

Abstract
SUMMARY
Initially discovered in the context of immunomodulation, peptidyl-prolyl cis/trans isomerases (PPIases) were soon identified as enzymes catalyzing the rate-limiting protein folding step at peptidyl bonds preceding proline residues. Intense searches revealed that PPIases are a superfamily of proteins consisting of three structurally distinguishable families with representatives in every described species of prokaryote and eukaryote and, recently, even in some giant viruses. Despite the clear-cut enzymatic activity and ubiquitous distribution of PPIases, reports on solely PPIase-dependent biological roles remain scarce. Nevertheless, they have been found to be involved in a plethora of biological processes, such as gene expression, signal transduction, protein secretion, development, and tissue regeneration, underscoring their general importance. Hence, it is not surprising that PPIases have also been identified as virulence-associated proteins. The extent of contribution to virulence is highly variable and dependent on the pleiotropic roles of a single PPIase in the respective pathogen. The main objective of this review is to discuss this variety in virulence-related bacterial and protozoan PPIases as well as the involvement of host PPIases in infectious processes. Moreover, a special focus is given to Legionella pneumophila macrophage infectivity potentiator (Mip) and Mip-like PPIases of other pathogens, as the best-characterized virulence-related representatives of this family. Finally, the potential of PPIases as alternative drug targets and first tangible results are highlighted.
INTRODUCTION
Bacterial virulence is a multilayered phenomenon, and only in some exceptional cases is it determined by a single factor. Virulence factors rather exert their function additively or in varying interdependence so that they promote optimal colonization, production of offspring, and propagation of pathogens. Most virulence factors are highly specialized determinants shaped by the adaptation of the pathogen to its host niche, e.g., toxins, secretion systems, and their effectors or receptor binding proteins. In contrast, some others, such as hydrolases, transporters, or chelators, are of a more general nature in their biological function and distribution among pathogenic and nonpathogenic organisms.
Peptidyl-prolyl cis/trans isomerases (PPIases) belong to the latter group of proteins. These ubiquitously distributed proteins catalyze the cis/trans isomerization of peptide bonds preceding prolyl residues, thereby assisting protein folding (1). Nevertheless, growing evidence from recent years points at virulence-associated functions of members of this protein family in pathogens. In this review, we give an overview of PPIases as such and their contributions to virulence in bacterial and protozoan parasites, with a focus on the group of macrophage infectivity potentiator (Mip)-like PPIases and their potential as drug targets.
A SHORT RETROSPECTIVE ON PPIases
The discovery of PPIases (EC 5.2.1.8), a superfamily currently comprising three distinct families, dates back to the 1980s and was driven by the observation of the relatively slow cis/trans conversion rate of peptidyl-prolyl bonds and the influence of this on protein folding and denaturation kinetics (2, 3). In almost all peptide bonds, the trans conformer is favored over the cis conformer, because of the steric hindrance of the side chains in the latter constellation and the resulting free energy difference of approximately 2.6 kcal/mol (Fig. 1A). However, due to the unique structure of proline, which results from the cyclization of the side chain with the α-amino group forming a pyrrolidine ring, the levels of steric hindrance in both isomers of a peptidyl-prolyl bond are comparable (Fig. 1B). Hence, the free energy difference between the trans conformer of a peptidyl-prolyl bond and its cis isomer is about 0.5 kcal/mol (4). This causes peptidyl-prolyl bonds, which have no significant energetic preference for one of the two conformations, to slow down protein dynamics by a factor of 100 during folding and denaturation (2). Accordingly, the effect of peptidyl-prolyl isomerization on folding could be observed in small peptides and also during the refolding of denatured RNase A, which exists in an equilibrium of fast- and slow-folding forms due to its four peptidyl-prolyl bonds (3, 5, 6).
FIG 1.

Conformations of peptide bonds. (A) In almost all peptide bonds, the trans conformation is energetically favored because the steric hindrance between the side chains (R1 and R2) of the two consecutive amino acids is the lowest in this conformation. (B) In peptidyl-prolyl bonds, where the α-amino group is part of the side chain, and thus an imide peptide bond is formed, the difference in free energy of the two conformers is comparably smaller. Hence, about 5 to 6% of peptidyl-prolyl bonds are in cis conformation, and the isomerization rate, with half-lives ranging from seconds to minutes, is noticeably long (4, 295).
An enzymatic activity which accelerates the cis/trans isomerization of peptidyl-prolyl bonds was detected for the first time in porcine kidney extracts, initially on peptide substrates and later also on RNase A and some other proteins (7–10). Consecutive efforts to isolate the first PPIase resulted in the finding that it was identical to cyclophilin A (CypA) (11, 12). This 17-kDa protein had previously been named after its interaction with the undecapeptide cyclosporine (CsA), a natural product with immunosuppressive properties isolated from the fungus Trichoderma polysporum (13–15). Coincidentally, another immunosuppressant secondary metabolite, the macrolide FK506, isolated from the soil bacterium Streptomyces tsukabensis, soon enabled the identification of the next representative of PPIases, which was named, accordingly, FK506 binding protein (FKBP) (16, 17). These studies also confirmed that CypA and FKBP were distinct proteins with distinct ligand specificities, because FK506 and CsA did not inhibit the PPIase activities of CypA and FKBP, respectively. To this point, inhibitor specificity was one of the major discriminatory properties of different PPIase families and was decisive for the discovery of the third family of PPIases, which did not respond to CsA or FK506. A small protein of approximately 10 kDa was discovered and named parvulin (18–20). Later it was shown that parvulins can be inhibited by the small 1,4-naphthoquinone derivative juglone (21).
Now, almost 30 years after their initial discovery, PPIases constitute a biochemically well-defined superfamily of proteins. However, in many instances, their cellular and physiological functions remain enigmatic. This is mainly due to the fact that despite their clear-cut enzymatic activity, which can be measured in established biochemical assays, many of the in vitro or in vivo observable phenotypes of mutants and PPIase-protein interactions seem to be independent of the enzymatic property. In many instances, deletion of the PPIase domain or diminishment of its activity by amino acid substitutions had no or little impact on protein-protein interactions and chaperoning activities (22–24). Hence, the initial idea that PPIases are primarily facilitators of protein folding by specifically influencing the isomerization of peptidyl-prolyl bonds has now shifted to a broader interpretation of their cellular functions from acting as chaperones to being accessory proteins in multiprotein complexes. As such, they are involved in diverse processes, from signal transduction to gene regulation on the transcriptional and translational levels (1, 25). Also, there are many reported deletion mutants which show no or very subtle phenotypic changes under laboratory conditions (26, 27). In a seminal work in which 12 PPIases were deleted in Saccharomyces cerevisiae, no prominent effect could be observed (28). Similarly, deletion of all known periplasmic PPIases of Yersinia pseudotuberculosis did not result in a measurable phenotype under laboratory growth conditions; however, the generated quadruple mutant was impaired during colonization of mice (29). Thus, the contribution of PPIases to cellular physiology is thought to be very specific and dependent on growth conditions and the niche in which a cell resides.
GENERAL PROPERTIES OF PPIases AND THEIR ENZYMATIC ACTIVITIES
PPIases exert a very general enzymatic activity, and their phenotypic impact is experimentally difficult to define. But several characteristics of PPIases can be linked to their in vivo roles, as follows.
Composition of a Peptidyl-Prolyl Bond
Although not consequently approved for in vivo PPIase-protein interactions, peptide-based enzyme assays clearly show that not every peptidyl-prolyl bond is equally targeted and isomerized by a PPIase (30). The amino acid preceding the proline (i.e., position P1) is often crucial for the enzymatic activity and selectivity. Human FKBP12 (hFKBP12) preferentially isomerizes peptides containing a leucine or a phenylalanine residue at P1, whereas the bacterial homolog Mip of Legionella pneumophila, despite high conservation within the enzymatically active cavity, lacks the preference for leucine and can isomerize peptides with a lysine residue at P1 comparably well (31). Other examples of selectivity are the human parvulin Pin1 and its homologs in several pathogenic and nonpathogenic yeast species, which specifically recognize phosphotyrosine or phosphoserine residues at P1 (32, 33). Other than sequence specificity, steric accessibility of a peptidyl-prolyl bond is also a factor determining the action of PPIases (1, 34).
Modular Structure
The smallest representatives of peptidyl-prolyl isomerases, e.g., the 12-kDa hFKBP12, the 18-kDa human CypA protein, and the 10-kDa parvulin from Escherichia coli, reveal only a single PPIase domain and are accordingly small. However, all three classes also have members that possess additional domains. The most common among these are tetratricopeptide repeats (TPR), which form α-helical bundles and contribute to the assembly of multiprotein complexes by mediating protein-protein interactions (35, 36). Later, PPIases were found to contain the protein-protein interaction domains WD40 and RanBD1, as well as the RNA recognition motif RRM, zinc finger domains, and calcium-binding EF-hand motifs. These additional domains facilitate interactions with a multitude of partners and equip PPIases with chaperone activities (36, 37). Some FKBPs have multiple PPIase domains, most probably due to gene duplication events (38, 39). Recently, a novel type of PPIase (FCBP) which contains an FKBP and a cyclophilin domain was also discovered in protozoan parasites, such as Toxoplasma gondii, as well as in the bacteria Flavobacterium johnsonii and Treponema denticola. In the dual PPIase of T. gondii, both enzymatic domains are separated by three TPR domains. The exact in vivo role of this new type of isomerases and their distribution in nature still need to be explored (40, 41).
The influence of the modularity of PPIases on their enzymatic properties was nicely demonstrated by combining the archetypical hFKBP12 with the chaperone domains of unrelated PPIases. Upon fusion with the chaperone domain of SlyD, an FKBP of E. coli, hFKBP12 lost its specificity for leucine or phenyalanine residues on P1 and was able to isomerize Lys-Pro, Ala-Pro, and Glu-Pro bonds equally well, and with high efficiency (42). The same observation could be made even when chaperone domains of unrelated protein families, such as the protein disulfide isomerase (PDI) of yeast, the chaperonin GroEL, or parvulin SurA of E. coli, were used (43).
Cellular Localization
Besides their modular architecture, the action of peptidyl-prolyl isomerases can be identified by their cellular localization. In eukaryotes, PPIases are located in the cytosol, nucleus, mitochondria, chloroplasts, or endoplasmic reticulum (ER), and they can even be secreted (36, 44–46). In bacteria, PPIases can be divided roughly into soluble and membrane-bound subgroups. Soluble PPIases are either cytosolic or, in Gram-negative bacteria, periplasmic (47). Among soluble PPIases, some, including the parvulins HP0175 of Helicobacter pylori and SurA of Brucella abortus, as well as two cyclophilins of Legionella pneumophila, PpiB (Lcy) and Lpg1962, are known to be secreted into the supernatant (48–50). Membrane-bound bacterial PPIases can be facing the outside or the periplasm, depending on the membrane where they are anchored (51–54). Depending on their localization, PPIases have access to only a subset of proteins, by which their substrate spectrum is defined.
Differential Expression
An additional feature of PPIases that helps to determine their physiological role is their differential expression. In prokaryotes and eukaryotes, several PPIases are upregulated by stress factors, such as heat or cold shock, due to the accumulation of misfolded proteins under these conditions (49, 55–57). Some others, such as the parvulin PrsA2 of the Gram-positive intestinal pathogen Listeria monocytogenes or the FKBP12 homolog of the plant-pathogenic fungus Botrytis cinerea, are upregulated during infection and significantly contribute to infection (58, 59). Additionally, in metazoans, differential expression can also be regulated developmentally or be tissue specific. The ER-resident protein FKBP65, for example, is highly expressed during embryonic development, especially in the lungs. Its expression ceases during adulthood but increases after lung injury, as part of the repair process (60, 61). Many FKBPs, although initially associated with immunomodulatory processes, were later found to be expressed at much higher degrees in brain tissue, with neuroprotective or neuroregenerative properties, and hence are considered important factors in neurological diseases (25, 62). Differential expression in tissues and during development was also observed for plant PPIases, such as in the case of maize or wheat (56, 63). All these data are indicative of the physiologic versatility of this protein family.
MICROBIAL PPIases
Bacterial PPIases
Exploration of eukaryotic PPIases has been driven mainly by their drug-related immunomodulatory properties, their potential in therapeutic applications, especially in transplantation medicine, and, later on, the neuroprotective action of mammalian FKBPs in neurodegenerative diseases (1, 25). In contrast, research on bacterial PPIases has been concentrated more on the maturation of proteins during translation and secretion. Similarly, in eukaryotic systems, much research has been done to identify interaction partners within signal transduction pathways where PPIases seem to play pivotal roles (64, 65). Interaction studies are rather scarce in cases where the general approach is more phenotype oriented, with the exception of studies of the cytosolic FKBP trigger factor (TF) and the periplasmic parvulin SurA in bacterial systems. But even in these two cases, the focus of research lies on protein folding during translation and secretion, respectively.
No major structural discrimination can be made between the enzymatic domains of eukaryotic and prokaryotic PPIases of the same family. Structural differences can, however, be found between the families (1, 36). In general, PPIase domains are globular and comprised of 100 to 120 amino acids that form a central structure composed of β-folds (Fig. 2). In FKBPs (Fig. 2A), this central structure is an amphipathic five-stranded β-sheet that wraps around an α-helix, forming a hydrophobic cavity where inhibitors and possible substrates bind (66). In cyclophilins (Fig. 2B), an eight-stranded antiparallel β-barrel is flanked by an α-helix on each side (67). Finally, in parvulins (Fig. 2C), the β-sheet core is surrounded by four α-helices (68).
FIG 2.

Representative protein structures for bacterial PPIases. Displayed are structures of the representative members of the three PPIase families of Escherichia coli. (A) PPIase domain of the FKBP FkpA (PDB entry 1Q6U) showing the characteristic α-helix wrapped by a five-stranded β-sheet. The space between the helix and the β-sheet forms the enzymatically active hydrophobic pocket. (B) PPIase domain of the cyclophilin PpiB (PDB entry 2NUL), consisting of the centrally located β-barrel flanked by one α-helix on each side. (C) PPIase domain of the parvulin Par10 (PDB entry 1JNT), organized in the form of a central antiparallel sheet of 4 β-strands with 4 α-helices surrounding it.
Representatives of all three families can be found to different extents in bacteria. In the genome of the Gram-negative model organism E. coli K-12, four FKBPs, two cyclophilins, and three parvulins are encoded. In contrast, the model organism of Gram-positive bacteria, Bacillus subtilis, has one cyclophilin, two parvulins, and no FKBP-type PPIase except for TF (Table 1). Considering that some of the additional PPIases in E. coli, i.e., FkpA, PpiA, and FklB, are periplasmic leads to the assumption that in Gram-negative bacteria the presence of the outer membrane and the additional compartment (periplasm) requires a larger repertoire of accessory folding proteins. Despite this high variation in the number of PPIases between single species, the presence of at least one is apparently of vital importance, as even the free-living bacterium with the smallest genome, Mycoplasma genitalium, possesses TF (69).
TABLE 1.
PPIases of E. coli and B. subtilisb
| Family | Protein (gene accession no.a) | Homolog in B. subtilis (gene accession no.a) | % Identity/% similarity |
|---|---|---|---|
| FKBPs | FkpA (947850) | ND | ND |
| FkpB (944807) | ND | ND | |
| FklB (948726) | ND | ND | |
| SlyD (947859) | ND | ND | |
| Trigger factor/Tig (945081) | Tig (14770493) | 37/55 | |
| Cyclophilins | PpiA (947870) | PpiB (2634771) | 39/49 |
| PpiB (949038) | 29/39 | ||
| Parvulins | SurA (944812) | PrsA (14768595) | 33/51 |
| PpiD (946056) | YacD (14767585) | 32/55 |
As annotated for E. coli K-12 strain MG1655 and B. subtilis strain 168.
ND, no homolog present.
TF is a modular protein with a central FKBP domain flanked by an N-terminal ribosome binding domain with chaperone activity and a C-terminal domain of unknown function (70). It associates with ribosomes via its N-terminal domain, in a 1:1 ratio, whereby the association with nascent peptides lasts longer than the association with ribosomes, indicating that multiple TF proteins are involved in the maturation of newly formed proteins (24, 70). Initially, this association was thought to be the indication for an ultimate physiological function of a PPIase, since during translation, nascent polypeptides exit the ribosomes unfolded, with great accessibility of isomerization-prone peptidyl-prolyl bonds. But although binding of the PPIase domain to nascent peptides could be demonstrated, this property was not dependent on the enzymatic activity (71).
Further FKBPs in E. coli are the periplasmic FkpA and FklB proteins, both of which contain N-terminal dimerization domains, are known to be involved in the transport of some secreted proteins, and also act as chaperones (72–75). The cytoplasmic PPIase SlyD of Gram-negative bacteria consists of an N-terminal PPIase domain and a C-terminal nickel binding domain. Interestingly, in E. coli, SlyD mutations confer resistance toward the phage φX174. Apart from that, in E. coli and Helicobacter pylori, SlyD is also involved in the maturation of nickel-containing hydrogenases independently of its PPIase activity (76–79).
Bacteria typically possess two cyclophilins: PpiA and -B. In Gram-negative bacteria, PpiA is periplasmic, whereas in Gram-positive bacteria, it is associated with the cytoplasmic membrane facing the outside and involved in the transport of secreted proteins (47, 54, 80). PpiB is a cytoplasmic PPIase and is upregulated in B. subtilis upon cold shock (81, 82).
Parvulins, including SurA in E. coli, PrsA in B. subtilis, and PrsA2 in L. monocytogenes, are some of the most investigated bacterial PPIases. SurA is a periplasmic protein central to the transport into and correct assembly of many β-barrel proteins in the outer membranes of Gram-negative bacteria (83). It has a modular structure with one or two parvulin domains, depending on the bacterial species, as well as a longer N- and a shorter C-terminal chaperone domain. In E. coli, SurA has two domains with parvulin folds, among which only the C-terminal one has enzymatic activity. However, the PPIase domain is very often negligible for in vivo activity, and no major defects in outer membrane maturation can be observed in its absence (23). In E. coli and many Gram-negative bacteria, there is also a second periplasmic parvulin, named PpiD (52). Its sole parvulin domain seems to be devoid of PPIase activity, and its contribution to protein secretion remains difficult to disentangle (84–86). PrsA can be considered the functional homolog of SurA in B. subtilis and other Gram-positive bacteria, since it is localized to the cytoplasmic membrane facing outside (19, 87–89). Hence, it facilitates the maturation of secreted proteins. Since it is the only PPIase in the secretory pathway, PrsA is an essential protein in B. subtilis, in contrast to the case in L. monocytogenes, which has the additional membrane-localized parvulin PrsA2 (87, 90, 91). Similar to the case of SurA, for B. subtilis PrsA the PPIase activity itself is dispensable for secretion and the majority of in vitro phenotypes, as shown for PPIase-negative single amino acid mutants. However, interestingly enough, the PPIase domain still seems to be important for full action, since deletion of the parvulin domain prevented the secretion of the alpha-amylase AmyQ (87).
PPIases of Parasitic Protozoa
The extent of knowledge about PPIases in protozoa is more limited than that for metazoans. In protozoa, the main focus of research regarding PPIases lies on the human-pathogenic species. This was also driven by the observation that CsA and FK506 have antiparasitic properties (92–94). Accordingly, the first characterized protozoan PPIases were cyclophilins of Toxoplasma gondii and Plasmodium falciparum (95–97). As a matter of fact, most of the characterized protozoal PPIases are cyclophilins, while FKBPs and parvulins are less represented in the literature.
An extensive in silico genome survey revealed that apicomplexans possess large numbers of cyclophilins (from 7 in Cryptosporidium to 15 in Toxoplasma), which are diverse in their cellular localization and domain composition (41). All seven cyclophilins of Cryptosporidium hominis have homologs in T. gondii or P. falciparum. Interestingly, among the analyzed organisms, the diarrhea-causing species C. hominis has the simplest infection cycle. Infected individuals release fastidious oocysts into the environment that are then taken up via food or drinking water. Theileria parva and Babesia bovis, both of which are parasites of cattle, are transmitted by insect vectors to their final hosts, and each carries 10 cyclophilins and one hybrid protein containing FKBP and cyclophilin (FCBP) domains. Finally, P. falciparum and T. gondii possess the most complicated infection cycles, with either broad-tissue tropism in the respective host, as in the case of the blood and liver stages of P. falciparum (11 cyclophilins), or a higher diversity in intermediate hosts, as in the case of T. gondii (14 cyclophilins and 1 FCBP). Accordingly, it can be speculated that the occurrence of additional cyclophilins in a parasite may reflect the complexity of the corresponding life cycle.
Understandably, this diversity is far from being explored to its fullest extent, and details about physiological functions, roles during infection, and parasite infection cycles are known for only a very few representatives. For example, 8 of the 11 putative cyclophilins and cyclophilin-like proteins of P. falciparum were analyzed after recombinant production. Only two of those, P. falciparum Cyp19A (PfCyp19A) and PfCyp19B, bound CsA and displayed in vitro PPIase activity, while all had chaperone functions preventing thermal aggregation of the model protein substrates rhodanese and citrate synthase (98, 99). This alone can be taken as an indication of the unexplored functional diversity of cyclophilins in parasitic protozoa, depending on their developmental or infection cycle stage.
Among the 15 putative cyclophilins of T. gondii, Cyp18 (TgCyp18), with an approximate molecular mass of 18 kDa, is the only one that has been examined in detail so far. It is secreted by infectious tachyzoites into the culture supernatant and was identified as an inducer of interleukin-12 (IL-12) production by dendritic cells. This induction was due to the interaction of TgCyp18 with cysteine-cysteine chemokine receptor 5 (CCR5) and was inhibited by the addition of CsA (100). Nevertheless, the direct involvement of the PPIase activity in this interaction was considered rather unlikely, because this effect could not be reproduced when human or plasmodial cyclophilin homologs were used. A follow-up study with site-directed amino acid substitutions revealed that binding to CCR5 was independent of the PPIase activity, since only mutants with substitutions at the N- and C-terminal parts of the protein were affected. Curiously, mutants with diminished PPIase activity failed to induce IL-12 production by dendritic cells (101). However, how the PPIase activity itself contributes to IL-12 production is still not clear. A possible explanation is that additional secreted TgCyp18-specific substrates of T. gondii contribute to the induction of IL-12 production. Inhibition by CsA may also be due to structural changes in TgCyp18 caused by binding of the drug (100). The decoupling of IL-12 production from CCR5 binding and the fact that TgCyp18 was not able to induce IL-12 production in neutrophils also suggest that T. gondii has multiple immunomodulatory mechanisms that are complementary to each other (102).
A similar immunomodulatory effect was also observed in the case of a secreted 19-kDa cyclophilin of the coccidian parasite Neospora caninum, which causes spontaneous abortions in canids and cattle. N. caninum Cyp (NcCyp) has 86% sequence similarity to TgCyp18 and is able to elicit a gamma interferon (IFN-γ) response in peripheral blood monocytes and CD4+ T cells. These effects could be inhibited by CsA in a dose-dependent manner. But, again, whether this was due to the inhibition of the enzymatic activity or to overall structural changes in the protein-drug complex is not known (103). As in N. caninum, TgCyp18 homologs are present in all analyzed apicomplexan organisms (41), but whether they contribute to the infection cycles of the respective parasites needs further experimental evaluation, as is the case for all the remaining members of the cyclophilin protein family.
In this respect, three different groups of cyclophilins, as identified by Krücken et al. (41), may be extremely interesting for future antiparasitic therapy strategies (Table 2). The first group is the small apicomplexan-specific cyclophilins, which seem to be present in all genera except Cryptosporidium (41). Although these PPIases segregate clearly in a phylogenetic analysis and have no mammalian orthologs, their monophylogeny is still questionable. All identified representatives are predicted to be cytoplasmic, except for TgCyp38, which possesses an N-terminal mitochondrial targeting signal. Among the cytosolic members, some, such as Cyp20 of T. parva (TpCyp20), consist of a sole PPIase domain, while PfCyp26 and B. bovis Cyp28 (BbCyp28) have C-terminal extensions with no homologous sequences in current databases (41). The second group consists of cyclophilins with an N-terminal apicoplast-targeting signal, as found in the genus Theileria. The apicoplast, together with the single mitochondrion, contributes to the specific metabolic needs of apicomplexan parasites, depending on the host niche to which they are adapted (104). It is puzzling that this group is currently restricted to one genus, despite the central role of this organelle. One explanation for this may be that the algorithms searching for transport signals are optimized for P. falciparum (41). The third group contains Plasmodium-specific large cyclophilins with multiple nuclear localization signals and coiled-coil protein interaction domains, which suggests that they might be involved in RNA processing. Single representatives of this group were identified in P. falciparum and its close relative Plasmodium yoelii (41).
