

Abstract
The PR promoter of mycobacteriophage BPs directs early lytic gene expression and is under the control of the BPs repressor, gp33. Reporter gene fusions showed that PR has modest activity in an extrachromosomal context but has activity that is barely detectable in an integrated context, even in the absence of its repressor. Mutational dissection of PR showed that it uses a canonical −10 hexamer recognized by SigA, and mutants with mutations to the sequence 5′-TATAMT had the greatest activities. It does not contain a 5′-TGN-extended −10 sequence, although mutants with mutations creating an extended −10 sequence had substantially increased promoter activity. Mutations in the −35 hexamer also influenced promoter activity but were strongly context dependent, and similar substitutions in the −35 hexamer differentially affected promoter activity, depending on the −10 and extended −10 motifs. This warrants caution in the construction of synthetic promoters or the bioinformatic prediction of promoter activity. Combinations of mutations throughout PR generated a calibrated series of promoters for expression of stably integrated recombinant genes in both Mycobacterium smegmatis and M. tuberculosis, with maximal promoter activity being more than 2-fold that of the strong hsp60 promoter.
INTRODUCTION
Transcriptional regulation in mycobacteria is complex, with many different sigma factors and promoter sequences (1–5). Mycobacterium smegmatis and Mycobacterium tuberculosis encode 28 and 13 different sigma factors, respectively, but SigA, the sole group 1 member in each species, is essential and is presumably responsible for expression of all or most housekeeping genes (4). Multiple types of promoter specificities have also been reported, and although the correspondence with specific sigma factors is incomplete, the SigA promoters have similarity to the Sig-70 promoters of Escherichia coli. A consensus sequence derived from promoters with mapped transcription start sites (TSSs) identified a 5′-TTGACW −35 motif and a 5′-TATAMT −10 motif, spaced 16 to 21 bp apart (3, 6–9). Mutational analysis of the −35 motif in a synthetic promoter indicated that optimal activity was provided by mutations in the sequence 5′-TTGCGA (10). Spacing between the −10 and −35 hexamers is typically 16 to 19 bp, and optimal spacing in the synthetic promoter was 18 bp (10), although 17 bp is most common (8, 9). It has also been shown that many mycobacterial promoters contain a 5′-TGN-extended −10 motif, which plays a major role in promoter strength and facilitates open complex formation with RNA polymerase even in the absence of −35 sequences (11).
Analysis of a large collection of sequenced mycobacteriophages showed that they are genomically diverse, and there are at least 28 types that share few or no nucleotide sequences with each other (12). Many of the phage genes are organized into long operons, and although the average number of genes per genome is about 100, there are anticipated to be a much smaller number of promoters (13). Little is known about the patterns of gene expression in most of the phages, although there are distinct groups of early- and late-expressed proteins in phage Bxb1, L5, and TM4 lytic growth (14–16), and transcriptomic analysis shows early and late patterns in phage Giles lytic growth (17). None of the phages are predicted to carry a viral RNA polymerase, and they presumably use the host RNA polymerase for transcription of the phage genome. A relatively small number of mycobacteriophage promoters has been described (18–23), including several L5 promoters, such as the strong early lytic promoter, Pleft, and three promoters upstream of the repressor, all of which are generally similar to SigA promoters (21, 24). In contrast, although the locations of transcription initiation have been defined in phage Giles, the promoters have not been identified and SigA-like recognition sequences are not predicted (17). Presumably, the phage utilizes other host sigma factors or perhaps encodes its own factor for promoting transcription initiation. However, the only putative sigma factor genes carried by mycobacteriophages are restricted to the phages of cluster A, which does not include Giles (13).
Mycobacteriophage BPs is a cluster G phage that utilizes an unusual integration-dependent immunity system for establishing and maintaining lysogeny (25, 26). The phage attachment site (attP) for prophage integration is unusually located within the repressor gene, such that the 3′ end of the gene is separated from the 5′ end of the gene by site-specific recombination during prophage establishment (Fig. 1A). The truncated prophage form of the protein is required for lysogenic maintenance, but the longer virally encoded form of the repressor fails to confer immunity, due to a C-terminally located tag that targets it for proteolytic degradation (25). The repressor acts by binding to a 12-bp operator (OR) that overlaps an early lytic promoter, PR (Fig. 1B). PR contains sequence features suggesting that it directs transcription using SigA, with the −35 and −10 hexamers of 5′-TTTCCA and 5′-TATGTT, respectively, being separated by 18 bp (Fig. 1B). The +1 site of transcription from PR corresponds to the first base of the ATG initiation codon, generating a leaderless mRNA (Fig. 1B) (25).
FIG 1.
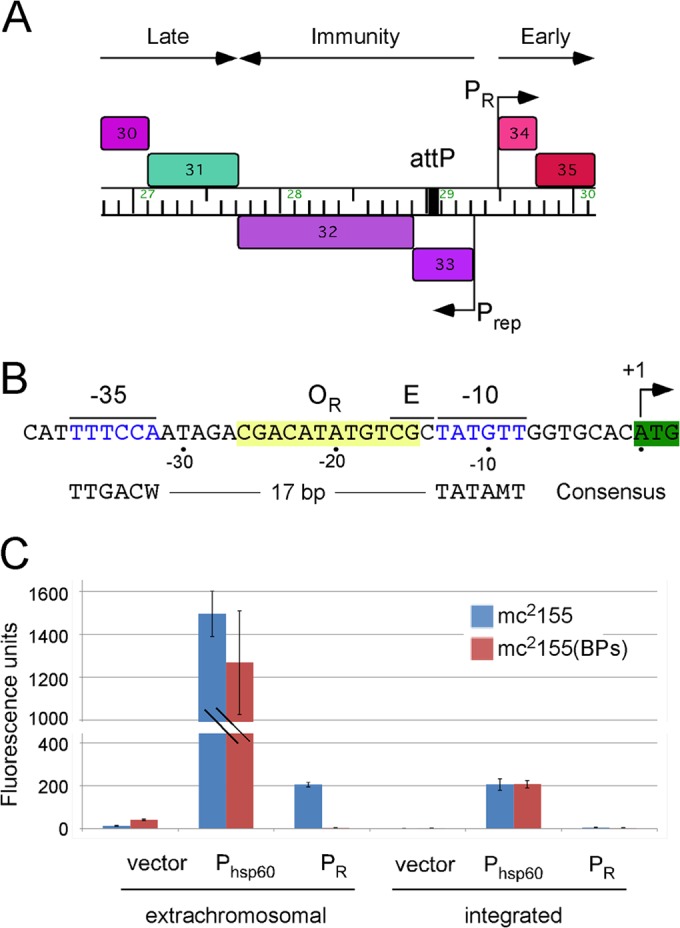
Location of the phage BPs PR promoter. (A) Map of a segment of the BPs genome (coordinates 26,790 to 30,150) showing the locations of the early lytic promoter PR and the promoter Prep that expresses the repressor gene (gene 33) and the integrase gene (gene 32). The putative expression patterns are included above. Note that attP is located within the repressor gene (25). The map was generated using the program Phamerator (48). (B) Sequence of the BPs PR promoter (coordinates 29,470 to 29,512). The +1 site for PR transcription initiation corresponds to the first base of the gene 34 initiation codon (25), and putative hexamers at the −10 and −35 regions are shown in blue type. The three bases to the left of the −10 hexamer (labeled E) correspond to the position where a 5′-TGN-extended −10 motif is located in some promoters. The putative OR operator is highlighted in yellow. The consensus sequences for the −10 and −35 regions of mycobacterial SigA promoters reported previously are shown (8, 9). (C) Promoter activities are shown as fluorescence units resulting from expression of an mCherry reporter gene in M. smegmatis using both extrachromosomal and integration-proficient vectors. Expression of the BCG hsp60 promoter and the BPs PR promoter is compared to that of a promoterless vector in both M. smegmatis mc2155 and a BPs lysogen.
