivative with full-length sleB (pDPV346) (23). In order to ensure efficient plasmid partitioning into the forespore during spore formation and thus achieve more-complete complementation of the deletion phenotype, pDPV346 was integrated into the ΔsleB chromosome by shifting the temperature to 42°C. Plasmid integration through homologous recombination just upstream of the sleB deletion in the chromosome was verified with PCR, and this new sleB complementation strain was designated DPBa134. To create a ypeB complementation plasmid that includes the native promoter, PCR was performed using the ΔsleB chromosome (23) as the template. The resulting PCR product contained the promoter region for the sleB operon, followed by the ΔsleB region, ypeB, and approximately 500 bp downstream of ypeB. The DNA fragment was cloned into pBKJ236 by digestion with NotI and ligation to form pDPV416. This ypeB complementation plasmid was introduced into the ΔypeB strain of B. anthracis via conjugation as done in the initial stages of the markerless gene replacement procedure (41). Subsequent plasmid integration within the 500-bp homologous region downstream of the ypeB deletion in the chromosome was achieved by shifting the temperature to 42°C and was verified by PCR.
YpeB truncation and internal deletion mutations were designed by using InterPro (42) to predict the locations of PepSY domains and the Protean application of Lasergene, version 10.0 (DNAStar), to choose truncation/deletion sites on the basis of high residue surface probability and residues not predicted to disrupt protein secondary structure. For the truncation mutations, inverse PCR of the ypeB complementation plasmid was performed using sets of two primers. The tails of the forward primers contained codons encoding a hexahistidine tag followed by a stop codon and a BglII restriction enzyme recognition sequence. The tail of the reverse primer used to create all the truncation mutants also had a BglII site added. The linear PCR products from inverse PCRs were digested with BglII and were ligated in order to recircularize the plasmids, generating pDPV421 through pDPV424. The internal deletion mutations were constructed using overlap extension PCR (43), followed by restriction-free cloning (44) to insert the PCR products into the plasmid encoding full-length YpeB with a C-terminal His6 tag (pDPV424). The YpeB truncation and internal deletion plasmids were moved into the ΔypeB strain of B. anthracis and were integrated into the chromosome as described above. The 500-bp homologous region following ypeB in pDPV416 was removed during inverse PCR to create pDPV421 through -424; thus, these plasmids were inserted into the ΔypeB chromosome through homologous recombination upstream of sleB, as confirmed by PCR.
In order to predict residues of the YpeB PepSY domains that might be important for protein-protein interactions, the PepSY domain sequences from B. anthracis YpeB were aligned with a PepSY domain consensus sequence (45), other PepSY domain sequences (40, 45, 46), and the YpeB PepSY domain sequences from other Bacillus species by using Clustal W sequence alignment software (47). Based on these alignments, highly conserved residues were identified as targets for mutagenesis. Site-directed mutagenesis by overlap extension PCR (43) was used to create point mutations in ypeB. The resulting PCR products were cloned into the ypeB complementation plasmid pDPV416 by using restriction-free cloning (44). Successful plasmid construction was verified by screening for the gain or loss of a restriction site in ypeB that was designed as part of the mutagenic primers, followed by DNA sequencing. Plasmids were mobilized into the ΔypeB strain of B. anthracis and were integrated into the chromosome downstream of the ypeB deletion as described above for the ypeB complementation plasmid.
Spore preparation and decoating.
B. anthracis strains were sporulated in modified G broth (48) with appropriate antibiotics at 37°C with aeration. After 3 to 4 days, dormant spores were harvested by centrifugation and were repeatedly washed with deionized water. Any remaining vegetative cells were heat killed at 65°C for 25 min. Spores were further purified with a 50% sodium diatrizoate (Sigma) gradient as described previously (49). Decoated spores were prepared by suspending up to 30 optical density (OD) units of spores in 1 ml decoating solution (50 mM Tris-HCl [pH 8], 8 M urea, 1% SDS, 50 mM dithiothreitol [DTT]). The spores were incubated at 37°C for 1 h and were then centrifuged at 8,000 × g for 1 min to remove the decoating solution. The decoating procedure was repeated, followed by five washes with 1 ml deionized water at room temperature. Untreated (native) and decoated spores were stored in deionized water at 4°C until analysis.
Spore and sporangium sample preparation for Western blotting.
Following spore purification, 7.5 to 10 OD units of dormant, native spores were pelleted, frozen at −80°C, and lyophilized. Dried spores were mechanically disrupted with 100 mg 0.1 mm glass beads and 20 pulses of 30 s each at 4,200 rpm by using a Wig-L-Bug bead beater. Samples were placed on ice between pulses. Proteins were extracted from the broken material by adding 10 μl/OD unit of 1× sample loading buffer (62.5 mM Tris-HCl [pH 6.8], 2% SDS, 10% glycerol, 5% β-mercaptoethanol, 0.05% bromophenol blue) and heating to 100°C for 5 min. Extracts were centrifuged at 15,800 × g for 1 min, and volumes of supernatants derived from equal starting OD units of spores were used for Western blot analysis.
Strains grown in modified G broth with appropriate antibiotics were monitored spectrophotometrically until the OD at 600 nm (OD600) indicated that the cells had entered stationary phase. This time point, termed t0, coincides with the initiation of sporulation, and t2 through t6 designate 2 through 6 h, respectively, past t0. At t2 through t6, 10-ml sporangium samples were collected and were centrifuged at 10,000 × g for 10 min at 4°C. Pellets were resuspended in 1 ml of 8 mM NaPO4 (pH 7.0) and were centrifuged at 15,800 × g for 1 min. The resulting pellets were flash frozen in liquid N2 and were lyophilized. Lyophilized sporangia were broken with glass beads, and proteins were extracted as described above, except that 100 μl of 1× sample loading buffer was used. Supernatants of protein extracts were used for Western blotting. The average OD600 measured for a particular strain from t2 through t6 was used to adjust sample loading between strains.
Germination assays.
To assess the rate of spore germination and outgrowth, decoated spores were heat activated at 70°C for 30 min and were quenched on ice for 5 min. Heat-activated spores were diluted to an OD600 of 0.2 in liquid BHI medium at 37°C to initiate germination, and the change in OD600 was measured over time. For germination efficiency assays, decoated spores at an OD600 of 0.2 were heat activated at 70°C for 20 min and were quenched on ice. Heat-activated spores were serially diluted in deionized water, plated onto BHI medium without antibiotics, and incubated at 37°C overnight. Colonies were counted to determine CFU/OD unit values. Unpaired, two-tailed Student t tests with unequal variance were used for statistical analyses of germination assays.
Protein expression and purification.