TABLE 2.
Apicomplexan-specific cyclophilins
| Group | Organism | Protein (gene accession no.) | Localizationa | Additional domain(s) and function(s)b |
|---|---|---|---|---|
| Small apicomplexan-specific cyclophilins | B. bovis | BbCyp28.6 (5477723) | Cytoplasm | C-terminal extension with no known homology |
| P. falciparum | PfCyp26.4 (811077) | Cytoplasm | C-terminal extension with no known homology | |
| T. parva | TpCyp20.3 (3502670) | Cytoplasm | ND | |
| T. gondii | TgCyp31.8 (052840) | Mitochondrion | N-terminal mitochondrial localization signal, C-terminal extension with no known homology | |
| Apicoplast-specific cyclophilins | Theileria annulata | TaCyp25.7 (3864051) | Apicoplast | N-terminal apicoplast localization signal |
| T. parva | TpCyp25.5 (3502505) | Apicoplast | N-terminal apicoplast localization signal | |
| Plasmodium-specific cyclophilins | P. falciparum | PfCyp72.5 (813578) | Nucleus | Multiple nuclear localization signals, N-terminal RNP motif for RNA binding, internal coiled-coil protein interaction domains |
| P. yoelii | PyCyp69.8 (3830381) | Nucleus | Multiple nuclear localization signals, internal coiled-coil protein interaction domains |
Predicted.
Putative. ND, not determined.
Cyclophilins are also highly represented in trypanosomal parasites, as exemplified by the agent of Chagas disease, Trypanosoma cruzi, whose genome encodes 15 paralogs of this family, in the mass range of 19 to 110 kDa. While orthologs in other genomes could be identified for eight of the genes, the sequence diversity of the remaining seven was too high, preventing a solid classification (105). Although the smallest representative in T. cruzi, Cyp19, and its homolog in other Tryponosoma species, CypA, have been expressed recombinantly and characterized enzymatically, not many biological functions have been described (106–108).
In trypanomastoid parasites of the genus Leishmania, the main cyclophilin is the homolog of CypA and has a molecular mass of approximately 18 kDa. LmCypA, the CypA protein of Leishmania major, which causes cutaneous lesions after replicating in macrophages and dendritic cells, also responds to CsA, but the parasites are extracellularly more resistant to CsA than others. The reason for this was found to be an arginine-to-asparagine substitution at the calcineurin docking site in LmCypA, which is crucial for the interaction when the protein is in complex with CsA (109). Hence, the antileishmanial activity of CsA observed in mice or in in vitro macrophage infections is due to the inhibition of host calcineurin (110). In addition, in Leishmania donovani, the pathogen causing visceral leishmaniasis, the unusually low cytosolic concentration of L. donovani Cyp (LdCyp) is thought to be a reason for increased CsA tolerance (111). Apart from this, biological functions appointed to LdCyp include the disaggregation and reactivation of adenosine kinase, a crucial enzyme of purine metabolism and a key to parasite differentiation processes (112, 113). However, this function is not sensitive to CsA and is independent of the PPIase activity (114).
In Entamoeba histolytica, the causative agent of amoebal diarrhea, a CsA-responsive cyclophilin could be isolated and biochemically analyzed (115). Also, a PPIase, which was not further characterized, was upregulated in trophozoites of E. histolytica after a 2-h exposure to oxygen stress (116). Since E. histolytica is an obligate anaerobe that prevails in the environment in the form of stress-resistant and infective cysts, the upregulated PPIase might be an important factor for adaptation to hostile conditions during the infection cycle, hence representing a relevant virulence factor.
In contrast to the case for cyclophilins, the numbers of identified and analyzed FKBPs and parvulins in protozoan parasites are small. The only FKBPs analyzed in detail are PfFKBP35 of P. falciparum and Plasmodium vivax FKBP35 (PvFKBP35) (117–119). FKBP35 contains, in addition to its PPIase domain, a TPR domain, which facilitates interaction with Hsp90. It can inhibit plasmodial calcineurin without FK506, which is unique within this family of proteins (117). However, no strong colocalization of FKBP35 with calcineurin could be observed in cells (118). FKBP35 seems to be shuttled between the nucleus and the cytoplasm in a stage-dependent manner. While in the ring stage it is localized mainly to the cytosol, in the trophozoite and schizont stages it is translocated into the nucleus. Interestingly, in the last two stages, the concentration of calcineurin in the cytoplasm increases measurably. This differential localization of the two proteins probably prevents an inhibitory action of FKBP35 on calcineurin (118). While FKBP35 seems to be the only PPIase in P. falciparum, in the other important human plasmodial pathogen, P. vivax, a second representative of this family, FKBP25, was recently identified. This PPIase is predicted to be nuclear and contains, in contrast to human FKBP25, a calmodulin binding domain (120).
The first identified parvulin-like protein was an immunodominant antigen of T. gondii, TgMIC5, which was localized in the secretory organelle (microneme) of the infectious tachyzoites (121). It was shown to be secreted into the surrounding medium. Although TgMIC5 seems to be involved in the maturation of other surface proteins by facilitating their proteolysis, the type of involvement of the parvulin-like domain in those processes is not clear (122, 123). Besides that protein, TcPin1 and TcPar45 of T. cruzi, as well as their homologs TbPin1 and TbPar42 of Trypanosoma brucei, are the only parvulins described for protozoan parasites (124–127). TcPin1 is able to complement the temperature-related phenotypes of its homolog Ess1 in S. cerevisiae, suggesting similar physiological functions (124). Interestingly, unlike its metazoan homologs, Pin1 is localized in the cytoplasm and not in the nucleus. Knocking down pin1 by RNA interference (RNAi) had no detrimental effects on cell proliferation. Accordingly, it was speculated that Pin1 of T. cruzi might participate in signaling mechanisms in protozoan parasites that are different from those in metazoans (126). The 45-kDa protein TcPar45 displayed more similarities to human Par14, since it was located in the nucleus and did not isomerize Glu-Pro bonds. RNAi knockdowns of TcPar45 and TbPar42 resulted in growth defects in procyclic parasites (125, 127). For TbPar42, a preference for peptides containing phosphorylated serine or threonine residues preceding the target proline could be proven, which is also known for other parvulins (127, 128).
PPIases IN VIRULENCE
Bacterial PPIases in Virulence
The role of PPIases during pathogenic processes and their contribution to the virulence of bacteria and protozoa are still not fully understood. Research on bacterial virulence-associated PPIases focuses mainly on periplasmic or membrane-bound PPIases. Also, many studies attribute a rather secondary role in virulence to PPIases, mostly caused by improper folding or secretion of virulence factors, such as adhesins or degradative enzymes, in the absence of a PPIase (83, 129–132). Again, typical representatives of PPIases with rather accessory roles in virulence are the periplasmic parvulin SurA and the cytoplasmic FKBP TF.
The involvement of SurA in pathogenic processes was analyzed most extensively in the Enterobacteriaceae (83). Being a key player in periplasmic transport, together with the chaperones DegP and DnaK as well as the β-barrel assembly (BAM) complex, SurA facilitates the correct assembly of many outer membrane proteins (OMPs), including outer membrane protein A (OmpA), type I pilus components, the maltose transporter (LamB), and the two siderophore receptors FhuA and FecA, which are known virulence factors (133–135). A reduction in type I pilus assembly in the ΔsurA mutant of uropathogenic E. coli (UPEC) results in decreased invasion of bladder cells. Furthermore, after invasion, the bacterium cannot form intracellular bacterial colonies, a hallmark of persistent infection (133, 136). Fimbriae of UPEC are translocated by the chaperone-usher mechanism, where the usher facilitating the transport of the pilin subunit across the outer membrane is a β-barrel protein, such as FimD in the case of type I fimbriae (137). Accordingly, the folding and assembly of FimD were shown to be dependent on SurA together with BamB, a nonessential component of the BAM complex (138). In enteropathogenic E. coli (EPEC), the secretion of the major adhesin, intimin, depends on SurA, as deletion of surA results in a 4-fold decrease in surface exposure of intimin. Hereby, SurA mediates the insertion of the N-terminal β-barrel domain into the outer membrane, in concert with the BAM complex and the Skp-DegP chaperone-protease system (139). However, at least in UPEC, the PPIase domains of SurA are negligible for the in vitro pleiotropic effects (140).
Yersinia pseudotuberculosis causes self-limiting gastrointestinal disorders. Host colonization is mediated by several adhesins on the cell surface. The assembly of one of these, the invasin, is strongly dependent on chaperone and PPIase activities of SurA, since complementation of surA deletion with chaperone or PPIase domains only could not entirely restore this deficiency (141). This study is also interesting because it points to the fact that in the absence of SurA, major OMPs, such as OmpF, or components of the BAM complex, i.e., BamA, -C, and -E, are affected in their targeting to the outer membrane. Considering that OMPs stabilize the outer membrane and the BAM complex is central to translocation of many β-barrel proteins, SurA can be considered crucially important to bacterial transport processes and cell integrity. In the enteropathogenic organism Salmonella enterica serovar Enteritidis, SurA affects the expression of OMPs without influencing the amounts of secreted substrates of the type III secretion system, the main virulence determinant (142). In Shigella flexneri, deletion of surA causes a defect in the correct localization of IcsA, which facilitates the polymerization of actin at the bacterial pole so that the bacterium can use the actin tail for intercellular spread (130).
An interesting finding regarding SurA-mediated transport comes from the plant pathogen Dickeya dadantii, which has a broad host spectrum. In the economically important crop potato, it causes soft rot disease due to maceration of host tissue by degrading enzymes (143). One such enzyme is the pectin-degrading lyase PnlH, which is targeted to the outer membrane after Tat-mediated translocation into the periplasm. PnlH is devoid of a β-barrel structure typical for OMPs or an acylation signal. Its targeting to the outer membrane was instead shown to be dependent on its highly hydrophobic N-terminal signal sequence, which most probably is recognized by SurA and, by this, protected from degradation (144). The authors of that study showed that the chaperone activity of SurA is not restricted to β-barrel proteins.
Parvulins have also been described for further Gram-negative pathogens in the context of virulence. One of these is HP0175, which was isolated from the culture supernatants of in vitro-grown Helicobacter pylori, an epsilonproteobacterium that causes chronic gastritis and is widely distributed among humans. Interestingly, this secreted PPIase is also one of the five immunodominant antigens detected in gastroduodenal ulcers (48). As shown in a follow-up study, HP0175 induces apoptosis in the gastric epithelial cell line AGS by binding to Toll-like receptor 4 (TLR4) on the cell surface and, by this, activating ASK1, a kinase upstream of mitogen-activated protein (MAP) kinase (145). This PPIase is of particular interest not only because it is one of the few reported soluble extracellular bacterial PPIases but also because, together with Mip, it is the only PPIase with a reported eukaryotic binding partner. Thus, further evaluation of the interaction mechanisms of HP0175 with TLR4 will help our understanding of the mechanisms of this chronic disease with high prevalence in the human population.
The search for binding partners of filamentous hemagglutinin, the main adhesin of the betaproteobacterium Bordetella pertussis, which causes whooping cough, led to the identification of Par27, a novel type of parvulin, by affinity chromatography (146). In contrast to other parvulins, Par27 possesses atypical N- and C-terminal extensions that surround the PPIase domain, confer structural flexibility to the protein, and allow dimerization (147). It was shown that Par27 has PPIase and chaperone activities, which probably have pleiotropic effects on virulence by facilitating secretion processes in the periplasm (146). A homolog of Par27, in contrast, was discovered in the periplasm of the epsilonproteobacterium and intestinal pathogen Campylobacter jejuni and was named PEB4, without any further implication for virulence (148).
The functional homolog of SurA of Gram-negative bacteria is PrsA of Gram-positive bacteria, which is associated with the membrane, reaching into the compartment between the cell membrane and the peptidoglycan wall. In contrast to the nonpathogenic species B. subtilis, L. monocytogenes possesses two PrsA-like parvulins, among which PrsA2 (lmo2219) is virulence related and under the control of the global virulence gene regulator PrfA (59, 90). PrsA2-related pathogenic properties include, among others, hemolytic activity, flagellation, lecithinase activity, cell-to-cell spreading, and, eventually, virulence in the mouse model (149, 150). Similar to the case with SurA, all the measured in vitro phenotypes were not dependent on the presence of the PPIase domain. However, it is interesting that in mouse infections, a functional PPIase domain was needed in order to regain full virulence in the complemented deletion strain (151).
Streptococcus pneumoniae has, alongside the parvulin PpmA, the cyclophilin SlrA, both of which are surface-exposed lipoproteins (152, 153). Although they belong to different protein families, the achieved phenotypes of the single knockout mutants of these proteins are astonishingly similar. For both mutants, adherence to epithelial and endothelial cells was significantly reduced, while the uptake by macrophages was increased, which resulted in a significant colonization deficiency in murine pneumonia models (129, 154). For none of the proteins could a direct binding to host cells or selected extracellular matrix (ECM) components be demonstrated, which suggests the indirect influence of these proteins on as yet unknown bacterial adhesins. For PpmA, it was shown that PPIase activity had no effect on the deficiencies, thus making the contributions of these proteins to pathogenicity less interpretable. However, the answer to the increased bacterial clearance by phagocytes in the absence of PpmA and SlrA, in particular, might be derived from observations made with PpiA, the homolog of SlrA in Streptococcus mutans. Like the case for the ΔslrA mutant, deleting ppiA increased the phagocytosis of bacteria and was found to be due to the upregulation of the macrophage-specific phagocytic receptor MARCO (80). Although this upregulation was not via the typical route of TLR2 activation and PpiA is supposedly not the direct ligand for MARCO, this finding certainly narrows the possible mechanistic explanations for an important immune evasion mechanism of streptococci.
Interestingly, all the current virulence-related reports on TF are from Gram-positive pathogens. The deletion of the TF homolog RopA in Streptococcus mutans, a pathogen associated with dental caries, resulted in the differential expression of about 33 proteins, with 22 being up- and 11 downregulated. Among the upregulated proteins were the general chaperones DnaK, GroEL, and GroES, indicating that the bacterium attempts to compensate for the loss of the chaperone activity of TF. In contrast, several glycosyltransferase genes responsible for the production of glucans which mediate cell-cell and cell-surface adhesion were downregulated. Accordingly, a decrease in in vitro biofilm formation was observed when glucose was used as the sole carbon source. In addition, further virulence traits, such as genetic competence as well as acid and peroxide tolerance, suffered by the deletion of ropA (155).
The secreted cysteine proteinase (SCP) of Streptococcus pyogenes, also known as streptococcal pyogenic exotoxin B (SpeB), with diverse implications in the extracellular virulence of this pathogen, was shown to be dependent on a functional RopA protein for its secretion and processing (156). While in other trigger factors the PPIase activity was shown to play a rather marginal role, in RopA a clear relationship between the PPIase activity and the generation of active SpeB could be shown. The proline residue at position 78 of SpeB resembles the target of RopA (157). In Streptococcus suis, mainly an animal and, occasionally, human pathogen, TF deficiency resulted in the suppression of many virulence genes. Along with this, several virulence phenotypes, including adherence to host cells, hemolytic activity, resistance to several stress factors, and virulence in the CD1 mouse infection model, also diminished (158). In L. monocytogenes, the deletion of TF negatively affected the tolerance of the bacterium toward heat and ethanol, as well as its persistence in the organs of infected mice. However, the exact mechanisms behind these phenotypes are still not known, and as in the previous examples, a rather indirect contribution of TF is most likely (159).
Protozoan PPIases in Virulence
The mode of action of PPIases of parasitic protozoa during infectious processes is, in many instances, insufficiently described. This is also related to the fact that very few PPIases have been analyzed in detail, as in, for example, the case of apicomplexan cyclophilins, where a large variety has been detected in every analyzed species. Besides being primary actors of host-parasite interactions, PPIases are certainly essential, as shown for leishmanial cyclophilins, for the development and differentiation of parasitic protozoans, which show a high degree of plasticity in their cellular organization and metabolic status during their infection cycles (160). In the following, the most prominent examples of virulence-associated phenotypes of PPIases of parasitic protozoa are depicted.
In T. gondii, TgCyp18 was shown to be responsible for activation of dendritic cells via the receptor CCR5. While binding does not seem to be dependent on the PPIase domain of TgCyp18, the induction of IL-12 production partly requires a functional PPIase active site (101). In addition, the interpretation of the immunomodulatory effect in the context of Toxoplasma infection is debatable. On the one hand, TgCyp18 leads to the activation of dendritic cells and macrophages, which produce the proinflammatory signals IL-12, tumor necrosis factor alpha (TNF-α), nitric oxide, and IL-6 and prime the host for defense. On the other hand, the parasite adapts to this by differentiating from tachyzoites into bradyzoites (161). Apart from this, TgCyp18 at high concentrations enhances the proliferation of macrophages and spleen cells, in a CCR5-independent manner, while the migration of these cells toward the infection site is dependent on the interaction between CCR5 and TgCyp18 (162).
The transition of T. cruzi from replicating, noninfectious epimastigotes to the infective, metacyclic form is crucial for transmission from its insect vector to mammalian hosts. In a recent study, it was shown that cationic antimicrobial peptides (CAMPs), part of the innate defense system of the reduviid bug vector of T. cruzi, can stimulate this transition rather than having an antiparasitic action. Intriguingly, this activation is dependent on the presence of the secreted cyclophilin Cyp19 of the parasite (163). For their study, the authors focused on the model CAMP P6, which is the biologically active N-terminal part of trialysin of Triatoma infestans. P6 is 32 amino acids long and contains a single proline residue as a potential target for Cyp19. Replacing this proline with alanine abolished the binding of Cyp19 as well as the activation of the parasitic transition and, in contrast, restored the antiparasitic activity of P6 on T. cruzi. The authors were also able to show that Cyp19 and P6 in concert are able to activate the calcium-dependent phosphatase calcineurin, by an as yet unknown mechanism, and thereby increase the infectivity of the parasites. Moreover, the activating and protective effects of the Cyp19-P6 complex could be transferred successfully to L. major and Leishmania amazonensis strains when conditioned medium of either species was used, indicating that these are general virulence mechanisms of trypanosomatid parasites (163). It is still an open question whether the binding of Cyp19 to P6 or other proline-containing CAMPs resembles a simple receptor-ligand interaction mediated by a proline residue or whether the PPIase activity contributes to neutralization of P6 by isomerizing it from an active to an inactive form. Also, the mode of action of the proposed Cyp19-P6 complex and whether it is recognized by a cognate receptor on the parasite membrane are not yet known.
Host PPIases Implicated in Microbial Disease
Not only are infectious processes influenced by PPIases of pathogenic organisms, but PPIases of the host may also participate in the development of the disease. The most prominent group of infection-related host PPIases are, again, the cyclophilins. The earliest observation of a cyclophilin in an infection-related process was made in lipopolysaccharide (LPS)-stimulated RAW264.7 murine macrophages. Cyclophilin A accumulated in the culture supernatant of these cells, and it promoted an in vivo inflammatory response and in vitro chemotactic activity in peripheral blood mononuclear cells and monocytes (164). Although this process was CsA sensitive, the direct involvement of the PPIase activity has still not been proved. Later studies showed that extracellular CypB can also have similar effects and that both proteins elicit these effects by binding to the ubiquitous surface protein CD147 via their heparan binding domains (165–167). As mentioned before, during infection, many parasites secrete cyclophilins which exert proinflammatory and chemotactic activities on host cells. However, at least for L. major, molecular mimicry could be excluded, since no direct interaction between CD147 and LmCypA could be observed due to the absence of heparin binding domains in this protein, indicating that cyclophilins may act by different means (168).
The main focus of research on host cyclophilins and infection certainly lies on viruses, because of the early discovery that human CypA and CypB bind the capsid protein of HIV-1 (169). Although observations were also made that extracellular CypA may mediate binding of virus particles to cellular receptors on CD4+ cells (164), the majority of the research points to an intracellular role of cyclophilins during viral infections. Accordingly, for many pathogens, including vesicular stomatitis virus, influenza A virus, hepatitis C virus, Japanese encephalitis virus, severe acute respiratory syndrome (SARS) coronavirus, human cytomegalovirus, and rotavirus, host cyclophilins were proven to be important for efficient infection (170–176). The literature on this topic was recently reviewed elsewhere in detail, and hence, in the following, only key points of these interactions, with several examples, are provided (177, 178).
In HIV-1, binding of CypA to three Gly-Pro sites, at amino acids 90, 157, and 224, could be seen, among which the first one seemed to be the primary interaction site. Binding of CypA and -B to the capsid protein p24 was about 10-fold better after maturational cleavage by the viral protease. It was concluded that CypA may promote capsid disassembly after host infection (179). Binding of capsid protein by CypA was further shown to protect the virus from the host restriction factor Ref-1 in humans. In nonhuman primate cells, however, the interaction between CypA and capsid protein leads to restriction, explaining the host specificity (180, 181). While CypA interaction is dependent on peptidyl-prolyl sites in the capsid and is susceptible to CsA, control of infection by restriction is dependent on the presence of a histidine residue at position 87 (182). Although CypA causes conformational changes in the premature capsid protein, this activity is not PPIase dependent and is mediated by two cysteine residues, at positions 198 and 218 (183). All these observations point to intricate and multilayered roles of CypA in the infection process of HIV-1. Capsid association of host cyclophilins was also observed for many of the aforementioned viruses. Most interestingly, the newly discovered giant virus mimivirus, a parasite of free-living amoeba, encodes a cyclophilin without measurable PPIase activity that localizes to the surface of the mature virion (184).
Another HIV-1 protein binding to a host PPIase is the viral integrase, which catalyzes the recombination of the cDNA genome of the virus with the host genome. The viral integrase is first phosphorylated by the host kinase Jun N-terminal protein kinase (JNK), at a conserved serine residue, after which it is recognized by the human parvulin Pin1 and changes its conformation. This stabilizes the integrase and increases HIV-1 integration into the host genome (185). Pin1 also interacts with the catalytic subunit of the DNA polymerase of Epstein-Barr virus. This interaction is dependent on the phosphorylation of a threonine residue and improves replication of viral DNA (186).
The first interaction between a bacterial effector and a host PPIase was shown for the plant-pathogenic species Agrobacterium tumefaciens. This bacterium transforms its host by translocating a nucleoprotein complex into the cytosol via its type IV secretion system. This complex consists of a single-stranded DNA and the covalently bound endonuclease VirD2. Using a yeast two-hybrid system, three Arabidopsis cyclophilins, CYPA, ROC1, and ROC4, were identified. This interaction could be verified at the protein level, and a distinct domain in VirD2 could be spotted as important for cyclophilin binding. The interactions were sensitive to CsA treatment, which also inhibited the transformation of the host cells with the T complex. However, a detailed analysis of this interaction and the role of the PPIase activity was not performed (187).
For Pseudomonas syringae, with its 28 pathovars probably the most important plant pathogen (143), it was shown that a host cyclophilin is essential for activation of the cysteine protease AvrRpt2 (188). This bacterial effector is secreted in an unfolded and inactive state into the plant cell cytoplasm by a type III secretion system. After its translocation, AvrRpt2 mounts a hypersensitive response in Arabidopsis thaliana, via the host resistance protein RPS2. This is due to the proteolytic cleavage of another host protein, RIN4, by AvrRpt2 (189). Degraded RIN4 is then recognized by RPS2 (189, 190). Transition of AvrRpt2 from an inactive to an autoproteolytic and active state requires eukaryotic cyclophilins, such as CRP1 of S. cerevisiae and ROC1 of A. thaliana, which are homologs of human Cyp18 (188). Systematic analysis conducted by employing site-directed amino acid substitutions revealed that all four probable cyclophilin target sites with the consensus sequence Gly-Pro-Xaa-Leu are important for binding of ROC1 and subsequent activation (191). Interestingly, three of these sites surround the catalytic triad of Cys122, His208, and Asp226. Hence, it was proposed that ROC1 serves as a molecular switch that isomerizes the prolines from trans to cis conformation and, by this, activates AvrRpt2 (191). However, not only the activation of AvrRpt2 but also its proteolytic activity in general seems to be dependent on the presence of a eukaryotic cyclophilin, as shown for autoprocessed AvrRpt72–255 (192). Besides these nice studies, recent reports indicate that plant PPIases are crucial actors of plant immunity and infectious processes, and these areas deserve further examination. For example, overexpression of GhCyp1 of the cotton plant in tobacco resulted in increased salt tolerance and resistance toward P. syringae pv. tabaci (193). In line with this, in A. thaliana, deletion of the cyclophilins AtCYP19 and AtCYP57, as well as the FKBP AtFKBP65, led to increased susceptibility toward P. syringae. Also, expression of these genes at sites of infection could be demonstrated to be linked to additional defense reactions, such as production of reactive oxygen species or callose accumulation (194).