A variety of experimental approaches, including the development of recombinant mycobacterial vaccines, require systems for efficient gene expression in mycobacteria (27, 28). Several strong promoters for expression of foreign antigens, including the M. bovis BCG hsp60 and phage L5 Pleft promoters, have been described (24, 27, 29), although optimal expression levels are accomplished using extrachromosomal vectors (27, 28). Integration vectors exploiting phage integration systems provide enhanced vector maintenance and single-copy attributes, but expression, even from the strong hsp60 promoter, is relatively poor (27). However, expression can be elevated by using multiple integration vectors (30). A variety of inducible expression systems have also been described (31–33), but no systems for providing a calibrated level of expression from mycobacterial integrative or extrachromosomal vectors have been described.
Here, we describe a detailed mutational dissection of the BPs PR promoter. The PR promoter shows barely detectable levels of activity when integrated into the M. smegmatis chromosome but is strongly activated when extrachromosomal, although absolute activity is 7-fold lower than that of the strong hsp60 promoter. Mutations in the −10 hexamer are consistent with PR being recognized by SigA, although substitutions within the −35 hexamer show that a substitution with G or C at four of the six positions increases promoter strength. Although several base substitutions increase promoter strength, as well as change the interhexamer spacing from 18 bp to 17 bp, they also impair repression, consistent with a polymerase retention model for repression and a compromise between promoter activity and regulation. Combining mutations generates a series of promoters with calibrated strengths, with the strongest promoter being more active than the hsp60 promoter in an integrated context.
MATERIALS AND METHODS
Bacterial strains and cultures.
Mycobacterium smegmatis mc2155, mc2155(BPs), and M. tuberculosis mc27000 were grown as described previously (34–36). Antibiotics (kanamycin at 20 μg/ml) were added as needed. Plasmids were transformed into electrocompetent M. smegmatis and M. tuberculosis cells, prepared as previously described (37), by electroporation at 1,000 Ω, 2.5 kV, and 25 μF.
Plasmid construction.
Plasmids containing single point mutations at PR were generated by site-directed mutagenesis (SDM) of pLO07 (see Table S1 in the supplemental material) using the protocol for the QuikChange SDM kit (Stratagene). Briefly, SDM primers in both orientations (see Table S2 in the supplemental material) containing the desired mutations were used to amplify the parental plasmid by PCR with Pfu polymerase (Agilent). Cycling conditions were as follows: 95°C for 30 s; 95°C for 30 s, 55°C for 1 min, and 68°C for 1 min/1 kb of plasmid for between 12 and 18 cycles; 68°C for 7 min; and hold at 4°C. The PCR products were digested with DpnI (New England BioLabs) to eliminate the parental plasmid and leave only the amplified DNAs. The digested PCR was transformed into Escherichia coli NEB5α (New England BioLabs), and the plasmids were isolated (MiniPrep Fermentas). The promoter regions were sequenced (GeneWiz, NJ) using primers LMO51 and LMO52 (see Table S2 in the supplemental material).
Phage Tweety integration vectors with promoter-reporter transcriptional fusions (pLO73, pLO74, pLO75, and pLO76) were made by cloning PCR products from pLO86, pLO87, pLO07, and pLO244 (primers LMO350 and LMO351) into pTTP1b (38) at the XbaI site. Further mutations were added by site-directed mutagenesis as described above with the primers listed in Table S2 in the supplemental material.
Fluorescence assays.
Cultures of M. smegmatis were grown with shaking at 37°C for 48 h. Standing cultures of M. tuberculosis were grown at 37°C for 4 weeks, fixed with a final concentration of 2% paraformaldehyde (Sigma), and stored at 4°C for less than 1 week. Experiments were performed with biological triplicates, and 50-μl aliquots were dispensed into 96-well plates (Falcon). Fluorescence was quantified at 532 nm (FLA-5100; FujiFilm) using ImageGauge software and analyzed with MultiGauge software. The optical density at 595 nm (OD595) of the culture was measured (EL800 Universal Microplate Reader; Bio-Tek Instruments) to normalize the fluorescence readings for differences in the approximate numbers of cells. Fluorescence units are reported as the amount of fluorescence per area (linear arbitrary units/mm2) per OD595 or in relative fluorescence units, where the number of fluorescence units from the hsp60 promoter in a nonlysogen was set equal to 1.0. Graphs display the mean ± 95% confidence interval. Fluorescence microscopy was performed on fixed M. tuberculosis strains at ×1,000 magnification with an Axiostar Plus microscope (Carl Zeiss) and photographed with controlled exposure times (100 ms) using an AxioCam MRc5 camera (Carl Zeiss) and Carl Zeiss AxioVision (release 4.6) software.
RESULTS
Expression of BPs PR promoter in extrachromosomal and integrated contexts.
The BPs PR promoter is located immediately upstream of the early lytic operon that begins at gene 34 and is regulated by the BPs repressor, gp33 (Fig. 1A). The start site for transcription has been mapped and corresponds to the first base of the initiation codon of gene 34 (Fig. 1B) (25). Putative −10 and −35 motifs with similarity to the mycobacterial consensus sequences have been predicted (Fig. 1B) and are separated by 18 bp. PR lacks the extended −10 5′-TGN reported to strongly influence promoter activity in some sigma factor A (SigA) promoters (11). A 12-bp operator, OR, is located in the spacer region between the −35 and −10 motifs and is the binding site for gp33 (25). When fused to an mCherry reporter gene in an extrachromosomal vector, PR has moderate activity about 7-fold lower than that of the strong hsp60 promoter (Fig. 1C), although when integrated into the chromosome using an integration vector derived from phage Tweety (38), PR activity is barely detectable above the background activity of a promoterless vector (Fig. 1C). The ratio of activities of extrachromosomal to integrated plasmids for PR of 43-fold is considerably greater than the ratio of 7-fold for the hsp60 promoter (Fig. 1C) (30); the copy numbers of pAL5000-derived extrachromosomal vectors is 23 but may vary when carrying strong promoters (30). Activation of PR in the extrachromosomal context is consistent with a model in which its reduced chromosomal activity contributes to its repression in the prophage state but it is fully active when extrachromosomal during lytic growth. DNA supercoiling could play a role in this activation, as described previously for other mycobacterial promoters (39). We note that the instability of extrachromosomal plasmids with high-level expression of mCherry (such as the hsp60 plasmid pLO87) likely contributes to the relatively high variance in reporter gene activity (Fig. 1C). Although it was propagated with antibiotic selection for plasmid maintenance, other studies show that plasmid pLO87 is rapidly lost in the absence of selection (M. Olm, L. M. Oldfield, and G. F. Hatfull, unpublished observations). This instability may thus contribute to the variation in the assays of all extrachromosomal constructs with high-level mCherry expression, and the mean levels reported may somewhat underestimate promoter strength.