The ypeB gene lacking the first 20 codons was amplified by PCR and was inserted into a modified version of pDEST-HisMBP (50) containing a tobacco etch virus (TEV) cleavage site (pDEST-HisMBP-T) by using restriction-free cloning (44). The resulting plasmid encoded an N-terminally His6-tagged maltose binding protein (MBP) and YpeB21–446, separated by a TEV cleavage site. Successful plasmid construction was verified by sequencing. The His6-MBP-YpeB21–446 fusion protein was overexpressed in E. coli BL21(DE3) [λ(DE3) pLysS Cmr] (Novagen) grown at 37°C until the OD600 reached 1.0, at which point isopropyl-β-d-thiogalactopyranoside was added to a final concentration of 1 mM and the culture was incubated at 10°C for an additional 16 h. Cells were harvested by centrifugation at 10,000 × g for 10 min at 4°C, and the pellet was resuspended in 5 ml/g buffer A (50 mM NaCl, 50 mM Tris-HCl [pH 7.5], 5% glycerol, 30 mM imidazole). Resuspended cells were lysed by sonication for 15 min and were then centrifuged at 117,000 × g for 1 h at 4°C. The fusion protein in the soluble fraction was purified using a Ni-Sepharose HisTrap HP affinity column (GE Healthcare) equilibrated with buffer A. The protein was eluted with a linear gradient of 30 to 500 mM imidazole in buffer A, and the elution fractions were dialyzed in buffer A. The fusion protein was digested with 1 mg His6-tagged TEV(S219V) protease (51) per 7 mg fusion protein at 15°C for 16 h, and the mixture was centrifuged at 117,000 × g for 20 min at 4°C to remove any precipitated protein. The cleavage of the fusion protein was verified by SDS-PAGE. YpeB21–446 was purified from His6-MBP and His6-TEV by using a second Ni-Sepharose HisTrap HP affinity column as described above. SleB was overexpressed and purified as described previously (21).
Antibody preparation and Western blot analysis.
Polyclonal anti-SleB and anti-YpeB antibodies were raised in rabbits (Open Biosystems) using purified SleB33–253 (21) or YpeB21–446. SleB, YpeB, and derivatives were detected in Western blots on Amersham Hybond-P (polyvinylidene difluoride [PVDF]) membranes (GE Healthcare) by using BM Blue POD substrate, precipitating (Roche), for colorimetric detection. Primary anti-SleB and anti-YpeB antibodies were generally used at 1:1,000 and 1:3,000 dilutions, respectively. Horseradish peroxidase (HRP)-conjugated secondary goat anti-rabbit antibodies (Bio-Rad) were used at a 1:200,000 dilution. Western blot quantification was performed using Image Lab software (Bio-Rad). While efforts were made to ensure that the total protein load was identical for each strain, there is the inherent possibility of slight variations stemming from unequal protein extraction between samples and unequal sample loading. To account for experimental error, relative values were normalized using a highly reproducible non-YpeB background band detected in all strains, including the ΔypeB strain. Similarly, SleB band intensities were normalized using a background band present on anti-SleB immunoblots.
RESULTS
Germination of ΔypeB mutant spores.
As previously reported by Heffron et al., a homolog of the B. subtilis ypeB gene (57% identity; 77% similarity) exists as part of a putative tricistronic operon at locus BAS2561 in the B. anthracis chromosome (23). The ypeB homolog is the second gene in the operon; it is preceded by sleB (BAS2562) and followed by the BAS2560 open reading frame (23). BAS2560 shows homology to the B. subtilis genes encoding the lipoproteins YlaJ and YhcN; YlaJ is an uncharacterized spore protein, and YhcN plays an unknown role in spore germination or outgrowth (23, 52, 53). In this study, a deletion of the ypeB gene (BAS2561) in the B. anthracis chromosome was made using the markerless gene replacement strategy (41). The effects of this ypeB deletion, and those of an sleB deletion, are not due to polar effects on the expression of BAS2560, because (i) a BAS2560 deletion results in no change in spore germination, and specifically in SleB activity (data not shown); (ii) as shown below, all effects of a ypeB deletion can be complemented by the ypeB gene alone; and (iii) as shown below and by Heffron et al. (23), all effects of an sleB deletion can be complemented by sleB alone.
In assays in which purified spores were germinated in BHI medium and the change in the OD600 measured over time, the germination phenotype of ΔypeB spores was essentially identical to that of ΔsleB spores (23). This could be seen in assays using native spores, in which ΔypeB and ΔsleB spores produced a shallower germination curve, indicative of a slightly less efficient germination response, and also showed a lower rate of outgrowth (data not shown). The germination defect in ΔypeB and ΔsleB spores alike was more pronounced in assays using decoated spores, where the other GSLEs localized to the outer periphery of the spore are removed or inactivated, and SleB alone is responsible for cortex hydrolysis during germination (Fig. 1A). Both ΔypeB and ΔsleB decoated spores proceeded through stage I of germination similarly to wild-type spores, where the initial OD decreases as the spores take up water and release their large deposits of Ca2+-DPA. Decoated wild-type spores continued to lose nearly half of their initial OD as the cortex PG was degraded during stage II of germination, followed by an increase in OD as germination was completed and outgrowth into vegetative cells began. Conversely, germination was arrested for decoated ΔypeB and ΔsleB spores at the stage of cortex hydrolysis; ΔypeB and ΔsleB spores never lost more than 34% of their initial OD and did not proceed to outgrowth (Fig. 1A). This germination deficiency is also illustrated in spore plating efficiency assays, where decoated ΔypeB and ΔsleB spores showed >104-fold reductions in the ability to germinate and form colonies on a rich medium from that of wild-type spores (Table 2). A wild-type germination phenotype in terms of both germination rate and plating efficiency assays was achieved using spores from a ypeB complementation strain (DPBa113 [ypeB+]), in which ypeB, provided on a plasmid downstream of its native promoter, was integrated into the ΔypeB chromosome. High-performance liquid chromatography (HPLC) analyses of muropeptides collected during spore germination revealed the complete absence of the SleB-specific lytic transglycosylase products identified by Heffron et al. (23) in both ΔsleB and ΔypeB spores, while these peaks were restored during the germination of ypeB+ spores (data not shown). In agreement with the results observed from a ypeB deletion in B. subtilis (34, 36), these results indicate that YpeB is necessary for the lytic activity of SleB on spore cortex PG in B. anthracis.
FIG 1.