Pseudomonas aeruginosa exoenzyme S, a type III secretion system effector with GTPase and ADP-ribosyltransferase activities, was shown to ADP-ribosylate host cyclophilins at distinct arginine residues. This had a moderate effect on the PPIase activity of these proteins but efficiently inhibited the interaction with calcineurin, explaining its effects on host signaling processes (195). Another exotoxin that interacts with a host PPIase is leukotoxin (LKT) of Mannheimia haemolytica, the causative agent of fibrinous and necrotizing pleuropneumonia in cattle. LKT binds to mitochondrial CypD of bovine cells, which results in its enrichment in the mitochondrial matrix. CypD is a core protein of the mitochondrial permeability transition pore (mPTP), together with adenine nucleotide translocase (ANT). In case of stress or Ca2+ overload, mPTP is activated by CypD in a PPIase-dependent way, which results in mitochondrial swelling, loss of membrane potential, and, eventually, necrotic or apoptotic cell death (196, 197). Accordingly, LKT most probably induces cell death by disturbing mitochondrial homeostasis after binding to CypD and activating mPTP, but the details of the molecular interactions still need to be explored (196, 198).
Involvement of host CypD was also shown in Shigella-infected nonmyeloid cells, which were killed after induction of mitochondrial damage by necrotic cell death. Shigella flexneri, a facultative intracellular pathogen and the causative agent of dysentery, activates prosurvival mechanisms in the host cell via Nod1 and NF-κB signaling. In the absence of Nod1, however, infection with Shigella induces necrosis by the activation of mPTP, which can be inhibited by CsA. In this case, no direct interaction between a bacterial effector and host CypD was reported (199). These reports show that CypD, being a central regulator of mitochondrial integrity, can influence the outcome of infections caused by intra- or extracellular pathogens by several possible ways, and it might be an interesting target for developing novel strategies of treatment.
An interesting aspect of host PPIase-mediated disease progression was uncovered when translocation of bacterial binary toxins was studied. Typically, these toxins consist of a receptor subunit which binds to host cells and triggers the uptake of the binary toxin complex into the host cell by endocytosis. Upon acidification of the endosome during endosome maturation, the receptor domain is activated and forms a pore for the translocation of the enzymatically active subunit into the cytosol. This translocated subunit then modifies host proteins and exerts the actual toxic activity (200). In initial studies, a protective effect of CsA on intoxication by the experimentally constructed chimeric toxin LFNDTA could be observed (201). This chimera consists of the lethal factor (LF; a metalloproteinase of MAP kinase kinases) of Bacillus anthracis and the A subunit of diphtheria toxin (DTA; an ADP-ribosyltransferase) of Corynebacterium diphtheriae. Intriguingly, CsA was not able to exert its protective effect on LF, and only DTA, not LF, was able to bind to immobilized CypA (201). DTA is the enzymatically active subunit of diphtheria toxin, which belongs to the group of bacterial binary toxins. In accordance with this, the translocation of the enzymatically active subunits of other binary toxins, such as botulinum toxin of Clostridium botulinum, CDT of Clostridium difficile, and iota toxin of Clostridium perfringens, was shown to be facilitated by host CypA (202). By using the C2I subunit of botulinum toxin in coprecipitation experiments, FKBP51 was identified as a further PPIase capable of binding binary toxins and promoting their translocation into the cytosol. While FK506 was able to delay the intoxication of the cells by binary toxins, it could not protect the cells against the large Rho-glucosylating toxin of C. difficile, again indicating that the PPIase involvement is specific to binary toxins (203). It is certainly of major interest whether host PPIases facilitate the translocation of these proteins via their enzymatic activity or, rather, their chaperone-like interaction.
Mip-LIKE PPIases—A SPECIAL CLASS OF VIRULENCE FACTORS
Generally speaking, Mip-like PPIases are virulence-associated, secreted, and, typically, outer membrane-localized FKBPs of Gram-negative bacteria. They are widely distributed among Gram-negative pathogens, but in the majority of cases, only the presence of a Mip-like PPIase and its immunogenic character are reported, whereas the mechanism of action remains unexplored (Table 3).
TABLE 3.
Virulence-associated Mip-like proteins of bacterial and protozoan pathogens
| Organism | Protein | Commentsa | Reference(s) |
|---|---|---|---|
| Aeromonas hydrophila | FkpA | Cross-reactive with L. longbeachae anti-Mip antibodies, no significant attenuation in suckling mouse model | 222 |
| Aggregatibacter actinomycetemcomitans | Mip-like protein | Upregulated in primary isolates, antigenic in periodontitis patients, decreased viability in HeLa cell culture | 221 |
| Burkholderia pseudomallei | BpML1 | Severe defects in cell culture and murine infection model, flagellation (swarming) and survival affected at pHs of <5, X-ray structure suggests alternative inhibitor structures | 261, 263 |
| Chlamydia psittaci | Mip-like protein | Immunodominant in convalescent guinea pigs | 212 |
| Chlamydia trachomatis | Mip | Present on elementary bodies, involved in initial infection stages | 211, 215–217, 296 |
| Chlamydophila abortus | Mip (CAB080) | Immunodominant antigen in infected sheep | 214 |
| Chlamydophila pneumoniae | CpMip | Failure of formation of inclusion bodies in the presence of FK506 | 213 |
| Coxiella burnetii | CbMip | Cross-reactive with polyclonal anti-Mip for LpMip | 210 |
| Legionella longbeachae | LlMip | Replication defect in macrophage cell line HL-60 and in Acanthamoeba castellanii, attenuation in guinea pigs | 208, 209 |
| Legionella micdadei | LmMip | Decreased survival in the initial hours of infection in Hartmanella vermiformis, attachment defect in macrophage cell line U937 | 207 |
| Legionella pneumophila | Mip | Impaired in infection of macrophages, blood monocytes, and amoebae in the initial time points of infection, significant attenuation in guinea pigs | 31, 233, 235, 243–246 |
| Neisseria gonorrhoeae | NgMip | Surface exposed, decreased persistence in macrophages | 218 |
| Neisseria meningitidis | NmMip | Upregulated in blood stages, drastic attenuation in ex vivo blood-stage septicemia model, potential serogroup B vaccine candidate | 219, 220 |
| Salmonella enterica serovar Typhimurium | FkpA | Probable attenuation in Caco-2 epithelial and J774 macrophage cells, extracellular localization not confirmed | 223, 224 |
| Xanthomonas campestris | Mip | Influences protease activity and exopolysaccharide production, smaller lesions on infected leafs | 229, 230 |
| Trypanosoma cruzi | TcMip | Secreted into the supernatant, involved in infection of HeLa cells | 44, 232 |
| Leishmania infantum | Mip | Secreted virulence factor | 231 |
Phenotypes of deletion mutants.
Due to its first description in L. pneumophila and its connection to virulence, the distribution of Mip was of primary interest, and the mip gene was found to be ubiquitously present in the genus Legionella. This led to the employment of the mip gene as a molecular marker for differential detection of Legionella species in environmental and patient samples, in a method which is still valid (204–206). In addition, Mip was analyzed under the aspect of virulence only in the two other Legionella species relevant to human disease. For Legionella micdadei, deleting mip decreased the viability of bacteria in the initial hours of infection in the amoeba Hartmanella vermiformis and in U937 macrophage-like cells, though an attachment defect cannot be excluded in the case of U937 cells (207). In Legionella longbeachae, the dominant causative agent of Legionnaires' disease in Australia, similar effects were observed in the natural host amoeba Acanthamoeba castellanii and in the macrophage cell line HL-60. Additionally, the isogenic deletion mutant was drastically attenuated in the guinea pig infection model (208, 209). A Mip protein was also identified and cloned from Coxiella burnetii, another intracellular pathogen and a close relative of L. pneumophila, by the cross-reaction of polyclonal anti-Mip antibodies toward a genomic library (210). However, the contribution to virulence was not evaluated.
Several Mip-like PPIases were also identified in the obligate intracellular pathogens of the genera Chlamydia and Chlamydophila (211–214). The first reported among these, the Mip protein of Chlamydia trachomatis, is a lipoprotein which is mainly present on elementary bodies (215, 216). Its PPIase activity is needed for full virulence, since preincubating bacteria with FK506 resulted in irregularities during inclusion body formation inside the host cell (217). A similar observation was made for the Mip protein of Chlamydophila pneumoniae, for which surface localization on inclusion bodies and preferential secretion from 48 to 72 h postinfection were demonstrated by immunofluorescence (213).
Surface-exposed Mip-like proteins are also present in the important human pathogens Neisseria gonorrhoeae and Neisseria meningitidis. In the case of the sexually transmitted organism N. gonorrhoeae, N. gonorrhoeae Mip (NgMip) is involved in persistence in macrophages (218). For N. meningitidis, which is the main cause of meningitis in children under the age of 3 years and is endemic in sub-Saharan Africa, N. meningitidis Mip (NmMip) is important for survival of bacteria in the blood (219). Recombinant NmMip induced serum bactericidal activity that provided protection against different serogroups due to its high degree of sequence conservation. By this, NmMip meets one of the most important criteria for development of a global meningococcal vaccine, which is hampered mostly by the huge level of antigenic diversity of the different N. meningitidis serogroups (220).
Further Mip-like PPIases were characterized in the periodontal pathogen Aggregatibacter actinomycetemcomitans, the nosocomial pathogen Aeromonas hydrophila, and the enteropathogen Salmonella enterica serovar Typhimurium. In A. actinomycetemcomitans, Mip was found to be upregulated in primary isolates, and a deletion of the gene caused a significant reduction in the efficiency of bacteria to invade HeLa cells (221). While for A. hydrophila no noteworthy effect was seen in a suckling mouse model when a Mip-like protein was missing, for S. Typhimurium the reports differ (222–224). Initially, deleting the periplasmic Mip homolog FkpA in S. Typhimurium was reported to cause attenuation in cell culture infections of Caco-2 epithelial and J774 macrophage cells (223). However, in a more recent study, this finding was questioned, and attenuation was observed only when, in addition to FkpA, the periplasmic parvulin SurA or the periplasmic chaperone DegP was missing (224). This suggests that FkpA might be functionally replaced by SurA.
Functional replacement between these two PPIases was very recently shown in E. coli lacking SurA and the periplasmic chaperone Skp (86). Deleting both leads to a decrease in the amount of folded outer membrane proteins and is deleterious at 37°C (225, 226). However, under heat shock conditions, this phenotype disappears due to the elevated chaperone activity of FkpA (86). Accordingly, it was suggested that FkpA is as important as SurA under certain conditions. Similarly, the secretion of the virulence-associated autotransporter protease EspP of enterohemorrhagic E. coli is dependent mainly on SurA and the periplasmic chaperone/protease DegP (227). Again, under heat shock conditions, part of the translocation process can be taken over by FkpA, as it shows affinity to EspP in surface plasmon resonance experiments (228).
Mip-like PPIases are not restricted to human or animal pathogens but are also present in plant pathogens, such as the gammaproteobacterium Xanthomonas campestris, which causes black rot on all cultivated brassicas (143, 229). This periplasmic PPIase influences exopolysaccharide production and causes smaller lesions on infected leaves. The latter observation is probably caused by the inefficient transport of a type II secreted extracellular protease, PrtA (230). Mip-like PPIases are also not restricted to bacteria alone but have been reported for major protozoan parasites, such as T. cruzi and Leishmania infantum (44, 231). For the Mip-like protein of T. cruzi, it is known that it facilitates invasion of mammalian epithelial cells (44). Interestingly, this protein is secreted into the culture supernatant and exerts its role in this free form. The structural similarity between TcMip and the Mip protein of L. pneumophila is so high that the invasion deficiency of the knockout mutant can be compensated to a certain degree by adding recombinant Legionella Mip (232).
The exact role of this protein family during infection is now being unraveled successively, beginning with L. pneumophila and some other pathogens. In the following, we summarize the progress made in regard to virulence properties of Mip-like PPIases and their utilization as drug targets by focusing on L. pneumophila and Burkholderia pseudomallei.
Mip of Legionella pneumophila
The FKBP Mip was the first genetically identified virulence factor of L. pneumophila shown to be relevant for intracellular infection. Deletion of the genetic locus harboring mip resulted in reduced intracellular replication rates in human alveolar macrophages and protozoa, especially in the initial time points of infection, hence its name, macrophage infectivity potentiator (233–235). Soon after its discovery, Mip was proven to be an FKBP-type PPIase, which was a novel finding within this protein family (31). It is differentially expressed under the control of LetA, which is a global regulator of virulence-associated genes (236, 237).
Structural deciphering of Mip.
The structural properties of Mip were analyzed extensively using X-ray crystallography and nuclear magnetic resonance (NMR), with the protein being alone or in complex with FK506 or rapamycin (238–240). Mip is a 24-kDa basic protein which exists as a homodimer on the outer membranes of bacteria (53, 241). Besides an N-terminal signal sequence that is cleaved off during transport across the cytoplasmic membrane, Mip can be divided into three distinct regions: an N-terminal domain consisting of two α-helices, a long connecting α-helix in the center, and a C-terminal FKBP domain (238). The N-terminal domain is globular and facilitates dimerization via a methionine knot, in which Met38 and Met42 of one monomer interact with Met42 and Met38, respectively, of the other monomer. This dimerization mechanism via the mirror image-like interaction of hydrophobic amino acid pairs is typical for dimeric bacterial PPIases and is repeated as a Leu-Val knot in FkpA of E. coli and a Val-Leu knot in FKBP22 of Shewanella sp. SIB1. The dimeric nature is probably decisive for the in vivo action of Mip-like PPIases, as suggested by the example of FKBP22. A monomeric mutant, although similarly active in a peptide-based isomerization assay, exerted far less enzymatic activity in an RNase T1 folding assay (242). The connecting α-helix between the N and C termini has an as yet unknown function, but it might have some important role regarding protein interactions and virulence, as discussed below. NMR studies assigned this helix considerable structural flexibility, while the helical structure is sustained (239). Thus, it is currently thought that this helix confers Mip the flexibility it needs for binding different substrates as a dimer. Finally, the C-terminal domain reveals a globular β-fold structure typical of FKBPs.
Subsequent studies after the discovery of Mip and its characterization as a PPIase focused on the enzymatic feature of this novel virulence factor. The fact that the PPIase domain has highly conserved amino acids within its FK506 binding pocket led to the generation of single substitution mutants where Asp142 was replaced by leucine and Tyr185 by alanine. The purified recombinant proteins exhibited a pronounced loss of PPIase activity in in vitro peptide folding assays (5.3% for the Asp142Leu mutant and 0.6% for the Tyr185Ala mutant). However, when the same variants of mip were used to complement Δmip mutants, they behaved in infection studies of amoeba or human macrophages and blood monocytes as if wild-type mip had been used (243). It was concluded that either additional properties other than PPIase activity are important for Mip action during intracellular infection or the residual enzymatic activity of the mutant proteins was still sufficient for exerting PPIase-dependent phenotypes.
These considerations were further evaluated in a follow-up study where, in addition to the full-length single amino acid mutants, two variants were used, which were generated by truncating Mip in the middle of the long connecting α-helix. This resulted in a monomeric C-terminal variant containing the FKBP domain and a variant containing the N-terminal dimerization domain. The monomer retained enzymatic activity comparable to that of wild-type Mip in peptide folding assays. But in protein folding experiments with denatured RCM-T1 as the substrate, a significant loss in activity was observed, whereas full-length Mip was as efficient as TF from E. coli (244). Interestingly, in cell culture and guinea pig infections, complementing the mutant with monomeric Mip reverted the phenotype only to a limited degree. Moreover, of the two previously described full-length substitution variants, only the Tyr185Ala mutant, with 1 to 2% PPIase activity, failed to complement the phenotype back to wild-type levels (244).
Similar to the case of FkpA of E. coli (242), a probable in vitro chaperone activity of Mip is dependent on dimerization via its N terminus as the major facilitator of interactions associated with protein folding, which is typical for modular PPIases. In accordance with this, the results from infection studies suggest that for complete Mip action in vivo, the dimeric nature of the protein is substantial and that the PPIase activity and N-terminal accessory chaperone activity, which is not yet described in detail, need to act in a concerted fashion (244).
Interaction of Mip with human collagen IV.
The impact of Mip on systemic infection could be demonstrated in guinea pigs, where Mip contributes to the dissemination of bacteria within the lung tissue and to the spleen (245, 246). This also suggests a role for Mip in the extracellular virulence of L. pneumophila. In the extracellular milieu within the lung, bacteria are confronted mainly with epithelial cells and, even more, with components of the extracellular matrix (ECM). Accordingly, when single ECM components were tested for Mip binding, collagens were found to be the primary target of Mip. Among the potential collagens, Mip binds mainly to collagen IV (246). By doing so, it facilitates the interaction with lung epithelial cells and promotes the in vitro transmigration of bacteria across the epithelial cell barrier. This interaction can be inhibited by rapamycin and was less pronounced in the Asp142Leu and Tyr185Ala substitution mutants, showing the involvement of the PPIase active site (246).
Upon identifying human collagen IV as a Mip target, we were recently able to prove that Mip binds to a specific target sequence within the globular noncollagenous NC1 domain of the α1 isomer of collagen IV (247). This 13-mer peptide (127IPPCPSGWSSLWI139; P290) was identified with the help of a peptide array consisting of overlapping peptides resembling the complete sequences of the two most dominant isomers of collagen IV in human lung tissue, α1 and α2. (Collagens are trimeric proteins, wherein the monomers wind around each other to form a superhelix. In lung tissue, collagen IV is typically built with two α1 isomers and one α2 isomer.) According to the published crystal structure of the NC1 domain (PDB entry 1LI1), P290 is completely localized on the surface, and hence ideally situated for interacting with Mip. As assessed by competitive binding to immobilized collagen IV and coprecipitation studies using rapamycin as an inhibitor, P290 binds to the PPIase active domain of Mip. These biochemical data are supported by molecular docking models generated according to NMR measurements of the complex of the C-terminal Mip fragment with P290 in solution. Finally, P290 and an optimized cyclic derivative thereof inhibited bacterial transmigration at micromolar concentrations (247).
Identification of P290 as the Mip-binding sequence on the surface of collagen IV α1 brought up the question of whether Mip acts as a PPIase or, rather, as an adhesin binding its receptor, making it a typical moonlighting protein. The first indication that the PPIase activity is probably of secondary significance was gained from NMR studies of the P290-Mip complex in solution. Computer models generated from these measurements affirmed that P290 indeed binds to the enzymatic cleft of Mip, forming close interactions with key residues. However, the bottom of the binding pocket is occupied by Trp134 and not one of the prolines, which are positioned more in the periphery, hence questioning a PPIase action of Mip on P290 or collagen IV (247). In addition, these measurements enabled simulation of the Mip-NC1 interaction over a period of 40 ns by use of molecular docking algorithms. This simulation clearly showed (i) that the Mip dimer is able to interact with two NC1 domains, via their P290 sequences within the trimeric NC1 complex; and (ii) that the tertiary and quaternary structures of NC1 are not relevantly influenced by this interaction (247). Considering the rigid globular structure of the NC1 domains, due to several disulfide bonds, the latter finding is comprehensible and again supports the idea that Mip acts primarily as an adhesin within the context of Legionella-ECM interaction.
Implications of further functions of Mip during infection.
Certainly, Mip-collagen IV interaction, with its implications for extracellular virulence of bacteria within human lung tissue, delivers important hints for understanding the progression and outcome of Legionnaires' disease. Furthermore, this is substantial for the targeted design of alternative treatments, as discussed later. Nevertheless, the contribution of Mip to the course of intracellular infection requires alternative explanations, since the Legionella-containing vacuole is a “collagen-free” environment.
Identification of an intracellular host target is an important prerequisite for explaining the effect of Mip on cellular infection. Immunogold labeling studies proved the presence of Mip on intracellular membranes within infected host cells (53). Additionally, Mip is present on outer membrane vesicles (OMVs), which are shed by L. pneumophila under all tested growth conditions, including intracellular stages (248). Thus, OMVs can serve as vehicles to deliver Mip into the host cell cytoplasm, where it might mimic host PPIases, such as hFKBP12, due to the high sequence similarity. Accordingly, in light of the immunomodulatory involvement of hFKBP12, it was initially proposed that Mip might interact with calcineurin in a similar way (235).
Together with calcineurin, hFKBP12 also binds the inositol-1,4,5-trisphosphate receptor (IP3R), forming a ternary complex and regulating its activity (249, 250). IP3R forms a Ca2+ channel within the ER membrane, which releases Ca2+ into the cytoplasm upon binding the messenger IP3, and it is involved in regulation of autophagy in the model protozoan Dictyostelium discoideum (251, 252). Autophagy is a general host response toward intracellular bacteria and is manipulated by L. pneumophila in macrophages and D. discoideum (253, 254). Although this is highly speculative, it can be proposed that the contribution of Mip to intracellular infection might be an interference with the host autophagy machinery via an hFKBP12-dependent signaling pathway.
Another relevant observation for alternative Mip actions was made when the supernatants of the wild-type strain and its Mip-deficient mutant were fractionated and their phospholipase C activities compared. The supernatant derived from the Δmip mutant lacked a distinct phospholipase C activity which was independent of the major phospholipase C (PlcA) (255). This protein, which has not yet been identified definitively, is the only reported potential bacterial substrate of Mip to date. C-type phospholipases hydrolyze phospholipids into 1,2-diacylglycerol (DAG) and a phosphorylated residue, such as inositol phosphate, ethanolamine phosphate, or choline phosphate. DAG and inositol phosphates act as second messengers in signal transduction pathways regulating vesicle trafficking and autophagy (252, 256). L. pneumophila has several type IV secretion system-dependent effectors that bind host phosphoinositides and actively participate in the host phosphoinositol metabolism (257, 258). Also, secreted phospholipases of L. pneumophila are active in the lung tissue, as shown for phospholipase A in the guinea pig infection model (259). Hence, it may be speculated that the Mip-dependent phospholipase C activity contributes to the course of infection by destroying host tissue. Considering all this, it can be concluded that Mip most probably executes extra- and intracellular functions on multiple levels during the progression of Legionnaires' disease.
Mip-like PPIase of Burkholderia pseudomallei (BpML1).
B. pseudomallei, the causative agent of melioidosis and a potential bioterror agent, is a saprophytic betaproteobacterium that is widely distributed in tropical habitats. It is a major threat due to its intrinsic resistance to a broad range of antibiotics. The infection route is typically contact mediated via skin lesions but can also be by inhalation or ingestion. Once inside the human host, the bacterium can replicate within many different cell types and is able to escape the phagosome and spread from cell to cell. The disease can progress acutely, remain chronic, or, in some cases, even be asymptomatic (260, 261). The genome of B. pseudomallei encodes at least two FKBPs: BpML1 and BPSL0918. Interestingly, the latter lacks any measurable PPIase activity due to mutation of several key residues in the enzymatic cavity. But its deletion has severe effects on the intracellular replication capacity of the bacterium. Although the specific molecular mechanisms are not known, it was assumed that, like the case for many PPIases, BPSL0918 functions as a chaperone, with probable impacts on outer membrane integrity and stress tolerance (262).