Influence of single base substitutions in the −10 motif on PR activity.
The similarity of the −10 hexamer of PR (positions −8 to −13; Fig. 1B) to the previously reported 5′-TATAMT consensus strongly suggests that PR is recognized by SigA (1, 8, 9). To examine the sequence contributions at the −10 motif, we constructed a series of base substitution mutants and measured their promoter activity using an mCherry gene reporter (Fig. 2A). Positions −8 and −13 showed a strong dependence on the wild-type T at those locations, and the wild-type A base was preferred at position −12 (although the A−12T mutation had a relatively mild influence); most of the substitutions at these three positions were severely deleterious to PR activity. At the −9 position, substitution of the wild-type T base with either A, C, or G substantially enhanced activity, and both the G−10A and G−10C mutants had elevated activities (Fig. 2A). Thus, a −10 hexamer combining bases that contribute to maximal activity at each position can be expressed as 5′-TATAMT (Fig. 2A), identical to the consensus sequence.
FIG 2.
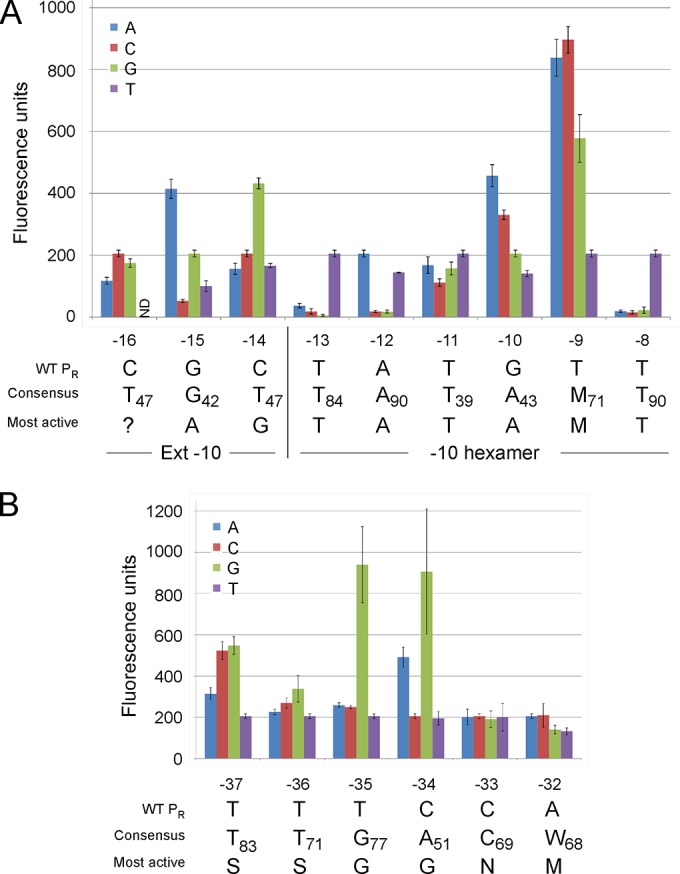
Mutational analysis of the PR promoter. (A) The fluorescence of PR-mCherry transcriptional fusions containing single point mutations in the extended (Ext) −10 motif (positions −16 to −14) and the −10 hexamer (positions −13 to −8) of PR is shown; all mutants were derived from the extrachromosomal plasmid carrying PR (pLO07), for which the results are shown in Fig. 1C. The wild-type (WT) PR sequence and the substitutions with the highest activities are shown below. The consensus −10 bases for mycobacterial SigA promoters and their respective frequencies (as percentage from a total of over 80 promoters with mapped transcription start sites), as reported previously (8, 9), are included; the extended −10 base frequencies are as reported previously (1). The most active substitutions at the −16 position were not determined (?), as the C−16T mutant could not be transformed into M. smegmatis mc2155. M, A or C. (B) The activities of the single base substitutions in the −35 hexamer (−37 to −32) are shown, as described for panel A. S, C or G; N, any base; M, A or C.
A subset of SigA promoters has an extension of the −10 sequence, 5′-TGN (Fig. 2B), that substantially elevates promoter activity and renders such promoters relatively insensitive to changes in the −35 motif (10, 11). The corresponding region of PR has the sequence 5′-CGC (positions −16, −15, and −14, respectively) and thus lacks an extended −10 motif. We examined the roles of bases in these positions and found that several substitutions, including C−14G and G−15A, increased promoter activity more than 2-fold. However, we were unable to transform the C−16T mutant PR-mCherry fusion into M. smegmatis, even after multiple attempts. This was the only mutant out of the more than 70 that we have constructed that displayed this phenotype. Because strong promoter activity is known to interfere with plasmid replication, we favor the interpretation that the C−16T mutation strongly enhances promoter activity by generating an extended −10 sequence. We were able to transform the C−16T mutant into a BPs lysogen, and it had substantial activity (see Fig. S1 in the supplemental material), consistent with this interpretation (the C−16T substitution also alters OR, but this does not itself substantially impair repression; see below).
Influence of single base substitutions in the −35 motif on PR activity.
To determine the role of individual base pairs in the −35 region of PR (positions −32 to −37) (Fig. 1B), we constructed a similar set of mutant derivatives in which each base in the −35 hexamer was changed and the promoter activities were determined (Fig. 2B). At the −33 position, none of the substitutions influenced activity, and at the −32 position, replacement of the A with C had no effect and A−32G and A−32T mutations modestly reduced activity (Fig. 2B). In contrast, at positions −34, −35, −36, and −37, several of the substitutions increased activity, some substantially so, especially substitutions of G at positions −34 and −35 (Fig. 2B). Although T is well conserved at PR positions −36 and −37, it is somewhat surprising that either C or G substitutions increased promoter activity at these positions. We note that in a prior study, any substitution at these positions resulted in at least a 2-fold reduction in transcription (10). The bases within the −35 hexamer of PR that contribute to optimal promoter strength thus differ from the mycobacterial SigA consensus sequence (5′-TTGACW) and the most active sequence (5′-TTGCGA) derived by mutagenesis (10). It seems unlikely that PR uses a sigma factor other than SigA, and these observations suggest a strong context dependence of promoter function. The activities of the mutant promoters were also tested in a BPs lysogen (see Fig. S1 in the supplemental material), and moderate negative effects on repression were observed with some mutations, primarily T−36G and C−34G (see below).
Influences of PR mutations on repression.