Effects of a variety of ypeB mutations on the germination and outgrowth of decoated B. anthracis spores. Decoated spores were heat activated and were germinated in BHI medium at 37°C. The data shown are averages of results from three independent spore preparations; error bars are omitted for clarity. (A) Germination and outgrowth are blocked in decoated ΔypeB spores. Wild-type (●), ΔsleB (□), ΔypeB (▲), and ypeB complementation (ypeB+) (⬥) strains were analyzed. The germination rates of ΔsleB and ΔypeB spores were statistically indistinguishable (P > 0.23), as were the germination rates of wild-type and ypeB+ spores (P > 0.08). Both ΔsleB and ΔypeB spores were significantly different (P < 0.04) from wild-type and ypeB+ spores during stage II of germination, from 45 to 95 min. (B) Truncations removing YpeB PepSY domains block decoated-spore germination. ΔypeB (▲), YpeB1–208-His6 (△), YpeB1–283-His6 (+), YpeB1–368-His6 (□), YpeB1–446-His6 (—), and ypeB+ (⧫) strains were analyzed. The germination rates of ypeB+ and YpeB1–446-His6 spores were not statistically different (P > 0.08); likewise, the germination rates of ΔypeB and YpeB1–283-His6 spores were statistically indistinguishable (P > 0.49). YpeB1–208-His6 and YpeB1–368-His6 spores did not differ (P > 0.05) from ΔypeB spores during germination, except at 55 min and from 45 to 55 min, respectively (P < 0.05). The germination rates of YpeB1–208-His6, YpeB1–283-His6, and YpeB1–368-His6 spores were not statistically different (P > 0.11) from 4 min onward. (C) Point mutations in YpeB PepSY domain conserved residues slow decoated-spore germination. ΔypeB (▲), YpeBY254A (■), YpeBY329A (×), YpeBT377A (○), YpeBY410A (●), YpeBG430A (◇), and ypeB+ (⬥) strains were analyzed. Statistical analysis of ypeB point mutants is complex due to the greater variability of germination rates of decoated-spore preparations, as well as the fact that spores of some strains are initiating outgrowth while others are still germinating. During stage II of germination, between 45 and 95 min, YpeBY329A and YpeBT377A spores were not significantly different from ypeB+ spores (P > 0.38). The germination rates of YpeBY254A spores from 80 to 90 min, YpeBY410A spores from 45 to 55 min and 70 to 95 min, and YpeBG430A spores at 50 min and from 75 to 95 min were significantly different from that of ypeB+ spores (P < 0.05).
TABLE 2.
Decoated-spore plating efficiencies for sleB and ypeB mutants
| Strain | Genotype/phenotype | CFU/OD600 valuea |
|---|---|---|
| 34F2 | Wild type | 8.4 × 107 |
| DPBa38 | ΔsleB | 8.3 × 103* |
| DPBa89 | ΔypeB | <5.8 × 103* |
| DPBa113 | ypeB+ | 8.7 × 107 |
| DPBa124 | YpeB1–208-His6 | 9.7 × 103* |
| DPBa125 | YpeB1–283-His6 | <3.5 × 103* |
| DPBa126 | YpeB1–368-His6 | <4.5 × 103* |
| DPBa127 | YpeB1–446-His6 | 1.1 × 108 |
Values are averages for three independent spore preparations. Asterisks indicate values significantly different from those of wild-type and ypeB+ spores (P, <0.006 and <0.02, respectively).
Stability of SleB and YpeB in spores and sporangia.
Not only is YpeB needed for SleB activity, but Western blotting of ΔypeB spore extracts demonstrated that YpeB is also required for the incorporation of SleB into the spore (Fig. 2). SleB was essentially not detected in ΔypeB spore extracts, and this could be complemented. As in B. subtilis (36), this relationship is mutual: SleB was also needed for the stable incorporation of YpeB into the spore (Fig. 2). Immunoblots of ΔsleB spore extracts revealed significantly diminished levels of YpeB when SleB was absent, which could also be complemented. Miniscule levels of SleB and YpeB detected in Western blots of ΔypeB and ΔsleB spores, respectively, could be attributed either to expression of the proteins during spore formation followed by rapid proteolysis in the absence of the stabilizing partner or to escape of the proteins from the developing spore. The former proposal is supported by Western blot analyses of whole-sporangium samples taken throughout sporulation (Fig. 3). In wild-type and ypeB+ strains, significant amounts of SleB and YpeB were detected beginning 3 h after the initiation of sporulation (t3) (Fig. 3A and B). This is consistent with the findings of previous β-galactosidase activity assays performed in B. anthracis, which show that sleB transcription reaches its peak near t3.5 (23). During spore formation in a ΔsleB or ΔypeB strain, some YpeB or SleB, respectively, could be seen at t3 and t4 but was not strongly detected thereafter (Fig. 3A and B). The expression of YpeB at t3 and t4 coincided with the production of large quantities of two YpeB-specific degradation products in ΔsleB sporangia (and to a lesser degree in wild-type and ypeB+ sporangia [data not shown]), indicating that the protein was degraded immediately following its expression (Fig. 3C). Specific SleB degradation products were not observed during the sporulation of any of the strains tested (data not shown). The protease(s) responsible for the degradation of SleB and YpeB during spore formation is unknown. Elimination of SpoIVB, which is known to be active in the forespore intermembrane space in B. subtilis (54, 55), had no effect on the stability of SleB in the absence of YpeB (see Fig. S1 in the supplemental material). Elimination of BAS5314 (HtrC), which in B. subtilis is expressed in the forespore and possesses a signal sequence/membrane anchor (56), had no effect on the stability of SleB and YpeB in the absence of the partner protein (C. B. Bernhards, Y. Chen, H. Toutkoushian, and D. L. Popham, unpublished data).
FIG 2.
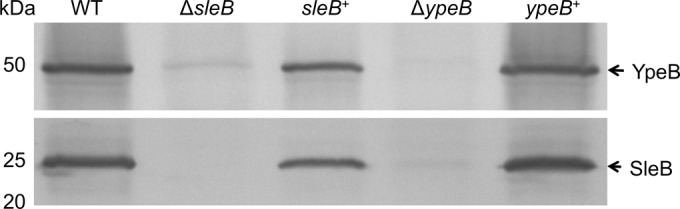
B. anthracis SleB and YpeB are codependent for incorporation into dormant spores. Dormant wild-type (WT), ΔsleB, sleB complementation (sleB+), ΔypeB, and ypeB complementation (ypeB+) spores were mechanically disrupted, and proteins were extracted with sample loading buffer for Western blot analysis. Blots were probed with anti-YpeB (top) and anti-SleB (bottom) antibodies. The predicted molecular sizes of mature SleB lacking its signal sequence and YpeB are 24.1 and 50.0 kDa, respectively. The positions of molecular size marker proteins (not shown) are indicated on the left.
FIG 3.

B. anthracis SleB and YpeB are each degraded during spore formation in the absence of the partner protein. Wild-type (WT), ΔsleB, ΔypeB, and ypeB complementation (ypeB+) strains were grown in modified G broth at 37°C, and samples collected during sporulation were used for Western blot analysis. The designations t2 through t6 represent the numbers of hours since the initiation of sporulation. (A) Sporangia probed with anti-SleB antibodies. (B) Sporangia probed with anti-YpeB antibodies. (C) Sporangia from the ΔsleB strain probed with anti-YpeB antibodies. The positions of molecular size marker proteins (not shown), in kilodaltons, are indicated on the left.