In contrast, BpML1 is enzymatically active and is the second most thoroughly analyzed Mip-like PPIase. However, unlike Mip, BpML1 consists mainly of the PPIase domain and lacks the long α-helix and the N-terminal dimerization domain. Accordingly, localization to the outer membrane or secretion is not reported. But the PPIase domains of both proteins reveal 40% identity, with strong conservation in the enzymatically active groove. BpML1 influences several virulence-related traits, including resistance to low pH, survival in macrophage-like and epithelial cells, swarming due to flagellation, and extracellular protease activity. Additionally, a BpML1-deficient mutant was significantly attenuated in a mouse infection model (261). That study assigned BpML1 a central role in the virulence of B. pseudomallei and can be instructive for the analysis of other known Mip-like PPIases. Furthermore, the major contribution of BpML1 described so far suggests that it is primarily a facilitator for other primary virulence determinants. Thus, it would be interesting if attenuation in the mouse model is also due to the direct participation of BpML1, as it is for Mip.
A first step toward understanding the action of BpML1 was made by analyzing its crystal structure (263). This revealed high analogy to the FKBP domain of Legionella Mip, especially in the hydrophobic cleft binding FK506 and rapamycin. Due to the high structural resolution of 0.91 Å, the researchers were able to observe pronounced flexibility of the crucial active side residues Asp44 and Tyr89 (corresponding to Asp142 and Tyr185, respectively, in Mip), which was not obvious in a crystal structure of hFKBP12 with 0.92-Å resolution. This was also confirmed in NMR-derived solution structures, in which Asp44 showed a wide range of conformations, while Tyr89 adopted two alternative conformations and the rest of the ligand-binding residues remained in one conformation. From this structural flexibility, it can be speculated that BpML1 possesses some functional flexibility regarding its substrate binding. Another interesting finding derived from the crystal structure was that during crystallization, the purification tag of one protein molecule underwent close interaction with the active site of the next protein molecule. Similar to the observation made in the case of the P290-Mip complex, the hydrophobic pocket is not occupied by the proline within this tag but by a preceding valine residue, in a manner that is not typical for known peptide-PPIase interactions (263). This again suggests that Mip-like PPIases can utilize a variety of substrates.
Mip-LIKE PPIases AS DRUG TARGETS
Current anti-infective chemotherapeutics target physiologically relevant pathways and, by this approach, create marked selection pressure on microorganisms, which in turn has caused the quick rise of resistances toward the existing antibiotic classes (264). Virulence factors are generally considered nonessential and benefit pathogens only in the aspects of host colonization and disease. Dampening the virulence of a pathogen should therefore prevent, or at least slow down, the fast emergence of resistant strains after a new drug is introduced. Hence, anti-infective drug design approaches are undergoing a rethinking process and are focusing more and more on virulence factors, such as secretion systems, adhesins, or quorum sensing, as targets (265–267).
Up to this point, we have given an overview of the virulence potential of Mip-like PPIases in pathogenic bacteria, with a focus on the best-analyzed representatives. Although for most of the reported Mip-like PPIases, understanding the exact mode of action during infection still requires extensive research, their noticeable association with pathogenic microorganisms renders this new group of virulence factors an attractive drug target. In addition, considerable research on derivatives of FK506 and rapamycin, as well as alternative structures of FKBP inhibitors, has already been done, which can be of great use for the identification or design of specific inhibitors of Mip-like PPIases. However, one major obstacle remains, i.e., the high degrees of sequence and structural similarity among the bacterial and host FKBPs, which confers the risk of immunomodulatory side effects. This necessitates the design of structures with high specificity toward the Mip-like PPIase of interest. One crucial advantage is that there are structural data for FK506 and rapamycin as well as for a number of derivatives of these in complex with FKBP12 (268, 269). Furthermore, crystallographic and NMR data on Mip, BpML1, and the secreted protein TcMip enable comparison to human PPIases and structure-based design of specific inhibitors (232, 238, 240, 263). Thus, the dilemma with structural conservation among FKBPs requires the integration of information derived from structural data with information about PPIase-protein interactions in order to discover novel and specific inhibitors. In the following, we give a short overview of existing approaches for identifying FKBP inhibitors.
A Short History of PPIase Inhibitor Design
All initial PPIase inhibitors were identified during the screening of natural compounds for biological activity (13, 270, 271). In the case of cyclophilins and FKBPs, secondary metabolites that had been discovered even helped to isolate and characterize the first representatives of these families of proteins. Although a specific inhibitor of parvulin-like PPIases was missing at the time of their discovery, juglone, a secondary metabolite of chestnut trees, was also identified by screening of a library of natural compounds (21). For FKBPs, with rapamycin, ascomycin, and FK523, there are already natural analogs present (272–274). Hence, nature remains one of the most important sources for looking for PPIase inhibitors. Complementary to this, novel FKBP inhibitors were identified by structure-activity relationship (SAR) approaches using computer modeling or screening of compound libraries (275).
FK506 and its derivatives.
The early, puzzling finding that the macrolides FK506 and rapamycin exert their immunosuppressant action by binding to human FKBP12 (16, 17) was soon followed by the more surprising discovery that this effect was not due to inhibition of the PPIase activity by either of them (276, 277). Instead, as shown in subsequent studies, FK506 or rapamycin, upon binding to FKBP12, facilitates its association with calcineurin or mTOR, respectively (278–280). Interaction with FKBP12 occurs via a “binding domain” of FK506 (Fig. 3), which contains a pipecolinyl ring that mimics a peptidyl-prolyl bond and occupies the hydrophobic cleft of the PPIase formed by the aromatic side chains of Trp55, Phe46, Tyr26, and Phe99 (268, 281). Further on, the ester carbonyl group forms a hydrogen bond with the backbone amine of Ile56, as does the amide carbonyl with Tyr82, and the hemiketal hydroxyl interacts with Asp37. By this, half of FK506 is buried in FKBP12, while the other, mainly hydrophobic part is solvent exposed. This creates a unique “effector domain” that enables FKBP12 to bind to calcineurin (269, 282).
FIG 3.

Natural FKBP inhibitors and their chemical derivatives. Shown are the structures of FK506 and rapamycin, the two naturally occurring macrolides. Extensive research was done in order to exploit the FKBP binding domain of FK506 (highlighted in yellow) as a lead structure for generating acyclic inhibitors without immunomodulatory side effects. Representatives of each approach with the strongest Ki (app), i.e., 7 nM for SB3 (283), 0.5 nM for VA-10,367 (284), and 2.8 nM for 19d (285), are depicted.
The bifunctional nature of FK506 inevitably opened the way for the design of more specific drugs with only PPIase-inhibitory properties. Most of the synthetic structures were generated under the SAR point of view, which assumes that there is a pronounced relationship between the chemical structure of a molecule and its biological activity (37, 269). As a result, the core of the binding domain of FK506, consisting of the pipecolinyl ring and the keto amide group, which interact with the hydrophobic pocket of FKBP12 and its relatives, was kept as the scaffold for acyclic derivatives of FK506 (Fig. 4). Screenings showed that the pyran ring could be replaced by a (dimethylethyl)methyl group, as in SB3, where the macrocyclic part and the cyclohexan ring were replaced by aromatic groups. The resulting compound had a Ki (app) of 7 nM. The crystal structure of the FKBP12-SB3 complex (PDB entry 1FKG) revealed that the substitutes undertake similar hydrogen bonds with the respective amino acids, confirming the good affinity (283). An even better substitution for the pyran ring turned out to be 3,4,5-trimethoxyphenyl, as in the case of VA-10,367. This compound, in combination with hydrophobic residues replacing the cyclohexan ring and the macrocycle, achieved a Ki (app) of 0.5 nM, and thus was as good as FK506, with a Ki (app) of 0.4 nM (284). Similar efficiencies, with a Ki (app) of 2.8 nM, were reached with a cyclohexan derived from (R)-(−)-carvone substituting for the pyran residue (285).
FIG 4.

Novel FKBP inhibitors identified by synthetic approaches (A to D) and by screening of databases and compound libraries (E to H), as well as pipecolinic acid derivatives as inhibitors of Mip (I to K). (A) The best alternative to the keto amide group was shown to be a sulfonamide group with a Ki (app) of 160 nM, proving the importance of the keto amide for the protein-ligand interaction (269). (B) In the structurally most divergent PPIase inhibitor, the pipecolinic ring was replaced by a bicyclic ring system which binds to the same hydrophobic cleft, as shown by cocrystallization. Further on, the aromatic ring replaces the pyran, the sulfur atom fills in the pocket usually occupied by the ketone carbonyl group, and the carbonyl group forms a hydrogen bond with Ile56, like the carbonyl of the macrocycle of FK506 does. This compound has a Ki (app) of 7.9 μM (286). (C) A step-by-step approach resulted in the identification of a peptidomimetic inhibitor, β-naphthyl-Pro-Phe-Val-phenyl tetrapeptide, with an IC50 of 0.3 μM (287). (D) Peptidomimetics were generated in a mechanism-based approach. Among the derivatives, a compound resembling a Leu-Pro-tyrosyl tripeptide displayed the highest affinity to FKBP12, with a Ki (app) value of 8.6 μM (288). (E) Using the molecular docking software SANDOCK, N-benzyl-oxycarbonyl-Pro-Pro was identified as the best inhibitor, with a Kd value of 0.8 μM. (F) Steroids such as 5β-pregnan-3,20-dione, with a Kd value of 7 μM, were also detected as novel lead structures for FKBP inhibitors (289). (G) Screening of a library of secondary metabolites resulted in the discovery of cycloheximide as an FKBP12 inhibitor, with an IC50 of 3.6 μM. (H) The cytotoxic effects of cycloheximide were reduced substantially, without remarkably affecting its PPIase-inhibitory property, in the case of the derivative cycloheximide-N-ethylethanoate, with an IC50 of 4.4 μM (290). (I and J) Two lead structures, consisting of a pipecolinic acid ring with either a diketone (I) or a sulfonamide linker (J), were used for further derivatization, and the derivatives were tested in protease-coupled PPIase assays. In accordance with the molecular docking analyses performed in advance, the (S)-enantiomer of structure B showed promising results as an inhibitor, with an IC50 of 6 μM. (K) Replacing the phenyl residue with a benzyl residue led to reduction of the inhibitory capacity (IC50 of 6 μM), underlining the importance of the structural details for a future Mip inhibitor (291).
Identification of novel structures by synthetic approaches.
Further efforts basically affirmed the importance of the keto amide bond for the ligand-FKBP interaction. Modifications at this position were fruitful only with a sulfonamide substitution, which gave a Ki (app) of 160 nM (Fig. 4A) (269). A novel class of FKBP inhibitors was introduced in which a bicyclic ring system coupled via a thioester bond to a pyridine was employed (Fig. 4B). Crystallographic data showed that the new ring system was able to bind to the same hydrophobic pocket as the pipecolinyl ring of FK506. However, the efficiency of this type of compound, with a Ki (app) of 7.9 μM, was clearly below those of FK506 and VA-10,367 (286).
Novel structures were also generated with the aim of designing peptidomimetic inhibitors. In a very early effort, a step-by-step approach was used to determine the best structural characteristics of a peptidomimetic inhibitor (287). It was concluded, in contrast to later reports, that a pipecoline ring with an amino ketone instead of an amido ketone constituted the minimal binding moiety. Through successive testing, a peptidomimetic inhibitor resembling a β-naphthyl-Pro-Phe-Val-phenyl tetrapeptide was determined to be the best inhibitor, with a 50% inhibitory concentration (IC50) of 0.3 μM (Fig. 4C). In another study following a mechanism-based approach, several peptidomimetics containing a trans-substituted alkene in place of a proline were produced and tested for PPIase-inhibitory activity. The most promising compound out of this study was a Leu-Pro-tyrosyl tripeptide, which had a Ki (app) of 8.6 μM (Fig. 4D) (288).
Identification of novel structures by screening of compound libraries.
Some of the most innovative structures with FKBP-inhibitory capacity were identified by two different screening strategies. The first strategy involved using the docking program SANDOCK to evaluate the structures in compound libraries for their suitability as FKBP inhibitors. By use of a fluorescence binding assay, N-benzyl-oxycarbonyl-Pro-Pro was isolated as the best candidate, with a Kd value of 0.8 μM (Fig. 4E). In the same study, steroids, such as 5β-pregnan-3,20-dione, with a Kd value of 7 μM, were detected as a completely novel structure (Fig. 4F) (289).
The second strategy employed the screening of a compound library containing structurally diverse secondary metabolites to identify new lead structures. Surprisingly, this study yielded cycloheximide (Fig. 4G), an inhibitor of eukaryotic protein synthesis, as a new FKBP12 inhibitor (IC50 = 3.6 μM) (290). The authors of the study generated derivatives of this new lead structure in order to reduce its cytotoxic properties. Among those derivatives, cycloheximide-N-(ethylethanoate) was the most promising candidate, since it was about 200 times less cytotoxic, without losing its inhibitory capacity (IC50 = 4.4 μM) (Fig. 4H).
First Attempts toward Specific Inhibitors of FKBPs of Pathogens
As presented above, the search for inhibitors of human FKBPs with primarily no immunomodulatory side effects has led to the discovery and identification of a wide variety of FK506 derivatives and unrelated structures. In contrast to this, the search for specific inhibitors of FKBPs of pathogens has just recently come into focus and is in its infancy. Nevertheless, first attempts were already made and yielded promising candidate precursors for bacterial Mip-like PPIases and FKBP35 of Plasmodium species.
Small-molecule inhibitors of Mip-like PPIases.
In two recent publications, small-molecule inhibitors of Mip and BpML1 were identified and tested for PPIase-inhibitory activity (263, 291). Juli et al. (291) combined data from structural studies with computer-assisted docking methods in order to find novel inhibitors of Mip. For this, they used two previously reported acyclic inhibitors as lead structures A and B, with Ki (app) values of 10 nM and 0.23 μM, respectively. Both structures had the pipecoline ring in common (Fig. 4I and J). However, while structure A contained the previously mentioned keto amide moiety, in structure B this group was replaced by a sulfonamide anchor (283, 292). The suitability of both structures for Mip was analyzed by molecular docking. Interestingly, in these analyses, structure A yielded docking scores which were significantly lower than the top-ranked score for the model human FKBP12. In contrast, the (S)-enantiomer of structure B, with its less bulky sulfonamide, fitted much better into the hydrophobic pocket of Mip (291).
Subsequently, several derivatives of each structure were generated. As predicted by the molecular docking analyses, none of the derivatives of structure A inhibited Mip. In the case of structure B, it was confirmed that the compound can inhibit Mip at low micromolar concentrations and that the stereochemistry influences the activity, since a pure (S)-enantiomer of structure B (S-4c) had a slightly higher inhibitory activity toward Mip than that of its racemic mixture, 4c (IC50 of 6 μM versus 9 μM). It was also shown, with the help of the derivative 4d (IC50 = 28 μM), in which the phenyl sulfonamide group was replaced by a benzyl sulfonamide group (Fig. 4K), that an extended aromatic group improved the inhibitory potential.
Compound S-4c was also tested in an infection assay and compared to rapamycin. Interestingly, when cells of the macrophage-like cell line U937 were infected with L. pneumophila JR32 (wild type) and were simultaneously treated with 50 μM S-4c or rapamycin, there was no detrimental effect of S-4c on the outcome of the infection, in contrast to the case with rapamycin treatment. It was concluded from this that properties other than the PPIase activity of the Mip protein determine its virulence characteristics (291).
A study dealing with the Mip-like PPIase BpML1 of B. pseudomallei reported that cycloheximide-N-ethylethanoate, which was previously identified as an inhibitor of FKBP12, is also a potent inhibitor of BpML1 (263). The study also showed, by NMR measurements, that the inhibitor interacted with BpML1 in a way similar to that with a probable protein substrate. Although the inhibitor was not tested in infection studies, this study again confirms that existing structures can be recycled to give valuable lead structures.
Structure-based inhibitors.
Designing specific inhibitors of virulence-associated Mip proteins can also greatly benefit from existing structural data. To date, crystal structures of Mip (PDB entry 1FD9), TcMip (PDB entry 1JVW), and BpML1 (PDB entry 2Y78) have been generated (232, 238, 263). Additionally, for Mip and BpML1, NMR structures of respective complexes with rapamycin or cycloheximide have been published (240, 263). Furthermore, structural data are available for many of the human FKBPs, among which hFKBP12 (PDB entry 1FKG) is considered primordial and is used for comparative structure-based approaches (283).
Comparing all three microbial structures to the hFKBP12 structure reveals the high structural conservation of the catalytic cleft, which is formed by Trp59 at the base of the cleft. The residues Tyr26, Phe46, Glu54, Val55, Ile56, Tyr82, and Phe99 line up along the sides of this hydrophobic pocket. The only variations observed in this constellation among the different Mip proteins are at residues Phe46 and Glu54 of hFKBP12. Glu54 is replaced by Gln157 in Mip and Met61 in BpML1, without any major structural consequences. The same is true for Phe46, which aligns with Ala151 of Mip and Thr112 of TcMip but is functionally replaced by Phe153 and by Phe114, respectively (Fig. 5). This degree of conservation points out that targeting only the enzymatically active pocket will not deliver sufficiently discriminating drug candidates. Besides the divergence in the remote parts of the proteins, the most striking structural differences between hFKBP12 and its microbial orthologs are, interestingly, in the direct vicinity of the binding pocket, as seen in the structural superimposition of hFKBP12 and Mip (Fig. 6). While in the case of microbial representatives, this pocket is more accessible from the outside, in hFKBP12, the hydrophobic cleft is sterically occupied by residues His87, Pro88, Gly89, and Ile90. Another structural variation is observed in the cleft base, formed by the residues between Phe46 and Glu54. These differences led Pereira et al. to suggest adding a large hydrophobic group, such as a benzyl ring to C-43 and an ethyl group to C-44 of rapamycin, to produce a derivative which is too bulky to bind to hFKBP12 but still able to bind to TcMip (232). Since the same structural deviations also apply for Mip, and to a certain degree for BpML1, it would be an interesting undertaking to modify a nonimmunosuppressive derivative of FK506 or rapamycin accordingly in order to evaluate the broadband activity of such a compound.
FIG 5.

A close view into the catalytic pocket reveals a high level of structural conservation. Shown is the superimposition of the main amino acid residues building up the catalytically active, hydrophobic pocket of hKBP12 (PDB entry 1FKG; yellow), Mip (PDB entry 1FD9; red), TcMip (PDB entry 1JVW; cyan), and BpML1 (PDB entry 2Y78; green). A remarkable conservation on the amino acid and structural levels can be seen.
FIG 6.

Surface topological comparison of Mip and hFKBP12 reveals discriminatory features. Space-filling structural models of hFKBP12 (yellow) (A) and the C-terminal PPIase domain of Legionella Mip (purple) (B) are shown, with views into the respective catalytic pockets. (C) Overlaying both models reveals that the majority of the differences are outside the active site, with a marked difference right in the vicinity of the pocket (highlighted in blue). In hFKBP12, the accessibility of the pocket is sterically hindered, allowing the design of smaller inhibitory molecules with Mip-discriminatory capacity.
Substrate-based approach.
The identification of a 13-mer peptide (127IPPCPSGWSSLWI139; P290) as the Mip binding sequence within human collagen IV prompted our group to follow a substrate-based approach to design a potential peptide-based drug precursor for this bacterial PPIase. (The enumeration in the peptide sequence is according to the amino acid sequence of the published crystal structure of only the NC1 domain [PDB entry 1LI1]. Within the complete sequence of collagen IV α1, this peptide spans residues 1517 to 1529.) NMR measurements were conducted to evaluate the peptide-protein interaction in solution. A model based on molecular docking, generated with the help of these measurements, suggested that P290 interacts over its full length with Mip, whereby the catalytic cleft is occupied mainly by Trp134 and partially by Pro131. While these central residues are engaged in hydrophobic interactions with Mip, the rest of P290 performs a turn and projects out of the enzymatic cleft, aligning along both sides of the topological landscape created by residues Val190 to Pro193. This conformation of P290 is stabilized by an intramolecular hydrogen bond between Pro131CO and Trp134NH, as well as by intermolecular hydrogen bonds between P290 and Mip along the interaction surface (Fig. 7). The average B factor, reflecting the mean square displacement of residues within a complex, and hence the degree of stability, was calculated as 59.08 for P290 in complex with Mip. Among all the residues, Pro131 and Leu137 were the most-defined positions, with B factors ranging around 30. When the interaction between P290 and hFKBP12 was simulated by molecular docking with the same settings, Trp134 and Pro131 of P290 again occupied the hydrophobic cleft, but the average B factor was 200 due to the high degree of flexibility of the terminal residues, which suggested a much weaker binding of P290 to hFKBP12. This again hints to the importance of the areas around the catalytic cleft for the design of target discriminating drugs (247).
FIG 7.

Proposed structural model for the interaction between the PPIase domain of Mip and P290, derived from collagen IV. (A) Average structural model of the Mip77–213-P290 complex derived from NMR measurements. Mip is shown as a blue surface, with residues that were classified as being part of the interaction surface (Γ2 > 12 Hz) colored red. P290 is shown as a stick model. The color coding of P290 reflects the flexibility of the residues according to their calculated B factors, with yellow being rigid and blue being very flexible. (B) Enlarged cutaway view of the Mip binding pocket, where P290 is shown as yellow sticks and Mip is depicted as a blue cartoon, with residues forming the hydrophobic cavity highlighted in green. (Reprinted from reference 247 with permission of the publisher [copyright 2011 Blackwell Publishing Ltd.].)
Having observed the indications that a substrate-derived peptide inhibitor of Mip is feasible, we went on further to work out ways of improving P290's action. For this purpose, single amino acid substitutions, cyclization of P290, and use of d-amino acids for peptide synthesis were chosen as strategies. Interestingly, none of the variants synthesized by using d-amino acids were bound by Mip, suggesting that it stereoselectively binds its target. For amino acid substitution analysis, Cys130 and Trp134 were picked. Cys130 occupies position P1 with respect to Pro131, which is the proline residue closest to the enzymatically active site. Assuming that P290 represents an enzymatic substrate for Mip, Cys130 would be the discriminating amino acid during an eventual PPIase-substrate interaction (31). Trp134 was chosen because it is the central residue interacting with the enzymatic cleft. Testing respective P290 variants in a peptide microarray setup yielded Cys130Leu and Trp134Phe mutants as the most promising alternatives at these positions, with 2- and 1.8-fold better Mip binding, respectively. Strikingly, molecular docking simulations performed with the Cys130Leu variant of P290 indicated that the stability of the complex can be improved even more by replacing Pro131 instead of Cys130 by a leucine residue (247).
When P290 was cyclized over its full length by replacing the terminal isoleucine residues with cysteines, a 2-fold improvement in binding could be achieved. In contrast, a shorter cyclic version containing only the core residues 131PSGW134 flanked by two cysteines had a binding strength of only 5% on the peptide microarray, compared to P290. In combination, these results allowed us to conclude that an improved variant should be (i) cyclic, (ii) approximately 13 amino acids long, and (iii) optimized at positions 131 and 134. In light of these conclusions, a new cyclic variant of P290, CPPCLSGFSSLWC (mutations are shown in italics), was tested in a molecular docking simulation. It was found that the B factor of this variant was improved by 5% and that its solvent-exposed surface was reduced by 7%. In accordance with these findings, we were also able to observe improved inhibition efficiency of this cyclic variant compared to its lead structure, P290, in an in vitro bacterial transmigration assay (247). The activity of P290 and its derivatives opens up the possibility of inhibiting Mip-like FKBPs of other pathogens, which needs further evaluation.
Identification of inhibitors of plasmodial FKBP35 by structure-based in silico screening.
Malaria is the most devastating parasite-related disease worldwide, and the constant increase in resistance against current therapeutics, including the most recent antimalarial drug, artemisinin, necessitates the identification of new druggable targets. It has been shown that nonimmunosuppressive derivatives of FK520, which can inhibit the PPIase or chaperone activity of plasmodial FKBP35, also have antiparasitic activity on P. falciparum cultures (293). In light of these findings, Harikishore et al. (294) applied a structure-based in silico screening approach using the crystallographic data on human FKBP12 in complex with FK506. Structural comparison of the PPIase domains of the FKBP35 proteins of P. falciparum and P. vivax with this complex revealed that the amino acids His87 and Ile91, in the β5-β6 loop of human FKBP12, are replaced by Cys106/105 and Ser109/108, respectively, in these plasmodial species. Taking this variance into consideration, together with further predicted features for strong interactions, the authors of that study screened a three-dimensional (3D) virtual database and identified, after molecular docking, scoring, and prioritizing, the substance D44 (Fig. 8), consisting of a purine-like ring system and a phenyl ring linked by a thio-acetamide linkage (294).
FIG 8.