The mutants described above were also tested in a BPs lysogen (see Fig. S1 in the supplemental material), in which the wild-type PR promoter is tightly repressed (Fig. 1). Many of the mutants showed modest increases in activity, especially those with increased activities in the nonlysogen (Fig. 2). The most notable difference, however, was that the mutant with the C−16T substitution was able to transform a BPs lysogen and had substantial activity, about 4-fold lower than that of hsp60 (Fig. 1). Other mutational data (see below) showed that the mutation had little impact on repression (i.e., the mutant was repressed >40-fold), such that the predicted activity of the C−16T mutant in wild-type M. smegmatis was almost 10-fold that of hsp60, readily accounting for the nontransformable phenotype. Mutants with some other substitutions, such as C−34G, were substantially derepressed (that mutant had 4-fold lower activity in the lysogen than the nonlysogen), even though the C−34 position is distal to the operator.
Impact of OR substitutions on activity and repression.
The OR operator lies between the −10 and −35 hexamers of PR and overlaps the position that an extended −10 motif would occupy (Fig. 1B); we showed previously that phage base substitution mutants that respond poorly to repressor expression map in OR and show mild derepression (25). Base substitutions at each position of the 12-bp operator showed that many had small changes in promoter activity (<2-fold) (see Fig. S2 in the supplemental material) but were also mildly derepressed. The greatest impairments reduced the level of repression to 3- to 4-fold, magnitudes similar to the magnitude for the C−34G mutation (see Fig. S1 in the supplemental material). Many of the symmetry-related substitutions had similar impacts, such that the T−21A and A−20T substitutions and the G−25C and C−16G substitutions had the least effect on the repression of changes at those positions, reflecting the palindromic nature of OR.
Role of spacer length on promoter activity.
Promoter strength is strongly influenced by interhexamer spacing (40), and we therefore examined the effect of this spacing on PR activity. The interhexamer spacing in PR is 18 bp (Fig. 1B), and we altered this by inserting a base (between G−28 and A−27) to increase it to 19 bp or deleting either one base (ΔA−29) or two bases (ΔA−29/ΔG−28) to decrease it to 17 bp and 16 bp, respectively (Fig. 3A). We found that a spacing of 17 bp is optimal for PR activity and that a mutant with this spacing is somewhat more active than the wild type, which has an 18-bp spacer (Fig. 3B). We also tested PR activity in a strain lysogenic for BPs, in which wild-type PR is tightly downregulated by the repressor. Although the 17-bp-spacer mutant had elevated activity in the absence of repressor, there was substantial derepression in the lysogen. Because the spacer mutation lies outside the OR operator (Fig. 1B), it is not expected to influence the binding of the repressor directly (25), and poor repression presumably is the consequence of the altered configuration of OR and PR. We note that poor repression was also seen in the 16-bp-spacer mutant and normal repression was observed in the 19-bp-spacer mutant (Fig. 3). Thus, the architecture of the PR promoter presumably represents a compromise between promoter strength in the absence of repressor and the ability to tightly repress PR in the prophage.
FIG 3.
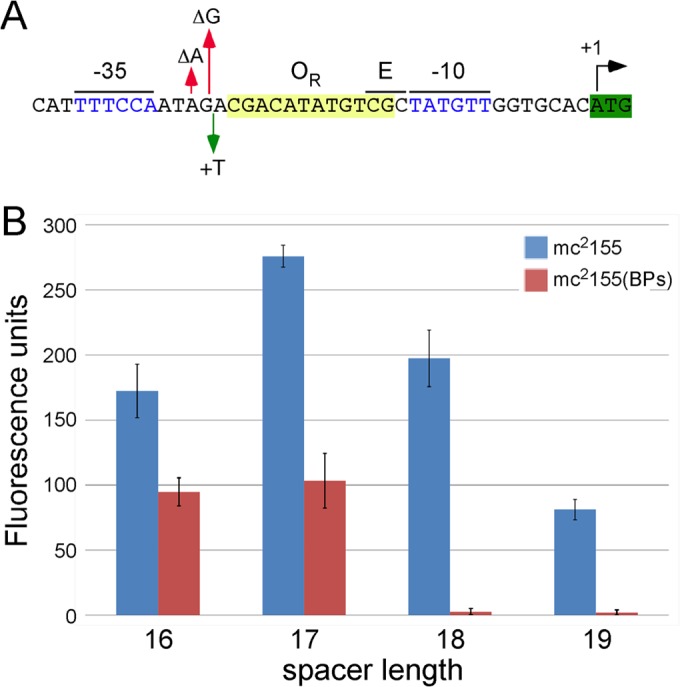
Impact of interhexamer spacer length on PR promoter activity. (A) Sequence of PR showing mutations altering the interhexameric spacing of PR. Spacing was increased to 19 bp by insertion of a T base between positions A−27 and G−28, reduced to 17 bp by removal of A−29, or reduced to 16 bp by removal of both A−29 and G−28. (B) Fluorescence activities of PR promoter spacer mutants in M. smegmatis mc2155 and in a BPs lysogen.
Transformation of the C−16T mutant by chromosomal integration.
As noted above, the extrachromosomal C−16T mutant does not transform M. smegmatis but does transform a BPs lysogen and is quite active (see Fig. S1 in the supplemental material). Assuming that the high activity of the C−16T substitution is responsible for these phenotypes, we transferred the C−16T substitution into an integration-proficient vector derived from phage Tweety (38) and tested transformation in a nonlyosgen. The integration vectors are generally more stable than the extrachromosomal vectors, such that high-level expression of mCherry should be better tolerated. We observed that integrating plasmid pLO76 (C−16T) efficiently transformed M. smegmatis, had activity that was nearly 15-fold greater than that of wild-type PR, and was fully repressed in a BPs lysogen (Fig. 4). Although the integrated C−16T PR promoter had activity about 3-fold lower than that of the integrated hsp60 promoter, if it is stimulated in the extrachromosomal context to the same extent as the wild-type PR promoter, its activity would be more than 2-fold that of hsp60. We note that even though C−16 is within OR, the C−16T mutation had little impact on repression (Fig. 4).
FIG 4.

Activities of PR promoter mutations in genomically integrated contexts. (A) Expression of mCherry by the BPs PR promoter and mutant variants integrated into either M. smegmatis mc2155 or a BPs lysogen. Plasmids pLO73, pLO74, and pLO75 are integrating plasmids containing no promoter insert, the hsp60 promoter, and the wild-type PR promoter, respectively. Other plasmids correspond to four series beginning with pLO76, pLO77, pLO78, or pLO92 and contain increasing numbers of mutations in PR, as shown in panel B. (B) The positions of mutations in the PR promoter mutants are shown.
Combinatorial effects of PR mutations.
The behaviors of mutants with single base substitutions are consistent with PR being recognized as a SigA promoter, but with base preferences that reflect the particular context of PR. Because base changes in both the −35 and −10 motifs and changes in spacing can result in increases in promoter activity, we constructed several series of mutants with combinations of different mutations, using the integrated forms of the promoters. We first sequentially introduced −10 hexamer mutations that enhanced activity as single base substitutions (Fig. 4). Addition of the T−9C substitution (pLO77) to the C−16T (pLO76) mutant gave a further 2-fold increase in activity (Fig. 4), in agreement with the results obtained with the single substitutions in the extrachromosomal context (Fig. 2). Small additional increases in activity resulted from sequential addition of the G−10A and C−14G mutations (pLO78 and pLO92, respectively; Fig. 4), with levels of activity close to 90% of that of the hsp60 promoter (pLO74) being achieved.