Analyses of YpeB PepSY domain truncation mutants.
InterPro (42) predicted the presence of three PepSY domains in B. anthracis YpeB from residues 218 to 282, 291 to 358, and 375 to 436. To assess the individual contributions of the three putative PepSY domains located at the C terminus of YpeB, strains expressing truncated forms of YpeB with a C-terminal His6 tag were constructed. Plasmids expressing YpeB from its native promoter were integrated into the ΔypeB chromosome; the resulting strains expressed YpeB with no PepSY domains (YpeB1–208-His6), one PepSY domain (YpeB1–283-His6), two PepSY domains (YpeB1–368-His6), or all three PepSY domains (full-length YpeB) (YpeB1–446-His6). In decoated-spore germination rate assays in liquid BHI medium and in decoated-spore plating efficiency assays, spores containing YpeB with no, one, or two PepSY domains were similar to ΔypeB spores (Fig. 1B and Table 2). Like ΔypeB spores, these spores containing truncated versions of YpeB were arrested during germination and showed a >103-fold reduction in plating efficiency in the case of the YpeB1–208-His6 mutant, but generally a >104-fold decrease. The reason for the similarity was revealed in Western blots of dormant spore extracts, which show that YpeB is unstable in many of the truncated forms (Fig. 4A). In fact, YpeB and SleB were both stable only in strains possessing all three YpeB PepSY domains. In spores with highly unstable YpeB (YpeB1–208-His6 and YpeB1–368-His6), SleB was also unstable, as anticipated (Fig. 4A). Interestingly, although YpeB with only the first PepSY domain (YpeB1–283-His6) appeared somewhat stable in the anti-YpeB Western blot, SleB was unstable (Fig. 4A), and these spores had germination phenotypes like those of ΔypeB spores (Fig. 1B and Table 2). This suggests that the C-terminal region of YpeB, more specifically a region beyond the first PepSY domain, is critical for SleB stability and thus for spore germination.
FIG 4.

Effects of ypeB truncation and internal deletion mutations on the stability of YpeB and SleB. Dormant spores were mechanically disrupted, and proteins were extracted with sample loading buffer for Western blot analysis. Samples were probed with anti-YpeB (top) and anti-SleB antibodies (bottom) antibodies. Right arrows indicate YpeB derivatives. The positions of molecular mass marker proteins (not shown) are indicated on the left. (A) Truncations removing YpeB PepSY domains destabilize both YpeB and SleB. YpeB1–208-His6, YpeB1–283-His6, YpeB1–368-His6, and YpeB1–446-His6 have predicted molecular sizes of 24.3, 32.5, 42.1, and 50.9 kDa, respectively. (B) Internal deletions in the YpeB N-terminal domain destabilize SleB. Lanes intervening between the 5th and 6th lanes were removed for clarity. YpeBΔ25–203-His6, YpeBΔ67–203-His6, YpeBΔ119–203-His6, YpeBΔ156–203-His6, and YpeB1–446-His6 have predicted molecular sizes of 30.6, 35.5, 41.2, 45.6, and 50.9 kDa, respectively.
Analysis of YpeB internal deletion mutants.
In order to determine what other areas of YpeB may be necessary for YpeB function in stabilizing SleB and SleB activity during germination, internal deletions of different lengths were made between the N-terminal signal sequence and the C-terminal PepSY domains. Deletion endpoints were chosen based on software predictions of transitions between secondary structural elements. The strains created expressed YpeB without residues 25 to 203 (YpeBΔ25–203-His6), 67 to 203 (YpeBΔ67–203-His6), 119 to 203 (YpeBΔ119–203-His6), or 156 to 203 (YpeBΔ156–203-His6). Immunoblots of dormant spore extracts revealed that YpeBΔ25–203-His6 and YpeBΔ67–203-His6 are at least moderately stable, while YpeBΔ119–203-His6 and YpeBΔ156–203-His6 are not; however, SleB was not stably incorporated into spores of any of these internal deletion strains (Fig. 4B). Even YpeBΔ25–203-His6, which appears to accumulate in dormant spores to levels similar to those of full-length YpeB (YpeB1–446-His6), failed to stabilize SleB. Thus, the N-terminal half of YpeB, likely a region between residues 67 and 203, must also play a role in stabilizing SleB.
Analyses of YpeB PepSY domain point mutants.
As an alternative approach to studying the PepSY domains while attempting to maintain YpeB stability, site-directed mutagenesis was performed on selected highly conserved residues within the three PepSY domains. Y254 within the first PepSY domain, Y329 in the second PepSY domain, and T377, Y410, and G430, all in the third and last PepSY domain, were selected to be individually changed to alanine. Spores with YpeBY329A or YpeBT377A showed no significant decrease either in the germination rate in liquid BHI medium or in plating efficiency from ypeB+ spores (Fig. 1C and 5). Spores with YpeBY254A or YpeBG430A showed a delay in germination, reaching their lowest ODs 40 min and 50 min later than ypeB+ spores, respectively (Fig. 1C). While YpeBY254A and ypeB+ spores each lost 59 to 60% of their initial ODs, YpeBG430A spores had a shallower germination curve, losing 51% of their initial OD (Fig. 1C). Both YpeBY254A and YpeBG430A spores showed slight but significant (P < 0.007) decreases in plating efficiency, but <10-fold reductions in the CFU/OD values were seen (Fig. 5). The most dramatic effect on spore germination was seen for YpeBY410A spores, which showed a significant delay in germination and a shallower germination curve (Fig. 1C). YpeBY410A spores lost only 50% of their initial OD, and this point was reached 105 min after ypeB+ spores lost 59% of their initial OD (Fig. 1C). YpeBY410A spores also showed a significant decrease (P < 0.004) in their ability to form colonies on BHI plates, with a nearly 100-fold reduction (Fig. 5).
FIG 5.

Protein stability and plating efficiency in YpeB point mutant spores. Dried, dormant ΔypeB, YpeBY254A, YpeBY329A, YpeBT377A, YpeBY410A, YpeBG430A, and ypeB complementation (ypeB+) spores were broken, and extracted proteins were analyzed by Western blotting with anti-SleB (α-SleB) or anti-YpeB (α-YpeB) antibodies. Band intensities were quantified and are depicted as the levels of YpeB (open bars) and SleB (shaded bars) present in spores, relative to the native protein levels found in ypeB+ spores, which were set at 100%. Spores of the same strains were heat activated, serially diluted, and plated on BHI medium. Following incubation, colonies were counted, and the percentages of native CFU/OD values (striped bars) were determined by comparison to the CFU/OD value for ypeB+ spores, which was set at 100%. Plating efficiency and Western blot quantification data shown are averages from three independent spore preparations; error bars represent the standard deviations. The Western blots below the graph show representative results from one of the replicates.