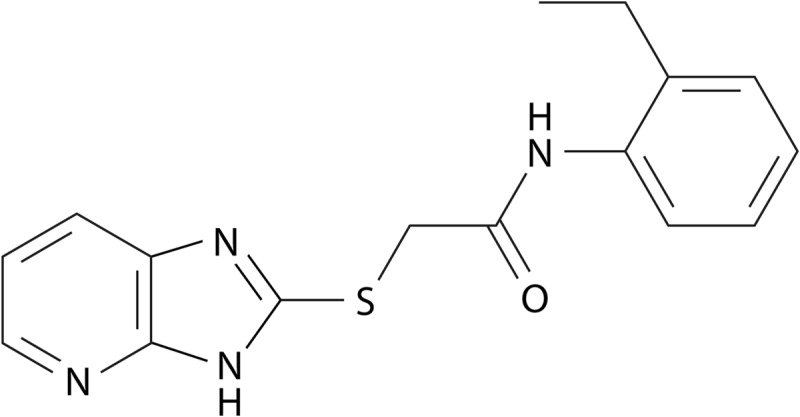
Structure of D44, a novel inhibitor of plasmodial FKBP35. D44 was identified using structure-based in silico screening of a small-molecule database and verified by in vitro tests.
The efficacy of D44 could be confirmed in PPIase assays, where it exhibited inhibition of FKBP35 proteins of both species, with IC50s of around 130 nM, whereas human FKBP12 and the phosphatase activity of calcineurin were not affected at concentrations as high as 10 μM. In accordance with the biochemical data, D44 effectively suppressed parasitemia of several P. falciparum strains and arrested the infection cycle at the schizont stage so that reinfection of erythrocytes was prohibited. Proliferation of human cell lines was inhibited at substantially higher concentrations, with IC50s of around 400 μM, underlining the discriminative action of D44 (294). The interaction of D44 with FKBP35 of P. falciparum and P. vivax was also analyzed by NMR and revealed that D44 is able to undergo stronger interactions with the residues Cys106/105 and Ser109/108 than those seen with FK506. These interactions seem to cause structural movements of Phe65/64 and Trp78/77, which are further away but form the basis of the enzymatically active hydrophobic pocket of FKBP35 (294). As a result, it was elegantly shown that structure-based approaches yield valuable alternative drug candidates, among them small-molecule inhibitors targeting the vicinity of the hydrophobic pocket of PPIases.
CONCLUSIONS
Almost 30 years of PPIase research has taught us that this protein superfamily is ubiquitous, diverse, and abundant among living organisms. Members of this family are connected to many physiological phenomena, such as protein secretion, gene regulation, immunomodulation, and neurodegeneration. Since the initial discovery of Mip of L. pneumophila, the first microbial FKBP proven to be involved in infection, the number of reports about virulence-associated PPIases has constantly been increasing. In many instances, the contribution of the isomerase activity to the observed phenotypes remains elusive and enigmatic. Enzymatically, as accessories or in a pleiotropic way, by whatever means PPIases are involved in the observed phenotypes, they are and will remain an interesting target for anti-infective therapies, despite the presumably high similarity of their PPIase active domains. Accordingly, a lead anti-infective targeting PPIases should restrict its inhibitory activity to bacterial PPIases in order to avoid immunosuppressive side effects. Although targeting as many representatives of the same class as possible would be advantageous for the broad clinical setting, the applicability of this strategy could turn out to be challenging. One reason for this is that selectivity that discriminates between host and pathogen PPIases builds on structural nuances around the catalytically active cleft and may yield inhibitors with a narrow target range. However, the fact that peptide derivatives, small synthetic molecules, and derivatives of known natural products have been identified as nonimmunosuppressive inhibitors shows the versatility of structural possibilities for novel anti-infective lead structures. In line with this, efforts toward selective inhibitors are starting to yield initial promising candidates for alternative therapeutics and emphasize the potential of structure-based approaches in this matter. It should be pointed out that not only PPIases of pathogens but also host PPIases are interesting targets for therapeutic approaches. This is already being put into practice in regard to viral infections and human cyclophilins. Furthermore, the findings regarding activation of bacterial binary toxins by host PPIases also open new perspectives in this respect and support the need for a thorough understanding of the involvement of PPIases in infectious processes. Not only inhibiting but also activating host PPIases can be a novel approach for combating infections, as demonstrated by recent studies with plant PPIases.
ACKNOWLEDGMENTS
We apologize to all our colleagues who were not cited despite our strongest efforts to give credit to the excellent work done in the field of microbial PPIases.
Can M. Ünal is supported by a 2219 (2012/2 term) grant from TÜBITAK and the State of Lower Saxony, Niedersächsisches Vorab (grant VWZN2889), within the joint research project “Epidemiology and systems biology of the bacterial pathogen Clostridium difficile (CDiff).” Michael Steinert is funded by DFG grant STE838/8-1.
We thank Kenton Barnes for critically reading the manuscript.
REFERENCES
- 1.Göthel SF, Marahiel MA. 1999. Peptidyl-prolyl cis-trans isomerases, a superfamily of ubiquitous folding catalysts. Cell. Mol. Life Sci. 55:423–436. 10.1007/s000180050299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brandts JF, Halvorson HR, Brennan M. 1975. Consideration of the possibility that the slow step in protein denaturation reactions is due to cis-trans isomerism of proline residues. Biochemistry (Mosc.) 14:4953–4963. 10.1021/bi00693a026 [DOI] [PubMed] [Google Scholar]
- 3.Cook KH, Schmid FX, Baldwin RL. 1979. Role of proline isomerization in folding of ribonuclease A at low temperatures. Proc. Natl. Acad. Sci. U. S. A. 76:6157–6161. 10.1073/pnas.76.12.6157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stewart DE, Sarkar A, Wampler JE. 1990. Occurrence and role of cis peptide bonds in protein structures. J. Mol. Biol. 214:253–260. 10.1016/0022-2836(90)90159-J [DOI] [PubMed] [Google Scholar]
- 5.Schmid FX, Baldwin RL. 1978. Acid catalysis of the formation of the slow-folding species of RNase A: evidence that the reaction is proline isomerization. Proc. Natl. Acad. Sci. U. S. A. 75:4764–4768. 10.1073/pnas.75.10.4764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fischer G, Heins J, Barth A. 1983. The conformation around the peptide bond between the P1- and P2-positions is important for catalytic activity of some proline-specific proteases. Biochim. Biophys. Acta 742:452–462. 10.1016/0167-4838(83)90261-3 [DOI] [PubMed] [Google Scholar]
- 7.Fischer G, Bang H, Mech C. 1984. Determination of enzymatic catalysis for the cis-trans-isomerization of peptide binding in proline-containing peptides. Biomed. Biochim. Acta 43:1101–1111 [PubMed] [Google Scholar]
- 8.Fischer G, Bang H, Berger E, Schellenberger A. 1984. Conformational specificity of chymotrypsin toward proline-containing substrates. Biochim. Biophys. Acta 791:87–97. 10.1016/0167-4838(84)90285-1 [DOI] [PubMed] [Google Scholar]
- 9.Fischer G, Bang H. 1985. The refolding of urea-denatured ribonuclease A is catalyzed by peptidyl-prolyl cis-trans isomerase. Biochim. Biophys. Acta 828:39–42. 10.1016/0167-4838(85)90006-8 [DOI] [PubMed] [Google Scholar]
- 10.Lang K, Schmid FX, Fischer G. 1987. Catalysis of protein folding by prolyl isomerase. Nature 329:268–270. 10.1038/329268a0 [DOI] [PubMed] [Google Scholar]
- 11.Fischer G, Wittmann-Liebold B, Lang K, Kiefhaber T, Schmid FX. 1989. Cyclophilin and peptidyl-prolyl cis-trans isomerase are probably identical proteins. Nature 337:476–478. 10.1038/337476a0 [DOI] [PubMed] [Google Scholar]
- 12.Takahashi N, Hayano T, Suzuki M. 1989. Peptidyl-prolyl cis-trans isomerase is the cyclosporin A-binding protein cyclophilin. Nature 337:473–475. 10.1038/337473a0 [DOI] [PubMed] [Google Scholar]
- 13.Dreyfuss M, Härri E, Hofmann H, Kobel H, Pache W, Tscherter H. 1976. Cyclosporin A and C. Appl. Microbiol. Biotechnol. 3:125–133 [Google Scholar]
- 14.Handschumacher RE, Harding MW, Rice J, Drugge RJ, Speicher DW. 1984. Cyclophilin: a specific cytosolic binding protein for cyclosporin A. Science 226:544–547. 10.1126/science.6238408 [DOI] [PubMed] [Google Scholar]
- 15.Harding MW, Handschumacher RE, Speicher DW. 1986. Isolation and amino acid sequence of cyclophilin. J. Biol. Chem. 261:8547–8555 [PubMed] [Google Scholar]
- 16.Harding MW, Galat A, Uehling DE, Schreiber SL. 1989. A receptor for the immunosuppressant FK506 is a cis-trans peptidyl-prolyl isomerase. Nature 341:758–760. 10.1038/341758a0 [DOI] [PubMed] [Google Scholar]
- 17.Siekierka JJ, Hung SH, Poe M, Lin CS, Sigal NH. 1989. A cytosolic binding protein for the immunosuppressant FK506 has peptidyl-prolyl isomerase activity but is distinct from cyclophilin. Nature 341:755–757. 10.1038/341755a0 [DOI] [PubMed] [Google Scholar]
- 18.Rahfeld JU, Schierhorn A, Mann K, Fischer G. 1994. A novel peptidyl-prolyl cis/trans isomerase from Escherichia coli. FEBS Lett. 343:65–69. 10.1016/0014-5793(94)80608-X [DOI] [PubMed] [Google Scholar]
- 19.Rahfeld JU, Rücknagel KP, Schelbert B, Ludwig B, Hacker J, Mann K, Fischer G. 1994. Confirmation of the existence of a third family among peptidyl-prolyl cis/trans isomerases. Amino acid sequence and recombinant production of parvulin. FEBS Lett. 352:180–184 [DOI] [PubMed] [Google Scholar]
- 20.Scholz C, Rahfeld J, Fischer G, Schmid FX. 1997. Catalysis of protein folding by parvulin. J. Mol. Biol. 273:752–762. 10.1006/jmbi.1997.1301 [DOI] [PubMed] [Google Scholar]
- 21.Hennig L, Christner C, Kipping M, Schelbert B, Rücknagel KP, Grabley S, Küllertz G, Fischer G. 1998. Selective inactivation of parvulin-like peptidyl-prolyl cis/trans isomerases by juglone. Biochemistry (Mosc.) 37:5953–5960. 10.1021/bi973162p [DOI] [PubMed] [Google Scholar]
- 22.Timerman AP, Wiederrecht G, Marcy A, Fleischer S. 1995. Characterization of an exchange reaction between soluble FKBP-12 and the FKBP-ryanodine receptor complex. Modulation by FKBP mutants deficient in peptidyl-prolyl isomerase activity. J. Biol. Chem. 270:2451–2459 [DOI] [PubMed] [Google Scholar]
- 23.Behrens S, Maier R, de Cock H, Schmid FX, Gross CA. 2001. The SurA periplasmic PPIase lacking its parvulin domains functions in vivo and has chaperone activity. EMBO J. 20:285–294. 10.1093/emboj/20.1.285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kramer G, Patzelt H, Rauch T, Kurz TA, Vorderwülbecke S, Bukau B, Deuerling E. 2004. Trigger factor peptidyl-prolyl cis/trans isomerase activity is not essential for the folding of cytosolic proteins in Escherichia coli. J. Biol. Chem. 279:14165–14170. 10.1074/jbc.M313635200 [DOI] [PubMed] [Google Scholar]
- 25.Gerard M, Deleersnijder A, Demeulemeester J, Debyser Z, Baekelandt V. 2011. Unraveling the role of peptidyl-prolyl isomerases in neurodegeneration. Mol. Neurobiol. 44:13–27. 10.1007/s12035-011-8184-2 [DOI] [PubMed] [Google Scholar]
- 26.Göthel SF, Scholz C, Schmid FX, Marahiel MA. 1998. Cyclophilin and trigger factor from Bacillus subtilis catalyze in vitro protein folding and are necessary for viability under starvation conditions. Biochemistry (Mosc.) 37:13392–13399. 10.1021/bi981253w [DOI] [PubMed] [Google Scholar]
- 27.Justice SS, Hunstad DA, Harper JR, Duguay AR, Pinkner JS, Bann J, Frieden C, Silhavy TJ, Hultgren SJ. 2005. Periplasmic peptidyl prolyl cis-trans isomerases are not essential for viability, but SurA is required for pilus biogenesis in Escherichia coli. J. Bacteriol. 187:7680–7686. 10.1128/JB.187.22.7680-7686.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dolinski K, Muir S, Cardenas M, Heitman J. 1997. All cyclophilins and FK506 binding proteins are, individually and collectively, dispensable for viability in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U. S. A. 94:13093–13098. 10.1073/pnas.94.24.13093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Obi IR, Nordfelth R, Francis MS. 2011. Varying dependency of periplasmic peptidylprolyl cis-trans isomerases in promoting Yersinia pseudotuberculosis stress tolerance and pathogenicity. Biochem. J. 439:321–332. 10.1042/BJ20110767 [DOI] [PubMed] [Google Scholar]
- 30.Harrison RK, Stein RL. 1990. Substrate specificities of the peptidyl prolyl cis-trans isomerase activities of cyclophilin and FK-506 binding protein: evidence for the existence of a family of distinct enzymes. Biochemistry (Mosc.) 29:3813–3816. 10.1021/bi00468a001 [DOI] [PubMed] [Google Scholar]
- 31.Fischer G, Bang H, Ludwig B, Mann K, Hacker J. 1992. Mip protein of Legionella pneumophila exhibits peptidyl-prolyl-cis/trans isomerase (PPIase) activity. Mol. Microbiol. 6:1375–1383. 10.1111/j.1365-2958.1992.tb00858.x [DOI] [PubMed] [Google Scholar]
- 32.Yaffe MB, Schutkowski M, Shen M, Zhou XZ, Stukenberg PT, Rahfeld JU, Xu J, Kuang J, Kirschner MW, Fischer G, Cantley LC, Lu KP. 1997. Sequence-specific and phosphorylation-dependent proline isomerization: a potential mitotic regulatory mechanism. Science 278:1957–1960. 10.1126/science.278.5345.1957 [DOI] [PubMed] [Google Scholar]
- 33.McNaughton L, Li Z, Van Roey P, Hanes SD, LeMaster DM. 2010. Restricted domain mobility in the Candida albicans Ess1 prolyl isomerase. Biochim. Biophys. Acta 1804:1537–1541. 10.1016/j.bbapap.2010.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Scholz C, Mücke M, Rape M, Pecht A, Pahl A, Bang H, Schmid FX. 1998. Recognition of protein substrates by the prolyl isomerase trigger factor is independent of proline residues. J. Mol. Biol. 277:723–732. 10.1006/jmbi.1997.1604 [DOI] [PubMed] [Google Scholar]
- 35.D'Andrea LD, Regan L. 2003. TPR proteins: the versatile helix. Trends Biochem. Sci. 28:655–662. 10.1016/j.tibs.2003.10.007 [DOI] [PubMed] [Google Scholar]
- 36.Schiene-Fischer C, Aumüller T, Fischer G. 2013. Peptide bond cis/trans isomerases: a biocatalysis perspective of conformational dynamics in proteins. Top. Curr. Chem. 328:35–67. 10.1007/128_2011_151 [DOI] [PubMed] [Google Scholar]
- 37.Blackburn EA, Walkinshaw MD. 2011. Targeting FKBP isoforms with small-molecule ligands. Curr. Opin. Pharmacol. 11:365–371. 10.1016/j.coph.2011.04.007 [DOI] [PubMed] [Google Scholar]
- 38.Pahl A, Keller U. 1994. Streptomyces chrysomallus FKBP-33 is a novel immunophilin consisting of two FK506 binding domains; its gene is transcriptionally coupled to the FKBP-12 gene. EMBO J. 13:3472–3480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Somarelli JA, Lee SY, Skolnick J, Herrera RJ. 2008. Structure-based classification of 45 FK506-binding proteins. Proteins 72:197–208. 10.1002/prot.21908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Adams B, Musiyenko A, Kumar R, Barik S. 2005. A novel class of dual-family immunophilins. J. Biol. Chem. 280:24308–24314. 10.1074/jbc.M500990200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Krücken J, Greif G, von Samson-Himmelstjerna G. 2009. In silico analysis of the cyclophilin repertoire of apicomplexan parasites. Parasit. Vectors 2:27. 10.1186/1756-3305-2-27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jakob RP, Zoldák G, Aumüller T, Schmid FX. 2009. Chaperone domains convert prolyl isomerases into generic catalysts of protein folding. Proc. Natl. Acad. Sci. U. S. A. 106:20282–20287. 10.1073/pnas.0909544106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geitner A-J, Schmid FX. 2012. Combination of the human prolyl isomerase FKBP12 with unrelated chaperone domains leads to chimeric folding enzymes with high activity. J. Mol. Biol. 420:335–349. 10.1016/j.jmb.2012.04.018 [DOI] [PubMed] [Google Scholar]
- 44.Moro A, Ruiz-Cabello F, Fernández-Cano A, Stock RP, González A. 1995. Secretion by Trypanosoma cruzi of a peptidyl-prolyl cis-trans isomerase involved in cell infection. EMBO J. 14:2483–2490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Himukai R, Kuzuhara T, Horikoshi M. 1999. Relationship between the subcellular localization and structures of catalytic domains of FKBP-type PPIases. J. Biochem. 126:879–888. 10.1093/oxfordjournals.jbchem.a022530 [DOI] [PubMed] [Google Scholar]
- 46.Arora K, Gwinn WM, Bower MA, Watson A, Okwumabua I, MacDonald HR, Bukrinsky MI, Constant SL. 2005. Extracellular cyclophilins contribute to the regulation of inflammatory responses. J. Immunol. 175:517–522. 10.4049/jimmunol.175.1.517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hayano T, Takahashi N, Kato S, Maki N, Suzuki M. 1991. Two distinct forms of peptidylprolyl-cis-trans-isomerase are expressed separately in periplasmic and cytoplasmic compartments of Escherichia coli cells. Biochemistry (Mosc.) 30:3041–3048. 10.1021/bi00226a009 [DOI] [PubMed] [Google Scholar]
- 48.Kim N, Weeks DL, Shin JM, Scott DR, Young MK, Sachs G. 2002. Proteins released by Helicobacter pylori in vitro. J. Bacteriol. 184:6155–6162. 10.1128/JB.184.22.6155-6162.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Söderberg MA, Cianciotto NP. 2008. A Legionella pneumophila peptidyl-prolyl cis-trans isomerase present in culture supernatants is necessary for optimal growth at low temperatures. Appl. Environ. Microbiol. 74:1634–1638. 10.1128/AEM.02512-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Delpino MV, Comerci DJ, Wagner MA, Eschenbrenner M, Mujer CV, Ugalde RA, Fossati CA, Baldi PC, Delvecchio VG. 2009. Differential composition of culture supernatants from wild-type Brucella abortus and its isogenic virB mutants. Arch. Microbiol. 191:571–581. 10.1007/s00203-009-0484-9 [DOI] [PubMed] [Google Scholar]
- 51.Kontinen VP, Sarvas M. 1993. The PrsA lipoprotein is essential for protein secretion in Bacillus subtilis and sets a limit for high-level secretion. Mol. Microbiol. 8:727–737. 10.1111/j.1365-2958.1993.tb01616.x [DOI] [PubMed] [Google Scholar]
- 52.Dartigalongue C, Raina S. 1998. A new heat-shock gene, ppiD, encodes a peptidyl-prolyl isomerase required for folding of outer membrane proteins in Escherichia coli. EMBO J. 17:3968–3980. 10.1093/emboj/17.14.3968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Helbig JH, Lück PC, Steinert M, Jacobs E, Witt M. 2001. Immunolocalization of the Mip protein of intracellularly and extracellularly grown Legionella pneumophila. Lett. Appl. Microbiol. 32:83–88. 10.1046/j.1472-765x.2001.00861.x [DOI] [PubMed] [Google Scholar]
- 54.Trémillon N, Morello E, Llull D, Mazmouz R, Gratadoux J-J, Guillot A, Chapot-Chartier M-P, Monlezun L, Solé V, Ginisty H, Poquet I. 2012. PpiA, a surface PPIase of the cyclophilin family in Lactococcus lactis. PLoS One 7:e33516. 10.1371/journal.pone.0033516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kandror O, Goldberg AL. 1997. Trigger factor is induced upon cold shock and enhances viability of Escherichia coli at low temperatures. Proc. Natl. Acad. Sci. U. S. A. 94:4978–4981. 10.1073/pnas.94.10.4978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kurek I, Stöger E, Dulberger R, Christou P, Breiman A. 2002. Overexpression of the wheat FK506-binding protein 73 (FKBP73) and the heat-induced wheat FKBP77 in transgenic wheat reveals different functions of the two isoforms. Transgenic Res. 11:373–379. 10.1023/A:1016374128479 [DOI] [PubMed] [Google Scholar]
- 57.Fasseas MK, Dimou M, Katinakis P. 2012. The Caenorhabditis elegans parvulin gene subfamily and their expression under cold or heat stress along with the fkb subfamily. Biochem. Biophys. Res. Commun. 423:520–525. 10.1016/j.bbrc.2012.05.157 [DOI] [PubMed] [Google Scholar]
- 58.Gioti A, Simon A, Le Pêcheur P, Giraud C, Pradier JM, Viaud M, Levis C. 2006. Expression profiling of Botrytis cinerea genes identifies three patterns of up-regulation in planta and an FKBP12 protein affecting pathogenicity. J. Mol. Biol. 358:372–386. 10.1016/j.jmb.2006.01.076 [DOI] [PubMed] [Google Scholar]
- 59.Port GC, Freitag NE. 2007. Identification of novel Listeria monocytogenes secreted virulence factors following mutational activation of the central virulence regulator, PrfA. Infect. Immun. 75:5886–5897. 10.1128/IAI.00845-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Patterson CE, Schaub T, Coleman EJ, Davis EC. 2000. Developmental regulation of FKBP65. An ER-localized extracellular matrix binding-protein. Mol. Biol. Cell 11:3925–3935. 10.1091/mbc.11.11.3925 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Patterson CE, Abrams WR, Wolter NE, Rosenbloom J, Davis EC. 2005. Developmental regulation and coordinate reexpression of FKBP65 with extracellular matrix proteins after lung injury suggest a specialized function for this endoplasmic reticulum immunophilin. Cell Stress Chaperones 10:285–295. 10.1379/CSC-118R.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Steiner JP, Dawson TM, Fotuhi M, Glatt CE, Snowman AM, Cohen N, Snyder SH. 1992. High brain densities of the immunophilin FKBP colocalized with calcineurin. Nature 358:584–587. 10.1038/358584a0 [DOI] [PubMed] [Google Scholar]
- 63.Wang W-W, Ma Q, Xiang Y, Zhu S-W, Cheng B-J. 2012. Genome-wide analysis of immunophilin FKBP genes and expression patterns in Zea mays. Genet. Mol. Res. 11:1690–1700. 10.4238/2012.June.25.2 [DOI] [PubMed] [Google Scholar]
- 64.Kang CB, Feng L, Chia J, Yoon HS. 2005. Molecular characterization of FK-506 binding protein 38 and its potential regulatory role on the anti-apoptotic protein Bcl-2. Biochem. Biophys. Res. Commun. 337:30–38. 10.1016/j.bbrc.2005.09.023 [DOI] [PubMed] [Google Scholar]
- 65.Wang L. 2011. FKBP51 regulation of AKT/protein kinase B phosphorylation. Curr. Opin. Pharmacol. 11:360–364. 10.1016/j.coph.2011.03.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kang CB, Hong Y, Dhe-Paganon S, Yoon HS. 2008. FKBP family proteins: immunophilins with versatile biological functions. Neurosignals 16:318–325. 10.1159/000123041 [DOI] [PubMed] [Google Scholar]