Although it has been reported previously that −10 extended promoters are relatively insensitive to substitutions in the −35 region (10, 11), we tested the influence of −35 substitutions on the C−16T mutant (pLO76 and derivatives; Fig. 4). Addition of a T−35G substitution (pLO80), which gave a substantial increase in PR activity as a single base change (Fig. 2), increased the activity of the C−16T mutant over 3-fold, and its activity was greater than that of hsp60 (Fig. 4). Addition of the C−34G substitution and then the T−37C substitution gave only small further increases in activity (Fig. 4). We then tested the impacts of these −35 substitutions in the context of more complex substitutions in the −10 hexamer, with a view to further elevating PR activity. All of these combinations of −10 and −35 substitutions enhanced activity above that of hsp60 (Fig. 4), with the optimal combination being C−16T, T−9C, T−35G, C−34G, and T−37C (i.e., pLO85; Fig. 4). Although some of the mutant combinations showed greater effects of the benefits of the single substitutions, this was not always observed, illustrating the context dependency of the substitutions (Fig. 4). For example, in the context of PR containing the G−10A substitution (pLO78 and derivatives), fewer impacts were generally noted with changes in the −35 hexamer than without them (pLO77 and derivatives). Similarly, the same −35 hexamer changes had even fewer effects when they occurred in combination with the G−10A/C−14G mutations (pLO92 and derivatives) and generally caused behavior more like that in the context of a wild-type PR −10 hexamer with the C−16T extended −10 substitution (pLO76 derivatives; Fig. 4).
We note that repression in a BPs lysogen is compromised for many of the mutants, with the C−34G and T−37C substitutions having the greatest impact (Fig. 4), as was observed with the single base mutants (see Fig. S1 in the supplemental material). Removing a base to decrease the interhexameric spacer to 17 bp did not substantially enhance expression of any of the mutants with multiple −10/−35 mutations that we tested, although repression was weakened so that it was similar to that of wild-type PR (data not shown).
A calibrated series of M. smegmatis promoters.
The large number of promoters with substitutions in PR generated here represents a pool of promoters with different strengths in M. smegmatis mc2155 and various degrees of repression in a BPs lysogen. From among these, we selected six promoters, along with wild-type PR, that had activities spanning a range from activity barely greater than that of the promoterless vector (pLO75; wild-type PR) to activity about 2-fold stronger than that of hsp60 (Fig. 5A) and that were tightly repressed in the BPs lysogen. When tested together, they show a graduation of activities, with each contributing activity approximately one-fifth higher than that of the promoter before it in the series (Fig. 5A). The behaviors of these promoters are illustrated in the colors of the M. smegmatis colonies, reflecting expression of the mCherry reporter gene (Fig. 5B).
FIG 5.
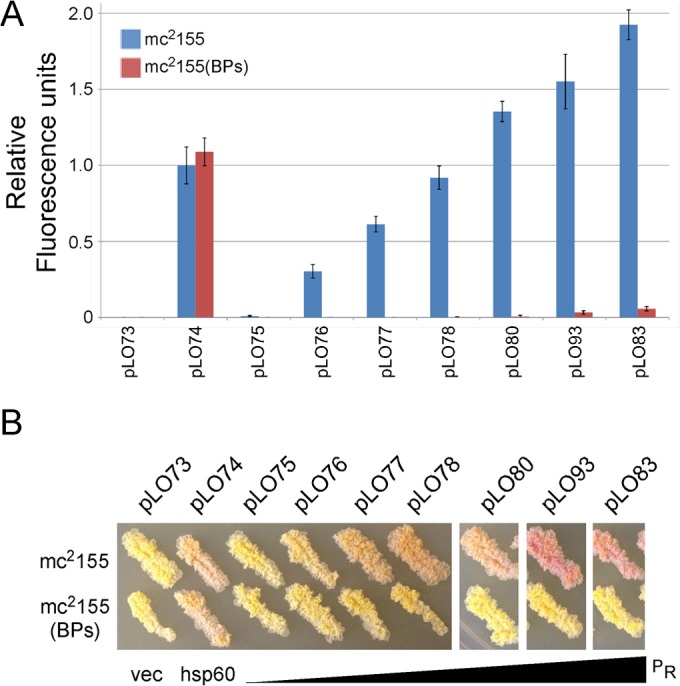
A calibrated series of promoters for M. smegmatis gene expression. (A) Fluorescence activities are shown in relative fluorescence units for a selection of promoters with increasing steps of activity in M. smegmatis mc2155 and in a BPs lysogen. Descriptions of the mutations in each plasmid are shown in Fig. 4. (B) Representative streaks of bacterial strains carrying the plasmids shown in panel A showing red fluorescence by mCherry. Notations below the photograph indicate the promoterless vector (vec), the hsp60 promoter (hsp60), and increasing mCherry fluorescence in PR derivatives.
A calibrated series of M. tuberculosis promoters.
We also tested a subset of the promoter mutants in M. tuberculosis mc27000, including most of those shown in Fig. 5 and some additional mutants (Fig. 6). We used three methods to observe the fluorescence in these strains: a quantitative measure of fluorescence from liquid cultures (Fig. 6A), fluorescence microscopy (Fig. 6B), and the visual colors of colonies (Fig. 6B). The general trend for the mutants tested in M. smegmatis (Fig. 5) was found in M. tuberculosis (Fig. 6), although none had activity greater than that of the hsp60 promoter using the quantitative assay on liquid cultures (Fig. 6A). The three additional mutants tested (which had spacing changes as well as additional hexamer mutations) did not have activity substantially better than that of pLO83 and pLO93 in the quantitative assay (Fig. 6A). We note, however, that strains with several of the PR mutants appeared as bright as the strain with hsp60 by fluorescence microscopy (Fig. 6B), and for some mutants colony colors appeared dark or even darker than those of colonies with hsp60 (Fig. 6B). The quantitative assay of mCherry in M. tuberculosis may underrepresent promoter strength. However, although the activities of this series of promoters may differ according to the assay used, the strength of the activity relative to each other generally reflects the activities in M. smegmatis (Fig. 5).
FIG 6.