The trend seen for the YpeB point mutants in germination rate and plating efficiency assays was mirrored by the levels of YpeB and SleB seen in Western blots of dormant spore extracts (Fig. 5). Relative YpeB band intensities for each strain were computed by comparison to the YpeB band from the ypeB+ strain. Similarly, SleB band intensities relative to the SleB band detected in ypeB+ spores were determined. The strains that appeared most similar to ypeB+ spores in germination rate and plating efficiency assays (the YpeBY329A and YpeBT377A strains) contained 101 and 99% of the YpeB protein levels and 102 and 93% of the SleB protein levels detected in ypeB+ spores, respectively. Likewise, YpeBY254A and YpeBG430A spores, which showed minor defects during the other assays tested, possessed 69 and 75% of native YpeB levels and 70 and 65% of native SleB levels, respectively. Immunoblots of dormant YpeBG430A spore extracts also revealed the presence of two stable YpeB-specific degradation products (not shown) similar to those observed during the sporulation of a ΔsleB strain (Fig. 3C). Among the point mutants, the lowest levels of the two proteins were seen for YpeBY410A, with YpeB levels equivalent to 60%, and SleB levels equivalent to 40%, of those found in ypeB+ spores. This corresponds well with the more severe deficit in germination phenotypes demonstrated by YpeBY410A spores. While the correlation between YpeB and SleB band intensities was not perfect, the trend was quite clear: YpeB abundance, SleB abundance, and the germination rate decreased in concert in all strains tested.
DISCUSSION
This study investigated the relationship between SleB and YpeB in B. anthracis for the first time. In agreement with findings for B. subtilis (34), the germination of decoated ΔsleB and ΔypeB spores of B. anthracis was blocked after the initial steps, these spores exhibited a >104-fold decrease in colony-forming ability, and lytic transglycosylase products were absent in germinating spores, confirming that YpeB is needed for the lytic activity of SleB on cortex PG in vivo. The current work clearly demonstrates that not only is YpeB required for SleB incorporation into the dormant spore, as in B. subtilis (26, 36), but YpeB has a reciprocal requirement for SleB. This mutual dependency was also discovered recently using a B. subtilis strain that expressed YpeB ectopically in a cwlJ sleB ypeB mutant background (36). In that strain, YpeB was detected in Western blots of spore inner membrane fractions only if SleB was also expressed (36). It should be noted that since only the inner membrane fraction was analyzed in those immunoblots, it is possible that the absence of SleB resulted in the mislocalization of YpeB. The Western blotting procedure in the present study utilized entire spore extracts derived from strains in which sleB or ypeB was deleted or expressed from the native operon. Thus, these results provide more-conclusive evidence that YpeB is indeed absent in ΔsleB spores. Furthermore, it is demonstrated that in the absence of the stabilizing partner, SleB and YpeB are expressed and degraded during early sporulation, rather than simply failing to localize within the developing spore.
Li et al. suggested that both the N- and C-terminal domains of YpeB (YpeBN and YpeBC) are necessary for SleB lytic activity during spore germination, since a B. subtilis strain deficient in cwlJ, sleB, and ypeB and ectopically expressing an individual ypeB domain (encoding YpeBN or YpeBC) with full-length sleB (encoding SleBFL) could not complement the germination defect in these spores (36). While these results were hampered by an inability to determine if YpeBN or YpeBC was stably incorporated into the spore, Li et al. did demonstrate that the C terminus of YpeB fused to its signal sequence (memseg-YpeBC) was stable yet could not complement the defect when expressed with SleBFL, indicating that the N terminus of YpeB is needed (36). The results obtained in the current study when stretches of residues between the YpeB signal sequence and the PepSY domains were deleted support the necessity of a region within the N-terminal portion of YpeB. This critical region occurs within the area spanning residues 67 to 203, since YpeBΔ67–203-His6 was moderately stable in the dormant spore yet could not stabilize SleB. Deletions of regions corresponding to the PepSY domains at the C terminus of YpeB also add to what is known about the portions of YpeB needed for SleB stability. While many of the truncated YpeB variants were unstable, YpeB1–283-His6, which contained the first predicted PepSY domain, was moderately stable, yet SleB was not detected in Western blots, and the germination phenotypes resembled those of ΔypeB spores. These results clearly show that a region beyond the first YpeB PepSY domain is also essential for SleB stabilization. Taken together with the findings by Li et al. (36), they demonstrate that both the N terminus and the PepSY domain-containing C terminus of YpeB are required for stable incorporation of SleB into the spore.
In studies of the effects of ypeB point mutations on SleB stability and function, the codependency between the two proteins observed in ΔsleB and ΔypeB spores was reinforced. There was a strong correlation between the SleB and YpeB protein levels within the spores of individual strains, and protein abundance matched well with observed germination phenotypes. This intrinsic codependency becomes a complicating factor in determining which YpeB residues are actually important for the interaction with SleB and which are just important for proper YpeB folding. In the former scenario, alteration of a residue mediating the interaction between YpeB and SleB would destabilize the interaction, resulting in decreased levels of both proteins. In the latter scenario, a mutation affecting YpeB protein folding would likely decrease YpeB stability, subsequently decreasing the stability of SleB. Residue Y410 of YpeB clearly plays a large role in YpeB and SleB stability, and this residue is one of the most highly conserved across PepSY domains (40, 45). While the Y410A substitution had a large effect on spore germination, the tyrosine residues at similar positions in the other two PepSY domains did not prove to be as important. The Y254A substitution in the first PepSY domain resulted in a milder spore germination defect, and the Y329A replacement in the second PepSY domain produced spores that germinated equally as well as ypeB+ spores. Additionally, of the three mutations made in the region encoding the third PepSY domain of YpeB, two resulted in observable phenotypes, indicating the relative importance of this domain. The three ypeB point mutations that produced phenotypic changes (Y254A, Y410A, G430A) affect residues that are 100% conserved in an alignment of YpeB proteins from 13 Bacillus species, while those mutations that produced no phenotypic changes affect less-conserved residues. This could suggest that the three PepSY domains are unequal, with the third being the most crucial but the first also playing a role. Without an alternative means of stabilizing YpeB and SleB in vivo, e.g., via inhibition of proteolysis within the developing sporangium, further in vivo genetic study of the relationship between these proteins is difficult.