- 67.Wang P, Heitman J. 2005. The cyclophilins. Genome Biol. 6:226. 10.1186/gb-2005-6-7-226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Heikkinen O, Seppala R, Tossavainen H, Heikkinen S, Koskela H, Permi P, Kilpeläinen I. 2009. Solution structure of the parvulin-type PPIase domain of Staphylococcus aureus PrsA—implications for the catalytic mechanism of parvulins. BMC Struct. Biol. 9:17. 10.1186/1472-6807-9-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bang H, Pecht A, Raddatz G, Scior T, Solbach W, Brune K, Pahl A. 2000. Prolyl isomerases in a minimal cell. Catalysis of protein folding by trigger factor from Mycoplasma genitalium. Eur. J. Biochem. 267:3270–3280. 10.1046/j.1432-1327.2000.01355.x [DOI] [PubMed] [Google Scholar]
- 70.Lakshmipathy SK, Gupta R, Pinkert S, Etchells SA, Hartl FU. 2010. Versatility of trigger factor interactions with ribosome-nascent chain complexes. J. Biol. Chem. 285:27911–27923. 10.1074/jbc.M110.134163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lakshmipathy SK, Tomic S, Kaiser CM, Chang H-C, Genevaux P, Georgopoulos C, Barral JM, Johnson AE, Hartl FU, Etchells SA. 2007. Identification of nascent chain interaction sites on trigger factor. J. Biol. Chem. 282:12186–12193. 10.1074/jbc.M609871200 [DOI] [PubMed] [Google Scholar]
- 72.Arié JP, Sassoon N, Betton JM. 2001. Chaperone function of FkpA, a heat shock prolyl isomerase, in the periplasm of Escherichia coli. Mol. Microbiol. 39:199–210. 10.1046/j.1365-2958.2001.02250.x [DOI] [PubMed] [Google Scholar]
- 73.Ramm K, Plückthun A. 2001. High enzymatic activity and chaperone function are mechanistically related features of the dimeric E. coli peptidyl-prolyl-isomerase FkpA. J. Mol. Biol. 310:485–498. 10.1006/jmbi.2001.4747 [DOI] [PubMed] [Google Scholar]
- 74.Hullmann J, Patzer SI, Römer C, Hantke K, Braun V. 2008. Periplasmic chaperone FkpA is essential for imported colicin M toxicity. Mol. Microbiol. 69:926–937. 10.1111/j.1365-2958.2008.06327.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rahfeld JU, Rücknagel KP, Stoller G, Horne SM, Schierhorn A, Young KD, Fischer G. 1996. Isolation and amino acid sequence of a new 22-kDa FKBP-like peptidyl-prolyl cis/trans-isomerase of Escherichia coli. Similarity to Mip-like proteins of pathogenic bacteria. J. Biol. Chem. 271:22130–22138 [DOI] [PubMed] [Google Scholar]
- 76.Bernhardt TG, Roof WD, Young R. 2002. The Escherichia coli FKBP-type PPIase SlyD is required for the stabilization of the E lysis protein of bacteriophage φX174. Mol. Microbiol. 45:99–108. 10.1046/j.1365-2958.2002.02984.x [DOI] [PubMed] [Google Scholar]
- 77.Zhang JW, Butland G, Greenblatt JF, Emili A, Zamble DB. 2005. A role for SlyD in the Escherichia coli hydrogenase biosynthetic pathway. J. Biol. Chem. 280:4360–4366. 10.1074/jbc.M411799200 [DOI] [PubMed] [Google Scholar]
- 78.Zhang JW, Leach MR, Zamble DB. 2007. The peptidyl-prolyl isomerase activity of SlyD is not required for maturation of Escherichia coli hydrogenase. J. Bacteriol. 189:7942–7944. 10.1128/JB.00922-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cheng T, Li H, Xia W, Sun H. 2012. Multifaceted SlyD from Helicobacter pylori: implication in [NiFe] hydrogenase maturation. J. Biol. Inorg. Chem. 17:331–343. 10.1007/s00775-011-0855-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mukouhara T, Arimoto T, Cho K, Yamamoto M, Igarashi T. 2011. Surface lipoprotein PpiA of Streptococcus mutans suppresses scavenger receptor MARCO-dependent phagocytosis by macrophages. Infect. Immun. 79:4933–4940. 10.1128/IAI.05693-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Herrler M, Bang H, Marahiel MA. 1994. Cloning and characterization of ppiB, a Bacillus subtilis gene which encodes a cyclosporin A-sensitive peptidyl-prolyl cis-trans isomerase. Mol. Microbiol. 11:1073–1083. 10.1111/j.1365-2958.1994.tb00384.x [DOI] [PubMed] [Google Scholar]
- 82.Graumann P, Schröder K, Schmid R, Marahiel MA. 1996. Cold shock stress-induced proteins in Bacillus subtilis. J. Bacteriol. 178:4611–4619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Behrens-Kneip S. 2010. The role of SurA factor in outer membrane protein transport and virulence. Int. J. Med. Microbiol. 300:421–428. 10.1016/j.ijmm.2010.04.012 [DOI] [PubMed] [Google Scholar]
- 84.Weininger U, Jakob RP, Kovermann M, Balbach J, Schmid FX. 2010. The prolyl isomerase domain of PpiD from Escherichia coli shows a parvulin fold but is devoid of catalytic activity. Protein Sci. 19:6–18. 10.1002/pro.277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Matern Y, Barion B, Behrens-Kneip S. 2010. PpiD is a player in the network of periplasmic chaperones in Escherichia coli. BMC Microbiol. 10:251. 10.1186/1471-2180-10-251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ge X, Lyu Z-X, Liu Y, Wang R, Zhao XS, Fu X, Chang Z. 2014. Identification of FkpA as a key quality control factor for the biogenesis of outer membrane proteins under heat shock conditions. J. Bacteriol. 196:672–680. 10.1128/JB.01069-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vitikainen M, Lappalainen I, Seppala R, Antelmann H, Boer H, Taira S, Savilahti H, Hecker M, Vihinen M, Sarvas M, Kontinen VP. 2004. Structure-function analysis of PrsA reveals roles for the parvulin-like and flanking N- and C-terminal domains in protein folding and secretion in Bacillus subtilis. J. Biol. Chem. 279:19302–19314. 10.1074/jbc.M400861200 [DOI] [PubMed] [Google Scholar]
- 88.Wahlström E, Vitikainen M, Kontinen VP, Sarvas M. 2003. The extracytoplasmic folding factor PrsA is required for protein secretion only in the presence of the cell wall in Bacillus subtilis. Microbiology 149:569–577. 10.1099/mic.0.25511-0 [DOI] [PubMed] [Google Scholar]
- 89.Drouault S, Anba J, Bonneau S, Bolotin A, Ehrlich SD, Renault P. 2002. The peptidyl-prolyl isomerase motif is lacking in PmpA, the PrsA-like protein involved in the secretion machinery of Lactococcus lactis. Appl. Environ. Microbiol. 68:3932–3942. 10.1128/AEM.68.8.3932-3942.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Milohanic E, Glaser P, Coppée J-Y, Frangeul L, Vega Y, Vázquez-Boland JA, Kunst F, Cossart P, Buchrieser C. 2003. Transcriptome analysis of Listeria monocytogenes identifies three groups of genes differently regulated by PrfA. Mol. Microbiol. 47:1613–1625. 10.1046/j.1365-2958.2003.03413.x [DOI] [PubMed] [Google Scholar]
- 91.Hyyryläinen H-L, Marciniak BC, Dahncke K, Pietiäinen M, Courtin P, Vitikainen M, Seppala R, Otto A, Becher D, Chapot-Chartier M-P, Kuipers OP, Kontinen VP. 2010. Penicillin-binding protein folding is dependent on the PrsA peptidyl-prolyl cis-trans isomerase in Bacillus subtilis. Mol. Microbiol. 77:108–127. 10.1111/j.1365-2958.2010.07188.x [DOI] [PubMed] [Google Scholar]
- 92.Nickell SP, Scheibel LW, Cole GA. 1982. Inhibition by cyclosporin A of rodent malaria in vivo and human malaria in vitro. Infect. Immun. 37:1093–1100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bell A, Wernli B, Franklin RM. 1994. Roles of peptidyl-prolyl cis-trans isomerase and calcineurin in the mechanisms of antimalarial action of cyclosporin A, FK506, and rapamycin. Biochem. Pharmacol. 48:495–503. 10.1016/0006-2952(94)90279-8 [DOI] [PubMed] [Google Scholar]
- 94.Perkins ME, Wu TW, Le Blancq SM. 1998. Cyclosporin analogs inhibit in vitro growth of Cryptosporidium parvum. Antimicrob. Agents Chemother. 42:843–848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.High KP, Joiner KA, Handschumacher RE. 1994. Isolation, cDNA sequences, and biochemical characterization of the major cyclosporin-binding proteins of Toxoplasma gondii. J. Biol. Chem. 269:9105–9112 [PubMed] [Google Scholar]
- 96.Reddy GR. 1995. Cloning and characterization of a Plasmodium falciparum cyclophilin gene that is stage-specifically expressed. Mol. Biochem. Parasitol. 73:111–121. 10.1016/0166-6851(95)00103-8 [DOI] [PubMed] [Google Scholar]
- 97.Hirtzlin J, Färber PM, Franklin RM, Bell A. 1995. Molecular and biochemical characterization of a Plasmodium falciparum cyclophilin containing a cleavable signal sequence. Eur. J. Biochem. 232:765–772. 10.1111/j.1432-1033.1995.tb20871.x [DOI] [PubMed] [Google Scholar]
- 98.Marín-Menéndez A, Bell A. 2011. Overexpression, purification and assessment of cyclosporin binding of a family of cyclophilins and cyclophilin-like proteins of the human malarial parasite Plasmodium falciparum. Protein Expr. Purif. 78:225–234. 10.1016/j.pep.2011.04.012 [DOI] [PubMed] [Google Scholar]
- 99.Marín-Menéndez A, Monaghan P, Bell A. 2012. A family of cyclophilin-like molecular chaperones in Plasmodium falciparum. Mol. Biochem. Parasitol. 184:44–47. 10.1016/j.molbiopara.2012.04.006 [DOI] [PubMed] [Google Scholar]
- 100.Aliberti J, Valenzuela JG, Carruthers VB, Hieny S, Andersen J, Charest H, Reis e Sousa C, Fairlamb A, Ribeiro JM, Sher A. 2003. Molecular mimicry of a CCR5 binding-domain in the microbial activation of dendritic cells. Nat. Immunol. 4:485–490. 10.1038/ni915 [DOI] [PubMed] [Google Scholar]
- 101.Yarovinsky F, Andersen JF, King LR, Caspar P, Aliberti J, Golding H, Sher A. 2004. Structural determinants of the anti-HIV activity of a CCR5 antagonist derived from Toxoplasma gondii. J. Biol. Chem. 279:53635–53642. 10.1074/jbc.M410550200 [DOI] [PubMed] [Google Scholar]
- 102.Del Rio L, Butcher BA, Bennouna S, Hieny S, Sher A, Denkers EY. 2004. Toxoplasma gondii triggers myeloid differentiation factor 88-dependent IL-12 and chemokine ligand 2 (monocyte chemoattractant protein 1) responses using distinct parasite molecules and host receptors. J. Immunol. 172:6954–6960. 10.4049/jimmunol.172.11.6954 [DOI] [PubMed] [Google Scholar]
- 103.Tuo W, Fetterer R, Jenkins M, Dubey JP. 2005. Identification and characterization of Neospora caninum cyclophilin that elicits gamma interferon production. Infect. Immun. 73:5093–5100. 10.1128/IAI.73.8.5093-5100.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sheiner L, Vaidya AB, McFadden GI. 2013. The metabolic roles of the endosymbiotic organelles of Toxoplasma and Plasmodium spp. Curr. Opin. Microbiol. 16:452–458. 10.1016/j.mib.2013.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Potenza M, Galat A, Minning TA, Ruiz AM, Duran R, Tarleton RL, Marín M, Fichera LE, Búa J. 2006. Analysis of the Trypanosoma cruzi cyclophilin gene family and identification of cyclosporin A binding proteins. Parasitology 132:867–882. 10.1017/S0031182005009558 [DOI] [PubMed] [Google Scholar]
- 106.Dao-Thi MH, Transue TR, Pellé R, Murphy NB, Poortmans F, Steyaert J. 1998. Expression, purification, crystallization and preliminary X-ray analysis of cyclophilin A from the bovine parasite Trypanosoma brucei brucei. Acta Crystallogr. D Biol. Crystallogr. 54:1046–1048. 10.1107/S0907444998001607 [DOI] [PubMed] [Google Scholar]
- 107.Búa J, Aslund L, Pereyra N, García GA, Bontempi EJ, Ruiz AM. 2001. Characterisation of a cyclophilin isoform in Trypanosoma cruzi. FEMS Microbiol. Lett. 200:43–47. 10.1111/j.1574-6968.2001.tb10690.x [DOI] [PubMed] [Google Scholar]
- 108.Pellé R, McOdimba F, Chuma F, Wasawo D, Pearson TW, Murphy NB. 2002. The African trypanosome cyclophilin A homologue contains unusual conserved central and N-terminal domains and is developmentally regulated. Gene 290:181–191. 10.1016/S0378-1119(02)00559-0 [DOI] [PubMed] [Google Scholar]
- 109.Rascher C, Pahl A, Pecht A, Brune K, Solbach W, Bang H. 1998. Leishmania major parasites express cyclophilin isoforms with an unusual interaction with calcineurin. Biochem. J. 334:659–667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hoerauf A, Rascher C, Bang R, Pahl A, Solbach W, Brune K, Röllinghoff M, Bang H. 1997. Host-cell cyclophilin is important for the intracellular replication of Leishmania major. Mol. Microbiol. 24:421–429. 10.1046/j.1365-2958.1997.3401716.x [DOI] [PubMed] [Google Scholar]
- 111.Dutta M, Delhi P, Sinha KM, Banerjee R, Datta AK. 2001. Lack of abundance of cytoplasmic cyclosporin A-binding protein renders free-living Leishmania donovani resistant to cyclosporin A. J. Biol. Chem. 276:19294–19300. 10.1074/jbc.M009379200 [DOI] [PubMed] [Google Scholar]
- 112.Chakraborty A, Das I, Datta R, Sen B, Bhattacharyya D, Mandal C, Datta AK. 2002. A single-domain cyclophilin from Leishmania donovani reactivates soluble aggregates of adenosine kinase by isomerase-independent chaperone function. J. Biol. Chem. 277:47451–47460. 10.1074/jbc.M204827200 [DOI] [PubMed] [Google Scholar]
- 113.Sen B, Chakraborty A, Datta R, Bhattacharyya D, Datta AK. 2006. Reversal of ADP-mediated aggregation of adenosine kinase by cyclophilin leads to its reactivation. Biochemistry (Mosc.) 45:263–271. 10.1021/bi0518489 [DOI] [PubMed] [Google Scholar]
- 114.Chakraborty A, Sen B, Datta R, Datta AK. 2004. Isomerase-independent chaperone function of cyclophilin ensures aggregation prevention of adenosine kinase both in vitro and under in vivo conditions. Biochemistry (Mosc.) 43:11862–11872. 10.1021/bi049490o [DOI] [PubMed] [Google Scholar]
- 115.Ostoa-Saloma P, César Carrero J, Petrossian P, Hérión P, Landa A, Pedro Laclette J. 2000. Cloning, characterization and functional expression of a cyclophilin of Entamoeba histolytica. Mol. Biochem. Parasitol. 107:219–225. 10.1016/S0166-6851(00)00190-0 [DOI] [PubMed] [Google Scholar]
- 116.Akbar MA, Chatterjee NS, Sen P, Debnath A, Pal A, Bera T, Das P. 2004. Genes induced by a high-oxygen environment in Entamoeba histolytica. Mol. Biochem. Parasitol. 133:187–196. 10.1016/j.molbiopara.2003.10.006 [DOI] [PubMed] [Google Scholar]
- 117.Monaghan P, Bell A. 2005. A Plasmodium falciparum FK506-binding protein (FKBP) with peptidyl-prolyl cis-trans isomerase and chaperone activities. Mol. Biochem. Parasitol. 139:185–195. 10.1016/j.molbiopara.2004.10.007 [DOI] [PubMed] [Google Scholar]
- 118.Kumar R, Adams B, Musiyenko A, Shulyayeva O, Barik S. 2005. The FK506-binding protein of the malaria parasite, Plasmodium falciparum, is a FK506-sensitive chaperone with FK506-independent calcineurin-inhibitory activity. Mol. Biochem. Parasitol. 141:163–173. 10.1016/j.molbiopara.2005.02.007 [DOI] [PubMed] [Google Scholar]
- 119.Alag R, Shin J, Yoon HS. 2009. NMR assignments of the FK506-binding domain of FK506-binding protein 35 from Plasmodium vivax. Biomol. NMR Assign. 3:243–245. 10.1007/s12104-009-9185-1 [DOI] [PubMed] [Google Scholar]
- 120.Rajan S, Austin D, Harikishore A, Nguyen QT, Baek K, Yoon HS. 2014. Crystal structure of Plasmodium vivax FK506-binding protein 25 reveals conformational changes responsible for its noncanonical activity. Proteins 82:1235–1244. 10.1002/prot.24487 [DOI] [PubMed] [Google Scholar]
- 121.Brydges SD, Sherman GD, Nockemann S, Loyens A, Däubener W, Dubremetz JF, Carruthers VB. 2000. Molecular characterization of TgMIC5, a proteolytically processed antigen secreted from the micronemes of Toxoplasma gondii. Mol. Biochem. Parasitol. 111:51–66. 10.1016/S0166-6851(00)00296-6 [DOI] [PubMed] [Google Scholar]
- 122.Brydges SD, Zhou XW, Huynh M-H, Harper JM, Mital J, Adjogble KDZ, Däubener W, Ward GE, Carruthers VB. 2006. Targeted deletion of MIC5 enhances trimming proteolysis of Toxoplasma invasion proteins. Eukaryot. Cell 5:2174–2183. 10.1128/EC.00163-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Saouros S, Dou Z, Henry M, Marchant J, Carruthers VB, Matthews S. 2012. Microneme protein 5 regulates the activity of Toxoplasma subtilisin 1 by mimicking a subtilisin prodomain. J. Biol. Chem. 287:36029–36040. 10.1074/jbc.M112.389825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Erben ED, Daum S, Téllez-Iñón MT. 2007. The Trypanosoma cruzi PIN1 gene encodes a parvulin peptidyl-prolyl cis/trans isomerase able to replace the essential ESS1 in Saccharomyces cerevisiae. Mol. Biochem. Parasitol. 153:186–193. 10.1016/j.molbiopara.2007.03.004 [DOI] [PubMed] [Google Scholar]
- 125.Erben ED, Valguarnera E, Nardelli S, Chung J, Daum S, Potenza M, Schenkman S, Téllez-Iñón MT. 2010. Identification of an atypical peptidyl-prolyl cis/trans isomerase from trypanosomatids. Biochim. Biophys. Acta 1803:1028–1037. 10.1016/j.bbamcr.2010.05.006 [DOI] [PubMed] [Google Scholar]
- 126.Erben ED, Nardelli SC, de Jesus TCL, Schenkman S, Tellez-Iñon MT. 2013. Trypanosomatid pin1-type peptidyl-prolyl isomerase is cytosolic and not essential for cell proliferation. J. Eukaryot. Microbiol. 60:101–105. 10.1111/jeu.12009 [DOI] [PubMed] [Google Scholar]
- 127.Goh JY, Lai C-Y, Tan LC, Yang D, He CY, Liou Y-C. 2010. Functional characterization of two novel parvulins in Trypanosoma brucei. FEBS Lett. 584:2901–2908. 10.1016/j.febslet.2010.04.077 [DOI] [PubMed] [Google Scholar]
- 128.Sun L, Wu X, Peng Y, Goh JY, Liou Y-C, Lin D, Zhao Y. 2012. Solution structural analysis of the single-domain parvulin TbPin1. PLoS One 7:e43017. 10.1371/journal.pone.0043017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hermans PWM, Adrian PV, Albert C, Estevão S, Hoogenboezem T, Luijendijk IHT, Kamphausen T, Hammerschmidt S. 2006. The streptococcal lipoprotein rotamase A (SlrA) is a functional peptidyl-prolyl isomerase involved in pneumococcal colonization. J. Biol. Chem. 281:968–976. 10.1074/jbc.M510014200 [DOI] [PubMed] [Google Scholar]
- 130.Purdy GE, Fisher CR, Payne SM. 2007. IcsA surface presentation in Shigella flexneri requires the periplasmic chaperones DegP, Skp, and SurA. J. Bacteriol. 189:5566–5573. 10.1128/JB.00483-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Alonzo F, III, Freitag NE. 2010. Listeria monocytogenes PrsA2 is required for virulence factor secretion and bacterial viability within the host cell cytosol. Infect. Immun. 78:4944–4957. 10.1128/IAI.00532-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Forster BM, Zemansky J, Portnoy DA, Marquis H. 2011. Posttranslocation chaperone PrsA2 regulates the maturation and secretion of Listeria monocytogenes proprotein virulence factors. J. Bacteriol. 193:5961–5970. 10.1128/JB.05307-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Justice SS, Lauer SR, Hultgren SJ, Hunstad DA. 2006. Maturation of intracellular Escherichia coli communities requires SurA. Infect. Immun. 74:4793–4800. 10.1128/IAI.00355-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Vertommen D, Ruiz N, Leverrier P, Silhavy TJ, Collet J-F. 2009. Characterization of the role of the Escherichia coli periplasmic chaperone SurA using differential proteomics. Proteomics 9:2432–2443. 10.1002/pmic.200800794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Confer AW, Ayalew S. 2013. The OmpA family of proteins: roles in bacterial pathogenesis and immunity. Vet. Microbiol. 163(3–4):207–222. 10.1016/j.vetmic.2012.08.019 [DOI] [PubMed] [Google Scholar]
- 136.Anderson GG, Palermo JJ, Schilling JD, Roth R, Heuser J, Hultgren SJ. 2003. Intracellular bacterial biofilm-like pods in urinary tract infections. Science 301:105–107. 10.1126/science.1084550 [DOI] [PubMed] [Google Scholar]
- 137.Remaut H, Tang C, Henderson NS, Pinkner JS, Wang T, Hultgren SJ, Thanassi DG, Waksman G, Li H. 2008. Fiber formation across the bacterial outer membrane by the chaperone/usher pathway. Cell 133:640–652. 10.1016/j.cell.2008.03.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Palomino C, Marín E, Fernández LÁ. 2011. The fimbrial usher FimD follows the SurA-BamB pathway for its assembly in the outer membrane of Escherichia coli. J. Bacteriol. 193:5222–5230. 10.1128/JB.05585-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bodelón G, Marín E, Fernández LA. 2009. Role of periplasmic chaperones and BamA (YaeT/Omp85) in folding and secretion of intimin from enteropathogenic Escherichia coli strains. J. Bacteriol. 191:5169–5179. 10.1128/JB.00458-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Watts KM, Hunstad DA. 2008. Components of SurA required for outer membrane biogenesis in uropathogenic Escherichia coli. PLoS One 3:e3359. 10.1371/journal.pone.0003359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Obi IR, Francis MS. 2013. Demarcating SurA activities required for outer membrane targeting of Yersinia pseudotuberculosis adhesins. Infect. Immun. 81:2296–2308. 10.1128/IAI.01208-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Fardini Y, Trotereau J, Bottreau E, Souchard C, Velge P, Virlogeux-Payant I. 2009. Investigation of the role of the BAM complex and SurA chaperone in outer-membrane protein biogenesis and type III secretion system expression in Salmonella. Microbiology 155:1613–1622. 10.1099/mic.0.025155-0 [DOI] [PubMed] [Google Scholar]
- 143.Mansfield J, Genin S, Magori S, Citovsky V, Sriariyanum M, Ronald P, Dow M, Verdier V, Beer SV, Machado MA, Toth I, Salmond G, Foster GD. 2012. Top 10 plant pathogenic bacteria in molecular plant pathology. Mol. Plant Pathol. 13:614–629. 10.1111/j.1364-3703.2012.00804.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Rondelet A, Condemine G. 2012. SurA is involved in the targeting to the outer membrane of a Tat signal sequence-anchored protein. J. Bacteriol. 194:6131–6142. 10.1128/JB.01419-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Basak C, Pathak SK, Bhattacharyya A, Pathak S, Basu J, Kundu M. 2005. The secreted peptidyl prolyl cis/trans-isomerase HP0175 of Helicobacter pylori induces apoptosis of gastric epithelial cells in a TLR4- and apoptosis signal-regulating kinase 1-dependent manner. J. Immunol. 174:5672–5680. 10.4049/jimmunol.174.9.5672 [DOI] [PubMed] [Google Scholar]