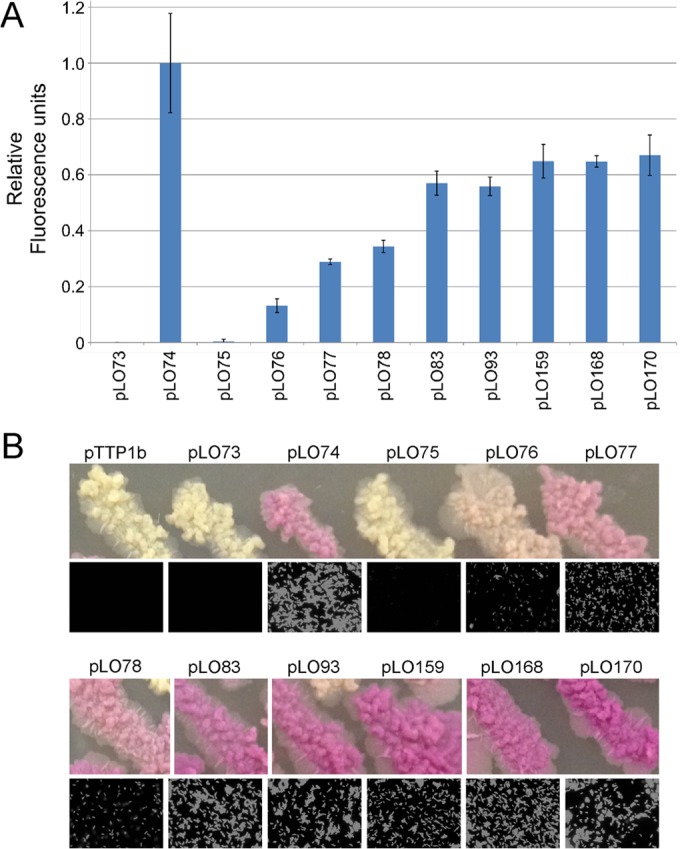
A calibrated series of promoters for M. tuberculosis gene expression. (A) Relative fluorescence activities are shown for a selection of promoters with increasing steps of activity in M. tuberculosis mc27000; fluorescence is relative to that in plasmid pLO74 carrying the hsp60 promoter, which is normalized to 1. Plasmids are the same as those described in Fig. 5 and also include pLO159 and pLO168, which contain the hexamer mutations of pLO93 but with 17-bp spacers resulting from ΔG−28 and ΔA−29 mutations, respectively (see Table S1 in the supplemental material); pLO170 is similar to pLO168 but also contains a C−34A mutation (see Table S1 in the supplemental material). (B) Representative streaks of bacterial strains carrying the plasmids in panel A showing red fluorescence by mCherry (top) and fluorescence microscopy of cells (bottom). Plasmid pTTP1b is a vector lacking the mCherry reporter gene.
DISCUSSION
Although there are a large number of sigma factors in M. smegmatis, mutational analysis strongly suggests that PR is recognized by SigA-associated RNA polymerase. Of the promoter consensus sequences described for other mycobacterial sigma factors, only SigA promoters contain a TA-rich −10 hexamer, and the strong negative impact of mutations at the −8, −12, and −13 positions is consistent with these being the most highly conserved in compilations of mycobacterial promoter consensus sequences (3, 8, 9); these are also critical for expression in Streptomyces (41). To our knowledge, this is the first analysis of the complete mutagenesis of the −10 hexamer of a mycobacterial promoter, and it is of interest that the nucleotides at the −9 and −10 positions have considerable variations in activity. Other reported mutagenesis studies of equivalent positions in mycobacterial promoters indicate that these positions are important for promoter activity, but the effects of particular bases are not consistent with the results for PR, demonstrating the effect of sequence context (42, 43). These positions may thus play a prominent role is modulating the activity of individual promoters in M. smegmatis and M. tuberculosis. The greatest sequence tolerance is at the −11 position, where, although T has the strongest activity, function is reduced by less than 2-fold by any of the substitutions.
Promoters with an extended −10 5′-TGN sequence are common in mycobacteria (11), but PR is not in this class. However, generating an extended −10 sequence with the C−16T mutation substantially elevates activity above that of the wild-type promoter in an integrated context (which is almost undetectable), and such a sequence appears to be too active in an extrachromosomal context to support plasmid replication. From the activities of the integrated C−16T mutant in a nonlysogen and the extrachromosomal C−16T mutant in a BPs lysogen, we predict that the activity of the C−16T mutant is 2- to 10-fold higher than that of hsp60 in an extrachromosomal plasmid.
Analysis of multiple combinations of mutations throughout the PR promoter illustrated that the impacts of base substitutions are highly context dependent. For example, the C−34G single base substitution increased PR activity about 4-fold (Fig. 2) but caused little change in promoter function in the context of C−16T and T−35G mutations (pLO80 versus pLO81; Fig. 4). Likewise, the same series of −35 substitutions clearly had different impacts depending on the specific mutations within the −10 and extended −10 motifs. Thus, although mutants with the single base mutations in the −35 hexamer sequence 5′-SSGGNM had the greatest increases in activity, this reflects the context of the wild-type PR sequence. Context variation thus likely explains the difference between the activities of PR −35 sequences and a synthetic promoter sequence (5′-TTGCGA) (10). This suggests that considerable caution is warranted when either constructing synthetic promoters or bioinformatically predicting promoter function.
Combining mutations in PR that elevate transcription enables construction of promoters that have greater strength than the hsp60 promoter in an integrated context and which may be of utility for high-level antigen expression in recombinant vaccines. Although the increase in activity in M. smegmatis is relatively modest (about 2-fold), this is greater than that which has been reported for other promoters, to our knowledge, and may be approaching maximal achievable levels in an integrated context. The strongest activity remains about 3-fold lower than that of the hsp60 promoter in an extrachromosomal vector, which benefits from a copy number of 23 (30). The relative activities of the promoter mutants in M. tuberculosis may be different from those in M. smegmatis, although expression levels are somewhat assay dependent, with colony color and fluorescence microscopy showing that several of the PR derivatives are more active than an integrated hsp60 promoter, contrary to the findings from fluorescence quantification derived from liquid cultures. We note that wild-type PR has only weak activity in E. coli and that the C−16T mutation does not substantially enhance expression (see Fig. S3 in the supplemental material). Mutations towards the E. coli Sig-70 consensus sequence generally enhanced activity, but the pattern was distinct from the pattern of activity achieved with promoters with mutations in M. smegmatis (see Fig. S3 in the supplemental material).
From the large collection of mutants constructed here, a set of promoters with suitable intervals of promoter activity which are calibrated in regard to expression of the mCherry reporter could be selected (Fig. 5 and 6). These may be of general utility in mycobacterial genetics to generate strains with different but predictable levels of recombinant gene expression. Although the absolute levels of expression are likely to differ for different genes, we expect expression among the promoter series to be consistent, with each having the same overall promoter context. Synthetic promoters have been similarly constructed for other bacterial systems, including E. coli and Streptomyces (41, 44–46).
Unlike many other phage early lytic promoters, the BPs PR promoter has only moderate activity in extrachromosomal vectors and its activity is nearly undetectable when it is integrated. As the integrated state reflects that of a BPs prophage where PR is downregulated by the BPs repressor (25), the behavior of PR may have evolved to avoid immediate prophage induction during the transient loss of repressor occupancy of OR. BPs excision results in synthesis of an inactive form of repressor and also activates PR in the extrachromosomal state, contributing to lytic gene expression. We observe that several promoter mutations, especially those changing the interhexameric spacing, interfere with repression, consistent with a model in which the BPs repressor acts by cobinding with RNA polymerase and retention at the promoter rather than polymerase occlusion (47).