Analysis of YpeB point mutants also indicated that reduced amounts of SleB in spores not only resulted in reduced germination rates but also hindered the ability of spores to complete outgrowth and form colonies. A similar phenomenon was evident in B. subtilis cwlJ sleB mutants expressing sleB ectopically in addition to a low level of ypeB expression through apparent readthrough of an sleB insertion mutation (36). While the amounts of SleB and YpeB present in the spores of these B. subtilis strains were not explicitly quantified, immunoblots clearly showed reductions in SleB and YpeB levels. The same spores exhibited a >50% decrease in colony-forming ability (36). One might expect that even limited quantities of SleB within a given spore would allow the spore to germinate, albeit at a lower rate. This finding may suggest heterogeneity within a population of dormant spores from an individual mutant strain with respect to the absolute SleB levels present or the portion of SleB capable of being activated.
The simplest explanation for how SleB and YpeB are able to stabilize each other is that the two proteins interact physically, yet no such direct interaction has been demonstrated. Li et al. did not observe an interaction using in vitro affinity pulldown assays involving various forms of B. cereus and B. megaterium SleB and YpeB purified from E. coli and Lactococcus lactis (36). Similarly, in conjunction with the work presented here, affinity pulldown assays were performed using His6-MBP-YpeB1–446 or His6-MBP-YpeB21–446 and untagged SleB33–253, and reciprocally, His6-MBP-SleB33–253 and untagged YpeB21–446. No interaction between SleB and YpeB was observed (data not shown). A logical explanation for the lack of in vitro interaction is that if SleB and YpeB do indeed physically interact, the interaction likely occurs during spore formation as the proteins are coexpressed and translocated in their unfolded forms across the inner forespore membrane via the Sec pathway. Thus, the proteins may need to be cotranslocated and/or cofolded, or, as postulated by Li et al., a membrane might be necessary for the interaction to occur (36). Protein coexpression is likely not the only variable missing from the equation, since assays performed using cell extracts from E. coli in which SleB and YpeB were coexpressed still failed to show an interaction between the two proteins (36), although it is feasible that the N-terminal affinity tags on both proteins impeded an interaction.
Since YpeB does not have any homologs of known function, one of the major clues for determining the precise nature of the relationship between SleB and YpeB may lie in the PepSY domains at the C terminus of YpeB, which this study has demonstrated are important. While these domains have an unknown function in many proteins, they have been found in the propeptide region of M4 class proteases and function as inhibitors of protease activity (40). Another potential model, where SleB and YpeB do not bind and YpeB instead forms an inhibitory interaction with the protease(s) responsible for SleB degradation in the absence of YpeB, fails to explain how SleB stabilizes YpeB. It is possible that PepSY domains are capable of inhibiting a broader range of enzymatic activities, since they belong to a superfamily of bacterial protein domains sharing a β-lactamase inhibitor protein (BLIP)-like fold (45). Members of this superfamily have been associated with various inhibitory roles, including inhibition of β-lactamases, and have also been proposed to mediate protein-protein interactions (45). The potential for PepSY domains to inhibit enzymatic activities outside their known niche makes YpeB a prime candidate for a way in which SleB is held inactive in the dormant spore. It has been demonstrated in B. subtilis that YpeB is cleaved during spore germination (26); such cleavage could release SleB from the inhibitory interaction with YpeB, thereby activating SleB for cortex degradation. Recent in vitro experiments using various forms of exogenous SleB and YpeB from B. cereus have demonstrated inhibition of SleB activity in the presence of YpeB (36). While the in vitro experiments performed by Li et al. (36) support an inhibitory role for YpeB, more work is needed to further elucidate the relationship between SleB and YpeB. Such studies should aim to clarify the function of the PepSY domains and the mechanism by which YpeB, and specifically YpeBN, is able to inhibit SleB activity. YpeB processing during germination should also be investigated to determine if this event is required for SleB activation or is merely part of the process whereby unneeded spore proteins are broken down and recycled for new protein synthesis during outgrowth.
Supplementary Material
ACKNOWLEDGMENTS
The research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under award AI060726.
The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Published ahead of print 14 July 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01899-14.
REFERENCES
- 1.Setlow P. 2003. Spore germination. Curr. Opin. Microbiol. 6:550–556. 10.1016/j.mib.2003.10.001 [DOI] [PubMed] [Google Scholar]
- 2.Setlow P. 2006. Spores of Bacillus subtilis: their resistance to and killing by radiation, heat and chemicals. J. Appl. Microbiol. 101:514–525. 10.1111/j.1365-2672.2005.02736.x [DOI] [PubMed] [Google Scholar]
- 3.Nicholson WL, Munakata N, Horneck G, Melosh HJ, Setlow P. 2000. Resistance of Bacillus endospores to extreme terrestrial and extraterrestrial environments. Microbiol. Mol. Biol. Rev. 64:548–572. 10.1128/MMBR.64.3.548-572.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gould GW. 2006. History of science—spores. J. Appl. Microbiol. 101:507–513. 10.1111/j.1365-2672.2006.02888.x [DOI] [PubMed] [Google Scholar]
- 5.Gerhardt P, Marquis RE. 1989. Spore thermoresistance mechanisms, p 43–63 In Smith I, Slepecky RA, Setlow P. (ed), Regulation of prokaryotic development. American Society for Microbiology, Washington, DC [Google Scholar]
- 6.Warth AD, Strominger JL. 1969. Structure of the peptidoglycan of bacterial spores: occurrence of the lactam of muramic acid. Proc. Natl. Acad. Sci. U. S. A. 64:528–535. 10.1073/pnas.64.2.528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Atrih A, Zöllner P, Allmaier G, Foster SJ. 1996. Structural analysis of Bacillus subtilis 168 endospore peptidoglycan and its role during differentiation. J. Bacteriol. 178:6173–6183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Popham DL, Helin J, Costello CE, Setlow P. 1996. Analysis of the peptidoglycan structure of Bacillus subtilis endospores. J. Bacteriol. 178:6451–6458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Atrih A, Bacher G, Körner R, Allmaier G, Foster SJ. 1999. Structural analysis of Bacillus megaterium KM spore peptidoglycan and its dynamics during germination. Microbiology 145:1033–1041. 10.1099/13500872-145-5-1033 [DOI] [PubMed] [Google Scholar]