- 146.Hodak H, Wohlkönig A, Smet-Nocca C, Drobecq H, Wieruszeski J-M, Sénéchal M, Landrieu I, Locht C, Jamin M, Jacob-Dubuisson F. 2008. The peptidyl-prolyl isomerase and chaperone Par27 of Bordetella pertussis as the prototype for a new group of parvulins. J. Mol. Biol. 376:414–426. 10.1016/j.jmb.2007.10.088 [DOI] [PubMed] [Google Scholar]
- 147.Clantin B, Leyrat C, Wohlkönig A, Hodak H, Ribeiro Ede A, Jr, Martinez N, Baud C, Smet-Nocca C, Villeret V, Jacob-Dubuisson F, Jamin M. 2010. Structure and plasticity of the peptidyl-prolyl isomerase Par27 of Bordetella pertussis revealed by X-ray diffraction and small-angle X-ray scattering. J. Struct. Biol. 169:253–265. 10.1016/j.jsb.2009.11.007 [DOI] [PubMed] [Google Scholar]
- 148.Kale A, Phansopa C, Suwannachart C, Craven CJ, Rafferty JB, Kelly DJ. 2011. The virulence factor PEB4 (Cj0596) and the periplasmic protein Cj1289 are two structurally related SurA-like chaperones in the human pathogen Campylobacter jejuni. J. Biol. Chem. 286:21254–21265. 10.1074/jbc.M111.220442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Alonzo F, III, Port GC, Cao M, Freitag NE. 2009. The posttranslocation chaperone PrsA2 contributes to multiple facets of Listeria monocytogenes pathogenesis. Infect. Immun. 77:2612–2623. 10.1128/IAI.00280-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zemansky J, Kline BC, Woodward JJ, Leber JH, Marquis H, Portnoy DA. 2009. Development of a mariner-based transposon and identification of Listeria monocytogenes determinants, including the peptidyl-prolyl isomerase PrsA2, that contribute to its hemolytic phenotype. J. Bacteriol. 191:3950–3964. 10.1128/JB.00016-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Alonzo F, III, Xayarath B, Whisstock JC, Freitag NE. 2011. Functional analysis of the Listeria monocytogenes secretion chaperone PrsA2 and its multiple contributions to bacterial virulence. Mol. Microbiol. 80:1530–1548. 10.1111/j.1365-2958.2011.07665.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Overweg K, Kerr A, Sluijter M, Jackson MH, Mitchell TJ, de Jong AP, de Groot R, Hermans PW. 2000. The putative proteinase maturation protein A of Streptococcus pneumoniae is a conserved surface protein with potential to elicit protective immune responses. Infect. Immun. 68:4180–4188. 10.1128/IAI.68.7.4180-4188.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Adrian PV, Bogaert D, Oprins M, Rapola S, Lahdenkari M, Kilpi T, de Groot R, Käyhty H, Hermans PWM. 2004. Development of antibodies against pneumococcal proteins alpha-enolase, immunoglobulin A1 protease, streptococcal lipoprotein rotamase A, and putative proteinase maturation protein A in relation to pneumococcal carriage and otitis media. Vaccine 22:2737–2742. 10.1016/j.vaccine.2004.01.042 [DOI] [PubMed] [Google Scholar]
- 154.Cron LE, Bootsma HJ, Noske N, Burghout P, Hammerschmidt S, Hermans PWM. 2009. Surface-associated lipoprotein PpmA of Streptococcus pneumoniae is involved in colonization in a strain-specific manner. Microbiology 155:2401–2410. 10.1099/mic.0.026765-0 [DOI] [PubMed] [Google Scholar]
- 155.Wen ZT, Suntharaligham P, Cvitkovitch DG, Burne RA. 2005. Trigger factor in Streptococcus mutans is involved in stress tolerance, competence development, and biofilm formation. Infect. Immun. 73:219–225. 10.1128/IAI.73.1.219-225.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lyon WR, Gibson CM, Caparon MG. 1998. A role for trigger factor and an Rgg-like regulator in the transcription, secretion and processing of the cysteine proteinase of Streptococcus pyogenes. EMBO J. 17:6263–6275. 10.1093/emboj/17.21.6263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lyon WR, Caparon MG. 2003. Trigger factor-mediated prolyl isomerization influences maturation of the Streptococcus pyogenes cysteine protease. J. Bacteriol. 185:3661–3667. 10.1128/JB.185.12.3661-3667.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Wu T, Zhao Z, Zhang L, Ma H, Lu K, Ren W, Liu Z, Chang H, Bei W, Qiu Y, Chen H. 2011. Trigger factor of Streptococcus suis is involved in stress tolerance and virulence. Microb. Pathog. 51:69–76. 10.1016/j.micpath.2010.10.001 [DOI] [PubMed] [Google Scholar]
- 159.Bigot A, Botton E, Dubail I, Charbit A. 2006. A homolog of Bacillus subtilis trigger factor in Listeria monocytogenes is involved in stress tolerance and bacterial virulence. Appl. Environ. Microbiol. 72:6623–6631. 10.1128/AEM.00624-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yau W-L, Blisnick T, Taly J-F, Helmer-Citterich M, Schiene-Fischer C, Leclercq O, Li J, Schmidt-Arras D, Morales MA, Notredame C, Romo D, Bastin P, Späth GF. 2010. Cyclosporin A treatment of Leishmania donovani reveals stage-specific functions of cyclophilins in parasite proliferation and viability. PLoS Negl. Trop. Dis. 4:e729. 10.1371/journal.pntd.0000729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Ibrahim HM, Bannai H, Xuan X, Nishikawa Y. 2009. Toxoplasma gondii cyclophilin 18-mediated production of nitric oxide induces bradyzoite conversion in a CCR5-dependent manner. Infect. Immun. 77:3686–3695. 10.1128/IAI.00361-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ibrahim HM, Xuan X, Nishikawa Y. 2010. Toxoplasma gondii cyclophilin 18 regulates the proliferation and migration of murine macrophages and spleen cells. Clin. Vaccine Immunol. 17:1322–1329. 10.1128/CVI.00128-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Kulkarni MM, Karafova A, Kamysz W, Schenkman S, Pelle R, McGwire BS. 2013. Secreted trypanosome cyclophilin inactivates lytic insect defense peptides and induces parasite calcineurin activation and infectivity. J. Biol. Chem. 288:8772–8784. 10.1074/jbc.M112.421057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sherry B, Zybarth G, Alfano M, Dubrovsky L, Mitchell R, Rich D, Ulrich P, Bucala R, Cerami A, Bukrinsky M. 1998. Role of cyclophilin A in the uptake of HIV-1 by macrophages and T lymphocytes. Proc. Natl. Acad. Sci. U. S. A. 95:1758–1763. 10.1073/pnas.95.4.1758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Allain F, Vanpouille C, Carpentier M, Slomianny M-C, Durieux S, Spik G. 2002. Interaction with glycosaminoglycans is required for cyclophilin B to trigger integrin-mediated adhesion of peripheral blood T lymphocytes to extracellular matrix. Proc. Natl. Acad. Sci. U. S. A. 99:2714–2719. 10.1073/pnas.052284899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Yurchenko V, O'Connor M, Dai WW, Guo H, Toole B, Sherry B, Bukrinsky M. 2001. CD147 is a signaling receptor for cyclophilin B. Biochem. Biophys. Res. Commun. 288:786–788. 10.1006/bbrc.2001.5847 [DOI] [PubMed] [Google Scholar]
- 167.Yurchenko V, Zybarth G, O'Connor M, Dai WW, Franchin G, Hao T, Guo H, Hung H-C, Toole B, Gallay P, Sherry B, Bukrinsky M. 2002. Active site residues of cyclophilin A are crucial for its signaling activity via CD147. J. Biol. Chem. 277:22959–22965. 10.1074/jbc.M201593200 [DOI] [PubMed] [Google Scholar]
- 168.Yurchenko V, Xue Z, Sherry B, Bukrinsky M. 2008. Functional analysis of Leishmania major cyclophilin. Int. J. Parasitol. 38:633–639. 10.1016/j.ijpara.2007.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP. 1993. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73:1067–1078. 10.1016/0092-8674(93)90637-6 [DOI] [PubMed] [Google Scholar]
- 170.Bose S, Mathur M, Bates P, Joshi N, Banerjee AK. 2003. Requirement for cyclophilin A for the replication of vesicular stomatitis virus New Jersey serotype. J. Gen. Virol. 84:1687–1699. 10.1099/vir.0.19074-0 [DOI] [PubMed] [Google Scholar]
- 171.Watashi K, Ishii N, Hijikata M, Inoue D, Murata T, Miyanari Y, Shimotohno K. 2005. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol. Cell 19:111–122. 10.1016/j.molcel.2005.05.014 [DOI] [PubMed] [Google Scholar]
- 172.Liu X, Sun L, Yu M, Wang Z, Xu C, Xue Q, Zhang K, Ye X, Kitamura Y, Liu W. 2009. Cyclophilin A interacts with influenza A virus M1 protein and impairs the early stage of the viral replication. Cell. Microbiol. 11:730–741. 10.1111/j.1462-5822.2009.01286.x [DOI] [PubMed] [Google Scholar]
- 173.Kambara H, Tani H, Mori Y, Abe T, Katoh H, Fukuhara T, Taguwa S, Moriishi K, Matsuura Y. 2011. Involvement of cyclophilin B in the replication of Japanese encephalitis virus. Virology 412:211–219. 10.1016/j.virol.2011.01.011 [DOI] [PubMed] [Google Scholar]
- 174.Pfefferle S, Schöpf J, Kögl M, Friedel CC, Müller MA, Carbajo-Lozoya J, Stellberger T, von Dall'Armi E, Herzog P, Kallies S, Niemeyer D, Ditt V, Kuri T, Züst R, Pumpor K, Hilgenfeld R, Schwarz F, Zimmer R, Steffen I, Weber F, Thiel V, Herrler G, Thiel H-J, Schwegmann-Wessels C, Pöhlmann S, Haas J, Drosten C, von Brunn A. 2011. The SARS-coronavirus-host interactome: identification of cyclophilins as target for pan-coronavirus inhibitors. PLoS Pathog. 7:e1002331. 10.1371/journal.ppat.1002331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Keyes LR, Bego MG, Soland M, St Jeor S. 2012. Cyclophilin A is required for efficient human cytomegalovirus DNA replication and reactivation. J. Gen. Virol. 93:722–732. 10.1099/vir.0.037309-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.He H, Mou Z, Li W, Fei L, Tang Y, Zhang J, Yan P, Chen Z, Yang X, Shen Z, Li J, Wu Y. 2013. Proteomic methods reveal cyclophilin A function as a host restriction factor against rotavirus infection. Proteomics 13:1121–1132. 10.1002/pmic.201100579 [DOI] [PubMed] [Google Scholar]
- 177.Frausto SD, Lee E, Tang H. 2013. Cyclophilins as modulators of viral replication. Viruses 5:1684–1701. 10.3390/v5071684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Liu X, Zhao Z, Liu W. 2013. Insights into the roles of cyclophilin A during influenza virus infection. Viruses 5:182–191. 10.3390/v5010182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Endrich MM, Gehrig P, Gehring H. 1999. Maturation-induced conformational changes of HIV-1 capsid protein and identification of two high affinity sites for cyclophilins in the C-terminal domain. J. Biol. Chem. 274:5326–5332. 10.1074/jbc.274.9.5326 [DOI] [PubMed] [Google Scholar]
- 180.Kootstra NA, Munk C, Tonnu N, Landau NR, Verma IM. 2003. Abrogation of postentry restriction of HIV-1-based lentiviral vector transduction in simian cells. Proc. Natl. Acad. Sci. U. S. A. 100:1298–1303. 10.1073/pnas.0337541100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Towers GJ, Hatziioannou T, Cowan S, Goff SP, Luban J, Bieniasz PD. 2003. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat. Med. 9:1138–1143. 10.1038/nm910 [DOI] [PubMed] [Google Scholar]
- 182.Ikeda Y, Ylinen LMJ, Kahar-Bador M, Towers GJ. 2004. Influence of gag on human immunodeficiency virus type 1 species-specific tropism. J. Virol. 78:11816–11822. 10.1128/JVI.78.21.11816-11822.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Bon Homme M, Carter C, Scarlata S. 2005. The cysteine residues of HIV-1 capsid regulate oligomerization and cyclophilin A-induced changes. Biophys. J. 88:2078–2088. 10.1529/biophysj.104.053298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Thai V, Renesto P, Fowler CA, Brown DJ, Davis T, Gu W, Pollock DD, Kern D, Raoult D, Eisenmesser EZ. 2008. Structural, biochemical, and in vivo characterization of the first virally encoded cyclophilin from the mimivirus. J. Mol. Biol. 378:71–86. 10.1016/j.jmb.2007.08.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Manganaro L, Lusic M, Gutierrez MI, Cereseto A, Del Sal G, Giacca M. 2010. Concerted action of cellular JNK and Pin1 restricts HIV-1 genome integration to activated CD4+ T lymphocytes. Nat. Med. 16:329–333. 10.1038/nm.2102 [DOI] [PubMed] [Google Scholar]
- 186.Narita Y, Murata T, Ryo A, Kawashima D, Sugimoto A, Kanda T, Kimura H, Tsurumi T. 2013. Pin1 interacts with the Epstein-Barr virus DNA polymerase catalytic subunit and regulates viral DNA replication. J. Virol. 87:2120–2127. 10.1128/JVI.02634-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Deng W, Chen L, Wood DW, Metcalfe T, Liang X, Gordon MP, Comai L, Nester EW. 1998. Agrobacterium VirD2 protein interacts with plant host cyclophilins. Proc. Natl. Acad. Sci. U. S. A. 95:7040–7045. 10.1073/pnas.95.12.7040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Coaker G, Falick A, Staskawicz B. 2005. Activation of a phytopathogenic bacterial effector protein by a eukaryotic cyclophilin. Science 308:548–550. 10.1126/science.1108633 [DOI] [PubMed] [Google Scholar]
- 189.Axtell MJ, Staskawicz BJ. 2003. Initiation of RPS2-specified disease resistance in Arabidopsis is coupled to the AvrRpt2-directed elimination of RIN4. Cell 112:369–377. 10.1016/S0092-8674(03)00036-9 [DOI] [PubMed] [Google Scholar]
- 190.Mackey D, Belkhadir Y, Alonso JM, Ecker JR, Dangl JL. 2003. Arabidopsis RIN4 is a target of the type III virulence effector AvrRpt2 and modulates RPS2-mediated resistance. Cell 112:379–389. 10.1016/S0092-8674(03)00040-0 [DOI] [PubMed] [Google Scholar]
- 191.Coaker G, Zhu G, Ding Z, Van Doren SR, Staskawicz B. 2006. Eukaryotic cyclophilin as a molecular switch for effector activation. Mol. Microbiol. 61:1485–1496. 10.1111/j.1365-2958.2006.05335.x [DOI] [PubMed] [Google Scholar]
- 192.Aumüller T, Jahreis G, Fischer G, Schiene-Fischer C. 2010. Role of prolyl cis/trans isomers in cyclophilin-assisted Pseudomonas syringae AvrRpt2 protease activation. Biochemistry (Mosc.) 49:1042–1052. 10.1021/bi901813e [DOI] [PubMed] [Google Scholar]
- 193.Zhu C, Wang Y, Li Y, Bhatti KH, Tian Y, Wu J. 2011. Overexpression of a cotton cyclophilin gene (GhCyp1) in transgenic tobacco plants confers dual tolerance to salt stress and Pseudomonas syringae pv. tabaci infection. Plant Physiol. Biochem. 49:1264–1271. 10.1016/j.plaphy.2011.09.001 [DOI] [PubMed] [Google Scholar]
- 194.Pogorelko GV, Mokryakova M, Fursova OV, Abdeeva I, Piruzian ES, Bruskin SA. 2014. Characterization of three Arabidopsis thaliana immunophilin genes involved in the plant defense response against Pseudomonas syringae. Gene 538:12–22. 10.1016/j.gene.2014.01.029 [DOI] [PubMed] [Google Scholar]
- 195.DiNovo AA, Schey KL, Vachon WS, McGuffie EM, Olson JC, Vincent TS. 2006. ADP-ribosylation of cyclophilin A by Pseudomonas aeruginosa exoenzyme S. Biochemistry (Mosc.) 45:4664–4673. 10.1021/bi0513554 [DOI] [PubMed] [Google Scholar]
- 196.Aulik NA, Hellenbrand KM, Kisiela D, Czuprynski CJ. 2011. Mannheimia haemolytica leukotoxin binds cyclophilin D on bovine neutrophil mitochondria. Microb. Pathog. 50:168–178. 10.1016/j.micpath.2011.01.001 [DOI] [PubMed] [Google Scholar]
- 197.Elrod JW, Molkentin JD. 2013. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ. J. 77:1111–1122. 10.1253/circj.CJ-13-0321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Atapattu DN, Albrecht RM, McClenahan DJ, Czuprynski CJ. 2008. Dynamin-2-dependent targeting of Mannheimia haemolytica leukotoxin to mitochondrial cyclophilin D in bovine lymphoblastoid cells. Infect. Immun. 76:5357–5365. 10.1128/IAI.00221-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Carneiro LAM, Travassos LH, Soares F, Tattoli I, Magalhaes JG, Bozza MT, Plotkowski MC, Sansonetti PJ, Molkentin JD, Philpott DJ, Girardin SE. 2009. Shigella induces mitochondrial dysfunction and cell death in nonmyeloid cells. Cell Host Microbe 5:123–136. 10.1016/j.chom.2008.12.011 [DOI] [PubMed] [Google Scholar]
- 200.Barth H, Aktories K, Popoff MR, Stiles BG. 2004. Binary bacterial toxins: biochemistry, biology, and applications of common Clostridium and Bacillus proteins. Microbiol. Mol. Biol. Rev. 68:373–402. 10.1128/MMBR.68.3.373-402.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Dmochewitz L, Lillich M, Kaiser E, Jennings LD, Lang AE, Buchner J, Fischer G, Aktories K, Collier RJ, Barth H. 2011. Role of CypA and Hsp90 in membrane translocation mediated by anthrax protective antigen. Cell. Microbiol. 13:359–373. 10.1111/j.1462-5822.2010.01539.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Kaiser E, Kroll C, Ernst K, Schwan C, Popoff M, Fischer G, Buchner J, Aktories K, Barth H. 2011. Membrane translocation of binary actin-ADP-ribosylating toxins from Clostridium difficile and Clostridium perfringens is facilitated by cyclophilin A and Hsp90. Infect. Immun. 79:3913–3921. 10.1128/IAI.05372-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Kaiser E, Böhm N, Ernst K, Langer S, Schwan C, Aktories K, Popoff M, Fischer G, Barth H. 2012. FK506-binding protein 51 interacts with Clostridium botulinum C2 toxin and FK506 inhibits membrane translocation of the toxin in mammalian cells. Cell. Microbiol. 14:1193–1205. 10.1111/j.1462-5822.2012.01788.x [DOI] [PubMed] [Google Scholar]
- 204.Mahbubani MH, Bej AK, Miller R, Haff L, DiCesare J, Atlas RM. 1990. Detection of Legionella with polymerase chain reaction and gene probe methods. Mol. Cell. Probes 4:175–187. 10.1016/0890-8508(90)90051-Z [DOI] [PubMed] [Google Scholar]
- 205.Jaulhac B, Nowicki M, Bornstein N, Meunier O, Prevost G, Piemont Y, Fleurette J, Monteil H. 1992. Detection of Legionella spp. in bronchoalveolar lavage fluids by DNA amplification. J. Clin. Microbiol. 30:920–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Svarrer CW, Uldum SA. 2012. The occurrence of Legionella species other than Legionella pneumophila in clinical and environmental samples in Denmark identified by mip gene sequencing and matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Clin. Microbiol. Infect. 18:1004–1009. 10.1111/j.1469-0691.2011.03698.x [DOI] [PubMed] [Google Scholar]
- 207.O'Connell WA, Bangsborg JM, Cianciotto NP. 1995. Characterization of a Legionella micdadei mip mutant. Infect. Immun. 63:2840–2845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Doyle RM, Steele TW, McLennan AM, Parkinson IH, Manning PA, Heuzenroeder MW. 1998. Sequence analysis of the mip gene of the soilborne pathogen Legionella longbeachae. Infect. Immun. 66:1492–1499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Arakaki N, Higa F, Koide M, Tateyama M, Saito A. 2002. Induction of apoptosis of human macrophages in vitro by Legionella longbeachae through activation of the caspase pathway. J. Med. Microbiol. 51:159–168 [DOI] [PubMed] [Google Scholar]
- 210.Mo YY, Cianciotto NP, Mallavia LP. 1995. Molecular cloning of a Coxiella burnetii gene encoding a macrophage infectivity potentiator (Mip) analogue. Microbiology 141:2861–2871. 10.1099/13500872-141-11-2861 [DOI] [PubMed] [Google Scholar]
- 211.Lundemose AG, Birkelund S, Fey SJ, Larsen PM, Christiansen G. 1991. Chlamydia trachomatis contains a protein similar to the Legionella pneumophila mip gene product. Mol. Microbiol. 5:109–115. 10.1111/j.1365-2958.1991.tb01831.x [DOI] [PubMed] [Google Scholar]
- 212.Rockey DD, Chesebro BB, Heinzen RA, Hackstadt T. 1996. A 28 kDa major immunogen of Chlamydia psittaci shares identity with Mip proteins of Legionella spp. and Chlamydia trachomatis—cloning and characterization of the C. psittaci mip-like gene. Microbiology 142:945–953. 10.1099/00221287-142-4-945 [DOI] [PubMed] [Google Scholar]
- 213.Herrmann M, Schuhmacher A, Mühldorfer I, Melchers K, Prothmann C, Dammeier S. 2006. Identification and characterization of secreted effector proteins of Chlamydophila pneumoniae TW183. Res. Microbiol. 157:513–524. 10.1016/j.resmic.2005.12.005 [DOI] [PubMed] [Google Scholar]
- 214.Marques PX, Souda P, O'Donovan J, Gutierrez J, Gutierrez EJ, Worrall S, McElroy M, Proctor A, Brady C, Sammin D, Basset HF, Whitelegge JP, Markey BE, Nally JE. 2010. Identification of immunologically relevant proteins of Chlamydophila abortus using sera from experimentally infected pregnant ewes. Clin. Vaccine Immunol. 17:1274–1281. 10.1128/CVI.00163-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Lundemose AG, Rouch DA, Penn CW, Pearce JH. 1993. The Chlamydia trachomatis Mip-like protein is a lipoprotein. J. Bacteriol. 175:3669–3671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Neff L, Daher S, Muzzin P, Spenato U, Gülaçar F, Gabay C, Bas S. 2007. Molecular characterization and subcellular localization of macrophage infectivity potentiator, a Chlamydia trachomatis lipoprotein. J. Bacteriol. 189:4739–4748. 10.1128/JB.01889-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Lundemose AG, Kay JE, Pearce JH. 1993. Chlamydia trachomatis Mip-like protein has peptidyl-prolyl cis/trans isomerase activity that is inhibited by FK506 and rapamycin and is implicated in initiation of chlamydial infection. Mol. Microbiol. 7:777–783. 10.1111/j.1365-2958.1993.tb01168.x [DOI] [PubMed] [Google Scholar]
- 218.Leuzzi R, Serino L, Scarselli M, Savino S, Fontana MR, Monaci E, Taddei A, Fischer G, Rappuoli R, Pizza M. 2005. Ng-MIP, a surface-exposed lipoprotein of Neisseria gonorrhoeae, has a peptidyl-prolyl cis/trans isomerase (PPIase) activity and is involved in persistence in macrophages. Mol. Microbiol. 58:669–681. 10.1111/j.1365-2958.2005.04859.x [DOI] [PubMed] [Google Scholar]