Supplementary Material
ACKNOWLEDGMENTS
We thank Carlos Guerrero for superb technical assistance and Gregory Broussard for comments on the manuscript.
This work was supported by National Institutes of Health grant GM093901.
Footnotes
Published ahead of print 4 August 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JB.01801-14.
REFERENCES
- 1.Gomez M, Smith I. 2000. Determinants of mycobacterial gene expression, p 111–130 In Hatfull GF, Jacobs WR., Jr (ed), Molecular genetics of the mycobacteria. ASM Press, Washington, DC [Google Scholar]
- 2.Smith I, Bishai WR, Nagaraja V. 2005. Control of mycobacterial transcription, p 219–231 In Cole ST, Eisenach KD, McMurray DN, Jacobs WR., Jr (ed), Tuberculosis and the tubercle bacillus. ASM Press, Washington, DC [Google Scholar]
- 3.Newton-Foot M, Gey van Pittius NC. 2013. The complex architecture of mycobacterial promoters. Tuberculosis (Edinb.) 93:60–74. 10.1016/j.tube.2012.08.003 [DOI] [PubMed] [Google Scholar]
- 4.Manganelli R. 2014. Sigma factors: key molecules in Mycobacterium tuberculosis physiology and virulence. Microbiol. Spectrum 2(1):MGM2-0007-2013. 10.1128/microbiolspec.MGM2-0007-2013 [DOI] [PubMed] [Google Scholar]
- 5.Sachdeva P, Misra R, Tyagi AK, Singh Y. 2010. The sigma factors of Mycobacterium tuberculosis: regulation of the regulators. FEBS J. 277:605–626. 10.1111/j.1742-4658.2009.07479.x [DOI] [PubMed] [Google Scholar]
- 6.Bashyam MD, Kaushal D, Dasgupta SK, Tyagi AK. 1996. A study of mycobacterial transcriptional apparatus: identification of novel features in promoter elements. J. Bacteriol. 178:4847–4853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mulder MA, Zappe H, Steyn LM. 1997. Mycobacterial promoters. Tuber. Lung Dis. 78:211–223. 10.1016/S0962-8479(97)90001-0 [DOI] [PubMed] [Google Scholar]
- 8.Unniraman S, Chatterji M, Nagaraja V. 2002. DNA gyrase genes in Mycobacterium tuberculosis: a single operon driven by multiple promoters. J. Bacteriol. 184:5449–5456. 10.1128/JB.184.19.5449-5456.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tare P, Nagaraja V. 2013. Regulation of transcription in mycobacteria. Curr. Sci. 105:632–642 [Google Scholar]
- 10.Agarwal N, Tyagi AK. 2006. Mycobacterial transcriptional signals: requirements for recognition by RNA polymerase and optimal transcriptional activity. Nucleic Acids Res. 34:4245–4257. 10.1093/nar/gkl521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Agarwal N, Tyagi AK. 2003. Role of 5′-TGN-3′ motif in the interaction of mycobacterial RNA polymerase with a promoter of ‘extended −10' class. FEMS Microbiol. Lett. 225:75–83. 10.1016/S0378-1097(03)00483-X [DOI] [PubMed] [Google Scholar]
- 12.Hatfull GF. 2012. The secret lives of mycobacteriophages. Adv. Virus Res. 82:179–288. 10.1016/B978-0-12-394621-8.00015-7 [DOI] [PubMed] [Google Scholar]
- 13.Hatfull GF. 2014. Molecular genetics of mycobacteriophages. Microbiol. Spectrum 2(2):MGM2-0032-2013. 10.1128/microbiolspec.MGM2-0032-2013 [DOI] [PubMed] [Google Scholar]
- 14.Hatfull GF, Sarkis GJ. 1993. DNA sequence, structure and gene expression of mycobacteriophage L5: a phage system for mycobacterial genetics. Mol. Microbiol. 7:395–405. 10.1111/j.1365-2958.1993.tb01131.x [DOI] [PubMed] [Google Scholar]
- 15.Mediavilla J, Jain S, Kriakov J, Ford ME, Duda RL, Jacobs WR, Jr, Hendrix RW, Hatfull GF. 2000. Genome organization and characterization of mycobacteriophage Bxb1. Mol. Microbiol. 38:955–970. 10.1046/j.1365-2958.2000.02183.x [DOI] [PubMed] [Google Scholar]
- 16.Ford ME, Stenstrom C, Hendrix RW, Hatfull GF. 1998. Mycobacteriophage TM4: genome structure and gene expression. Tuber. Lung Dis. 79:63–73. 10.1054/tuld.1998.0007 [DOI] [PubMed] [Google Scholar]
- 17.Dedrick RM, Marinelli LJ, Newton GL, Pogliano K, Pogliano J, Hatfull GF. 2013. Functional requirements for bacteriophage growth: gene essentiality and expression in mycobacteriophage Giles. Mol. Microbiol. 88:577–589. 10.1111/mmi.12210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chattopadhyay C, Sau S, Mandal NC. 2003. Cloning and characterization of the promoters of temperate mycobacteriophage L1. J. Biochem. Mol. Biol. 36:586–592. 10.5483/BMBRep.2003.36.6.586 [DOI] [PubMed] [Google Scholar]
- 19.Garcia M, Pimentel M, Moniz-Pereira J. 2002. Expression of mycobacteriophage Ms6 lysis genes is driven by two sigma(70)-like promoters and is dependent on a transcription termination signal present in the leader RNA. J. Bacteriol. 184:3034–3043. 10.1128/JB.184.11.3034-3043.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jain S, Hatfull GF. 2000. Transcriptional regulation and immunity in mycobacteriophage Bxb1. Mol. Microbiol. 38:971–985. 10.1046/j.1365-2958.2000.02184.x [DOI] [PubMed] [Google Scholar]
- 21.Nesbit CE, Levin ME, Donnelly-Wu MK, Hatfull GF. 1995. Transcriptional regulation of repressor synthesis in mycobacteriophage L5. Mol. Microbiol. 17:1045–1056. 10.1111/j.1365-2958.1995.mmi_17061045.x [DOI] [PubMed] [Google Scholar]
- 22.Ramesh G, Gopinathan KP. 1995. Cloning and characterization of mycobacteriophage I3 promoters. Indian J. Biochem. Biophys. 32:361–367 (Erratum, 33:83, 1996.) [PubMed] [Google Scholar]
- 23.Rybniker J, Plum G, Robinson N, Small PL, Hartmann P. 2008. Identification of three cytotoxic early proteins of mycobacteriophage L5 leading to growth inhibition in Mycobacterium smegmatis. Microbiology 154:2304–2314. 10.1099/mic.0.2008/017004-0 [DOI] [PubMed] [Google Scholar]