- 10.Dowd MM, Orsburn B, Popham DL. 2008. Cortex peptidoglycan lytic activity in germinating Bacillus anthracis spores. J. Bacteriol. 190:4541–4548. 10.1128/JB.00249-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Atrih A, Foster SJ. 2001. Analysis of the role of bacterial endospore cortex structure in resistance properties and demonstration of its conservation amongst species. J. Appl. Microbiol. 91:364–372. 10.1046/j.1365-2672.2001.01394.x [DOI] [PubMed] [Google Scholar]
- 12.Mock M, Fouet A. 2001. Anthrax. Annu. Rev. Microbiol. 55:647–671. 10.1146/annurev.micro.55.1.647 [DOI] [PubMed] [Google Scholar]
- 13.Sweeney DA, Hicks CW, Cui X, Li Y, Eichacker PQ. 2011. Anthrax infection. Am. J. Respir. Crit. Care Med. 184:1333–1341. 10.1164/rccm.201102-0209CI [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Setlow B, Melly E, Setlow P. 2001. Properties of spores of Bacillus subtilis blocked at an intermediate stage in spore germination. J. Bacteriol. 183:4894–4899. 10.1128/JB.183.16.4894-4899.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Makino S, Moriyama R. 2002. Hydrolysis of cortex peptidoglycan during bacterial spore germination. Med. Sci. Monit. 8:RA119–RA127 [PubMed] [Google Scholar]
- 16.Popham DL, Bernhards CB. Spore peptidoglycan. In Driks A, Eichenberger P. (ed), The bacterial spore: from molecules to systems, in press ASM Press, Washington, DC [Google Scholar]
- 17.Popham DL, Helin J, Costello CE, Setlow P. 1996. Muramic lactam in peptidoglycan of Bacillus subtilis spores is required for spore outgrowth but not for spore dehydration or heat resistance. Proc. Natl. Acad. Sci. U. S. A. 93:15405–15410. 10.1073/pnas.93.26.15405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Fukuoka S, Makino S. 2000. A novel spore peptidoglycan hydrolase of Bacillus cereus: biochemical characterization and nucleotide sequence of the corresponding gene, sleL. J. Bacteriol. 182:1499–1506. 10.1128/JB.182.6.1499-1506.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Y, Miyata S, Makino S, Moriyama R. 1997. Molecular characterization of a germination-specific muramidase from Clostridium perfringens S40 spores and nucleotide sequence of the corresponding gene. J. Bacteriol. 179:3181–3187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lambert EA, Sherry N, Popham DL. 2012. In vitro and in vivo analyses of the Bacillus anthracis spore cortex lytic protein SleL. Microbiology 158:1359–1368. 10.1099/mic.0.056630-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heffron JD, Sherry N, Popham DL. 2011. In vitro studies of peptidoglycan binding and hydrolysis by the Bacillus anthracis germination-specific lytic enzyme SleB. J. Bacteriol. 193:125–131. 10.1128/JB.00869-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lambert EA, Popham DL. 2008. The Bacillus anthracis SleL (YaaH) protein is an N-acetylglucosaminidase involved in spore cortex depolymerization. J. Bacteriol. 190:7601–7607. 10.1128/JB.01054-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heffron JD, Orsburn B, Popham DL. 2009. Roles of germination-specific lytic enzymes CwlJ and SleB in Bacillus anthracis. J. Bacteriol. 191:2237–2247. 10.1128/JB.01598-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Heffron JD, Lambert EA, Sherry N, Popham DL. 2010. Contributions of four cortex lytic enzymes to germination of Bacillus anthracis spores. J. Bacteriol. 192:763–770. 10.1128/JB.01380-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Giebel JD, Carr KA, Anderson EC, Hanna PC. 2009. The germination-specific lytic enzymes SleB, CwlJ1, and CwlJ2 each contribute to Bacillus anthracis spore germination and virulence. J. Bacteriol. 191:5569–5576. 10.1128/JB.00408-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chirakkal H, O'Rourke M, Atrih A, Foster SJ, Moir A. 2002. Analysis of spore cortex lytic enzymes and related proteins in Bacillus subtilis endospore germination. Microbiology 148:2383–2392 [DOI] [PubMed] [Google Scholar]
- 27.Paidhungat M, Ragkousi K, Setlow P. 2001. Genetic requirements for induction of germination of spores of Bacillus subtilis by Ca2+-dipicolinate. J. Bacteriol. 183:4886–4893. 10.1128/JB.183.16.4886-4893.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moriyama R, Hattori A, Miyata S, Kudoh S, Makino S. 1996. A gene (sleB) encoding a spore cortex-lytic enzyme from Bacillus subtilis and response of the enzyme to l-alanine-mediated germination. J. Bacteriol. 178:6059–6063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Setlow B, Peng L, Loshon CA, Li YQ, Christie G, Setlow P. 2009. Characterization of the germination of Bacillus megaterium spores lacking enzymes that degrade the spore cortex. J. Appl. Microbiol. 107:318–328. 10.1111/j.1365-2672.2009.04210.x [DOI] [PubMed] [Google Scholar]
- 30.Moriyama R, Fukuoka H, Miyata S, Kudoh S, Hattori A, Kozuka S, Yasuda Y, Tochikubo K, Makino S. 1999. Expression of a germination-specific amidase, SleB, of bacilli in the forespore compartment of sporulating cells and its localization on the exterior side of the cortex in dormant spores. J. Bacteriol. 181:2373–2378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christie G, Üstok FI, Lu Q, Packman LC, Lowe CR. 2010. Mutational analysis of Bacillus megaterium QM B1551 cortex-lytic enzymes. J. Bacteriol. 192:5378–5389. 10.1128/JB.00830-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Popham DL, Heffron JD, Lambert EA. 2012. Degradation of spore peptidoglycan during germination, p 121–142 In Abel-Santos E. (ed), Bacterial spores: current research and applications. Caister Academic Press, Norwich, United Kingdom [Google Scholar]
- 33.Moriyama R, Kudoh S, Miyata S, Nonobe S, Hattori A, Makino S. 1996. A germination-specific spore cortex-lytic enzyme from Bacillus cereus spores: cloning and sequencing of the gene and molecular characterization of the enzyme. J. Bacteriol. 178:5330–5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boland FM, Atrih A, Chirakkal H, Foster SJ, Moir A. 2000. Complete spore-cortex hydrolysis during germination of Bacillus subtilis 168 requires SleB and YpeB. Microbiology 146:57–64 [DOI] [PubMed] [Google Scholar]