- 219.Echenique-Rivera H, Muzzi A, Del Tordello E, Seib KL, Francois P, Rappuoli R, Pizza M, Serruto D. 2011. Transcriptome analysis of Neisseria meningitidis in human whole blood and mutagenesis studies identify virulence factors involved in blood survival. PLoS Pathog. 7:e1002027. 10.1371/journal.ppat.1002027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Hung M-C, Salim O, Williams JN, Heckels JE, Christodoulides M. 2011. The Neisseria meningitidis macrophage infectivity potentiator protein induces cross-strain serum bactericidal activity and is a potential serogroup B vaccine candidate. Infect. Immun. 79:3784–3791. 10.1128/IAI.05019-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Maeda T, Maeda H, Yamabe K, Mineshiba J, Tanimoto I, Yamamoto T, Naruishi K, Kokeguchi S, Takashiba S. 2010. Highly expressed genes in a rough-colony-forming phenotype of Aggregatibacter actinomycetemcomitans: implication of a mip-like gene for the invasion of host tissue. FEMS Immunol. Med. Microbiol. 58:226–236. 10.1111/j.1574-695X.2009.00624.x [DOI] [PubMed] [Google Scholar]
- 222.Wong CY, Heuzenroeder MW, Quinn DM, Flower RL. 1997. Cloning and characterization of two immunophilin-like genes, ilpA and fkpA, on a single 3.9-kilobase fragment of Aeromonas hydrophila genomic DNA. J. Bacteriol. 179:3397–3403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Horne SM, Kottom TJ, Nolan LK, Young KD. 1997. Decreased intracellular survival of an fkpA mutant of Salmonella typhimurium Copenhagen. Infect. Immun. 65:806–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Humphreys S, Rowley G, Stevenson A, Kenyon WJ, Spector MP, Roberts M. 2003. Role of periplasmic peptidylprolyl isomerases in Salmonella enterica serovar Typhimurium virulence. Infect. Immun. 71:5386–5388. 10.1128/IAI.71.9.5386-5388.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Rizzitello AE, Harper JR, Silhavy TJ. 2001. Genetic evidence for parallel pathways of chaperone activity in the periplasm of Escherichia coli. J. Bacteriol. 183:6794–6800. 10.1128/JB.183.23.6794-6800.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Sklar JG, Wu T, Kahne D, Silhavy TJ. 2007. Defining the roles of the periplasmic chaperones SurA, Skp, and DegP in Escherichia coli. Genes Dev. 21:2473–2484. 10.1101/gad.1581007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Ruiz-Perez F, Henderson IR, Leyton DL, Rossiter AE, Zhang Y, Nataro JP. 2009. Roles of periplasmic chaperone proteins in the biogenesis of serine protease autotransporters of Enterobacteriaceae. J. Bacteriol. 191:6571–6583. 10.1128/JB.00754-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Ruiz-Perez F, Henderson IR, Nataro JP. 2010. Interaction of FkpA, a peptidyl-prolyl cis/trans isomerase with EspP autotransporter protein. Gut Microbes 1:339–344. 10.4161/gmic.1.5.13436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Zang N, Tang D-J, Wei M-L, He Y-Q, Chen B, Feng J-X, Xu J, Gan Y-Q, Jiang B-L, Tang J-L. 2007. Requirement of a mip-like gene for virulence in the phytopathogenic bacterium Xanthomonas campestris pv. campestris. Mol. Plant Microbe Interact. 20:21–30. 10.1094/MPMI-20-0021 [DOI] [PubMed] [Google Scholar]
- 230.Meng Q-L, Tang D-J, Fan Y-Y, Li Z-J, Zhang H, He Y-Q, Jiang B-L, Lu G-T, Tang J-L. 2011. Effect of interactions between Mip and PrtA on the full extracellular protease activity of Xanthomonas campestris pathovar campestris. FEMS Microbiol. Lett. 323:180–187. 10.1111/j.1574-6968.2011.02377.x [DOI] [PubMed] [Google Scholar]
- 231.DebRoy S, Keenan AB, Ueno N, Jeronimo SMB, Donelson JE, Wilson ME. 2010. Leishmania infantum chagasi: a genome-based approach to identification of excreted/secreted proteins. Exp. Parasitol. 126:582–591. 10.1016/j.exppara.2010.06.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Pereira PJB, Vega MC, González-Rey E, Fernández-Carazo R, Macedo-Ribeiro S, Gomis-Rüth FX, González A, Coll M. 2002. Trypanosoma cruzi macrophage infectivity potentiator has a rotamase core and a highly exposed alpha-helix. EMBO Rep. 3:88–94. 10.1093/embo-reports/kvf009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Cianciotto NP, Eisenstein BI, Mody CH, Toews GB, Engleberg NC. 1989. A Legionella pneumophila gene encoding a species-specific surface protein potentiates initiation of intracellular infection. Infect. Immun. 57:1255–1262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Engleberg NC, Carter C, Weber DR, Cianciotto NP, Eisenstein BI. 1989. DNA sequence of mip, a Legionella pneumophila gene associated with macrophage infectivity. Infect. Immun. 57:1263–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Cianciotto NP, Fields BS. 1992. Legionella pneumophila mip gene potentiates intracellular infection of protozoa and human macrophages. Proc. Natl. Acad. Sci. U. S. A. 89:5188–5191. 10.1073/pnas.89.11.5188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Wieland H, Faigle M, Lang F, Northoff H, Neumeister B. 2002. Regulation of the Legionella mip-promotor during infection of human monocytes. FEMS Microbiol. Lett. 212:127–132. 10.1111/j.1574-6968.2002.tb11255.x [DOI] [PubMed] [Google Scholar]
- 237.Shi C, Forsbach-Birk V, Marre R, McNealy TL. 2006. The Legionella pneumophila global regulatory protein LetA affects DotA and Mip. Int. J. Med. Microbiol. 296:15–24. 10.1016/j.ijmm.2005.09.003 [DOI] [PubMed] [Google Scholar]
- 238.Riboldi-Tunnicliffe A, König B, Jessen S, Weiss MS, Rahfeld J, Hacker J, Fischer G, Hilgenfeld R. 2001. Crystal structure of Mip, a prolylisomerase from Legionella pneumophila. Nat. Struct. Biol. 8:779–783. 10.1038/nsb0901-779 [DOI] [PubMed] [Google Scholar]
- 239.Horstmann M, Ehses P, Schweimer K, Steinert M, Kamphausen T, Fischer G, Hacker J, Rösch P, Faber C. 2006. Domain motions of the Mip protein from Legionella pneumophila. Biochemistry (Mosc.) 45:12303–12311. 10.1021/bi060818i [DOI] [PubMed] [Google Scholar]
- 240.Ceymann A, Horstmann M, Ehses P, Schweimer K, Paschke A-K, Steinert M, Faber C. 2008. Solution structure of the Legionella pneumophila Mip-rapamycin complex. BMC Struct. Biol. 8:17. 10.1186/1472-6807-8-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Schmidt B, Rahfeld J, Schierhorn A, Ludwig B, Hacker J, Fischer G. 1994. A homodimer represents an active species of the peptidyl-prolyl cis/trans isomerase FKBP25mem from Legionella pneumophila. FEBS Lett. 352:185–190. 10.1016/0014-5793(94)00970-8 [DOI] [PubMed] [Google Scholar]
- 242.Budiman C, Bando K, Angkawidjaja C, Koga Y, Takano K, Kanaya S. 2009. Engineering of monomeric FK506-binding protein 22 with peptidyl prolyl cis-trans isomerase. Importance of a V-shaped dimeric structure for binding to protein substrate. FEBS J. 276:4091–4101. 10.1111/j.1742-4658.2009.07116.x [DOI] [PubMed] [Google Scholar]
- 243.Wintermeyer E, Ludwig B, Steinert M, Schmidt B, Fischer G, Hacker J. 1995. Influence of site specifically altered Mip proteins on intracellular survival of Legionella pneumophila in eukaryotic cells. Infect. Immun. 63:4576–4583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Köhler R, Fanghänel J, König B, Lüneberg E, Frosch M, Rahfeld J-U, Hilgenfeld R, Fischer G, Hacker J, Steinert M. 2003. Biochemical and functional analyses of the Mip protein: influence of the N-terminal half and of peptidylprolyl isomerase activity on the virulence of Legionella pneumophila. Infect. Immun. 71:4389–4397. 10.1128/IAI.71.8.4389-4397.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Cianciotto NP, Eisenstein BI, Mody CH, Engleberg NC. 1990. A mutation in the mip gene results in an attenuation of Legionella pneumophila virulence. J. Infect. Dis. 162:121–126. 10.1093/infdis/162.1.121 [DOI] [PubMed] [Google Scholar]
- 246.Wagner C, Khan AS, Kamphausen T, Schmausser B, Unal C, Lorenz U, Fischer G, Hacker J, Steinert M. 2007. Collagen binding protein Mip enables Legionella pneumophila to transmigrate through a barrier of NCI-H292 lung epithelial cells and extracellular matrix. Cell. Microbiol. 9:450–462. 10.1111/j.1462-5822.2006.00802.x [DOI] [PubMed] [Google Scholar]
- 247.Ünal C, Schwedhelm KF, Thiele A, Weiwad M, Schweimer K, Frese F, Fischer G, Hacker J, Faber C, Steinert M. 2011. Collagen IV-derived peptide binds hydrophobic cavity of Legionella pneumophila Mip and interferes with bacterial epithelial transmigration. Cell. Microbiol. 13:1558–1572. 10.1111/j.1462-5822.2011.01641.x [DOI] [PubMed] [Google Scholar]
- 248.Galka F, Wai SN, Kusch H, Engelmann S, Hecker M, Schmeck B, Hippenstiel S, Uhlin BE, Steinert M. 2008. Proteomic characterization of the whole secretome of Legionella pneumophila and functional analysis of outer membrane vesicles. Infect. Immun. 76:1825–1836. 10.1128/IAI.01396-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Cameron AM, Steiner JP, Roskams AJ, Ali SM, Ronnett GV, Snyder SH. 1995. Calcineurin associated with the inositol 1,4,5-trisphosphate receptor-FKBP12 complex modulates Ca2+ flux. Cell 83:463–472. 10.1016/0092-8674(95)90124-8 [DOI] [PubMed] [Google Scholar]
- 250.Cameron AM, Nucifora FC, Jr, Fung ET, Livingston DJ, Aldape RA, Ross CA, Snyder SH. 1997. FKBP12 binds the inositol 1,4,5-trisphosphate receptor at leucine-proline (1400–1401) and anchors calcineurin to this FK506-like domain. J. Biol. Chem. 272:27582–27588. 10.1074/jbc.272.44.27582 [DOI] [PubMed] [Google Scholar]
- 251.Lam D, Golstein P. 2008. A specific pathway inducing autophagic cell death is marked by an IP3R mutation. Autophagy 4:349–350. 10.4161/auto.5521 [DOI] [PubMed] [Google Scholar]
- 252.Lam D, Kosta A, Luciani M-F, Golstein P. 2008. The inositol 1,4,5-trisphosphate receptor is required to signal autophagic cell death. Mol. Biol. Cell 19:691–700. 10.1091/mbc.E07-08-0823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Amer AO, Swanson MS. 2005. Autophagy is an immediate macrophage response to Legionella pneumophila. Cell. Microbiol. 7:765–778. 10.1111/j.1462-5822.2005.00509.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Farbrother P, Wagner C, Na J, Tunggal B, Morio T, Urushihara H, Tanaka Y, Schleicher M, Steinert M, Eichinger L. 2006. Dictyostelium transcriptional host cell response upon infection with Legionella. Cell. Microbiol. 8:438–456. 10.1111/j.1462-5822.2005.00633.x [DOI] [PubMed] [Google Scholar]
- 255.Debroy S, Aragon V, Kurtz S, Cianciotto NP. 2006. Legionella pneumophila Mip, a surface-exposed peptidylproline cis-trans-isomerase, promotes the presence of phospholipase C-like activity in culture supernatants. Infect. Immun. 74:5152–5160. 10.1128/IAI.00484-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Shahnazari S, Yen W-L, Birmingham CL, Shiu J, Namolovan A, Zheng YT, Nakayama K, Klionsky DJ, Brumell JH. 2010. A diacylglycerol-dependent signaling pathway contributes to regulation of antibacterial autophagy. Cell Host Microbe 8:137–146. 10.1016/j.chom.2010.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Weber SS, Ragaz C, Reus K, Nyfeler Y, Hilbi H. 2006. Legionella pneumophila exploits PI(4)P to anchor secreted effector proteins to the replicative vacuole. PLoS Pathog. 2:e46. 10.1371/journal.ppat.0020046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Viner R, Chetrit D, Ehrlich M, Segal G. 2012. Identification of two Legionella pneumophila effectors that manipulate host phospholipids biosynthesis. PLoS Pathog. 8:e1002988. 10.1371/journal.ppat.1002988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Schunder E, Adam P, Higa F, Remer KA, Lorenz U, Bender J, Schulz T, Flieger A, Steinert M, Heuner K. 2010. Phospholipase PlaB is a new virulence factor of Legionella pneumophila. Int. J. Med. Microbiol. 300:313–323. 10.1016/j.ijmm.2010.01.002 [DOI] [PubMed] [Google Scholar]
- 260.Gan Y-H. 2005. Interaction between Burkholderia pseudomallei and the host immune response: sleeping with the enemy? J. Infect. Dis. 192:1845–1850. 10.1086/497382 [DOI] [PubMed] [Google Scholar]
- 261.Norville IH, Harmer NJ, Harding SV, Fischer G, Keith KE, Brown KA, Sarkar-Tyson M, Titball RW. 2011. A Burkholderia pseudomallei macrophage infectivity potentiator-like protein has rapamycin-inhibitable peptidylprolyl isomerase activity and pleiotropic effects on virulence. Infect. Immun. 79:4299–4307. 10.1128/IAI.00134-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Norville IH, Breitbach K, Eske-Pogodda K, Harmer NJ, Sarkar-Tyson M, Titball RW, Steinmetz I. 2011. A novel FK-506-binding-like protein that lacks peptidyl-prolyl isomerase activity is involved in intracellular infection and in vivo virulence of Burkholderia pseudomallei. Microbiology 157:2629–2638. 10.1099/mic.0.049163-0 [DOI] [PubMed] [Google Scholar]
- 263.Norville IH, O'Shea K, Sarkar-Tyson M, Zheng S, Titball RW, Varani G, Harmer NJ. 2011. The structure of a Burkholderia pseudomallei immunophilin-inhibitor complex reveals new approaches to antimicrobial development. Biochem. J. 437:413–422. 10.1042/BJ20110345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Lynch SV, Wiener-Kronish JP. 2008. Novel strategies to combat bacterial virulence. Curr. Opin. Crit. Care 14:593–599. 10.1097/MCC.0b013e32830f1dd5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Cusumano CK, Hultgren SJ. 2009. Bacterial adhesion—a source of alternate antibiotic targets. IDrugs 12:699–705 [PubMed] [Google Scholar]
- 266.Baron C. 2010. Antivirulence drugs to target bacterial secretion systems. Curr. Opin. Microbiol. 13:100–105. 10.1016/j.mib.2009.12.003 [DOI] [PubMed] [Google Scholar]
- 267.Sintim HO, Smith JAI, Wang J, Nakayama S, Yan L. 2010. Paradigm shift in discovering next-generation anti-infective agents: targeting quorum sensing, c-di-GMP signaling and biofilm formation in bacteria with small molecules. Future Med. Chem. 2:1005–1035. 10.4155/fmc.10.185 [DOI] [PubMed] [Google Scholar]
- 268.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. 1991. Atomic structure of FKBP-FK506, an immunophilin-immunosuppressant complex. Science 252:839–842. 10.1126/science.1709302 [DOI] [PubMed] [Google Scholar]
- 269.Babine RE, Bender SL. 1997. Molecular recognition of protein-ligand complexes: applications to drug design. Chem. Rev. 97:1359–1472. 10.1021/cr960370z [DOI] [PubMed] [Google Scholar]
- 270.Kino T, Hatanaka H, Hashimoto M, Nishiyama M, Goto T, Okuhara M, Kohsaka M, Aoki H, Imanaka H. 1987. FK-506, a novel immunosuppressant isolated from a Streptomyces. I. Fermentation, isolation, and physico-chemical and biological characteristics. J. Antibiot. (Tokyo) 40:1249–1255 [DOI] [PubMed] [Google Scholar]
- 271.Kino T, Hatanaka H, Miyata S, Inamura N, Nishiyama M, Yajima T, Goto T, Okuhara M, Kohsaka M, Aoki H. 1987. FK-506, a novel immunosuppressant isolated from a Streptomyces. II. Immunosuppressive effect of FK-506 in vitro. J. Antibiot. (Tokyo) 40:1256–1265 [DOI] [PubMed] [Google Scholar]
- 272.Arai T, Kouama Y, Suenaga T, Honda H. 1962. Ascomycin, an antifungal antibiotic. J. Antibiot. (Tokyo) 15:231–232 [PubMed] [Google Scholar]
- 273.Sehgal SN, Baker H, Vézina C. 1975. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J. Antibiot. (Tokyo) 28:727–732 [DOI] [PubMed] [Google Scholar]
- 274.Hatanaka H, Kino T, Miyata S, Inamura N, Kuroda A, Goto T, Tanaka H, Okuhara M. 1988. FR-900520 and FR-900523, novel immunosuppressants isolated from a Streptomyces. II. Fermentation, isolation and physico-chemical and biological characteristics. J. Antibiot. (Tokyo) 41:1592–1601 [DOI] [PubMed] [Google Scholar]
- 275.Wang XJ, Etzkorn FA. 2006. Peptidyl-prolyl isomerase inhibitors. Biopolymers 84:125–146. 10.1002/bip.20240 [DOI] [PubMed] [Google Scholar]
- 276.Bierer BE, Somers PK, Wandless TJ, Burakoff SJ, Schreiber SL. 1990. Probing immunosuppressant action with a nonnatural immunophilin ligand. Science 250:556–559. 10.1126/science.1700475 [DOI] [PubMed] [Google Scholar]
- 277.Bierer BE, Mattila PS, Standaert RF, Herzenberg LA, Burakoff SJ, Crabtree G, Schreiber SL. 1990. Two distinct signal transmission pathways in T lymphocytes are inhibited by complexes formed between an immunophilin and either FK506 or rapamycin. Proc. Natl. Acad. Sci. U. S. A. 87:9231–9235. 10.1073/pnas.87.23.9231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Liu J, Farmer JD, Jr, Lane WS, Friedman J, Weissman I, Schreiber SL. 1991. Calcineurin is a common target of cyclophilin-cyclosporin A and FKBP-FK506 complexes. Cell 66:807–815. 10.1016/0092-8674(91)90124-H [DOI] [PubMed] [Google Scholar]
- 279.Chiu MI, Katz H, Berlin V. 1994. RAPT1, a mammalian homolog of yeast Tor, interacts with the FKBP12/rapamycin complex. Proc. Natl. Acad. Sci. U. S. A. 91:12574–12578. 10.1073/pnas.91.26.12574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, Abraham RT. 1995. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem. 270:815–822. 10.1074/jbc.270.2.815 [DOI] [PubMed] [Google Scholar]
- 281.Van Duyne GD, Standaert RF, Karplus PA, Schreiber SL, Clardy J. 1993. Atomic structures of the human immunophilin FKBP-12 complexes with FK506 and rapamycin. J. Mol. Biol. 229:105–124. 10.1006/jmbi.1993.1012 [DOI] [PubMed] [Google Scholar]
- 282.Aldape RA, Futer O, DeCenzo MT, Jarrett BP, Murcko MA, Livingston DJ. 1992. Charged surface residues of FKBP12 participate in formation of the FKBP12-FK506-calcineurin complex. J. Biol. Chem. 267:16029–16032 [PubMed] [Google Scholar]
- 283.Holt DA, Luengo JI, Yamashita DS, Oh HJ, Konialian AL, Yen HK, Rozamus LW, Brandt M, Bossard MJ. 1993. Design, synthesis, and kinetic evaluation of high-affinity FKBP ligands and the X-ray crystal structures of their complexes with FKBP12. J. Am. Chem. Soc. 115:9925–9938. 10.1021/ja00075a008 [DOI] [Google Scholar]
- 284.Armistead DM, Badia MC, Deininger DD, Duffy JP, Saunders JO, Tung RD, Thomson JA, DeCenzo MT, Futer O, Livingston DJ, Murcko MA, Yamashita MM, Navia MA. 1995. Design, synthesis and structure of non-macrocyclic inhibitors of FKBP12, the major binding protein for the immunosuppressant FK506. Acta Crystallogr. D Biol. Crystallogr. 51:522–528. 10.1107/S0907444994014502 [DOI] [PubMed] [Google Scholar]
- 285.Tatlock JH, Kalish VJ, Parge HE, Knighton DR, Showalter RE, Lewis CT, French JV, Villafranca JE. 1995. High-affinity FKBP-12 ligands derived from (R)-(−)-carvone. Synthesis and evaluation of FK506 pyranose ring replacements. Bioorg. Med. Chem. Lett. 5:2489–2494. 10.1016/0960-894X(95)00429-W [DOI] [Google Scholar]
- 286.Babine RE, Bleckman TM, Kissinger CR, Showalter R, Pelletier LA, Lewis C, Tucker K, Moomaw E, Parge HE, Villafranca JE. 1995. Design, synthesis and X-ray crystallographic studies of novel FKBP-12 ligands. Bioorg. Med. Chem. Lett. 5:1719–1724. 10.1016/0960-894X(95)00290-A [DOI] [Google Scholar]
- 287.Hauske JR, Dorff P, Julin S, DiBrino J, Spencer R, Williams R. 1992. Design and synthesis of novel FKBP inhibitors. J. Med. Chem. 35:4284–4296. 10.1021/jm00101a005 [DOI] [PubMed] [Google Scholar]
- 288.Andres CJ, Macdonald TL, Ocain TD, Longhi D. 1993. Conformationally defined analogs of prolylamides. trans-Prolyl peptidomimetics. J. Org. Chem. 58:6609–6613. 10.1021/jo00076a018 [DOI] [Google Scholar]
- 289.Burkhard P, Hommel U, Sanner M, Walkinshaw MD. 1999. The discovery of steroids and other novel FKBP inhibitors using a molecular docking program. J. Mol. Biol. 287:853–858. 10.1006/jmbi.1999.2621 [DOI] [PubMed] [Google Scholar]
- 290.Christner C, Wyrwa R, Marsch S, Küllertz G, Thiericke R, Grabley S, Schumann D, Fischer G. 1999. Synthesis and cytotoxic evaluation of cycloheximide derivatives as potential inhibitors of FKBP12 with neuroregenerative properties. J. Med. Chem. 42:3615–3622. 10.1021/jm991038t [DOI] [PubMed] [Google Scholar]
- 291.Juli C, Sippel M, Jäger J, Thiele A, Weiwad M, Schweimer K, Rösch P, Steinert M, Sotriffer CA, Holzgrabe U. 2011. Pipecolic acid derivatives as small-molecule inhibitors of the Legionella MIP protein. J. Med. Chem. 54:277–283. 10.1021/jm101156y [DOI] [PubMed] [Google Scholar]
- 292.Holt DA, Konialian-Beck AL, Oh H-J, Yen H-K, Rozamus LW, Krog AJ, Erhard KF, Ortiz E, Levy MA, Brandt M, Bossard MJ, Luengo JI. 1994. Structure-activity studies of synthetic FKBP ligands as peptidyl-prolyl isomerase inhibitors. Bioorg. Med. Chem. Lett. 4:315–320. 10.1016/S0960-894X(01)80135-9 [DOI] [Google Scholar]
- 293.Monaghan P, Fardis M, Revill WP, Bell A. 2005. Antimalarial effects of macrolactones related to FK520 (ascomycin) are independent of the immunosuppressive properties of the compounds. J. Infect. Dis. 191:1342–1349. 10.1086/428454 [DOI] [PubMed] [Google Scholar]
- 294.Harikishore A, Niang M, Rajan S, Preiser PR, Yoon HS. 2013. Small molecule Plasmodium FKBP35 inhibitor as a potential antimalaria agent. Sci. Rep. 3:2501. 10.1038/srep02501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Fischer G, Tradler T, Zarnt T. 1998. The mode of action of peptidyl prolyl cis/trans isomerases in vivo: binding vs. catalysis. FEBS Lett. 426:17–20. 10.1016/S0014-5793(98)00242-7 [DOI] [PubMed] [Google Scholar]
- 296.Lundemose AG, Rouch DA, Birkelund S, Christiansen G, Pearce JH. 1992. Chlamydia trachomatis Mip-like protein. Mol. Microbiol. 6:2539–2548 [DOI] [PubMed] [Google Scholar]