- 24.Brown KL, Sarkis GJ, Wadsworth C, Hatfull GF. 1997. Transcriptional silencing by the mycobacteriophage L5 repressor. EMBO J. 16:5914–5921. 10.1093/emboj/16.19.5914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Broussard GW, Oldfield LM, Villanueva VM, Lunt BL, Shine EE, Hatfull GF. 2013. Integration-dependent bacteriophage immunity provides insights into the evolution of genetic switches. Mol. Cell 49:237–248. 10.1016/j.molcel.2012.11.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sampson T, Broussard GW, Marinelli LJ, Jacobs-Sera D, Ray M, Ko CC, Russell D, Hendrix RW, Hatfull GF. 2009. Mycobacteriophages BPs, Angel and Halo: comparative genomics reveals a novel class of ultra-small mobile genetic elements. Microbiology 155:2962–2977. 10.1099/mic.0.030486-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stover CK, de la Cruz VF, Fuerst TR, Burlein JE, Benson LA, Bennett LT, Bansal GP, Young JF, Lee MH, Hatfull GF, Snapper SB, Barletta RG, Jacobs WR, Jr, Bloom BR. 1991. New use of BCG for recombinant vaccines. Nature 351:456–460. 10.1038/351456a0 [DOI] [PubMed] [Google Scholar]
- 28.Kaufmann SH, Gengenbacher M. 2012. Recombinant live vaccine candidates against tuberculosis. Curr. Opin. Biotechnol. 23:900–907. 10.1016/j.copbio.2012.03.007 [DOI] [PubMed] [Google Scholar]
- 29.Jain P, Hartman TE, Eisenberg N, O'Donnell MR, Kriakov J, Govender K, Makume M, Thaler DS, Hatfull GF, Sturm AW, Larsen MH, Moodley P, Jacobs WR., Jr 2012. phi(2)GFP10, a high-intensity fluorophage, enables detection and rapid drug susceptibility testing of Mycobacterium tuberculosis directly from sputum samples. J. Clin. Microbiol. 50:1362–1369. 10.1128/JCM.06192-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Huff J, Czyz A, Landick R, Niederweis M. 2010. Taking phage integration to the next level as a genetic tool for mycobacteria. Gene 468:8–19. 10.1016/j.gene.2010.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pandey AK, Raman S, Proff R, Joshi S, Kang CM, Rubin EJ, Husson RN, Sassetti CM. 2009. Nitrile-inducible gene expression in mycobacteria. Tuberculosis (Edinb.) 89:12–16. 10.1016/j.tube.2008.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parish T, Mahenthiralingam E, Draper P, Davis EO, Colston MJ. 1997. Regulation of the inducible acetamidase gene of Mycobacterium smegmatis. Microbiology 143:2267–2276. 10.1099/00221287-143-7-2267 [DOI] [PubMed] [Google Scholar]
- 33.Ehrt S, Schnappinger D. 2006. Controlling gene expression in mycobacteria. Future Microbiol. 1:177–184. 10.2217/17460913.1.2.177 [DOI] [PubMed] [Google Scholar]
- 34.Ojha AK, Baughn AD, Sambandan D, Hsu T, Trivelli X, Guerardel Y, Alahari A, Kremer L, Jacobs WR, Jr, Hatfull GF. 2008. Growth of Mycobacterium tuberculosis biofilms containing free mycolic acids and harbouring drug-tolerant bacteria. Mol. Microbiol. 69:164–174. 10.1111/j.1365-2958.2008.06274.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Kessel JC, Hatfull GF. 2007. Recombineering in Mycobacterium tuberculosis. Nat. Methods 4:147–152. 10.1038/nmeth996 [DOI] [PubMed] [Google Scholar]
- 36.van Kessel JC, Hatfull GF. 2008. Efficient point mutagenesis in mycobacteria using single-stranded DNA recombineering: characterization of antimycobacterial drug targets. Mol. Microbiol. 67:1094–1107. 10.1111/j.1365-2958.2008.06109.x [DOI] [PubMed] [Google Scholar]
- 37.Bibb LA, Hatfull GF. 2002. Integration and excision of the Mycobacterium tuberculosis prophage-like element, phi Rv1. Mol. Microbiol. 45:1515–1526. 10.1046/j.1365-2958.2002.03130.x [DOI] [PubMed] [Google Scholar]
- 38.Pham TT, Jacobs-Sera D, Pedulla ML, Hendrix RW, Hatfull GF. 2007. Comparative genomic analysis of mycobacteriophage Tweety: evolutionary insights and construction of compatible site-specific integration vectors for mycobacteria. Microbiology 153:2711–2723. 10.1099/mic.0.2007/008904-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Levin ME, Hatfull GF. 1993. Mycobacterium smegmatis RNA polymerase: DNA supercoiling, action of rifampicin and mechanism of rifampicin resistance. Mol. Microbiol. 8:277–285. 10.1111/j.1365-2958.1993.tb01572.x [DOI] [PubMed] [Google Scholar]
- 40.Jensen PR, Hammer K. 1998. The sequence of spacers between the consensus sequences modulates the strength of prokaryotic promoters. Appl. Environ. Microbiol. 64:82–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Seghezzi N, Amar P, Koebmann B, Jensen PR, Virolle MJ. 2011. The construction of a library of synthetic promoters revealed some specific features of strong Streptomyces promoters. Appl. Microbiol. Biotechnol. 90:615–623. 10.1007/s00253-010-3018-0 [DOI] [PubMed] [Google Scholar]
- 42.Chauhan S, Singh A, Tyagi JS. 2010. A single-nucleotide mutation in the −10 promoter region inactivates the narK2X promoter in Mycobacterium bovis and Mycobacterium bovis BCG and has an application in diagnosis. FEMS Microbiol. Lett. 303:190–196. 10.1111/j.1574-6968.2009.01876.x [DOI] [PubMed] [Google Scholar]
- 43.Kenney TJ, Churchward G. 1996. Genetic analysis of the Mycobacterium smegmatis rpsL promoter. J. Bacteriol. 178:3564–3571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blount BA, Weenink T, Vasylechko S, Ellis T. 2012. Rational diversification of a promoter providing fine-tuned expression and orthogonal regulation for synthetic biology. PLoS One 7:e33279. 10.1371/journal.pone.0033279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang W, Li X, Wang J, Xiang S, Feng X, Yang K. 2013. An engineered strong promoter for streptomycetes. Appl. Environ. Microbiol. 79:4484–4492. 10.1128/AEM.00985-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siegl T, Tokovenko B, Myronovskyi M, Luzhetskyy A. 2013. Design, construction and characterisation of a synthetic promoter library for fine-tuned gene expression in actinomycetes. Metab. Eng. 19:98–106. 10.1016/j.ymben.2013.07.006 [DOI] [PubMed] [Google Scholar]
- 47.Lopez PJ, Guillerez J, Sousa R, Dreyfus M. 1998. On the mechanism of inhibition of phage T7 RNA polymerase by lac repressor. J. Mol. Biol. 276:861–875. 10.1006/jmbi.1997.1576 [DOI] [PubMed] [Google Scholar]
- 48.Cresawn SG, Bogel M, Day N, Jacobs-Sera D, Hendrix RW, Hatfull GF. 2011. Phamerator: a bioinformatic tool for comparative bacteriophage genomics. BMC Bioinformatics 12:395. 10.1186/1471-2105-12-395 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.