- 35.Hu K, Yang H, Liu G, Tan H. 2007. Cloning and identification of a gene encoding spore cortex-lytic enzyme in Bacillus thuringiensis. Curr. Microbiol. 54:292–295. 10.1007/s00284-006-0430-x [DOI] [PubMed] [Google Scholar]
- 36.Li Y, Butzin XY, Davis A, Setlow B, Korza G, Üstok FI, Christie G, Setlow P, Hao B. 2013. Activity and regulation of various forms of CwlJ, SleB, and YpeB proteins in degrading cortex peptidoglycan of spores of Bacillus species in vitro and during spore germination. J. Bacteriol. 195:2530–2540. 10.1128/JB.00259-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Masayama A, Fukuoka H, Kato S, Yoshimura T, Moriyama M, Moriyama R. 2006. Subcellular localization of a germination-specific cortex-lytic enzyme, SleB, of bacilli during sporulation. Genes Genet. Syst. 81:163–169. 10.1266/ggs.81.163 [DOI] [PubMed] [Google Scholar]
- 38.Li Y, Jin K, Setlow B, Setlow P, Hao B. 2012. Crystal structure of the catalytic domain of the Bacillus cereus SleB protein, important in cortex peptidoglycan degradation during spore germination. J. Bacteriol. 194:4537–4545. 10.1128/JB.00877-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jing X, Robinson HR, Heffron JD, Popham DL, Schubot FD. 2012. The catalytic domain of the germination-specific lytic transglycosylase SleB from Bacillus anthracis displays a unique active site topology. Proteins 80:2469–2475. 10.1002/prot.24140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yeats C, Rawlings ND, Bateman A. 2004. The PepSY domain: a regulator of peptidase activity in the microbial environment? Trends Biochem. Sci. 29:169–172. 10.1016/j.tibs.2004.02.004 [DOI] [PubMed] [Google Scholar]
- 41.Janes BK, Stibitz S. 2006. Routine markerless gene replacement in Bacillus anthracis. Infect. Immun. 74:1949–1953. 10.1128/IAI.74.3.1949-1953.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hunter S, Jones P, Mitchell A, Apweiler R, Attwood TK, Bateman A, Bernard T, Binns D, Bork P, Burge S, de Castro E, Coggill P, Corbett M, Das U, Daugherty L, Duquenne L, Finn RD, Fraser M, Gough J, Haft D, Hulo N, Kahn D, Kelly E, Letunic I, Lonsdale D, Lopez R, Madera M, Maslen J, McAnulla C, McDowall J, McMenamin C, Mi H, Mutowo-Muellenet P, Mulder N, Natale D, Orengo C, Pesseat S, Punta M, Quinn AF, Rivoire C, Sangrador-Vegas A, Selengut JD, Sigrist CJ, Scheremetjew M, Tate J, Thimmajanarthanan M, Thomas PD, Wu CH, Yeats C, Yong SY. 2012. InterPro in 2011: new developments in the family and domain prediction database. Nucleic Acids Res. 40:D306–D312. 10.1093/nar/gkr948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. 1989. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene 77:51–59. 10.1016/0378-1119(89)90358-2 [DOI] [PubMed] [Google Scholar]
- 44.van den Ent F, Löwe J. 2006. RF cloning: a restriction-free method for inserting target genes into plasmids. J. Biochem. Biophys. Methods 67:67–74. 10.1016/j.jbbm.2005.12.008 [DOI] [PubMed] [Google Scholar]
- 45.Das D, Finn RD, Carlton D, Miller MD, Abdubek P, Astakhova T, Axelrod HL, Bakolitsa C, Chen C, Chiu HJ, Chiu M, Clayton T, Deller MC, Duan L, Ellrott K, Ernst D, Farr CL, Feuerhelm J, Grant JC, Grzechnik A, Han GW, Jaroszewski L, Jin KK, Klock HE, Knuth MW, Kozbial P, Krishna SS, Kumar A, Marciano D, McMullan D, Morse AT, Nigoghossian E, Nopakun A, Okach L, Puckett C, Reyes R, Rife CL, Sefcovic N, Tien HJ, Trame CB, van den Bedem H, Weekes D, Wooten T, Xu Q, Hodgson KO, Wooley J, Elsliger MA, Deacon AM, Godzik A, Lesley SA, Wilson IA. 2010. The structure of BVU2987 from Bacteroides vulgatus reveals a superfamily of bacterial periplasmic proteins with possible inhibitory function. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66:1265–1273. 10.1107/S1744309109046788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gao X, Wang J, Yu DQ, Bian F, Xie BB, Chen XL, Zhou BC, Lai LH, Wang ZX, Wu JW, Zhang YZ. 2010. Structural basis for the autoprocessing of zinc metalloproteases in the thermolysin family. Proc. Natl. Acad. Sci. U. S. A. 107:17569–17574. 10.1073/pnas.1005681107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignments through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680. 10.1093/nar/22.22.4673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim HU, Goepfert JM. 1974. A sporulation medium for Bacillus anthracis. J. Appl. Bacteriol. 37:265–267. 10.1111/j.1365-2672.1974.tb00438.x [DOI] [PubMed] [Google Scholar]
- 49.Nicholson WL, Setlow P. 1990. Sporulation, germination, and outgrowth, p 391–450 In Harwood CR, Cutting SM. (ed), Molecular biological methods for Bacillus. John Wiley and Sons Ltd., Chichester, England [Google Scholar]
- 50.Austin BP, Nallamsetty S, Waugh DS. 2009. Hexahistidine-tagged maltose-binding protein as a fusion partner for the production of soluble recombinant proteins in Escherichia coli. Methods Mol. Biol. 498:157–172. 10.1007/978-1-59745-196-3_11 [DOI] [PubMed] [Google Scholar]
- 51.Kapust RB, Tozser J, Fox JD, Anderson DE, Cherry S, Copeland TD, Waugh DS. 2001. Tobacco etch virus protease: mechanism of autolysis and rational design of stable mutants with wild-type catalytic proficiency. Protein Eng. 14:993–1000. 10.1093/protein/14.12.993 [DOI] [PubMed] [Google Scholar]
- 52.Kuwana R, Kasahara Y, Fujibayashi M, Takamatsu H, Ogasawara N, Watabe K. 2002. Proteomics characterization of novel spore proteins of Bacillus subtilis. Microbiology 148:3971–3982 [DOI] [PubMed] [Google Scholar]
- 53.Bagyan I, Noback M, Bron S, Paidhungat M, Setlow P. 1998. Characterization of yhcN, a new forespore-specific gene of Bacillus subtilis. Gene 212:179–188. 10.1016/S0378-1119(98)00172-3 [DOI] [PubMed] [Google Scholar]
- 54.Cutting S, Driks A, Schmidt R, Kunkel B, Losick R. 1991. Forespore-specific transcription of a gene in the signal transduction pathway that governs Pro-σK processing in Bacillus subtilis. Genes Dev. 5:456–466. 10.1101/gad.5.3.456 [DOI] [PubMed] [Google Scholar]
- 55.Wakeley PR, Dorazi R, Hoa NT, Bowyer JR, Cutting SM. 2000. Proteolysis of SpoIVB is a critical determinant in signalling of pro-σK processing in Bacillus subtilis. Mol. Microbiol. 36:1336–1348. 10.1046/j.1365-2958.2000.01946.x [DOI] [PubMed] [Google Scholar]
- 56.Fabret C, Hoch JA. 1998. A two-component signal transduction system essential for growth of Bacillus subtilis: implications for anti-infective therapy. J. Bacteriol. 180:6375–6383 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.